

Abstract
Premise of the Study
Lentil is an important legume crop with reduced genetic diversity caused by domestication bottlenecks. Due to its large and complex genome, tools for reduced representation sequencing are needed. We developed an exome capture array for use in various genetic diversity studies.
Methods
Based on the CDC Redberry draft genome, we developed an exome capture array using multiple sources of transcript resources. The probes were designed to target not only the cultivated lentil, but also wild species. We assessed the utility of the developed method by applying the generated data set to population structure and phylogenetic analyses.
Results
The data set includes 16 wild lentils and 22 cultivar accessions of lentil. Alignment rates were over 90%, and the genic regions were well represented in the capture array. After stringent filtering, 6.5 million high‐quality variants were called, and the data set was used to assess the interspecific relationships within the genus Lens.
Discussion
The developed exome capture array provides large amounts of genomic data to be used in many downstream analyses. The method will have useful applications in marker‐assisted breeding programs aiming to improve the quality of cultivated lentil.
Keywords: crop wild relatives, exome capture, genetic diversity, legume, Lens, wild lentil
Advances in next‐generation sequencing and bioinformatics tools have made whole genome sequencing a widely utilized resource for many organisms. Despite the decreasing cost and the advancement of whole genome sequencing methods, data storage and computation time are still issues for organisms with large and highly repetitive genomes. Exome capture is a cost‐effective sequencing method that generates reduced representation libraries by targeting the protein‐coding region of a genome (Hodges et al., 2007). The method starts with total genomic DNA sheared into fragments, and target‐specific probes hybridize with the specific regions of interest. The selected fragments are then pulled down and PCR amplified before sequencing.
One of the flexibilities of exome sequencing is the probe design, which allows targeting of a wide range of closely related taxa, making it possible to recover orthologous loci across a clade of interest (Bragg et al., 2016). The probes are designed to capture the coding sequences of the genome, thus focusing on the regions that are targeted by natural selection. Whereas whole genome sequencing does not require any a priori knowledge, for exome capture it is necessary to have some level of knowledge of the intron boundaries and gene content of the organism of interest. Because well‐annotated genomes improve probe design, high‐quality reference genomes and transcriptomes reduce the risk of false positives or missing important variants in the generated data set (Chamala et al., 2015; Warr et al., 2015).
When compared to whole genome sequencing, exome capture covers fewer variants, not only because of the smaller size of the sequenced region, but also because the noncoding regions tend to have higher variation (Weitemier et al., 2014). Even though it is challenging to link function to noncoding sequences of the genome, high variation within the introns and the intergenic spaces neighboring the exons make these regions desirable targets (Engelhardt and Brown, 2015; Zhou and Troyanskaya, 2015). Exome capture probes can be designed to expand the target regions flanking the exons, thereby capturing the variation within these noncoding regions without dramatically increasing the data coverage (Weitemier et al., 2014).
Lentil (Lens culinaris Medik.) is an annual self‐pollinating legume that forms a symbiotic relationship with rhizobia, nitrogen‐fixing bacteria that take up atmospheric nitrogen and convert it to a form that is available for other organisms. Due to this association, legume crops like lentil play a significant role in environmentally sustainable agricultural systems. Having legumes in crop rotations decreases the use of fertilizers to replace the replenished nitrogen in the soil, thus enhancing the productivity of non‐legume crops while reducing the environmental impact of agricultural practices on soil systems (Young et al., 2003).
As a member of the Vicieae tribe in the Papilionoideae subfamily of Fabaceae, the genus Lens Mill. (Fabaceae subfam. Papilionoideae, tribe Vicieae) consists of seven species, divided into four gene pools with respect to their ability to make crosses with the cultivated lentil (Wong et al., 2015). The crosses within the primary gene pool (L. culinaris, L. orientalis Popow, and L. tomentosus Ladiz.) produce viable hybrids with negligible sterility, and the secondary (L. odemensis Ladiz., L. lamottei Czefr.) and tertiary (L. ervoides Grande) gene pools can generally be crossed successfully using embryo rescue. The quaternary gene pool includes the most distant species, L. nigricans (M. Bieb.) Godr., which has not been confirmed to produce successful hybrids with cultivated lentils to date.
The domestication history of lentil dates to 11,000 BP in the Fertile Crescent, with potential bottlenecks that reduced the genetic diversity in cultivated lentil when compared to its wild relatives (Erskine et al., 1998; Sonnante et al., 2009). Crop wild relatives are currently underused in crop development programs, and they are poorly represented in most germplasm collections (Hajjar and Hodgkin, 2007; Maxted et al., 2012). Whereas the wild crop relatives usually lack essential domestication traits, they are a useful resource for a variety of adaptive traits including disease and pest resistance and abiotic stress tolerance (Warshefsky et al., 2014). Aiming to develop improved lentil varieties, breeding programs can utilize genetic material from wild lentil species if the necessary variability is not available within the cultivated gene pool.
Using exome capture in crop research allows the application of genomic tools in plant species with large and complex genomes, facilitating the identification of potential variants for marker‐assisted selection. The method has been used in a variety of crops, including investigation of environmental adaptation in barley (Russell et al., 2016), identification of disease‐resistance genes in wheat (Steuernagel et al., 2016), cataloging of deleterious mutations in rice (Henry et al., 2014), and detection of genomic variations among different cultivars in soybean (Haun et al., 2011). Lentil is a diploid (2n = 14) organism with an estimated genome size of 4063 Mbp (Arumuganathan and Earle, 1991), and 130 Mbp of the whole genome is identified as genic sequence (L. Ramsay, University of Saskatchewan, Saskatoon, Saskatchewan, Canada, unpublished data). A draft assembly of the L. culinaris (cv. CDC Redberry) genome is available (Bett, 2016; http://knowpulse.usask.ca), but gene duplications, chromosomal rearrangements, and large amounts of repetitive elements make this large genome difficult to study, especially across the wild species. In this paper, we describe the development of an 85 Mbp exome capture array and show that this versatile method can be applied to many aspects of lentil research, including assessing wild lentils as a source of genetic variability for improving cultivated lentil.
MATERIALS AND METHODS
Capture array design
The exome capture probes were designed from the CDC Redberry (a Canadian L. culinaris cultivar) genome version Lc1.2. To select the regions of interest in the genome for the array, we used several sources: (1) the coding DNA sequence from the Medicago truncatula Gaertn. genome version Mt4.0 (Tang et al., 2014); (2) Illumina RNA‐Seq reads from L. culinaris 2 × 250 MiSeq data (BioProject PRJNA434239); and (3) a collection of previously generated L. culinaris Sanger expressed sequence tags, 454 reads, and contigs (Sharpe et al., 2013 [BioProject PRJNA192531]; Kaur et al., 2011). RNA‐Seq reads were aligned to the reference genome Lc1.2 using TopHat 2.1.1 (Trapnell et al., 2009), and Cufflinks 2.2.1 (Trapnell et al., 2010) was used to determine the transcript coordinates. All other transcript data sets were aligned to the reference genome Lc1.2 using GMAP (Wu and Watanabe, 2005), allowing for a maximum intron size of 30 kbp. Sequences identified as rRNA, plastid, and mitochondrial sequences for lentil, as well as repetitive DNA elements from Viridiplantae (Repbase; Bao et al., 2015) were searched for with BLAST against the target sequences from the initial probe design, and any regions that hit at e‐10 were removed. As a conservative measure to reduce wasted sequencing of multi‐mapping reads, k‐mers greater than expected fragment length (401 bp) were counted, and any with more than three hits were excluded. The coordinates of the capture regions can be found in Appendix S1.
Design of the final array based on the identified genic sequences was performed with Roche NimbleGen's custom probe design pipeline (454 Life Sciences, a Roche Company, Branford, Connecticut, USA; http://www.nimblegen.com/products/seqcap/ez/designs/). The final selected set of probes contained up to 20 close matches in the genome containing five or fewer single nucleotide polymorphisms or insertion/deletion polymorphisms (indels) between the probe and the genomic sequence, as determined by the SSAHA algorithm (Ning et al., 2001). The vast majority of the probes are unique, with a few probes that have a greater degree of multi‐locus homology to allow for increased coverage in the desired genomic regions. Probes were also screened against the chloroplast genome, and regions smaller than 100 bp were excluded from the final pool.
