

Abstract
Tooth enamel, the hardest tissue in the human body, is formed after a complex series of interactions between dental epithelial tissue and the underlying ectomesenchyme. Nonsyndromic amelogenesis imperfecta (AI) is a rare genetic disorder affecting tooth enamel without other nonoral symptoms. In this study, we identified 2 novel ENAM mutations in 2 families with hypoplastic AI by whole exome sequencing. Family 1 had a heterozygous splicing donor site mutation in intron 4, NM_031889; c.123+2T>G. Affected individuals had hypoplastic enamel with or without the characteristic horizontal hypoplastic grooves in some teeth. Family 2 had a nonsense mutation in the last exon, c.1842C>G, p.(Tyr614*), that was predicted to truncate the protein by 500 amino acids. Participating individuals had at least 1 mutant allele, while the proband had a homozygous mutation. Most interestingly, the clinical phenotype of the individuals harboring the heterozygous mutation varied from a lack of penetrance to a mild hypoplastic enamel defect. We believe that these findings will broaden our understanding of the clinical phenotype of AI caused by ENAM mutations.
Keywords: amelogenesis imperfecta, enamel, tooth, enamelin, penetrance, whole exome sequencing
Introduction
Enamel-forming cells (ameloblasts) differentiate from the oral epithelium by a series of complex preprogrammed, sequential, and reciprocal epithelial mesenchymal interactions (Thesleff 2003). Ameloblasts secrete uncalcified enamel matrix onto the preexisting dentin matrix, which is secreted by dentin-forming cells (odontoblasts), thereby forming the initial enamel at the dentinoenamel junction. Ameloblasts continue secreting enamel matrix as they move away from the dentinoenamel junction to achieve full-thickness enamel. The enamel forms as long, thin mineral ribbons separated by enamel matrix proteins, and they grow predominantly by lengthening near the ameloblast membrane (Simmer et al. 2012). The matrix proteins are cleaved by proteases and reabsorbed by ameloblasts to allow the mineral ribbons to thicken and widen, achieving a high mineral content with minimal residual organic matter (<1% by weight; Simmer and Fincham 1995).
During enamel-forming processes (amelogenesis), environmental or genetic disturbances can interfere with amelogenesis and cause enamel defects. Hereditary enamel defects are generally diagnosed as amelogenesis imperfecta (AI), a collection of rare genetic conditions affecting tooth enamel formation (Hu et al. 2007). AI has clinically diverse phenotypes, depending on which stage of amelogenesis is disturbed. Based on the clinical phenotype, AI is generally classified into 3 categories (hypoplastic, hypocalcified, or hypomatured) or simply 2 (hypoplastic or hypomineralized; Prasad, Laouina, et al. 2016).
At least 15 genes have been identified that cause various types of nonsyndromic AI. Hypoplastic AI is caused by mutations in genes encoding amelogenin (AMELX; MIM *300391; Lagerstrom et al. 1991), enamelin (ENAM; MIM *606585; Rajpar et al. 2001), ameloblastin (AMBN; MIM *616270; Poulter, Murillo, et al. 2014), acid phosphatase 4 (ACP4; MIM *606362; Seymen et al. 2016), laminin alpha-3 (LAMA3; MIM *600805; Yuen et al. 2012), laminin beta-3 (LAMB3; MIM *150310; Poulter, El-Sayed, et al. 2014), and collagen type XVII alpha-1 (COL17A1; MIM *113811; McGrath et al. 1996). Hypocalcified AI is caused by autosomal dominant mutations in family with sequence similarity 83 member H (FAM83H; MIM *611927; Kim et al. 2008) and recessive mutations in odontogenesis associated phosphoprotein (ODAPH, also known as C4orf26; MIM *614829; Parry et al. 2012). Hypomaturation AI is caused by heterozygous mutation of amelotin (AMTN; MIM *610912; Smith et al. 2016) or by recessive mutations in genes encoding kallikrein-related peptidase 4 (KLK4; MIM *603767; Hart et al. 2004), matrix metallopeptidase 20 (MMP20; MIM *604629; Kim, Simmer, et al. 2005), WD repeat-containing protein 72 (WDR72; MIM *613214; El-Sayed et al. 2009), solute carrier family 24 member 4 (SLC24A4; MIM *609840; Parry et al. 2013), integrin beta-6 (ITGB6; MIM *147558; Wang et al. 2014; Poulter, Brookes, et al. 2014), and G protein–coupled receptor 68 (GPR68; MIM *601404; Parry et al. 2016).
