

Abstract
An unprecedented set of structurally diverse sulfonimidamides (47 compounds) has been prepared by various N‐functionalization reactions of tertiary =NH sulfonimidamide 2 aa. These N‐functionalization reactions of model compound 2 aa include arylation, alkylation, trifluoromethylation, cyanation, sulfonylation, alkoxycarbonylation (carbamate formation) and aminocarbonylation (urea formation). Small molecule X‐ray analyses of selected N‐functionalized products are reported. To gain further insight into the properties of sulfonimidamides relevant to medicinal chemistry, a variety of structurally diverse reaction products were tested in selected in vitro assays. The described N‐functionalization reactions provide a short and efficient approach to structurally diverse sulfonimidamides which have been the subject of recent, growing interest in the life sciences.
Keywords: drug design, medicinal chemistry, N-functionalization, structural diversity, sulfonimidamides
Introduction
The sulfonamide group 1 (Figure 1) is an important pharmacophore found in about 200 drugs currently on the market.1 In contrast, sulfonimidamides 2, the mono‐aza analogues of sulfonamides 1, have received little interest until recently, despite being first synthesized as early as 1962.2 The infrequent take‐up of the sulfonimidamide group as a pharmacophore is surprising since it seems to offer very interesting properties, including high stability, favorable physicochemical properties, multiple hydrogen‐bond acceptor/donor functionalities and structural diversity.3
Figure 1.
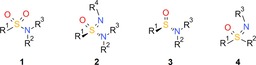
General structures of sulfonamides 1 (1° R2, R3=H; 2° R2=H, R3≠H; 3° R2≠H, R3≠H), sulfonimidamides 2 (1° R2, R3=H; 2° R2=H, R3≠H; 3° R2≠H, R3≠H), sulfinamides 3 (1° R2, R3=H; 2° R2=H, R3≠H; 3° R2≠H, R3≠H) and sulfoximines 4.
Possible reasons for the neglected use of the sulfonimidamide group are the lack of commercial availability and limited synthetic methods for its preparation,4 as well as an incomplete understanding of its medicinal chemistry properties. However, as recently pointed out by Arvidsson and co‐workers, the sulfonimidamide group is currently gaining popularity as a novel pharmacophore in the life sciences.3 Examples include the sulfonimidamide analogues 5 and 6 of the clinical sulfonamide‐containing anticancer agent tasisulam5 and non‐steroidal anti‐inflammatory drug celecoxib,6 respectively, as well as an increasing number of sulfonimidamides claimed in patent applications, for instance the sodium channel inhibitor 7 (Figure 2).7 However, to the best of our knowledge, a sulfonimidamide candidate for clinical testing has yet to be disclosed.
Figure 2.
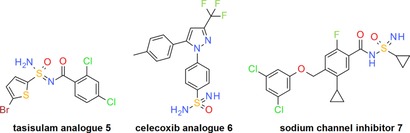
Structures of the sulfonimidamide analogues 5 and 6 of the clinical sulfonamide‐containing anticancer agent tasisulam5 and non‐steroidal anti‐inflammatory drug celecoxib,6 as well as of the sodium channel inhibitor 7 7 disclosed in a recent patent application.