Library preparation and sequencing
A single seed of each of 38 lentil accessions (16 wild and 22 cultivars; Table 1) was grown in controlled growth chambers in the Phytotron facility at the University of Saskatchewan (Saskatoon, Saskatchewan, Canada). Seeds had been obtained from gene banks or were our own cultivars as indicated in Table 1. Genomic DNA was extracted from fresh leaf tissue using a DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). DNA quantity and quality were checked using a NanoDrop 8000 spectrophotometer (Thermo Scientific, Wilmington, Delaware, USA). For library preparation, the SeqCap EZ HyperCap Workflow (Roche, Basel, Switzerland) using the HyperPrep protocol option was followed. For each library, 200 ng of genomic DNA was fragmented using a Bioruptor Pico sonication device (Diagenode, Liège, Belgium). The end‐repair and A‐tailing, adapter ligation, dual‐size selection, and ligation‐mediated–PCR steps were performed as stated in the protocol. A final average insert size was targeted to be between 350 and 380 bp. The concentration, size distribution, and quality of individual libraries were checked on an Agilent Bioanalyzer using DNA 1000 chips (Agilent, Santa Clara, California, USA). For post‐capture hybridization, the SeqCap EZ HyperCap Workflow was followed. For each post‐capture hybridization, six or 12 individual libraries were pooled (Table 1). Libraries were pooled based on the specific index combinations recommended by the supplier (Illumina, San Diego, California, USA) for low‐plex pooling. The hybridizations were performed at 47°C for 18 h. Sample washing, recovery, and amplification were performed as stated in the protocol. The concentration, size distribution, and quality of the captured, multiplexed DNA samples were checked on an Agilent Bioanalyzer using DNA 1000 chips. The samples were sent to the Genome Quebec Innovation Centre at McGill University (Montreal, Québec, Canada) for 2 × 125 paired‐end sequencing on an Illumina HiSeq 2500 instrument.
Table 1.
Exome capture data summary for the Lens samples used in the study
| Gene pool | Species | Samplea | Plex | Total reads | Aligned reads (%) | Single alignment (%) | Multiple alignment (%) | Uniquely mapped (%) | Multi mapped (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1° | L. culinaris | CDC Redberry 1 | 12 | 13,567,183 | 95.07 | 32.29 | 62.78 | 67.92 | 27.15 |
| 1° | L. culinaris | Indianhead 1 | 12 | 16,851,211 | 96.61 | 35.30 | 61.31 | 68.18 | 28.43 |
| 1° | L. culinaris | PI 178952 2 | 12 | 15,522,254 | 96.96 | 43.80 | 53.16 | 67.97 | 28.30 |
| 1° | L. culinaris | PI 299165 2 | 12 | 20,753,869 | 97.37 | 43.52 | 53.84 | 68.64 | 28.73 |
| 1° | L. culinaris | PI 468901 2 | 12 | 15,849,844 | 97.62 | 45.35 | 52.28 | 70.30 | 27.32 |
| 1° | L. culinaris | Shasta 1 | 12 | 12,691,222 | 95.01 | 28.87 | 66.15 | 63.05 | 31.96 |
| 1° | L. orientalis | BGE 016880 3 | 12 | 12,717,811 | 85.40 | 30.06 | 55.34 | 55.51 | 29.89 |
| 1° | L. orientalis | IG 72534 4 | 12 | 17,345,990 | 95.83 | 37.89 | 57.94 | 62.62 | 33.21 |
| 1° | L. orientalis | IG 72611 4 | 12 | 15,505,204 | 95.08 | 37.56 | 57.52 | 61.39 | 33.69 |
| 1° | L. tomentosus | IG 72614 4 | 12 | 21,754,287 | 93.55 | 35.87 | 57.68 | 56.73 | 36.83 |
| 1° | L. tomentosus | IG 72805 4 | 12 | 19,156,322 | 93.40 | 36.07 | 57.33 | 56.84 | 36.56 |
| 2° | L. odemensis | IG 72623 4 | 12 | 18,055,204 | 92.10 | 36.36 | 55.74 | 53.68 | 38.42 |
| 2° | L. odemensis | IG 72760 4 | 12 | 11,558,633 | 91.37 | 36.80 | 54.57 | 53.44 | 37.93 |
| 2° | L. lamottei | IG 110810 4 | 12 | 14,957,568 | 92.17 | 39.07 | 53.11 | 55.61 | 36.56 |
| 2° | L. lamottei | IG 110813 4 | 12 | 12,250,277 | 91.98 | 39.88 | 52.10 | 55.76 | 36.22 |
| 2° | L. lamottei | IG 72552 4 | 12 | 13,215,242 | 92.03 | 40.68 | 51.34 | 56.33 | 35.69 |
| 2° | L. lamottei | ILWL 29 4 | 12 | 14,065,720 | 88.78 | 30.81 | 57.97 | 50.08 | 38.70 |
| 3° | L. ervoides | IG 136620 4 | 12 | 14,905,955 | 92.13 | 38.64 | 53.48 | 54.54 | 37.59 |
| 3° | L. ervoides | IG 72815 4 | 12 | 17,179,746 | 91.92 | 38.24 | 53.68 | 54.17 | 37.75 |
| 3° | L. ervoides | L01‐827A 1 | 12 | 11,778,036 | 86.38 | 32.17 | 54.21 | 49.64 | 36.74 |
| 1° | L. culinaris | CN 105895 5 | 6 | 62,071,291 | 96.11 | 38.09 | 58.01 | 60.34 | 35.77 |
| 1° | L. culinaris | IG 1959 4 | 6 | 43,151,121 | 96.35 | 37.69 | 58.66 | 60.39 | 35.96 |
| 1° | L. culinaris | ILL 213 | 6 | 37,185,226 | 96.70 | 38.26 | 58.44 | 60.44 | 36.27 |
| 1° | L. culinaris | ILL 2507 4 | 6 | 42,707,717 | 96.58 | 36.09 | 60.50 | 59.58 | 37.00 |
| 1° | L. culinaris | ILL 4609 4 | 6 | 21,980,355 | 96.43 | 35.63 | 60.80 | 59.28 | 37.15 |
| 1° | L. culinaris | ILL 5722 4 | 6 | 41,432,535 | 97.39 | 38.51 | 58.88 | 62.72 | 34.66 |
| 1° | L. culinaris | ILL 7663 4 | 6 | 34,608,074 | 96.61 | 36.00 | 60.61 | 60.47 | 36.14 |
| 1° | L. culinaris | ILL 8007 4 | 6 | 27,894,290 | 96.66 | 36.74 | 59.92 | 59.58 | 37.08 |
| 1° | L. culinaris | ILL 9 4 | 6 | 57,429,161 | 96.93 | 38.53 | 58.40 | 61.79 | 35.15 |
| 1° | L. culinaris | PI 209858 2 | 6 | 69,589,951 | 96.53 | 35.95 | 60.58 | 59.95 | 36.58 |
| 1° | L. culinaris | PI 297285 LSP 2 | 6 | 41,070,927 | 97.06 | 37.30 | 59.76 | 63.03 | 34.03 |
| 1° | L. culinaris | PI 370481 LSP 2 | 6 | 52,612,438 | 96.89 | 36.39 | 60.50 | 61.13 | 35.75 |
| 1° | L. culinaris | PI 374118 2 | 6 | 34,753,926 | 96.82 | 36.48 | 60.33 | 60.92 | 35.89 |
| 1° | L. culinaris | PI 431710 2 | 6 | 38,337,889 | 96.37 | 36.24 | 60.13 | 59.41 | 36.97 |
| 1° | L. culinaris | PI 432245 LSP 2 | 6 | 33,097,715 | 97.02 | 37.26 | 59.77 | 61.90 | 35.12 |
| 1° | L. culinaris | PI 533693 LSP 2 | 6 | 47,350,007 | 97.42 | 35.42 | 62.00 | 61.92 | 35.50 |
| 4° | L. nigricans | IG 72539 4 | 6 | 19,064,179 | 73.95 | 28.42 | 45.53 | 41.34 | 32.62 |
| 4° | L. nigricans | IG 72541 4 | 6 | 12,618,403 | 69.80 | 24.87 | 44.94 | 37.67 | 32.14 |
Seed sources: 1 = Crop Development Centre, University of Saskatchewan, Saskatoon, Saskatchewan, Canada; 2 = USDA‐ARS Western Plant Introduction Station, Pullman, Washington, USA; 3 = Universidad de León, León, Spain; 4 = International Centre for Agricultural Research in the Dry Areas (ICARDA), Rabat, Morocco; 5 = Plant Gene Resources of Canada (PGRC), Saskatoon, Saskatchewan, Canada.