In this study, we performed a mutational analysis using whole exome sequencing for probands in 2 Turkish families presenting with hypoplastic AI, and we identified the disease-causing ENAM mutations. Both mutations were novel, and a splicing assay confirmed the effect of the splicing donor site mutation. Most importantly, the nonsense mutation in family 2 resulted in a highly variable clinical phenotype in individuals with a heterozygous mutant allele.
Materials and Methods
Enrollment of Human Subjects
The study was reviewed and approved by the Institution Review Board at the University of Istanbul and Seoul National University Dental Hospital. We recruited 2 unrelated Turkish families presenting nonsyndromic hypoplastic AI for the genetic studies. According to the Declaration of Helsinki, clinical examinations and collection of saliva samples were performed with the understanding and written consent of each participant.
Whole Exome Sequencing and Bioinformatics Analysis
Whole exome sequencing was performed with DNA samples from the probands of the 2 families with AI after exome capture with the Agilent SureSelect XT Human All Exon V5 Target Enrichment System. 101-bp paired-end sequencing reads were generated with the Illumina HiSeq 2500 (Theragen Etex Bio Institute). Sequencing reads were aligned to human reference genome hg19 with the Burrows-Wheeler Aligner. With a series of bioinformatics tools, such as Samtools, Genome Analysis Tool Kit, and Annovar, the sequence variants were annotated with dbSNP build 138 (Hintzsche et al. 2016; Appendix Table 1). The annotated variants were filtered with a cutoff value of 0.01 for the minor allele frequency.
Polymerase Chain Reaction and Sequencing
The identified variation in the ENAM gene and segregation within each family were confirmed by Sanger sequencing. The polymerase chain reaction (PCR) protocol and information on the primers were previously described (Kim, Seymen, et al. 2005). PCR amplifications were done with the HiPi DNA PCR premix (ElpisBio), and the amplification products were purified with a PCR purification kit (ElpisBio). Sanger sequencing was performed at a DNA sequencing center (Macrogen).
In Vitro Splicing Assay
PCR amplification with Hipi Super DNA polymerase (Elpis biotech) was performed with a DNA sample from the proband (forward primer: 5′-GCCCCATCCATTTCCATACT-3′; reverse primer: 5′-TGTGAGAGGATAGGGGCAAT-3′). The amplicon—a genomic fragment (1,306 bp) of ENAM exons 4, 5, and 6—was cloned into the pTop TA V2 vector (Enzynomics). Wild-type and mutant vectors were selected by plasmid sequencing and subsequently subcloned into the pSPL3 vector after double digestion with BamHI and XhoI restriction endonucleases. Wild-type and mutant pSPL3 vectors were transfected into COS-7 cells, and the total RNA was isolated after 36 h. Reverse transcriptase PCR was performed with the vector primer and exon 6–specific primer (SD6: 5′-TCTGAGTCACCTGGAC AACC-3′; reverse primer: 5′-GGGCCGTTCATAAAGTTGA A-3′). The amplification bands were excised from an agarose gel and characterized by DNA sequencing.
