

Abstract
Essentials.
von Willebrand Factor (VWF) and ADAMTS13 may affect early injury after subarachnoid hemorrhage (SAH).
Early brain injury was assessed in VWF−/−, ADAMTS13−/− and recombinant (r) ADAMTS13 treated mice.
VWF−/− and rADAMTS13 treated mice had less brain injury than ADAMTS13−/− and wild‐type mice.
Early administration of rADAMTS13 may improve outcome after SAH by reducing early brain injury.
Summary
Background
Early brain injury is an important determinant of poor functional outcome and case fatality after aneurysmal subarachnoid hemorrhage (SAH), and is associated with early platelet aggregation. No treatment exists for early brain injury after SAH. We investigated whether von Willebrand factor (VWF) is involved in the pathogenesis of early brain injury, and whether ultra‐early treatment with recombinant ADAMTS‐13 (rADAMTS‐13) reduces early brain injury after experimental SAH.
Methods
Experimental SAH in mice was induced by prechiasmatic injection of non‐anticoagulated blood from a littermate. The following experimental SAH groups were investigated: C57BL/6J control (n = 21), VWF −/− (n = 25), ADAMTS‐13−/− (n = 23), and C57BL/6J treated with rADAMTS‐13 (n = 26). Mice were killed at 2 h after SAH. Primary outcome measures were microglial activation (IBA‐1 surface area) and neuronal injury (number of cleaved caspase‐3‐positive neurons).
Results
As compared with controls, microglial activation was decreased in VWF −/− mice (mean difference of − 20.0%, 95% confidence interval [CI] − 4.0% to − 38.6%), increased in ADAMTS‐13−/− mice (mean difference of + 34.0%, 95% CI 16.2–51.7%), and decreased in rADAMTS‐13‐treated mice (mean difference of − 22.1%, 95% CI − 3.4% to − 39.1%). As compared with controls (185 neurons, interquartile range [IQR] 133–353), neuronal injury in the cerebral cortex was decreased in VWF −/− mice (63 neurons, IQR 25–78), not changed in ADAMTS‐13−/− mice (53 neurons, IQR 26–221), and reduced in rADAMTS‐13‐treated mice (45 neurons, IQR 9–115).
Conclusions
Our findings suggest that VWF is involved in the pathogenesis of early brain injury, and support the further study of rADAMTS‐13 as a treatment option for early brain injury after SAH.
Keywords: brain diseases, platelet aggregation, subarachnoid hemorrhage, thrombosis, von Willebrand factor
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a devastating subtype of stroke with a peak incidence between the ages of 40 years and 60 years. Although the prognosis after SAH has somewhat improved over the last few decades, the 90‐day in‐hospital case‐fatality rate in hospital‐based studies is still 30% 1. Major determinants of poor functional outcome after SAH include early brain injury and delayed cerebral ischemia (DCI) 2, 3. Early brain injury occurs immediately following the initial bleeding, whereas DCI is a complication that occurs in 30% of SAH patients 4–14 days after ictus. At present, no treatment exists for early brain injury. Although DCI can be prevented with oral nimodipine, its effect is only modest 4. Therefore, new treatment targets are needed to improve the prognosis after SAH.
Although the pathogenesis of early brain injury and DCI is poorly understood, accumulating data suggest that platelet aggregation is involved in both processes 5. Recently, we and others have shown that higher levels of von Willebrand factor (VWF) and lower levels of ADAMTS‐13 (which rapidly cleaves large VWF multimers) are associated with DCI in patients with SAH, and that treatment with recombinant ADAMTS‐13 (rADAMTS‐13) in a mouse model of SAH reduces microthrombosis and microglial activation 48 h after SAH 6, 7, 8, 9, 10, 11. However, it remains unclear whether VWF is also involved in brain injury shortly after SAH, and whether rADAMTS‐13 is a potential treatment option to prevent early brain injury.
In the current study, we assessed the extent of early brain injury in an SAH mouse model 2 h after SAH induction. We demonstrated that VWF is involved in the pathogenesis of early brain injury, and that ultra‐early treatment with rADAMTS‐13 reduces early brain injury after experimental SAH.
Materials and methods
Experimental animals and groups
All experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee at St Michael's Hospital, and conducted in accordance with the regulations outlined by the Canadian Council on Animal care. Wild‐type C57BL/6 mice were obtained from Charles River (Sherbrooke, Canada). The ADAMTS‐13−/− and VWF−/− mice were both generated on a C57BL/6 background, as reported in our previous studies 6, 12, 13. We used both male and female mice (⁓ 50% each), and planned the surgery according to the age (instead of the weight) of the mice, so that all mice used were aged 90 days. The mice weighed 18–30 g, had access to a standard pellet diet and water ad libitum, and were housed under a 12‐h : 12‐h light/dark cycle. Mice were randomly assigned to one of the following experimental SAH groups: (i) C57BL/6 wild‐type controls (n = 23, 11 males and 12 females); (ii) VWF−/− mice (n = 25, 12 males and 13 females); (iii) ADAMTS‐13−/− mice (n = 23, 12 males and 11 females); and (iv) C57BL/6 wild‐type mice (n = 26, 12 males and 14 females) with injection of rADAMTS‐13 (3460 U kg−1) in the tail vein 5 min after SAH induction 6. The sample sizes were established by an a priori formal power calculation, taking into account a minimum difference in extent of brain injury of 48% between groups, a standard deviation of ⁓ 45%, a 5% error, and 80% power. The expected difference and variance were based on our previous study on the effects of ADAMTS‐13 on microthrombosis 48 h after SAH 6. Experiments were performed in random order (generated by simple randomization technique) by a single surgeon (J.A.) who was blinded to type of mouse and treatment allocation. Surgeries were completed over a time period of 8 months.