Sequence alignment, variant calling, and filtering
Using FastQC (Andrews, 2010), we performed an initial quality control for the raw data. Samples were rejected as failing QC if they met any of the FastQC error conditions, with the following parameters adjusted for improved overall sequence quality: maximum N content error of 10%, base median quality minimum PHRED score of 28, and per‐sequence quality minimum of 25. Sequences were trimmed for quality and adapters using Trimmomatic 0.33 (Bolger et al., 2014), requiring quality scores to remain above 30 in a four‐base window and retaining no sequences shorter than 50 bp. We used Bowtie2 2.3.3.1 (Langmead and Salzberg, 2012) to perform end‐to‐end alignment with the reference genome, discarding discordant and mixed alignments. After the alignment, we filtered the data set for uniquely mapped reads based on alignment quality in cases with more than one hit and removed potential PCR duplicates using rmdup from SAMtools 1.3.1 (Li et al., 2009). Genome coverage was assessed using BedTools (Quinlan and Hall, 2010) and visualized using IGV 2.3.90 (Thorvaldsdóttir et al., 2013). We called variants using SAMtools 1.3.1 and set the minimum number of gapped reads required to call a potential indel to 10.
The initial variant call using the 38 samples resulted in 17,394,602 variants. By excluding the ones located on unanchored scaffolds, we reduced the number of variants to 13,286,870. We used VCFtools v0.1.14 (Danecek et al., 2011) for the filtering process with the following parameters: minimum read depth (min_DP): 3; maximum read depth (max_DP): 5000; and minimum Phred‐scaled quality score (min_QUAL): 20. We used the R package VcfR (Knaus and Grünwald, 2017) to visualize the distribution of the quality parameters. At the end of the filtering process, we kept 6,679,012 variants (38% of the initial set) to use in downstream analyses.
Population structure
Due to the large number of variants, we used a Bayesian clustering algorithm implemented in fastStructure 1.0 (Raj et al., 2014) in order to infer population structure in our sample group. The input files for fastStructure were generated using PLINK v1.9 (Chang et al., 2015). We executed the program using the default settings with simple prior and tested multiple K values ranging from 1 to 6. In order to infer the number of populations that best fit our data, we used the chooseK.py script provided with fastStructure. Bar plots were generated using Structure Plot v2.0 (Ramasamy et al., 2014).
We also performed a principal component analysis (PCA) to infer population stratification in our data set. Using VCFtools v0.1.14, we generated input files for PLINK 1.9, which was used to generate eigenvectors. We created PCA plots using basic plotting functions in R programming language 3.3.1 (R Core Team, 2016).
Phylogenetic analysis
In order to decrease the computation time for phylogenetic tree reconstruction, we generated three random subsets of 100,000 and 20,000 variants from the filtered VCF file. Subsets were generated in a purely random fashion using a custom script that selects a specified number of variants from a MAP file (a variant information file generated by PLINK 1.9) and extracts the randomly selected variants from the original VCF file (see Appendix S2 for details). After converting the VCF file to FASTA format using VCF‐kit (Cook and Andersen, 2017), we filtered out the monomorphic variants using the “remove invariant characters” option in Mesquite version 3.11 (Maddison and Maddison, 2018) to filter out the monomorphic variants. This filtering process further reduced the number of sites from 20,000 to around 11,600 and from 100,000 to around 58,300, which represented approximately 0.17% and 0.87% of the initial variant calls, respectively. Using Mesquite version 3.11, we converted the filtered FASTA files to PHYLIP format to be used as input in RaxML 8.0.0 (Stamatakis, 2014) for maximum likelihood (ML)–based phylogenetic reconstruction. We used a general time‐reversible (GTR) model (Tavaré, 1986) with gamma rate heterogeneity and implemented a likelihood correction for ascertainment bias in order to account for the use of variant‐only data. We performed an ML search with 1000 rapid bootstrapping and visualized the best‐scoring ML trees in FigTree v1.4.3 (Rambaut, 2009).
RESULTS
Capture design summary
Our exome capture data set includes 38 accessions from all seven species of the genus Lens (Table 1). Twenty of these samples were from 12‐plex pools, whereas the remaining 18 samples were from 6‐plex pools (Table 1). As expected, with an average of 39,830,845 reads, 6‐plex samples had higher read numbers than the 12‐plex samples, which had an average of 15,484,079 reads. The plex level did not affect the alignment success as the average alignment rate for the 6‐plex and 12‐plex samples were 93.98% and 93.04%, respectively. Because we used the L. culinaris cv. CDC Redberry as the reference genome, the wild Lens samples from the first three gene pools had slightly lower alignment rates (91.58%) when compared to the cultivars (96.66%). The two samples of L. nigricans, which is the most distant relative of L. culinaris, had the lowest alignment rates (71.88%) among the 38 samples. When we combine all 38 samples from both plex levels, the average single and multiple alignment rates were 36.40% and 57.09%, respectively. Of these aligned reads, 58.80% mapped to a unique region and 34.67% mapped to multiple regions on the reference genome.
The raw exome capture sequences for CDC Redberry total 85 Mbp, which roughly corresponds to 2% of the whole CDC Redberry genome (4063 Mbp). The median depth across target regions was 16.55 on average, ranging from 1.28 to 35.70 (Table 2). When the distribution of genes and the exome‐capture sequences across the lentil genome were compared, the exome‐capture sequences showed dense distribution around the genic regions of each chromosome (Fig. 1). On average, 78.23% of the sequence data were captured within 400 bp outside the probe regions, 76.05% within 200 bp outside the probe regions, and 73.78% within 100 bp outside the probe regions (Table 2). On average, 51.2% of the captured data correspond to exons, 22.58% correspond to introns, and 10.86% correspond to untranslated regions (Table 2).
Table 2.