Results
Family 1
The proband was a 19-year-old woman (II:1) from a nonconsanguineous nuclear family (Fig. 1A) presenting with generalized hypoplastic AI in all teeth (Fig. 1B). She had good occlusion without open bite or wide interdental spacing. It seemed that the enamel was slightly thicker along the cusps and marginal ridges of the posterior teeth than in the other areas (Fig. 1C, D). There were horizontal hypoplastic grooves in several teeth, especially in the buccal surfaces of the premolars (Fig. 1E, F). The panoramic radiograph clearly showed enamel thinness, the characteristic feature of hypoplastic AI (Fig. 1G). The father of the proband (I:1) had full coverage prosthetics on all teeth; therefore, his affection status could not be clearly established clinically (Fig. 1A, Appendix Fig. 1A). The 11-y-old younger brother (II:2) of the proband presented with similar clinical features, except for the defective regions in the labial side of the permanent maxillary central incisors (Appendix Fig. 1B–G).
Figure 1.

Pedigree and clinical photos of family 1. (A) Pedigree of family 1. The plus symbol (+) indicates members recruited for this study. The proband is indicated with a black arrow. The question mark (?) indicates that the affection status of this individual cannot be determined due to full-coverage prosthetics on all teeth. (B–F) Clinical photo of the proband (II:1) at age 19 y. Generalized hypoplastic enamel in all teeth can be seen, and characteristic horizontal hypoplastic grooves can be noticed in the buccal surfaces or mandibular premolars. (G) Panoramic radiograph of the proband at age 19 y.
Filtered annotation variants were screened for mutations in known AI-causing genes. The results showed 2 variants in KLK4 and ENAM. The heterozygous KLK4 variant was a missense mutation, NM_004917:exon5:c.680C>T:p.(Pro227Leu), with an allele frequency of 0.0016 in the Exome Aggregation Consortium database (rs144350395). In silico predictions of this variant were for a mild functional impact, with a Sift score of 0.12 (tolerated), a MutationTaster score of 1 (polymorphism), and a CADD 1.3 score of 10.91. Additionally, 1 individual in the European population had a homozygous variant, suggesting that it would be benign even if both KLK4 alleles were mutated (homozygous). Moreover, the clinical phenotype caused by a recessive KLK4 mutation is usually hypomaturation, not hypoplastic AI. As such, this variant in the proband did not seem to be related to her AI.
The heterozygous variant in ENAM was a splicing donor site mutation in intron 4 (NM_031889; c.123+2T>G). This variant was not listed in any genome sequence variation database, and the change was predicted to disrupt the canonical RNA intron border splicing sequence (GU-AG). Previously, an ENAM c.123+1G>A splicing mutation was reported to cause autosomal dominant AI (Prasad, Geoffroy, et al. 2016; Appendix Table 2). An in vitro splicing assay showed normal splicing with a strong single amplicon in the wild-type vector; however, there were 3 weak amplicons in the mutant vector (Fig. 2). The longest but weakest amplicon included the intronic sequence between exons 4 and 5. This transcript would introduce an early translation termination codon and be degraded by the nonsense-mediated decay system (NMDS). The weak amplicon having a similar size with the wild-type vector was a mixture of the normal and abnormal splicing products. The abnormal one used a cryptic splicing donor site in exon 4, which reduced the length of exon 4 by 12 bp. The shortest but strongest amplicon included only exons 5 and 6, skipping exon 4. In summary, the major effect of the mutation seemed to be the skipping of exon 4.
Figure 2.