SAH model
SAH was induced as reported previously 14, 15. Mice were anesthetized by inhalation of isofluorane (5% induction; 2–3% maintenance) carried by oxygen (1 L min−1). The body temperature was maintained at 37.0 ± 0.5 °C with a homeothermic heating pad and a rectal probe (Harvard Apparatus, Holliston, MA, USA). The head was fixed in a stereotactic frame equipped with a mouse adaptor (Harvard Apparatus). A 0.9‐mm burr hole was drilled 4.5 mm anterior to the bregma and angled ventrally at 40°. A Doppler flow meter (BLT21; Transonics Systems, New York, NY, USA) was used to monitor relative cerebral blood flow (CBF) for 7.5 min before and 15 min after blood injection. To induce SAH, 60 μL of non‐anticoagulated blood from a donor mouse was obtained by cardiac puncture and injected into the prechiasmatic cistern with a 27‐gauge spinal needle (BD Biosciences, San Jose, CA, USA) over a period of 9 s by use of a microsyringe pump (Model‐310; Stoelting, Wood Dale, IL, USA). In all four experimental groups, the donor mouse was of the same strain as the SAH mouse. A sharp drop in CBF indicated success of the SAH induction. Buprenorphine (0.2 mg kg−1) was given subcutaneously immediately after surgery for pain management. The mouse was allowed to recover in a heated chamber (Harvard Apparatus). Mice were killed 2 h after surgery, and perfused through the left cardiac ventricle with 0.9% NaCl, followed by 4% paraformaldehyde (PFA) in phosphate‐buffered saline (PBS) (pH 7.4), both under physiological blood pressure (60–80 mmHg). Any mice that were not properly perfused or died prior to the 2‐h time point were excluded from the analysis. Brains were removed and postfixed for 48 h in 4% PFA in PBS. Coronal cuts were made with a mouse brain matrix (Zivic Instruments, Pittsburgh, PA, USA) at − 2 mm from the bregma. Tissue blocks were processed, embedded in paraffin, and cut into 5‐μm sections with a microtome (Leica, Wetzlar, Germany).
Outcome measures
The primary outcome measures were: (i) the degree of microglial activation as reflected by the amount of IBA‐1 staining; and (ii) the degree of neuronal injury as reflected by active caspase‐3 staining in NeuN‐expressing cells. The secondary outcome measures were: (i) CBF 10 min after blood injection; and (ii) the amount of fibrin(ogen) staining.
Immunohistochemistry for IBA‐1 and fibrin(ogen)
For IBA‐1 and fibrin(ogen) staining, slides were deparaffinized and rehydrated through xylene and decreasing concentrations of ethanol. Antigen was retrieved in a 96 °C water bath for 30 min with Vector antigen unmasking solution (Vector Laboratories, Burlingame, CA, USA). Endogenous peroxidase activity was quenched with 0.3% hydrogen peroxide in water for 30 min. For IBA‐1 staining, samples were incubated with 0.3% Triton X‐100 for 1 h. Sections were blocked with 10% goat serum and 1% bovine serum albumin (BSA) in PBS for 30 min, and then incubated overnight at 4 °C with either 1 : 200 rabbit anti‐mouse fibrinogen (AB27913; Abcam, Cambridge, MA, USA) or 1 : 1000 rabbit anti‐IBA‐1 (Wako Chemicals, Cape Charles, VA, USA). Samples were then incubated with biotinylated secondary antibody (goat anti‐rabbit, 1 : 200 in 1% BSA/PBS; Vector Laboratories) for 30 min. Staining was visualized with VIP peroxidase substrate by use of the VECTASTAIN ABC Kit (Vector Laboratories), and nuclei were counterstained with 0.5% methyl green.
Immunofluorescence staining
For caspase‐3 immunohistological staining, after deparaffinization, dehydration, and antigen retrieval, sections were permeabilized with 0.3% Triton X‐100 for 1 h and blocked with 10% goat serum and 1% BSA in PBS for 1 h at room temperature. Samples were then incubated overnight at 4 °C with 1 : 200 rabbit anti‐active caspase‐3 antibody (BD Biosciences), and subsequently incubated for 1 h with Alexa Fluor 488‐conjugated goat anti‐rabbit IgG (1 : 1000; Life Technologies, Carlsbad, CA, USA) to visualize active caspase‐3. Samples were also counterstained with mouse anti‐NeuN (1 : 200; Millipore, Temecula, CA, USA) and Alexa Fluor 568‐conjugated goat anti‐mouse IgG (1 : 1000; Life Technologies) to visualize neurons, and 4,6‐diamidino‐2‐phenylindole to visualize nuclei.
CBF
For CBF quantification, digital data points of whole trace recorded (polyview; Natus, Pleasanton, CA, USA) were exported into Excel at a sampling frequency of 100 Hz. Every 1000th point of data was extracted from each of the traces to form a dataset with data points every 10 s. The first data point with CBF < 10% of the baseline was chosen as the peak point, and defined as time = 0 min. The baseline time was thereby defined as negative time in minutes from the peak point, and the recovery following the peak point was defined as positive time in minutes. Data were normalized to the average CBF value prior to the blood injection.