Summary of the regions represented in the exome capture for the Lens samples used in the study
| Speciesa | Sample | Median depth across target regions | <400 bp outside probe regions (%) | <200 bp outside probe regions (%) | <100 bp outside probe regions (%) | mRNA (%) | Exons (%) | Introns (%) | UTR (%) |
|---|---|---|---|---|---|---|---|---|---|
| cul | CDC Redberry | 10.36 | 67.88 | 66.64 | 65.27 | 62.45 | 41.69 | 20.76 | 9.25% |
| cul | CN 105895 | 35.70 | 76.17 | 75.07 | 73.50 | 71.03 | 47.97 | 23.06 | 10.49 |
| cul | IG 1959 | 25.77 | 76.64 | 75.52 | 73.97 | 71.36 | 48.12 | 23.24 | 10.67 |
| cul | ILL 213 | 19.89 | 75.75 | 74.69 | 73.45 | 70.72 | 48.51 | 22.21 | 10.55 |
| cul | ILL 2507 | 25.12 | 68.70 | 67.53 | 66.20 | 63.64 | 42.59 | 21.05 | 9.60 |
| cul | ILL 4609 | 4.77 | 75.53 | 74.47 | 73.25 | 70.46 | 48.10 | 22.37 | 10.58 |
| cul | ILL 5722 | 13.01 | 78.87 | 77.81 | 76.31 | 73.14 | 49.48 | 23.66 | 11.02 |
| cul | ILL 7663 | 22.79 | 73.50 | 72.35 | 70.97 | 68.41 | 46.39 | 22.02 | 10.16 |
| cul | ILL 8007 | 20.97 | 78.81 | 77.83 | 76.61 | 73.47 | 50.29 | 23.18 | 11.17 |
| cul | ILL 9 | 14.61 | 77.87 | 76.81 | 75.28 | 72.51 | 48.98 | 23.53 | 10.86 |
| cul | Indianhead | 12.91 | 72.57 | 71.48 | 70.27 | 67.56 | 46.08 | 21.48 | 10.03 |
| cul | PI 178952 | 26.34 | 87.45 | 86.83 | 85.97 | 82.76 | 59.37 | 23.39 | 12.12 |
| cul | PI 209858 | 29.18 | 72.93 | 71.86 | 70.56 | 68.32 | 46.52 | 21.80 | 10.18 |
| cul | PI 297285 LSP | 24.24 | 73.99 | 72.83 | 71.28 | 68.69 | 46.31 | 22.38 | 10.14 |
| cul | PI 299165 | 16.50 | 86.50 | 85.88 | 85.04 | 82.24 | 59.52 | 22.72 | 11.46 |
| cul | PI 370481 LSP | 32.77 | 73.08 | 72.01 | 70.64 | 68.18 | 46.21 | 21.97 | 10.14 |
| cul | PI 374118 | 21.76 | 73.84 | 72.75 | 71.31 | 68.81 | 46.62 | 22.18 | 10.06 |
| cul | PI 431710 | 23.08 | 74.46 | 73.37 | 71.92 | 69.61 | 47.21 | 22.40 | 10.11 |
| cul | PI 432245 LSP | 21.23 | 75.86 | 74.77 | 73.38 | 70.59 | 48.05 | 22.54 | 10.48 |
| cul | PI 468901 | 15.09 | 87.45 | 86.77 | 85.67 | 82.38 | 58.32 | 24.06 | 12.07 |
| cul | PI 533693 LSP | 29.44 | 72.08 | 71.00 | 69.71 | 67.19 | 45.63 | 21.56 | 9.85 |
| cul | Shasta | 18.53 | 65.39 | 64.25 | 63.13 | 61.83 | 43.48 | 18.35 | 7.69 |
| ori | BGE016880 | 4.17 | 70.08 | 68.77 | 67.09 | 64.87 | 43.40 | 21.47 | 9.66 |
| ori | IG 72534 | 15.17 | 80.39 | 79.46 | 78.09 | 75.07 | 51.41 | 23.66 | 11.28 |
| ori | IG 72611 | 13.29 | 81.52 | 80.57 | 79.19 | 76.17 | 52.17 | 24.01 | 11.51 |
| tom | IG 72614 | 17.14 | 81.96 | 81.07 | 79.79 | 77.14 | 53.23 | 23.91 | 11.76 |
| tom | IG 72805 | 14.98 | 82.62 | 81.71 | 80.44 | 77.82 | 53.79 | 24.03 | 11.82 |
| ode | IG 72623 | 14.12 | 85.11 | 84.40 | 83.47 | 81.43 | 57.40 | 24.03 | 12.44 |
| ode | IG 72760 | 8.63 | 85.54 | 84.78 | 83.62 | 81.51 | 56.88 | 24.63 | 12.55 |
| lam | IG 110810 | 12.44 | 86.05 | 85.45 | 84.59 | 82.49 | 57.99 | 24.51 | 12.79 |
| lam | IG 110813 | 10.06 | 86.91 | 86.27 | 85.20 | 83.17 | 57.88 | 25.29 | 12.79 |
| lam | IG 72552 | 10.85 | 87.15 | 86.50 | 85.35 | 83.53 | 58.05 | 25.48 | 12.76 |
| lam | ILWL 29 | 9.03 | 74.66 | 73.79 | 72.72 | 71.23 | 48.38 | 22.85 | 10.95 |
| erv | IG 136620 | 11.98 | 87.22 | 86.55 | 85.50 | 83.51 | 58.48 | 25.03 | 12.73 |
| erv | IG 72815 | 13.41 | 86.81 | 86.10 | 85.00 | 83.06 | 58.17 | 24.88 | 12.67 |
| erv | L01‐827A | 3.88 | 78.38 | 77.41 | 76.04 | 74.66 | 51.27 | 23.39 | 11.57 |
| nig | IG 72539 | 4.49 | 78.59 | 77.91 | 77.17 | 78.02 | 59.89 | 18.13 | 9.02 |
| nig | IG 72541 | 1.28 | 74.43 | 73.70 | 73.05 | 74.42 | 61.58 | 12.84 | 7.80 |
UTR = untranslated region.
cul = L. culinaris; ori = L. orientalis; tom = L. tomentosus; ode = L. odemensis; lam = L. lamottei; erv = L. ervoides; nig = L. nigricans.
Figure 1.
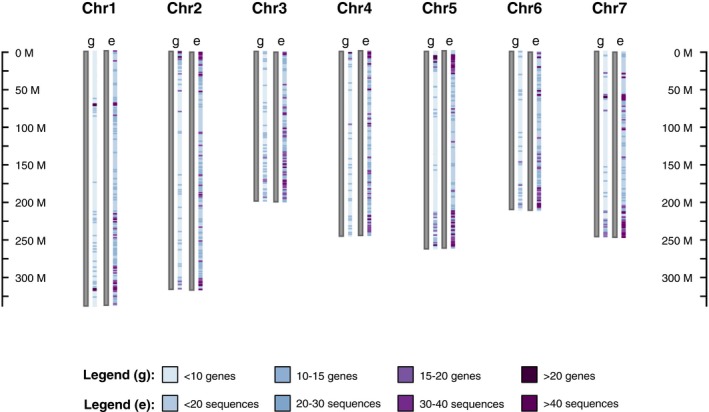
The distribution of genes (g) and the exome‐capture sequences (e) across the Lens culinaris (cv. CDC Redberry) genome. The scale shows the length (base pairs) of each chromosome, and the color‐coded legend shows the density of genes and exome‐capture sequences in each chromosome.
To demonstrate the coverage of our exome data within a gene, we examined glyceraldehyde 3‐phosphate dehydrogenase (GAPDH), which is a housekeeping gene that is expressed in all cells and consists of five exons (Fig. 2). The whole genic region and both 3′ and 5′ flanking regions were densely captured with similar read depth patterns in all the samples except for one: L. nigricans had very high read density (read depth values reaching up to 190) in the region comprising the first two exons of GAPDH but much lower density across the rest of the sequence. We also investigated a C2H2‐type zinc‐finger transcription factor family gene, which has a variant‐rich region in our exome‐capture data set (Fig. 3A). When we examined this region in two L. ervoides samples (IG 136620 and L01‐827A), we observed two alleles at three loci and a deletion in both samples (Fig. 3B). This, combined with the increased read depth observed for these two samples, suggests a gene duplication event in this species that can be detected by this technique.
Figure 2.
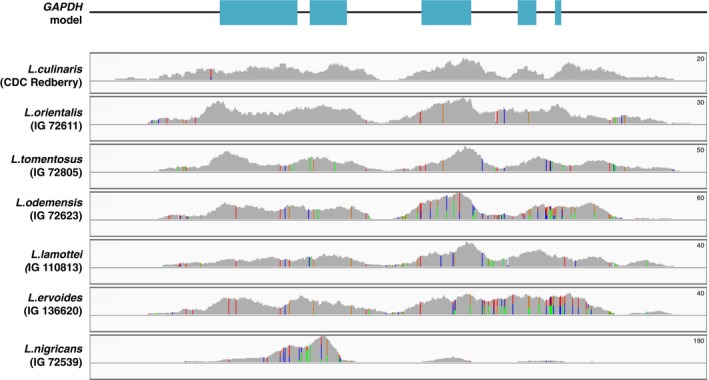
Exome coverage of GAPDH in Lens. GAPDH model shows the exons (cyan rectangles) and noncoding regions (black line). All sizes are proportional to the actual length of the genic region. Below the gene model are the exome coverage plots for seven Lens species. The peak sizes are proportional to the read depth, colored lines represent variant loci, and each color corresponds to a different allele. The maximum read depth is shown on the top right of each panel.