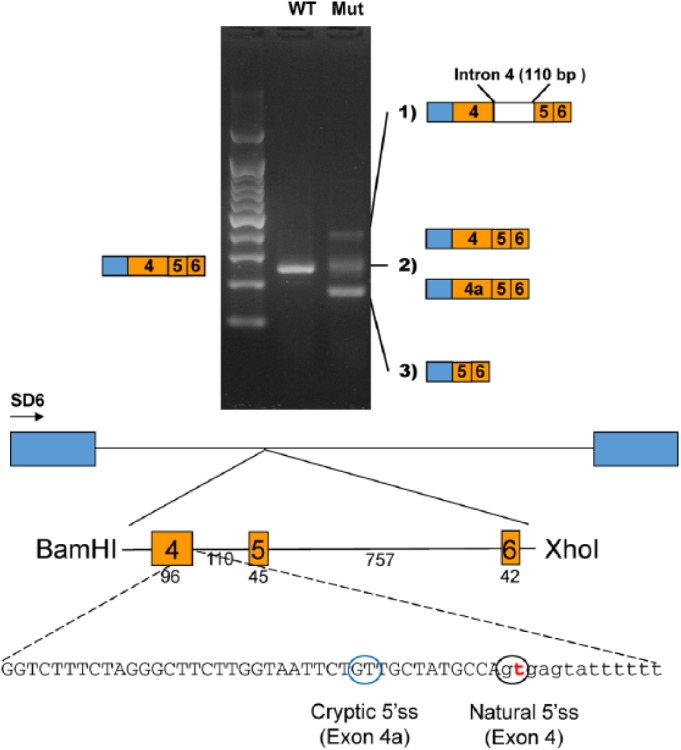
In vitro splicing assay. A genomic fragment, including exons 4, 5, and 6, was cloned into the pSPL3 splicing vector with the BamHI and XhoI restriction endonucleases. Boxes in the diagram indicate the exons in the pSPL3 vector. The number of base pairs (bp) are shown below the exons and the intron. Reverse transcriptase polymerase chain reaction revealed 3 weaker amplicons in the mutant vector, as compared with 1 in the wild-type vector. Sequencing identified abnormal splicing products in the mutant instead of the normal splicing product in the wild type. The mutation in the natural 5′ splicing site (5′ss) activated a cryptic 5′ss, and a new exon sequence (exon 4a) was included in some mutant transcripts instead of the normal exon 4. mut, mutant; WT, wild type.
Family 2
The proband was a 9-y-old female (V:3) who was the third daughter of a consanguineous marriage (Fig. 3A). She presented with very thin enamel in all permanent and deciduous teeth (Fig. 3B–D). She had no open bite but showed exogenous black pigmentation on almost all teeth (Fig. 3E, F). The panoramic radiograph showed almost no enamel coverage on her teeth (Fig. 3G). The 22-y-old older sister of the proband, the first daughter (V:1), exhibited enamel defects as well (Appendix Fig. 3). However, her enamel defects were mild. There was a single deep hypoplastic horizontal groove in the midbuccal surface of the permanent mandibular canines. Additionally, there were atypical attrition-like depressions on the incisal edges of the permanent mandibular incisors and the maxillary right lateral incisor. Horizontal hypoplastic defects were also seen on the lingual surfaces of the permanent maxillary and mandibular anterior teeth. The other family members were considered unaffected.
Figure 3.

Pedigree and clinical photos of family 2. (A) Pedigree of family 2. The plus symbol (+) indicates members recruited for this study. The proband is indicated with a black arrow. Number in the subject symbol indicates the number of siblings. (B–F) Clinical photo of the proband (V:3) at age 9 y. Generalized smooth hypoplastic enamel in all teeth can be seen, and there are external black pigmentations. (G) Panoramic radiograph of the proband at age 9 y.
Exome data analysis of the proband revealed a single variant among the known AI-causing genes, a nonsense ENAM mutation. This variant was a homozygous C-to-G transversion, which would introduce a premature amber stop codon in the last ENAM exon, NM_031889; c.1842C>G, p.(Tyr614*), that was predicted to truncate the protein by 500 amino acids. Because the mutation is located in the last exon, it is predicted that the mutant mRNA transcript would escape NMDS and be translated into a truncated ENAM protein (Miller and Pearce 2014). Sanger sequencing of the family members revealed that all other individuals were heterozygous for this ENAM mutation (Appendix Fig. 4).