Quantification of staining
For histology experiments, a single coronal slice taken 2 mm posterior to the bregma was used for evaluation. For evaluation of microglial activation, six images were obtained of the cerebral cortex at × 40 magnification (Olympus BX50; Olympus Canada, Richmond Hill, Canada). Images were then converted into 16‐bit binary images, and thresholded for positive staining by the use of imagej (NIH, Bethesda, MD, USA). The results are expressed as an area fraction with positive IBA‐1 staining. For fibrin(ogen) staining, both the total number of microclots in the cerebral cortex and arterioles and venules that had fibrin(ogen) deposition within the vessel wall were quantified from a single coronal slice. For quantification of neuronal injury, the total numbers of active caspase‐3‐positive neurons within the neocortex were quantified from a single coronal slice. Fluorescent images were obtained with a laser confocal microscope (Axiovert 200; Carl Zeiss, Thornwood, NY, USA).
Statistical analysis
All assessments were performed by two reviewers who were blinded to the type of experimental group. Data were assessed for normal distribution with the Shapiro–Wilk test. All normally distributed data are expressed as means ± standard deviation, except for the CBF data, which are expressed as means ± standard error of the mean for better visualization of the CBF traces. In cases of normally distributed data, we calculated mean differences between two groups with 95% confidence intervals (CIs), and for statistical comparison we used a one‐way anova followed by a Tukey's multiple comparison test. Non‐normally distributed data are expressed as medians ± interquartile range. For comparisons of two groups with non‐normally distributed data, we used a Kruskal–Wallis anova with Dunn's multiple comparison test. Differences were considered statistically significant at a P‐value of < 0.05.
Results
The mouse characteristics are summarized in Table 1. Mean CBF dropped to < 10% of baseline during blood injection in all four groups, representing the rapid increase in intracranial pressure during blood injection (Table 1; Fig. 1). As compared with the baseline CBF, the CBF values during peak drop were 6.2% ± 5.9% for wild‐type control mice, 4.2% ± 1.6% for VWF−/− mice, 7.1% ± 7.1% for ADAMTS‐13−/− mice, and 6.6% ± 2.5% for rADAMTS‐13‐treated wild‐type mice (P = 0.18; Table 1 and Fig. 1). Death occurred in two of 21 (10%) wild‐type mice, four of 25 (16%) VWF−/−mice, four of 23 (17%) ADAMTS‐13−/− mice, and two of 26 (8%) rADAMTS‐13‐treated mice (P = 0.71, Fisher's exact test). All mice that did not survive to the time of killing at 2 h died within 5 min of induction of SAH. Two mice were excluded because of incomplete perfusion (one in the wild‐type group, and one in the rADAMTS‐13‐treated group).