Figure 3.
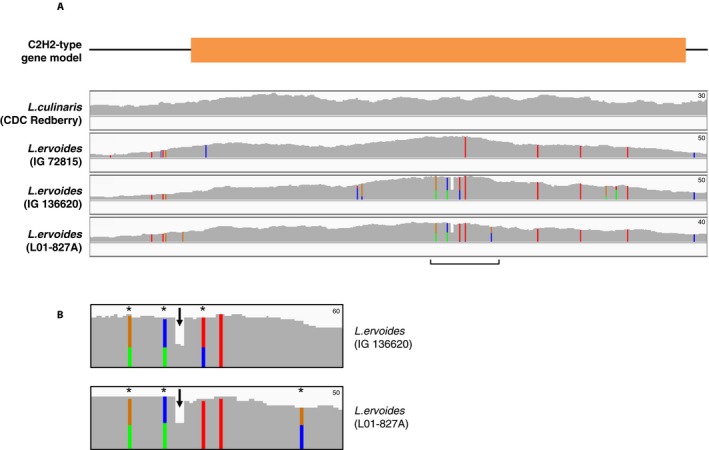
Exome coverage of the C2H2‐type zinc‐finger transcription factor family gene in Lens. (A) Gene model showing the coding (orange rectangle) and noncoding regions (black line). All sizes are proportional to the actual length of the region. Below the gene model are the exome coverage plots for the CDC Redberry and three L. ervoides samples. (B) The enlarged view of the variant‐rich region (marked with a bracket in A) in two L. ervoides samples. The arrows point to haplotypes with deletion, and the asterisks (*) indicate multiple alleles. The peak sizes are proportional to read depth, and colored lines represent variant loci. The maximum read depth values are shown on the top right of each panel.
Population structure
The top three principal components explain 47.93% of the total variance in our sample set, with PC1, PC2, and PC3 explaining 20.72%, 16.22%, and 10.99%, respectively. PCA plots using the combinations of the top three principal components show clear clustering of each species and larger‐scale grouping of the gene pools (Fig. 4). Overall, members of the primary and secondary gene pool are closer to each other than to the tertiary and quaternary gene pool members in all plots. PC1 distinctly separates the two L. nigricans samples (IG 72539 and IG 72541) representing the quaternary gene pool from the others, while PC3 isolates the tertiary gene pool species (L. ervoides) represented by three samples (IG 72815, IG 136620, and L01‐827A) from the rest of the samples. Despite their relatively close clustering, PC2 distinguishes the primary and secondary gene pools represented by three and two species, respectively.
Figure 4.
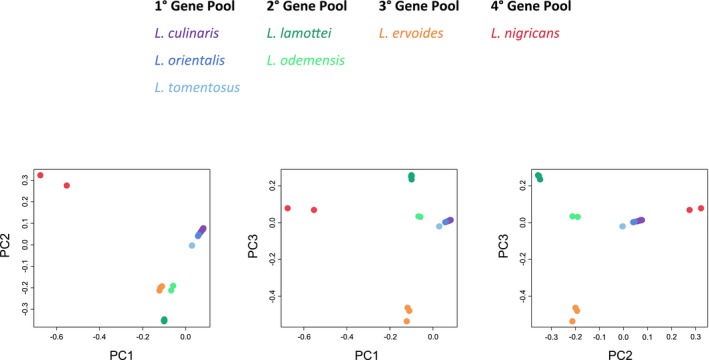
Composite PCA plot showing the clustering of samples using three combinations of the top three principal components (PC1, PC2, and PC3). Color scheme shows seven Lens species grouped under four gene pools with respect to their ability to cross with L. culinaris.
The fastStructure results show similar patterns of clustering (Fig. 5). The optimal number of populations (K value) is inferred to fall within the range of 2 to 4. In the K = 2 bar plot, the primary gene pool is distinctly separated from the rest of the samples. When the K value is increased to 3, the quaternary gene pool is isolated from the secondary and tertiary gene pools as a distinct cluster. At K = 4, the tertiary gene pool is separated from the secondary gene pool; therefore, each cluster clearly represents distinct gene pools.
Figure 5.
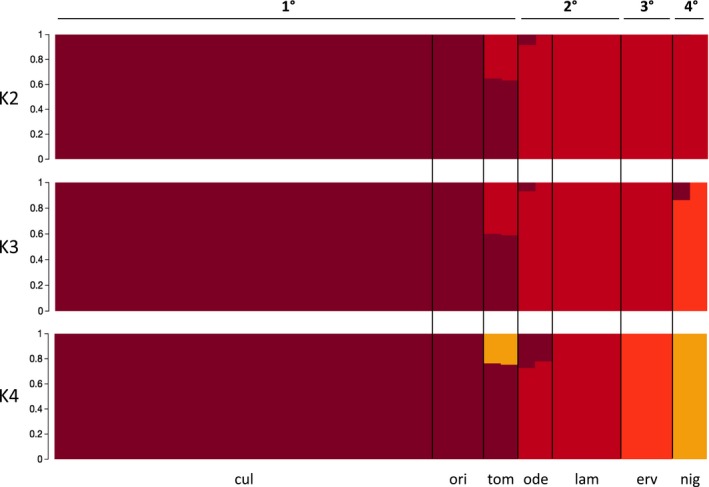
Bar plots showing the fastStructure results for K values 2, 3, and 4. 1° = primary gene pool; 2° = secondary gene pool; 3° = tertiary gene pool; 4° = quaternary gene pool; cul = L. culinaris; ori = L. orientalis; tom = L. tomentosus; ode = L. odemensis; lam = L. lamottei; erv = L. ervoides; nig = L. nigricans.
Lens phylogeny
The best‐scoring ML trees have similar topologies for all three random sets of 20,000‐variant and 100,000‐variant subsets. (For simplicity, only the best‐scoring ML tree from one of the 100,000‐variant subsets is shown [Fig. 6].) Overall, the 100,000‐variant subset phylogenies had higher bootstrap values than did the 20,000‐variant subset phylogenies.
Figure 6.
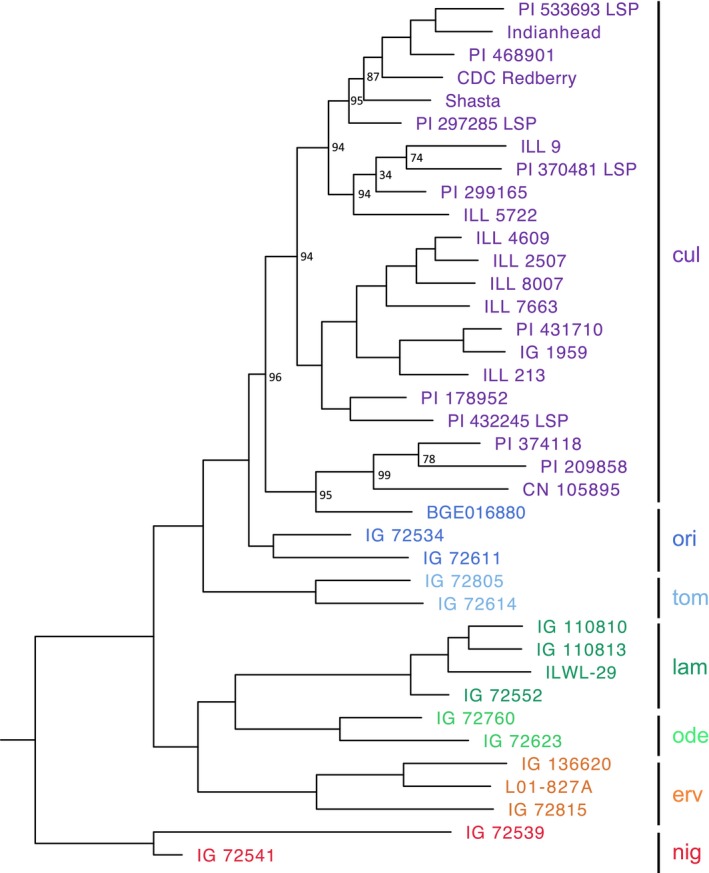
Top‐scoring maximum likelihood phylogenetic tree of the genus Lens. The color scheme for the species is the same as used in the PCA plot. Node labels represent bootstrap (BS) values. The nodes with BS = 100 are not labeled. cul = L. culinaris; ori = L. orientalis; tom = L. tomentosus; ode = L. odemensis; lam = L. lamottei; erv = L. ervoides; nig = L. nigricans.