After the ENAM mutation and its segregation within the family were found, the clinical phenotypes of the father, mother, and other sibling were carefully reexamined. The mother and father of the proband reported that they had no bad teeth. Even though the panoramic radiograph was available only for the father (IV:3), clinical examination did not identify any clinical phenotype related to hypoplastic AI. The remaining teeth in the panoramic radiograph showed normal enamel thickness (Appendix Fig. 5A). Clinical and panoramic radiograph examinations of the mother (IV:4) showed no evidence related to hypoplastic AI (Appendix Fig. 5B–E). The dentition of the proband’s 19-y-old sister (V:2) looked normal, but there were shallow horizontal hypoplastic defects on the lingual surfaces of the permanent maxillary and mandibular anterior teeth (Appendix Fig. 6).
Discussion
Among the 3 major enamel proteins secreted during the secretory stage of amelogenesis (amelogenin, ameloblastin, and enamelin), enamelin is the largest (>1,000 amino acids) with multiple glycosylations and phosphorylations, but its quantity is only 3% to 5% of the enamel matrix (Simmer and Fincham 1995). However, ENAM is essential for ameloblast integrity and proper enamel formation. Enam-knockout mice showed a thin pseudo enamel layer with an amorphous mineral deposition and occasional plate-like minerals over the normal dentin (Hu et al. 2014).
Variable expressivity of nonsyndromic AI has been reported in some cases, especially in LAMB3 and ENAM mutations. Heterozygous LAMB3 mutations with a dominant negative effect exhibit characteristic irregular hypoplastic pits and grooves in general, but there are only several hypoplastic pits and grooves in some teeth, even within the same family members (Lee et al. 2015). Variable expressivity from localized hypoplasia to generalized severe enamel loss has been reported with c.588+1delG mutational hotspot in ENAM, p.(Asn197Ilefs*81), among family members or between different families (Kida et al. 2002; Hart, Michalec, et al. 2003; Kim, Seymen, et al. 2005; Pavlic et al. 2007). The c.1259-1260insAG deletion mutation in ENAM, p.(Pro422Valfs*27), caused only small enamel pits on several teeth in heterozygotes (Hart, Hart, et al. 2003) but characteristic horizontal hypoplastic grooves in another family (Pavlic et al. 2007).
Only ENAM mutations have presented a lack of penetrance among AI-causing mutations to date. The c.211-2A>C splicing acceptor site mutation, p.(Met71-Gln157del) (Kim, Seymen, et al. 2005), and 2 nonsense mutations, c.358C>T, p.(Gln120*); c.454G>T, p.(Glu152*) (Seymen et al. 2014) were associated with a normal enamel phenotype in some individuals, even though the other family members with the same mutation presented with severe hypoplastic AI. In this study, 2 individuals in family 2 (IV:3 and IV:4) did not present with enamel defects in spite of a heterozygous ENAM mutation.
Could some degree of insufficient ENAM be compensated by other genetic factors regulating the expression of ENAM from a normal allele? FAM50A and beta-catenin/LEF1 were shown to regulate the differentiation of ameloblast or expression of enamelin expression (Tian et al. 2010; Kim et al. 2018). We tried to identify variants in these factors within the exome sequencing data in this and previous studies, but there were no variants. Furthermore, enamelin production can be influenced by environmental factors such as fluoride and vitamin D. A systematic study with a large cohort and whole genome–level analysis could identify a comprehensive regulating network for ENAM expression or other genes involved in enamel formation.
Postsecretion cleavage of enamelin from the C-terminus was demonstrated by immunohistochemistry with region-specific anti-enamelin antibodies (Hu and Yamakoshi 2003). The nonsense mutation identified in family 2, p.(Tyr614*), being located in the terminal exon, could escape from the NMDS and be translated into a truncated protein, which includes the 32-kDa enamel cleavage product (174 to 279 amino acids) that accumulates in secretory stage enamel (Tanabe et al. 1990) and might serve a partial enamelin function. This mutation could be less harmful than the more C-terminus mutation, p.(Leu998Trpfs*65), with additional novel amino acids introduced by frameshift, which seemed to be associated with a dominant negative effect. This could potentially explain the milder phenotype of the individuals with a heterozygous mutation—specifically, the horizontal hypoplastic grooves on only the lingual surface of the anterior teeth. This unique phenotype has never been previously reported. Interestingly, the other heterozygous individuals did not show any clinical phenotype, suggesting a lack of penetrance.