Table 1.
Baseline parameters of animals, cerebral blood flow (CBF) reduction and CBF at 10 min postoperatively
| Parameter | Wild type (n = 23) | VWF−/− (n = 25) | ADAMTS‐13−/− (n = 23) | Wild type treated with rADAMTS‐13 (n = 26) |
|---|---|---|---|---|
| Age (days), mean ± SD | 90 ± 0 | 90 ± 0 | 90 ± 0 | 90 ± 0 |
| Weight (g), mean, mean ± SD | 23.4 ± 2.4 | 20.1 ± 2.4 | 23.4 ± 3.7 | 23.3 ± 2.4 |
| Mortality, no. (%) | 2 (10) | 4 (16) | 4 (17) | 2 (8) |
| Male/female ratio | 11 : 12 | 12 : 13 | 12 : 11 | 12 : 14 |
| Peak CBF reduction (% of baseline), mean ± SD | 6.3 ± 5.9 | 4.2 ± 1.6 | 7.1 ± 7.1 | 6.6 ± 2.5 |
| CBF at 10 min (% of baseline), mean ± SD | 58 ± 15 | 68 ± 24 | 72 ± 22 | 59 ± 18 |
VWF, von Willebrand factor; r‐ADAMTS‐13, recombinant ADAMTS‐13; SD, standard deviation.
Figure 1.
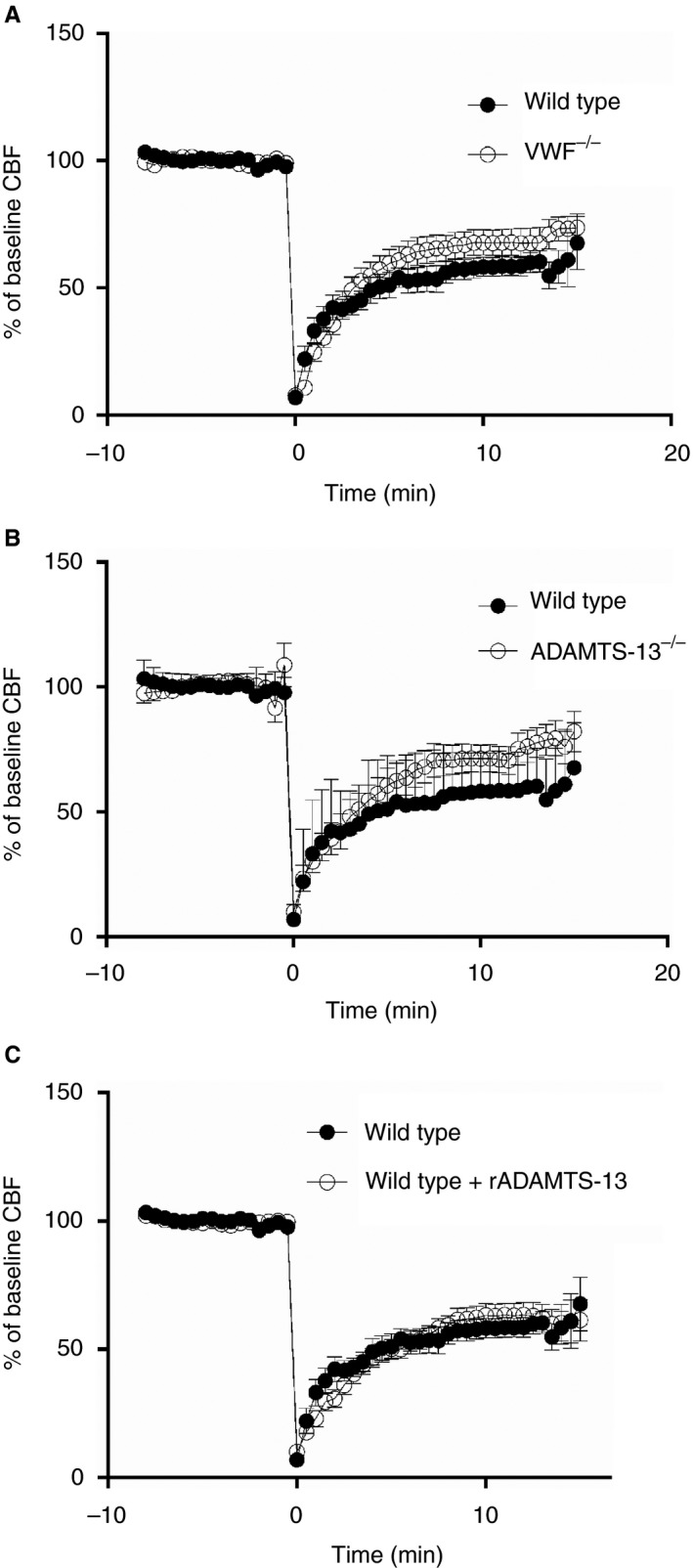
Cerebral blood flow (CBF) before and after subarachnoid hemorrhage (SAH) induction. Cerebral blood flow before and after SAH induction is compared between wild‐type controls and (A) von Willebrand factor (VWF)−/− mice, (B) ADAMTS‐13−/− mice, and (C) recombinant ADAMTS‐13 (rADAMTS‐13)‐treated mice. No differences were observed between groups. Data are displayed as means with standard error of the mean. Distances between points represent 10 s.
VWF deficiency or treatment with rADAMTS‐13 reduces microglial activation
VWF−/− mice had less microglial activation than wild‐type mice (3.0% ± 0.7% versus 3.7% ± 0.6%; mean difference of − 20%, 95% CI − 4% to − 39%), whereas ADAMTS‐13−/− mice had more microglial activation (5.0% ± 0.8% versus 3.7 ± 0.6%; mean difference of + 34%, 95% CI 16–52%), (Fig. 2). Similarly to VWF−/− mice, wild‐type mice treated with rADAMTS‐13 had less microglial activation than wild‐type mice without rADAMTS‐13 treatment (2.9% ± 0.9% versus 3.7% ± 0.6%; mean difference of − 22%, 95% CI − 3% to − 39%).
Figure 2.
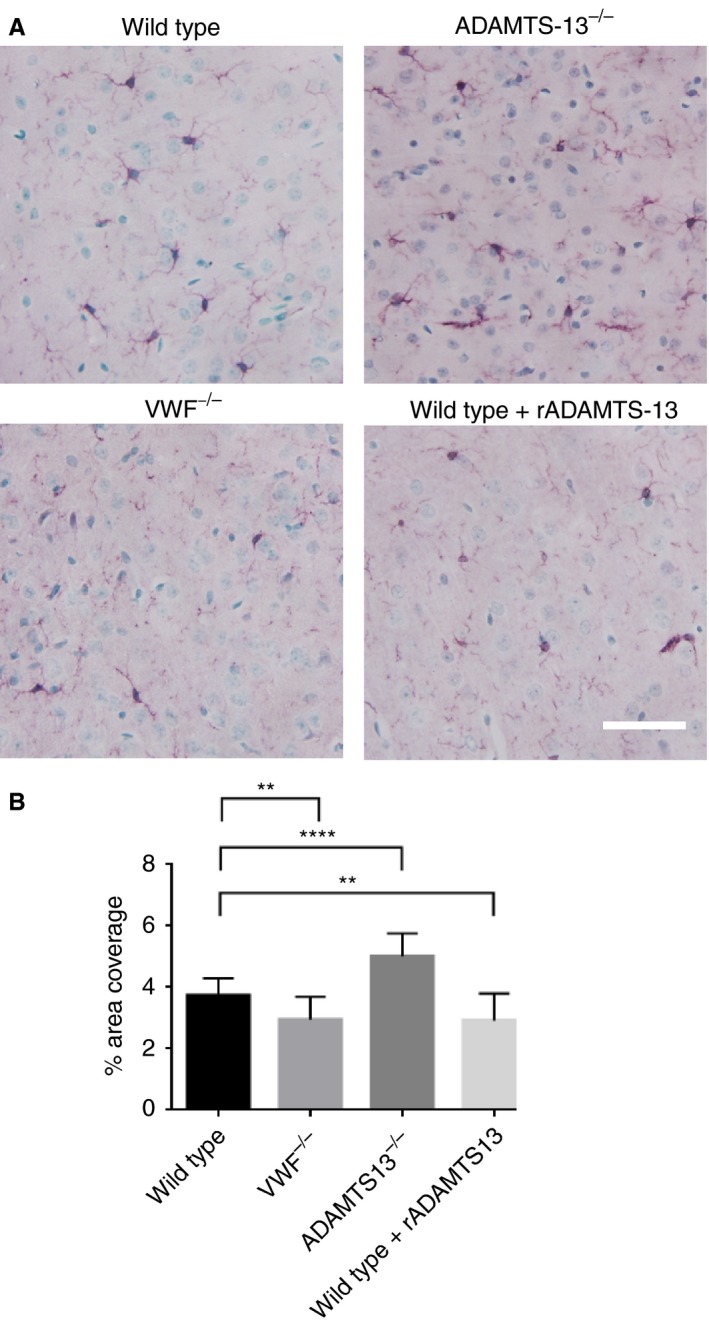