Five species (L. nigricans, L. ervoides, L. lamottei, L. odemensis, and L. tomentosus) are inferred to be monophyletic with high bootstrap support (BS = 100 for all five species). Lens culinaris is a paraphyletic species (BS = 100) in our analysis, with all three L. orientalis samples nested within its clade. The quaternary gene pool species L. nigricans is again the most divergent taxon within the genus. Tertiary (L. ervoides) and secondary gene pool species (L. lamottei and L. odemensis) form a sister clade to the primary gene pool species (L. tomentosus, L. orientalis, and L. culinaris) with high support (BS = 100).
DISCUSSION
In this paper, we describe the development of an exome capture method for lentil, and we present a brief showcase of applications for which the method can be used. The samples used in this paper represent a small subset of our lentil collection, which consists of accessions from six wild lentil species and hundreds of cultivars from a variety of environments and geographic regions. Our exome capture data represent the genic regions of this large genome well (Fig. 1) and include not only the exons but also introns, untranslated flanking regions, and some extent of intergenic space (Table 2). The amount of capture outside of the target regions depends on DNA fragment length, which was targeted to be 350–380 bp in this study, and this is consistent with previous studies (Henry et al., 2014; Suren et al., 2016). More than 86% of the sequence data are within 400 bp of the probe target regions. Overall, more than 50% of the sequences were exons and about 33% were introns and untranslated regions. The remaining captured sequences are largely spurious alignments along the repetitive regions of the genome and a trace amount of shotgun reads from the library. Despite the fact that the probes were designed for CDC Redberry, a Canadian cultivar, the low specificity of the probes allows this method to be used for wild lentil species as well. Probes with low specificity have been successfully utilized in studies on divergent taxa, but they usually produce fewer variants than the taxon‐specific probes (Bragg et al., 2016; Chau et al., 2018). However, in our stringently filtered data set, we were able to identify 6.5 million variants across the initial 38 samples tested. The alignment success for both plex levels and six out of seven lentil species was over 90% (over 70% for L. nigricans), and over 58% of the variants were uniquely mapped to a single locus. Taken together, these results demonstrate the ability of this capture array to identify large amounts of variation for further analyses.
Relationships within the genus Lens
We found strong support for the classification of seven Lens species into four gene pools, which is consistent with the cross‐compatibility of each species with cultivated lentil. Bayesian inference and PCA structure information as well as ML‐based phylogenetic analyses all demonstrated a consistent relationship among the species (Fig. 4, 5, 6). The major difference between the population structure and phylogenetic analyses was the former used the whole exome capture data, which constitutes about 6,680,000 variants, whereas less than 1% of the total variants were used in the latter. In either case, the results match well with what is known of the relationships from the previous studies (Mayer and Soltis, 1994; Sonnante et al., 2003; Wong et al., 2015).
Implementations in gene discovery
Crops with large genomes, such as wheat, corn, pea, and lentil, have a large amount of repetitive DNA, mainly due to the high number of transposable and repeat elements (Sudheesh et al., 2016). Along with the size and complexity issues, these repetitive elements make genome assembly challenging in these crops, thus limiting its utility for studies of large numbers of samples. An alternative to whole genome re‐sequencing, targeting a subset of the genome, is a more cost‐effective approach, and coding regions are common targets for most reduced‐representation methods (Hodges et al., 2007). In addition, as the significance of variation in noncoding regions is still unclear, capturing large numbers of variants in whole genome sequencing does not necessarily increase explanatory power (Warr et al., 2015). Because crop breeding programs concentrate on genes with already established functions, coding regions are high‐priority targets for crops with large genomes and limited resources (Bamshad et al., 2011).
When targeting coding regions, one challenge is the reliance on pre‐existing genomic resources for the study taxon. (Warr et al., 2015). To some extent, this issue has been alleviated by recently developed targeted sequencing methods that do not depend on reference genomes, but these methods still require extensive transcriptomic data (Chamala et al., 2015; Schott et al., 2017). As an alternative supplement, RNA sequencing can be used to generate a reference gene set, and these predicted genes can be incorporated into probe design (Sudheesh et al., 2016). The genes targeted in this array were taken not only from the genome annotation but also from RNA‐Seq data from various lentil experiments and from better‐characterized relatives such as the model legume M. truncatula. Having a detailed genotype for multiple accessions allows phenotypic associations to be made with a high likelihood of identifying the gene of interest.
Marker‐assisted selection (MAS) is an advanced breeding method where beneficial traits are tracked, identified, and selected during breeding generations using genetic markers. Combining MAS with interspecies hybridization opens up the possibility of using wild relatives for crop improvement in an efficient manner. Better understanding the genetic diversity and the alleles available in the wild lentil gene pool will aid in this effort. Exome capture focuses on the genic regions, which are the main targets of artificial selection of beneficial crop traits. This targeted approach makes exome capture an efficient method for screening more samples with less sequencing. Gene discovery in lentil can also benefit from studying other legume crops. Lens is closely related to Medicago L. and Cicer L., and conserved synteny has been demonstrated among these three genera (Gujaria‐Verma et al., 2014). Shared chromosomal organization can facilitate gene searches in lentil; having the exome sequence data available for a diverse set of lines makes it possible to search for useful variants based on knowledge from other species.
In searching for genetic variation across the different species, we noted an increased read depth in specific genes for certain lines that, upon closer examination, could be explained by the presence of a gene duplication. The C2H2‐type zinc‐finger transcription factor, for example, is a gene that is tandemly duplicated in the model legume M. truncatula (https://phytozome.jgi.doe.gov) and has been implicated in disease resistance (Shi et al., 2014). It will be interesting to follow up on these sorts of duplicated genes to determine if any are associated with the increased levels of resistance seen in some of the wild lentils relative to the cultivated types.
Potential applications in DNA barcoding
Exome capture can also be applied to DNA barcoding, which is a tool for fast and reliable species identification using a standardized DNA sequence. Genome skimming and target enrichment methods are promising for DNA barcoding studies as they are well suited for degraded DNA recovered from museum and herbarium specimens, and the collected data can also provide a powerful phylogenetic signal that is consistent across the plant kingdom (Coissac et al., 2016). If the developed barcoding system is applicable across all plant taxa, the use of different marker sets in different studies can be avoided. However, developing universal probes targeting loci conserved across the plant kingdom can be challenging. With the decreasing cost and increasing utility of high‐throughput sequencing, developing DNA barcodes specific to a plant group is a feasible alternative. The members of the genus Lens show high genetic similarity, and they are not readily distinguishable using standard chloroplast markers or other DNA barcodes (E. Ogutcen, University of Saskatchewan, Saskatoon, Saskatchewan, Canada, unpublished data). We often discover mis‐identified species in genebank collections when we try to make crosses with them. Developing a DNA barcoding system for lentil using exome capture and building a DNA barcode library will allow for identification of lentil species in a standardized fashion.