In this study, we identified 2 novel ENAM mutations, splicing and nonsense, in 2 families with hypoplastic AI. The splicing assay confirmed the effect of the mutation on the mRNA splicing. Furthermore, we presented for the first time a case of co-occurrence of the very mild AI defect and the lack of penetrance in a single family. Further genetic and biochemical studies will be needed to better understand the mechanism of variable expression and lack of penetrance.
Author Contributions
M. Koruyucu, J.W. Kim, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; J. Kang, Y.J. Kim, contributed to data analysis and interpretation, drafted the manuscript; F. Seymen, contributed to conception, design, and data acquisition, drafted the manuscript; Y. Kasimoglu, contributed to data acquisition, drafted the manuscript; Z.H. Lee, contributed to conception and design, critically revised the manuscript; T.J. Shin, H.K. Hyun, Y.J. Kim, S.H. Lee, contributed to conception, critically revised the manuscript; J.C.C. Hu, J.P. Simmer, contributed to conception, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, DS_10.1177_0022034518763152 for Hypoplastic AI with Highly Variable Expressivity Caused by ENAM Mutations by M. Koruyucu, J. Kang, Y.J. Kim, F. Seymen, Y. Kasimoglu, Z.H. Lee, T.J. Shin, H.K. Hyun, Y.J. Kim, S.H. Lee, J.C.C. Hu, J.P. Simmer and J.W. Kim in Journal of Dental Research
Acknowledgments
We thank the participants in this study for their cooperation.
Footnotes
This work was supported by grants from the National Research Foundation of Korea (funded by the Korean government; NRF-2017R1A2A2A05069281) and the National Institute of Dental and Craniofacial Research / National Institutes of Health (DE015846).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
A supplemental appendix to this article is available online.
ORCID iDs: M. Koruyucu  https://orcid.org/0000-0002-2077-5095
https://orcid.org/0000-0002-2077-5095
References
- El-Sayed W, Parry DA, Shore RC, Ahmed M, Jafri H, Rashid Y, Al-Bahlani S, Al Harasi S, Kirkham J, Inglehearn CF, et al. 2009. Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am J Hum Genet. 85(5):699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons D, Hong S, Wright JT. 2004. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet. 41(7):545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart PS, Michalec MD, Seow WK, Hart TC, Wright JT. 2003. Identification of the enamelin (g.8344delG) mutation in a new kindred and presentation of a standardized ENAM nomenclature. Arch Oral Biol. 48(8):589–596. [DOI] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Gorry MC, Michalec MD, Ryu OH, Uygur C, Ozdemir D, Firatli S, Aren G, Firatli E. 2003. Novel ENAM mutation responsible for autosomal recessive amelogenesis imperfecta and localised enamel defects. J Med Genet. 40(12):900–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintzsche JD, Robinson WA, Tan AC. 2016. A survey of computational tools to analyze and interpret whole exome sequencing data. Int J Genomics. 2016:7983236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JC, Chun YH, Al Hazzazzi T, Simmer JP. 2007. Enamel formation and amelogenesis imperfecta. Cells Tissues Organs. 186(1):78–85. [DOI] [PubMed] [Google Scholar]
- Hu JC, Hu Y, Lu Y, Smith CE, Lertlam R, Wright JT, Suggs C, McKee MD, Beniash E, Kabir ME, et al. 2014. Enamelin is critical for ameloblast integrity and enamel ultrastructure formation. PLoS One. 9(3):e89303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JC, Yamakoshi Y. 2003. Enamelin and autosomal-dominant amelogenesis imperfecta. Crit Rev Oral Biol Med. 14(6):387–398. [DOI] [PubMed] [Google Scholar]