Immunohistochemical staining of IBA‐1. (A) Representative images of positive IBA‐1 staining. Positive IBA‐1 staining is indicated by purple, and nuclei are green. Scale bar: 20 μm. (B) Quantification of positive IBA‐1 staining. Subarachnoid hemorrhage resulted in substantial activation of microglia, seen as positive IBA‐1 staining, in all groups. Both von Willebrand factor (VWF) deficiency (n = 21) and recombinant ADAMTS‐13 (rADAMTS‐13) treatment (n = 23) decreased microglial activation as compared with wild‐type mice (**P < 0.01, ****P < 0.0001, anova. Data are displayed as means ± standard deviation. [Color figure can be viewed at http://www.wileyonlinelibrary.com]
VWF deficiency or treatment with rADAMTS‐13 reduces neuronal injury
All data on neuronal injury were non‐normally distributed. Therefore, a Kruskal–Wallis anova was used for statistical comparison. VWF−/− mice had less neuronal injury than wild‐type mice (64 neurons, interquartile range [IQR] 25–78 versus 185 neurons, IQR 132–353; P < 0.01). Neuronal injury did not differ between ADAMTS‐13−/− mice and wild‐type mice, although there was a trend towards reduced neuronal injury in ADAMTS‐13−/− mice (53 neurons, IQR 26–221 versus 185 neurons, IQR 132–353; P > 0.05). Treatment with rADAMTS‐13 reduced neuronal injury (45 neurons, IQR 9–115 versus 185 neurons, IQR 133–353; P < 0.001) (Fig. 3).
Figure 3.
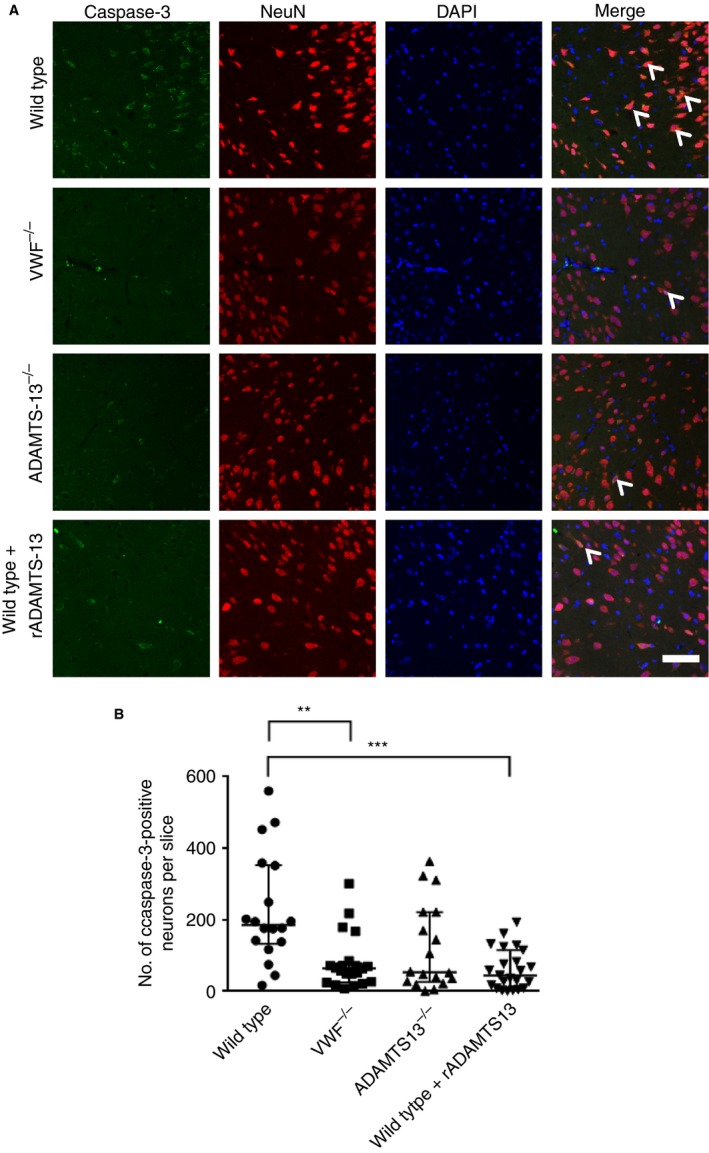
Immunofluorescent labeling of active caspase‐3. (A) Representative images of caspase‐3 staining, colabeled with NeuN and 4,6‐diamidino‐2‐phenylindole (DAPI). White arrowheads indicate neurons that are expressing active caspase‐3 in NeuN‐positive neurons. Scale bar: 20 μm. (B) Quantification of neurons with positive caspase‐3 staining. Subarachnoid hemorrhage induction resulted in a large number of neurons with positive staining for cleaved caspase‐3, which are presumed to undergo apoptosis. Wild‐type mice (n = 18) had a higher rate of cell death than von Willebrand factor (VWF)−/− mice (n = 21, **P < 0.01, ***P < 0.001, Kruskal–Wallis anova) and recombinant ADAMTS‐13 (rADAMTS‐13)‐treated mice (n = 23, P < 0.001, Kruskal–Wallis ANOVA). There was a trend towards reduced neuronal injury in ADAMTS‐13−/− mice as compared with wild‐type mice. Data are displayed as medians ± interquartile range. [Color figure can be viewed at http://www.wileyonlinelibrary.com]
CBF
At 10 min after blood injection, the mean CBF values were 58.3% ± 15.3% of the baseline CBF in wild‐type controls, 68.2% ± 24.1% in VWF−/− mice, 72.4% ± 21.7% in ADAMTS‐13−/− mice, and 59.4% ± 17.6% in rADAMTS‐13‐treated wild‐type mice (P = 0.08, anova; Table 1; Fig. 1).