This increasing interest in the life sciences is supported by the very recent advent of new and safe synthetic methods for the preparation of sulfonimidamides 2. Latest developments include the copper‐catalyzed conversion of sulfoximines 4 into sulfonimidamides 2,8 the preparation of trifluoromethylated sulfonimidamides9 and the one‐pot de novo synthesis of sulfonimidamides relying on the stable reagent N‐sulfinyltritylamine.6
Triggered by our experience that the use of uncommon functional groups, such as sulfoximines 4,10 can be crucial for overcoming hurdles in lead optimization,11 we also became interested in sulfonimidamides 2 as a neglected but potentially versatile pharmacophore for drug discovery. Due to the limited synthetic methodology available, we developed a new, practical one‐pot synthesis of tertiary =NH sulfonimidamides 2 a by transfer of electrophilic NH to sulfinamides 3 (Scheme 1).12 The reaction is mediated by commercially available (diacetoxyiodo)benzene and ammonium carbamate in methanol at room temperature and tolerates a wide range of functional groups. Originally, these reaction conditions had been newly reported by Bull, Luisi and co‐workers13 for the conversion of sulfoxides into sulfoximines 4. Moreover, we investigated a variety of in vitro properties, relevant to medicinal chemistry, of =NH sulfonimidamide 2 aa in comparison to its matched sulfonamide analogue 1 aa and did not identify any intrinsic flaw of the =NH sulfonimidamide group with respect to its application in the life sciences.12
Scheme 1.
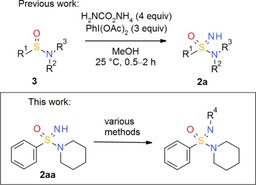
Synthesis of N‐functionalized tertiary sulfonimidamides by various methods using =NH sulfonimidamide 2 aa as a model compound.
However, in contrast to sulfonamides 1, =NH sulfonimidamides 2 a offer the possibility of exploration of novel chemical space via the introduction of substituents at the =NH position. Functionalization of this =NH position would also offer an additional handle to modulate the overall properties of the resulting molecules, for example with respect to conformational behavior, lipophilicity and physicochemical properties.
Therefore, we set course to investigate the N‐functionalization of tertiary =NH sulfonimidamides 2 a. To test the desired transformations, we selected =NH sulfonimidamide 2 aa as a model compound, which had been prepared from the corresponding sulfinamide 3 on a gram scale by the one‐pot NH‐transfer method (Scheme 1).12
With respect to the reactivity of the =NH position, we expected a certain similarity of sulfonimidamides and sulfoximines. Therefore, we elected to probe reaction conditions for the desired N‐functionalizations of =NH sulfonimidamides that we had already successfully employed for the analogous reactions of =NH sulfoximines, even on complex, drug‐like molecules.14
Results and Discussion
The palladium‐catalyzed coupling of =NH sulfoximines with aryl bromides was first described in 1998 by Bolm and Hildebrand who used catalytic amounts of Pd(OAc)2 and a chelating bisphosphine (e.g., BINAP) in the presence of Cs2CO3 as a base.15 In the meantime, a variety of modified reaction conditions and reagents have been described by both academic and industrial groups.16 In our drug discovery efforts, we had relied on the catalyst system Pd2(dba)3/BINAP14e, 14i or Pd2(dba)3/Xantphos17 in toluene in the presence of Cs2CO3 for this type of reaction. However, in an ongoing lead optimization approach, we recently switched to the combination of Pd(OAc)2 and Xantphos as a catalyst since the use of Pd2(dba)3 resulted in purification issues.18 In contrast to the broad variety of N‐arylation methods available for =NH sulfoximines, as far as we are aware there is only one report of the analogous palladium‐catalyzed coupling reaction of =NH sulfonimidamides. Thus, Arvidsson and co‐workers used RuPhos and 2nd generation RuPhos precatalyst in THF in the presence of NaOtBu under microwave conditions; nevertheless, only four structurally simple products, having no substituents at the aryl group, were described.19 The same research group also successfully investigated the copper‐catalyzed coupling of =NH sulfonimidamides with boronic acids.20 However, we were mainly interested in the N‐arylation of =NH sulfonimidamides using aryl halides, due to better availability and reduced costs. Hence, conditions that we had successfully applied for the analogous functionalization of =NH sulfoximines were tested. As a model reaction, tertiary =NH sulfonimidamide 2 aa (100 mg, 0.45 mmol, 1.1 equiv) was treated with bromobenzene (8 a, 1 equiv) in the presence of catalytic amounts of Pd(OAc)2 (5 mol %) and Xantphos (10 mol %), along with Cs2CO3 (1.5 equiv), in toluene at 100 °C overnight. To our delight, the desired coupling product 2 ba was isolated in 86 % yield after column chromatography (Table 1). Given the clean reaction and very good yield, we then elected to explore the substrate scope of this new process. A variety of substituted aryl bromides 8 b–l were subjected to the standard reaction conditions. Gratifyingly, the desired products 2 bb–2 bl were afforded in all cases, the majority in good to excellent yields (Table 1). The exception was the reaction of 2‐bromo‐1,3,5‐trimethylbenzene (8 h) which gave the coupling product 2 bh in very low yield (3 %). This is probably due to steric hindrance resulting from the two methyl groups at the positions ortho to the bromine, since the monosubstituted bromotoluenes 8 e–g all resulted in very good yields. Coupling product 2 bk was successfully recrystallized and the structure confirmed by X‐ray analysis (Figure 3) [for additional X‐ray analyses (compounds 2 bg, 2 bj and 2 bm), see the Supporting Information]. The reaction of various heteroaryl bromides 8 m–q also gave the desired heteroaromatic products 2 bm–2 bq in good yields.