Utilization for wild relatives
Exome capture is a versatile tool not only for cultivated lentil, but also for its wild relatives. The exome capture probes were designed to target genes identified in lentil, but under the hybridization protocol they only require an 80% match, allowing for a fair amount of non‐specificity. Even though going below the 80% threshold would allow the detection of more targets in L. nigricans, the most divergent relative of L. culinaris, it would also reduce the overall target efficiency across the rest of the samples. The alignment stringency and mapping parameters were kept high enough to reduce mapping highly similar sequences to a single locus, but low enough to allow for capturing the gene space in closely related species. Our results show the probes developed in this study are applicable to all Lens species with success.
The members of the genus Lens show high genetic similarity except for L. nigricans. As expected, L. nigricans has the lowest alignment rates, although still over 70%, when compared to the other Lens species, which had alignment rates of over 90%. The low alignment rates of L. nigricans samples are concluded to be due to the species' genetic distance from the other Lens species, because none of the samples in the same pool had such issues, and there were no major contaminants detected in any of the samples. Because L. nigricans is the only Lens species that has not produced successful hybrids with the cultivated lentil (Ladizinsky and Muehlbauer, 1993; Fiala et al., 2009; A. Vandenberg, University of Saskatchewan, Saskatoon, Saskatchewan, Canada, personal communication), its use in breeding programs is not feasible at this point. Therefore, the performance of our exome capture on this species is not a concern, and it is best to direct our efforts to the interbreeding species, as our exome capture can successfully be applied to these wild relatives. In order to assemble genomes successfully in taxa such as lentil that have very large genome sizes, it is necessary to use genetic linkage maps to order scaffolds into pseudomolecules. The exome capture array could be of benefit for developing these maps and will at the same time assist with comparing genome structure.
Crop wild relatives harbor a wide range of adaptive traits, and their use in breeding programs has been steadily increasing (Ford‐Lloyd et al., 2011; Maxted et al., 2012; Warshefsky et al., 2014; Dempewolf et al., 2017). Draft genomes of more than 30 crop wild relatives have been sequenced (see Brozynska et al., 2016 for a detailed review), and these numbers will dramatically increase with the decreasing cost of next‐generation sequencing. Access of these genomes, through the use of tools like exome capture arrays, will facilitate screening for potentially beneficial traits and studying genotype‐phenotype associations.
Conclusions
Despite the increasing use of high‐throughput sequencing resulting from reduced cost and effort, large and complex genomes still pose a challenge in crop genomics. Lentil has a genome size of over 4 Gbp, which makes exome capture an invaluable tool for a wide range of studies. The exome capture method we have developed for lentil will have immense utility in better understanding the genetic diversity in lentils, ultimately aiming to increase the productivity and quality of cultivated lentils through marker‐assisted breeding programs.
DATA ACCESSIBILITY
Exome capture sequences have been deposited in the National Center for Biotechnology Information Sequence Read Archive under BioProject PRJNA433205. The array can be accessed through Roche NimbleGen (http://www.nimblegen.com/products/seqcap/ez/designs/).
Supporting information
Appendix S1
Appendix S1
ACKNOWLEDGMENTS
This work is part of the project “Application of Genomics to Innovation in the Lentil Economy (AGILE),” funded by Genome Canada, the Saskatchewan Pulse Crop Development Board, Western Grains Research Foundation, the Saskatchewan Ministry of Agriculture, and the University of Saskatchewan. The support of the Vermont Agricultural Experimental Station is also gratefully acknowledged.
Ogutcen, E. , Ramsay L., von Wettberg E. B., and Bett K. E.. 2018. Capturing variation in Lens (Fabaceae): Development and utility of an exome capture array for lentil. Applications in Plant Sciences 6(7): e1165.
LITERATURE CITED
- Andrews, S. 2010. FastQC: A quality control tool for high throughput sequence data. Website http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [accessed 20 November 2017].
- Arumuganathan, K. , and Earle E. D.. 1991. Nuclear DNA content of some important plant species. Plant Molecular Biology Reporter 9: 208–218. [Google Scholar]
- Bamshad, M. J. , Ng S. B., Bigham A. W., Tabor H. K., Emond M. J., Nickerson D. A., and Shendure J.. 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nature Reviews Genetics 12: 745–755. [DOI] [PubMed] [Google Scholar]
- Bao, W. , Kojima K. K., and Kohany O.. 2015. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett, K. 2016. Lentil 1.0 and Beyond. PAG XXIV: Plant and Animal Genomics Conference, 8–13 January 2016, San Diego, California, USA.
- Bolger, A. M. , Lohse M., and Usadel B.. 2014. Trimmomatric: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg, J. G. , Potter S., and Moritz C.. 2016. Exon capture phylogenomics: Efficacy across scales of divergence. Molecular Ecology Resources 16: 1059–1068. [DOI] [PubMed] [Google Scholar]
- Brozynska, M. , Furtado A., and Henry R. J.. 2016. Genomics of crop wild relatives: Expanding the gene pool for crop improvement. Plant Biotechnology Journal 14: 1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamala, S. , García N., Godden G. T., Krishnakumar V., Jordon‐Thaden I. E., De Smet R., Barbazuk W. B., et al. 2015. MarkerMiner 1.0: A new application for phylogenetic marker development using angiosperm transcriptomes. Applications in Plant Sciences 3: 1400115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C. C. , Chow C. C., Tellier L. C. A. M., Vattikuti S., Purcell S. M., and Lee J. J.. 2015. Second‐generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau, J. H. , Rahfeldt W. A., and Olmstead R. G.. 2018. Comparison of taxon‐specific versus general locus sets for targeted sequence capture in plant phylogenomics. Applications in Plant Sciences 6(3): e1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coissac, E. , Hollingsworth P. M., Lavergne S., and Taberlet P.. 2016. From barcodes to genomes: Extending the concept of DNA barcoding. Molecular Ecology 25: 1423–1428. [DOI] [PubMed] [Google Scholar]
- Cook, D. E. , and Andersen E. C.. 2017. VCF‐kit: Assorted utilities for the variant call format. Bioinformatics 33: 1581–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., et al. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempewolf, H. , Baute G., Anderson J., Killian B., Smith C., and Guarino L.. 2017. Past and future use of wild relatives in crop breeding. Crop Science 57: 1070–1082. [Google Scholar]
- Engelhardt, B. E. , and Brown C. D.. 2015. Diving deeper to predict noncoding sequence function. Nature Methods 12(10): 925–926. [DOI] [PubMed] [Google Scholar]
- Erskine, W. , Chandra S., Chaudhry M., Malik I. A., Sarker A., Sharma B., Tufail M., et al. 1998. A bottleneck in lentil: Widening its genetic base in South Asia. Euphytica 101: 207–211. [Google Scholar]
- Fiala, J. V. , Tullu A., Banniza S., Séguin‐Swartz G., and Vandenberg A.. 2009. Interspecies transfer of resistance to anthracnose in lentil (Lens culinaris Medic.). Crop Science 49: 825–830. [Google Scholar]
- Ford‐Lloyd, B. V. , Schmidt M., Armstrong S. J., Barazani O., Engels J., Hadas R., Hammer K., et al. 2011. Crop wild relatives—Undervalued, underutilized and under threat? BioScience 61: 559–565. [Google Scholar]
- Gujaria‐Verma, N. , Vail S. L., Carrasquilla‐Garcia N., Penmetsa R. V., Cook D. R., Farmer A. D., Vandenberg A., et al. 2014. Genetic mapping of legume orthologs reveals high conservation of synteny between lentil species and the sequenced genomes of Medicago and chickpea. Frontiers in Plant Science 5: 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar, R. , and Hodgkin T.. 2007. The use of wild relatives in crop improvement: A survey of developments over the last 20 years. Euphytica 156: 1–13. [Google Scholar]
- Haun, W. J. , Hyten D. L., Xu W. W., Gerhardt D. J., Albert T. J., Richmond T., Jeddeloh J. A., et al. 2011. The composition and origins of genomic variation among individuals of the soybean reference cultivar Williams 82. Plant Physiology 155: 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, I. M. , Nagalakshmi U., Lieberman M. C., Ngo K. J., Krasileva K. V., Vasquez‐Gross H., Akhunova A., et al. 2014. Efficient genome‐wide detection and cataloging of EMS induced mutations using exome capture and next‐generation sequencing. Plant Cell 26: 1382–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges, E. , Xuan Z., Balija V., Kramer M., Molla M. N., Smith S. W., Middle C. M., et al. 2007. Genome‐wide in situ exon capture for selective resequencing. Nature Genetics 39: 1522–1527. [DOI] [PubMed] [Google Scholar]
- Kaur, S. , Cogan N. O., Pembleton L. W., Shinozuka M., Savin K. W., Materne M., and Forster J. W.. 2011. Transcriptome sequencing of lentil based on second‐generation technology permits large‐scale unigene assembly and SSR marker discovery. BMC Genomics 12: 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus, B. J. , and Grünwald N. J.. 2017. VCFR: A package to manipulate and visualize variant call format data in R. Molecular Ecology Resources 17: 44–53. [DOI] [PubMed] [Google Scholar]
- Ladizinsky, G. , and Muehlbauer F. J.. 1993. Wild lentils. Critical Reviews in Plant Sciences 12: 169–184. [Google Scholar]
- Langmead, B. , and Salzberg S. L.. 2012. Fast gapped‐read alignment with Bowtie 2. Nature Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison, W. P. , and Maddison D. R.. 2018. Mesquite: A modular system for evolutionary analysis. Version 3.40. Website http://mesquiteproject.org [accessed 18 June 2018].