VWF deficiency or treatment with rADAMTS‐13 reduces fibrin(ogen) deposition in parenchymal vessels
At 2 h after SAH, microthrombosis in capillaries was observed in three of 18 mice in the wild‐type group, none of 19 in the VWF−/− group, two of 17 in the ADAMTS‐13−/− group, and two of 23 in the wild‐type rADAMTS‐13‐treated group. Among these mice, wild‐type mice had 200 ± 90 clots, ADAMTS‐13−/− mice had 389 ± 62 clots, and rADAMTS‐13‐treated mice had 141 ± 104 clots. However, the small numbers of mice developing microthrombi in capillaries did not warrant statistical analysis. Apart from microthrombi, we analyzed fibrin(ogen) deposition within large parenchymal cortical vessels (Fig. 4). As compared with wild‐type controls, a trend was observed towards a lower total number of vessels with positive fibrin(ogen) staining in VWF−/− mice (16.1 ± 11.8 versus 23.8 ± 9.7; mean difference of − 34%, 95% CI − 74% to 5%) and a trend towards increased fibrin(ogen) vascular deposition was observed in ADAMTS‐13−/− mice (27.8 ± 12.5 versus 23.8 ± 9.7; mean difference of + 17%, 95% CI − 17% to 55%). Treatment with rADAMTS‐13 in wild‐type mice reduced the total number of vessels with positive fibrinogen staining (14.3 ± 6.8 versus 23.8 ± 9.7; mean difference of − 40%, 95% CI − 3% to − 76%) (Fig. 4).
Figure 4.
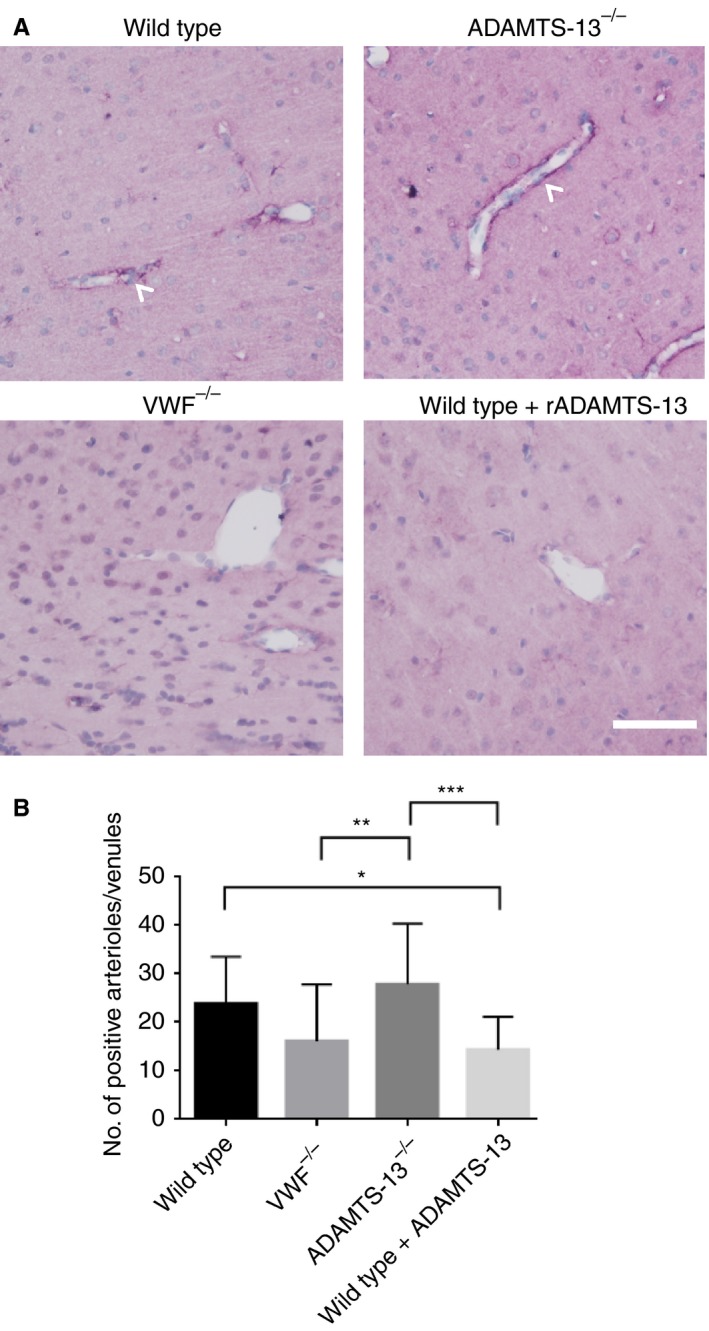
Fibrin(ogen) staining of vessels. (A) Representative images of large penetrating arterioles and venules stained by fibrin(ogen). Fibrin(ogen) staining is indicated by purple, and nuclei are green. Scale bar: 40 μm. (B) Quantification of the number of fibrin(ogen)‐stained vessels. Subarachnoid hemorrhage resulted in fibrin/fibrinogen deposition on large penetrating arterioles and venules in the mouse brain in all groups. As compared with wild‐type mice (n = 18), von Willebrand factor (VWF)−/− mice (n = 18) showed a trend towards having a lower number of fibrin(ogen)‐stained arterioles and venules, whereas ADAMTS‐13−/− mice (n = 17) showed a trend towards having a higher number of fibrin(ogen)‐stained arterioles and venules. Treatment with recombinant ADAMTS‐13 (rADAMTS‐13)‐treated mice (n = 21) reduced the number of fibrin(ogen)‐stained vessels as compared with wild‐type mice (*P < 0.05, **P < 0.01, ***P < 0.001, anova). Data are displayed as means ± standard deviation. [Color figure can be viewed at http://www.wileyonlinelibrary.com]
Discussion
Our study demonstrates that VWF plays an important role in mediating early brain injury after SAH. Animals that lacked endogenous VWF or received rADAMTS‐13 treatment had less microglial activation and neuronal injury than wild‐type controls at 2 h after SAH. In contrast, ADAMTS‐13−/− mice had increased microglial activation as compared with wild‐type mice, but not increased neuronal injury as compared with wild‐type mice. Furthermore, we found less fibrin(ogen) deposition within the vessel walls of VWF−/− or rADAMTS‐13‐treated mice than in the vessel walls of wild‐type controls, suggesting that rADAMTS‐13 may exert its beneficial effect at least partially by influencing thrombosis.