- Kida M, Ariga T, Shirakawa T, Oguchi H, Sakiyama Y. 2002. Autosomal-dominant hypoplastic form of amelogenesis imperfecta caused by an enamelin gene mutation at the exon-intron boundary. J Dent Res. 81(11):738–742. [DOI] [PubMed] [Google Scholar]
- Kim JW, Lee SK, Lee ZH, Park JC, Lee KE, Lee MH, Park JT, Seo BM, Hu JC, Simmer JP. 2008. FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am J Hum Genet. 82(2):489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Seymen F, Lin BP, Kiziltan B, Gencay K, Simmer JP, Hu JC. 2005. Enam mutations in autosomal-dominant amelogenesis imperfecta. J Dent Res. 84(3):278–282. [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hart TC, Hart PS, Ramaswami MD, Bartlett JD, Hu JC. 2005. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet. 42(3):271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Hur SW, Jeong BC, Oh SH, Hwang YC, Kim SH, Koh JT. 2018. The Fam50a positively regulates ameloblast differentiation via interacting with Runx2. J Cell Physiol. 233(2):1512–1522. [DOI] [PubMed] [Google Scholar]
- Lagerstrom M, Dahl N, Nakahori Y, Nakagome Y, Backman B, Landegren U, Pettersson U. 1991. A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1). Genomics. 10(4):971–975. [DOI] [PubMed] [Google Scholar]
- Lee KE, Ko J, Le CG, Shin TJ, Hyun HK, Lee SH, Kim JW. 2015. Novel LAMB3 mutations cause non-syndromic amelogenesis imperfecta with variable expressivity. Clin Genet. 87(1):90–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JA, Gatalica B, Li K, Dunnill MG, McMillan JR, Christiano AM, Eady RA, Uitto J. 1996. Compound heterozygosity for a dominant glycine substitution and a recessive internal duplication mutation in the type XVII collagen gene results in junctional epidermolysis bullosa and abnormal dentition. Am J Pathol. 148(6):1787–1796. [PMC free article] [PubMed] [Google Scholar]
- Miller JN, Pearce DA. 2014. Nonsense-mediated decay in genetic disease: friend or foe? Mutat Res Rev Mutat Res. 762:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry DA, Brookes SJ, Logan CV, Poulter JA, El-Sayed W, Al-Bahlani S, Al Harasi S, Sayed J, Raif EM, Shore RC, et al. 2012. Mutations in C4orf26, encoding a peptide with in vitro hydroxyapatite crystal nucleation and growth activity, cause amelogenesis imperfecta. Am J Hum Genet. 91(3):565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry DA, Poulter JA, Logan CV, Brookes SJ, Jafri H, Ferguson CH, Anwari BM, Rashid Y, Zhao H, Johnson CA, et al. 2013. Identification of mutations in SLC24A4, encoding a potassium-dependent sodium/calcium exchanger, as a cause of amelogenesis imperfecta. Am J Hum Genet. 92(2):307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry DA, Smith CE, El-Sayed W, Poulter JA, Shore RC, Logan CV, Mogi C, Sato K, Okajima F, Harada A, et al. 2016. Mutations in the pH-sensing G-protein-coupled receptor GPR68 cause amelogenesis imperfecta. Am J Hum Genet. 99(4):984–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlic A, Petelin M, Battelino T. 2007. Phenotype and enamel ultrastructure characteristics in patients with ENAM gene mutations g.13185-13186insAG and 8344deLG. Arch Oral Biol. 52(3):209–217. [DOI] [PubMed] [Google Scholar]