- Maxted, N. , Kell S., Ford‐Lloyd B., Dulloo E., and Toledo Á.. 2012. Toward the systematic conservation of global crop wild relative diversity. Crop Science 52: 774–785. [Google Scholar]
- Mayer, M. , and Soltis P. S.. 1994. Chloroplast DNA phylogeny of Lens (Leguminosae): Origin and diversity of cultivated lentil. Theoretical and Applied Genetics 87: 773–781. [DOI] [PubMed] [Google Scholar]
- Ning, Z. , Cox A. J., and Mullikin J. C.. 2001. SSAHA: A fast search method for large DNA databases. Genome Research 11(10): 1725–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A. R. , and Hall I. M.. 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2016. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
- Raj, A. , Stephens M., and Pritchard J. K.. 2014. fastSTRUCTURE: Variational inference of population structure in large variant data sets. Genetics 197: 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy, R. K. , Ramasamy S., Bindroo B. B., and Naik V. G.. 2014. STRUCTURE PLOT: A program for drawing elegant STRUCTURE bar plots in user friendly interface. Springerplus 3: 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. 2009. FigTree v1.3.1. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh, Scotland.
- Russell, J. , Mascher M., Dawson I. K., Kyriakidis S., Calixto C., Freund F., Bayer M., et al. 2016. Exome sequencing of geographically diverse barley landraces and wild relatives gives insights into environmental adaptation. Nature Genetics 48: 1024–1033. [DOI] [PubMed] [Google Scholar]
- Schott, R. K. , Panesar B., Card D. C., Preston M., Castoe T. A., and Chang B. S. W.. 2017. Targeted capture of complete coding regions across divergent species. Genome Biology and Evolution 9(2): 398–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe, A. G. , Ramsay L., Sanderson L. A., Fedoruk M. J., Clarke W. E., Rong L., Kagale S., et al. 2013. Ancient orphan crop joins modern era: Gene‐based variant discovery and mapping in lentil. BMC Genomics 14: 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, H. , Wang X., Ye T., Chen F., Deng J., Yang P., Zhang Y., et al. 2014. The Cysteine2/Histidine2‐Type transcription factor ZINC FINGER OF ARABIDOPSISTHALIANA6 modulates biotic and abiotic stress responses by activating salicylic acid‐related genes and C‐REPEAT‐BINDING FACTOR genes in Arabidopsis . Plant Physiology 165: 1367–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnante, G. , Galasso I., and Pignone D.. 2003. ITS sequence analysis and phylogenetic inference in the genus Lens Mill. Annals of Botany 91: 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnante, G. , Hammer K., and Pignone D.. 2009. From the cradle of agriculture a handful of lentils: History of domestication. Rendiconti Lincei 20: 21–37. [Google Scholar]
- Stamatakis, A. 2014. RAxML Version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuernagel, B. , Periyannan S. K., Hernández‐Pinzón I., Witek K., Rouse M. N., Yu G., Hatta A., et al. 2016. Rapid cloning of disease‐resistance genes in plants using mutagenesis and sequence capture. Nature Biotechnology 34: 652–655. [DOI] [PubMed] [Google Scholar]
- Sudheesh, S. , Verma P., Forster J. W., Cogan N. O. I., and Kaur S.. 2016. Generation and characterisation of a teference transcriptome for lentil (Lens culinaris Medik.). International Journal of Molecular Sciences 17: 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suren, H. , Hodgins K. A., Yeaman S., Nurkowski K. A., Smets P., Rieseberg L. H., Aitken S. N., et al. 2016. Exome capture from the spruce and pine giga‐genomes. Molecular Ecology Resources 16: 1136–1146. [DOI] [PubMed] [Google Scholar]
- Tang, H. , Krishnakumar V., Bidwell S., Rosen B., Chan A., Zhou S., Gentzbittel L., et al. 2014. An improved genome release (version Mt4.0) for the model legume Medicago truncatula . BMC Genomics 15: 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré, S. 1986. Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures on Mathematics in the Life Sciences 17: 57–86. [Google Scholar]
- Thorvaldsdóttir, H. , Robinson J. T., and Mesirov J. P.. 2013. Integrative Genomics Viewer (IGV): High‐performance genomics data visualization and exploration. Briefings in Bioinformatics 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter L., and Salzberg S. L.. 2009. TopHat: Discovering splice junctions with RNA‐Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Williams B. A., Perta G., Mortazavi A., Kwan G., van Baren M. J., Salzberg S. L., et al. 2010. Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr, A. , Robert C., Hume D., Archibald A., Deeb N., and Watson M.. 2015. Exome sequencing: Current and future perspectives. Genes Genomes Genetics 5: 1543–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshefsky, E. , Penmetsa R. V., and Cook D. R.. 2014. Back to the wilds: Tapping evolutionary adaptations for resilient crops through systematic hybridization with crop wild relatives. American Journal of Botany 101: 1791–1800. [DOI] [PubMed] [Google Scholar]
- Weitemier, K. , Straub S. C. K., Cronn R. C., Fishbein M., Schmickl R., McDonnell A., and Liston A.. 2014. Hyb‐Seq: Combining target enrichment and genome skimming for plant phylogenomics. Applications in Plant Sciences 2: 1400042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, M. M. L. , Gujaria‐Verma N., Ramsay L., Yuan H. Y., Caron C., Diapari M., Vandenberg A., et al. 2015. Classification and characterization of species within the genus Lens using genotyping‐by‐sequencing (GBS). PLoS ONE 10: e0122025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, T. D. , and Watanabe C. K.. 2005. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21: 1859–1875. [DOI] [PubMed] [Google Scholar]
- Young, N. D. , Mudge J., and Ellis T. H. N.. 2003. Legume genomes: More than peas in a pod. Current Opinion in Plant Biology 6: 199–204. [DOI] [PubMed] [Google Scholar]
- Zhou, J. , and Troyanskaya O. G.. 2015. Predicting effects of noncoding variants with deep learning–based sequence model. Nature Methods 12(10): 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Appendix S1
Data Availability Statement
Exome capture sequences have been deposited in the National Center for Biotechnology Information Sequence Read Archive under BioProject PRJNA433205. The array can be accessed through Roche NimbleGen (http://www.nimblegen.com/products/seqcap/ez/designs/).