Early brain injury is caused by the effects of the initial hemorrhage, resulting in microvascular constriction, platelet aggregation, blood–brain barrier disruption, and edema 3. Platelets have been shown to aggregate in parenchymal capillaries within 10 min in a rat model of SAH, and persist for up to 24 h 16. Similarly, in the pial microvasculature, platelet aggregates have been found 10 min after SAH, and persist for up to 3 days 17. The observed reductions in microglial activation and neuronal injury in VWF−/− mice and rADAMTS‐13‐treated mice in our study may be attributable to reduced microvascular plugging early after SAH, as these mice had less fibrin(ogen) deposition within parenchymal arterioles and venules than control mice. The effect of rADAMTS‐13 on microglial activation and microthrombosis was less pronounced than in our previous study on DCI when measured 2 h after SAH ictus 6. This suggests that there may be a cumulative effect of rADAMTS‐13 on early brain injury and DCI mechanisms after SAH.
The small number of mice that did develop microthrombosis did not warrant statistical analysis. As microthrombosis typically peaks at 48 h after SAH in mice, it is not surprising that few mice showed microthrombosis at 2 h after SAH. Another potential explanation for the low prevalence of microthrombosis is that microthrombi were washed out by the intracardiac perfusion fixation procedure, as these clots may not be as stable at this early time point as at 48 h after SAH. Furthermore, rADAMTS‐13 not only has an effect on platelet aggregation, but also reduces inflammation. In experimental intracerebral hemorrhage, rADAMTS‐13 reduced brain edema and neuronal injury as compared with wild‐type control mice by attenuating pro inflammatory cytokines, such as interleukin‐6 and interleukin‐1β, and promoting stabilization of the blood‐brain barrier 18. rADAMTS‐13 may also decrease early brain injury after SAH through these mechanisms. We found that there was no increased cellular injury in ADAMTS‐13−/− mice as compared with wild‐type and VWF−/− mice, despite an increase in microglial activation. We hypothesized that the strong early microglial activation observed in ADAMTS‐13−/− mice might suppress some neuronal injury, but, in the long term, becomes detrimental. Indeed, in previous studies, ADAMTS‐13−/− mice were found to have an enhanced microglial activation profile and increased neuronal cell death 48 h after ictus 6, 8.
There are several limitations of our current study. Two mice were excluded from the final analysis of our data, owing to improper tissue perfusion and/or fixation. Although all mice were of the same age, we found that VWF−/− mice were lighter than wild‐type or ADAMTS‐13−/− mice (Table 1). This suggests there may be some developmental differences in VWF−/− mice, which may introduce confounding. Furthermore, no functional assessment of mice was performed to determine whether VWF or ADAMTS‐13 had any effect on functional or behavioral outcome. rADAMTS‐13 was also administered early after SAH induction, which is not feasible in the clinical setting. An optimal and feasible time point for administration of rADAMTS‐13 in patients is required. It may also be important to explore whether additional infusions of rADAMTS‐13 may help to further reduce injury resulting from delayed cerebral ischemia. Finally, it remains to be investigated whether rADAMTS‐13 influences only primary hemostasis or also other processes involved in the pathogenesis of early brain injury after SAH.
In conclusion, our results support the further study of rADAMTS‐13 as a treatment option for the reduction of early brain injury after SAH. Further studies are required to determine whether there is improvement in CBF after rADAMTS‐13 treatment because of reductions in the number of thrombi in early brain injury and delayed brain injury. In addition, the mechanism of action of rADAMTS‐13 on platelet aggregation may be directly observed in vivo by using intravital microscopy, which may help to determine the optimal time window for administering rADAMTS‐13 to reduce early brain injury. Future studies are needed to investigate whether rADAMTS‐13 increases the risk of rebleeding of the aneurysm, and to determine the optimal time window for administering rADAMTS‐13 to reduce early brain injury.
Addendum
M. D. I. Vergouwen designed the study. H. Wan, Y. Li, and J. Ai performed the experiments. H. Wan, J. Ai, and S. Brathwaite analyzed the results. H. Wan wrote the first draft of the manuscript. Y. Li, J. Ai, S. Brathwaite, H. Ni, R. L. Macdonald, E. M. Hol, J. C. M. Meijers and M. D. I. Vergouwen performed critical revisions of the manuscript for important intellectual content.