- Poulter JA, Brookes SJ, Shore RC, Smith CE, Abi Farraj L, Kirkham J, Inglehearn CF, Mighell AJ. 2014. A missense mutation in ITGB6 causes pitted hypomineralized amelogenesis imperfecta. Hum Mol Genet. 23(8):2189–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulter JA, El-Sayed W, Shore RC, Kirkham J, Inglehearn CF, Mighell AJ. 2014. Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta. Eur J Hum Genet. 22(1):132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulter JA, Murillo G, Brookes SJ, Smith CE, Parry DA, Silva S, Kirkham J, Inglehearn CF, Mighell AJ. 2014. Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Hum Mol Genet. 23(20):5317–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad MK, Geoffroy V, Vicaire S, Jost B, Dumas M, Le Gras S, Switala M, Gasse B, Laugel-Haushalter V, Paschaki M, et al. 2016. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J Med Genet. 53(2):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad MK, Laouina S, El Alloussi M, Dollfus H, Bloch-Zupan A. 2016. Amelogenesis imperfecta: 1 family, 2 phenotypes, and 2 mutated genes. J Dent Res. 95(13):1457–1463. [DOI] [PubMed] [Google Scholar]
- Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ. 2001. Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum Mol Genet. 10(16):1673–1677. [DOI] [PubMed] [Google Scholar]
- Seymen F, Kim YJ, Lee YJ, Kang J, Kim TH, Choi H, Koruyucu M, Kasimoglu Y, Tuna EB, Gencay K, et al. 2016. Recessive mutations in ACPT, encoding testicular acid phosphatase, cause hypoplastic amelogenesis imperfecta. Am J Hum Genet. 99(5):1199–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymen F, Lee KE, Koruyucu M, Gencay K, Bayram M, Tuna EB, Lee ZH, Kim JW. 2014. Enam mutations with incomplete penetrance. J Dent Res. 93(10):988–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmer JP, Fincham AG. 1995. Molecular mechanisms of dental enamel formation. Crit Rev Oral Biol Med. 6(2):84–108. [DOI] [PubMed] [Google Scholar]
- Simmer JP, Richardson AS, Hu YY, Smith CE, Ching-Chun Hu J. 2012. A post-classical theory of enamel biomineralization . . . and why we need one. Int J Oral Sci. 4(3):129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Murillo G, Brookes SJ, Poulter JA, Silva S, Kirkham J, Inglehearn CF, Mighell AJ. 2016. Deletion of amelotin exons 3-6 is associated with amelogenesis imperfecta. Hum Mol Genet. 25(16):3578–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe T, Aoba T, Moreno EC, Fukae M, Shimuzu M. 1990. Properties of phosphorylated 32 kd nonamelogenin proteins isolated from porcine secretory enamel. Calcif Tissue Int. 46(3):205–215. [DOI] [PubMed] [Google Scholar]
- Thesleff I. 2003. Epithelial-mesenchymal signalling regulating tooth morphogenesis. J Cell Sci. 116(Pt 9):1647–1648. [DOI] [PubMed] [Google Scholar]
- Tian H, Lv P, Ma K, Zhou C, Gao X. 2010. Beta-catenin/LEF1 activated enamelin expression in ameloblast-like cells. Biochem Biophys Res Commun. 398(3):519–524. [DOI] [PubMed] [Google Scholar]
- Wang SK, Choi M, Richardson AS, Reid BM, Lin BP, Wang SJ, Kim JW, Simmer JP, Hu JC. 2014. ITGB6 loss-of-function mutations cause autosomal recessive amelogenesis imperfecta. Hum Mol Genet. 23(8):2157–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen WY, Pasmooij AM, Stellingsma C, Jonkman MF. 2012. Enamel defects in carriers of a novel lama3 mutation underlying epidermolysis bullosa. Acta Derm Venereol. 92(6):695–696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, DS_10.1177_0022034518763152 for Hypoplastic AI with Highly Variable Expressivity Caused by ENAM Mutations by M. Koruyucu, J. Kang, Y.J. Kim, F. Seymen, Y. Kasimoglu, Z.H. Lee, T.J. Shin, H.K. Hyun, Y.J. Kim, S.H. Lee, J.C.C. Hu, J.P. Simmer and J.W. Kim in Journal of Dental Research