Disclosure of Conflict of Interests
M. D. I. Vergouwen received a research grant, partly for this study, from Baxalta US Inc., now part of Shire. Recombinant ADAMTS‐13 was kindly provided by Baxalta under the same grant, and Baxalta US Inc., now part of Shire, had no further role in project planning, experiments, or data analysis. R. L. Macdonald reports receiving grants from the Brain Aneurysm Foundation, grants from the Canadian Institutes of Health Research, and grants from Ontario Genomics, and having stock, stock options and employment from Edge Therapeutics, all outside the submitted work. The other authors state that they have no conflict of interest.
Wan H, Wang Y, Ai J, Brathwaite S, Ni H, Macdonald RL, Hol EM, Meijers JCM, Vergouwen MDI. Role of von Willebrand factor and ADAMTS‐13 in early brain injury after experimental subarachnoid hemorrhage. J Thromb Haemost 2018; 16: 1413–22.
Manuscript handled by: X. Long Zheng
Final decision: P. H. Reitsma, 18 April 2018
References
- 1. Vergouwen MD, Jong‐Tjien‐Fa AV, Algra A, Rinkel GJ. Time trends in causes of death after aneurysmal subarachnoid hemorrhage: a hospital‐based study. Neurology 2016; 86: 59–63. [DOI] [PubMed] [Google Scholar]
- 2. Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, Zhang JH. Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Transl Stroke Res 2013; 4: 432–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol 2014; 10: 44–58. [DOI] [PubMed] [Google Scholar]
- 4. Liu GJ, Luo J, Zhang LP, Wang ZJ, Xu LL, He GH, Zeng YJ, Wang YF. Meta‐analysis of the effectiveness and safety of prophylactic use of nimodipine in patients with an aneurysmal subarachnoid haemorrhage. CNS Neurol Disord Drug Targets 2011; 10: 834–44. [DOI] [PubMed] [Google Scholar]
- 5. Friedrich V, Flores R, Muller A, Sehba FA. Luminal platelet aggregates in functional deficits in parenchymal vessels after subarachnoid hemorrhage. Brain Res 2010; 1354: 179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vergouwen MD, Knaup VL, Roelofs JJ, de Boer OJ, Meijers JC. Effect of recombinant ADAMTS‐13 on microthrombosis and brain injury after experimental subarachnoid hemorrhage. J Thromb Haemost 2014; 12: 943–7. [DOI] [PubMed] [Google Scholar]
- 7. Vergouwen MD, Bakhtiari K, van Geloven N, Vermeulen M, Roos YB, Meijers JC. Reduced ADAMTS13 activity in delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab 2009; 29: 1734–41. [DOI] [PubMed] [Google Scholar]
- 8. Muroi C, Fujioka M, Mishima K, Irie K, Fujimura Y, Nakano T, Fandino J, Keller E, Iwasaki K, Fujiwara M. Effect of ADAMTS‐13 on cerebrovascular microthrombosis and neuronal injury after experimental subarachnoid hemorrhage. J Thromb Haemost 2014; 12: 505–14. [DOI] [PubMed] [Google Scholar]
- 9. Tang QF, Lu SQ, Zhao YM, Qian JX. The changes of von Willebrand factor/a disintegrin‐like and metalloprotease with thrombospondin type I repeats‐13 balance in aneurysmal subarachnoid hemorrhage. Int J Clin Exp Med 2015; 8: 1342–8. [PMC free article] [PubMed] [Google Scholar]
- 10. Boluijt J, Meijers JC, Rinkel GJ, Vergouwen MD. Hemostasis and fibrinolysis in delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: a systematic review. J Cereb Blood Flow Metab 2015; 35: 724–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumar M, Cao W, McDaniel JK, Pham HP, Raju D, Nawalinski K, Frangos S, Kung D, Zager E, Kasner SE, Levine JM, Zheng XL. Plasma ADAMTS13 activity and von Willebrand factor antigen and activity in patients with subarachnoid haemorrhage. Thromb Haemost 2017; 117: 691–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reheman A, Yang H, Zhu G, Jin W, He F, Spring CM, Bai X, Gross PL, Freedman J, Ni H. Plasma fibronectin depletion enhances platelet aggregation and thrombus formation in mice lacking fibrinogen and von Willebrand factor. Blood 2009; 113: 1809–17. [DOI] [PubMed] [Google Scholar]
- 13. Ni H, Denis CV, Subbarao S, Degen JL, Sato TN, Hynes RO, Wagner DD. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J Clin Invest 2000; 106: 385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. D'Abbondanza JA, Ai J, Lass E, Wan H, Brathwaite S, Tso MK, Lee C, Marsden PA, Macdonald RL. Robust effects of genetic background on responses to subarachnoid hemorrhage in mice. J Cereb Blood Flow Metab 2016; 36: 1942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sabri M, Jeon H, Ai J, Shang X, Chen G, Macdonald RL. Anterior circulation mouse model of subarachnoid hemorrhage. Brain Res 2009; 1295: 179–85. [DOI] [PubMed] [Google Scholar]
- 16. Friedrich V, Flores R, Muller A, Sehba FA. Escape of intraluminal platelets into brain parenchyma after subarachnoid hemorrhage. Neuroscience 2010; 165: 968–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sehba FA, Mostafa G, Friedrich V Jr, Bederson JB. Acute microvascular platelet aggregation after subarachnoid hemorrhage. J Neurosurg 2005; 102: 1094–100. [DOI] [PubMed] [Google Scholar]
- 18. Cai P, Luo H, Xu H, Zhu X, Xu W, Dai Y, Xiao J, Cao Y, Zhao Y, Zhao BQ, Fan W. Recombinant ADAMTS 13 attenuates brain injury after intracerebral hemorrhage. Stroke 2015; 46: 2647–53. [DOI] [PubMed] [Google Scholar]