Table 1.
Exploration of the substrate scope of the palladium‐catalyzed N‐arylation of tertiary =NH sulfonimidamide 2 aa: Variation of aryl bromide 8.
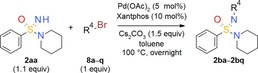
| |
|---|---|
| Aryl bromide (R4Br) | Isolated yield [%] |
| 8 a: bromobenzene | 2 ba: 86 |
| 8 b: 1‐bromo‐2‐fluorobenzene | 2 bb: 46 |
| 8 c: 1‐bromo‐3‐fluorobenzene | 2 bc: 97 |
| 8 d: 1‐bromo‐4‐fluorobenzene | 2 bd: 72 |
| 8 e: 2‐bromotoluene | 2 be: 97 |
| 8 f: 3‐bromotoluene | 2 bf: 84 |
| 8 g: 4‐bromotoluene | 2 bg: 99 |
| 8 h: 2‐bromo‐1,3,5‐trimethylbenzene | 2 bh: 3 |
| 8 i: 3‐bromoanisole | 2 bi: 65 |
| 8 j: 4‐bromobenzonitrile | 2 bj: 88 |
| 8 k: methyl 4‐bromobenzoate | 2 bk: 99 |
| 8 l: 4‐bromobenzotrifluoride | 2 bl: quant. |
| 8 m: 2‐bromopyridine | 2 bm: 71 |
| 8 n: 3‐bromopyridine | 2 bn: 77 |
| 8 o: 4‐bromopyridine | 2 bo: 72 |
| 8 p: 2‐bromopyrimidine | 2 bp: 77 |
| 8 q: 2‐bromo‐1,3‐thiazole | 2 bq: 45 |
Figure 3.
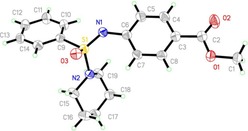
ORTEP plot (50 % thermal ellipsoids) of the crystal structure of N‐arylated sulfonimidamide 2 bk.
Another important option for the N‐functionalization of =NH sulfoximines and =NH sulfonimidamides is the corresponding N‐alkylation reaction. It is noteworthy that all recent sulfoximine clinical candidates, the kinase inhibitors roniciclib (BAY 1000394),11a, 21 atuveciclib (BAY 1143572),22 AZD 6738 (Figure 4)23 and BAY 1251152 (Figure 4),24 contain an unsubstituted =NH group. However, since for instance low Caco2 permeability and high efflux can be an issue with =NH sulfoximines,10e N‐alkylation may be a means of improving the permeability properties.10d Moreover, N‐alkylation is an interesting option for drug design to explore novel chemical space. Satzinger and Stoss, who pioneered the use of the sulfoximine group in drug discovery, were originally attracted to this functional group by this possibility of introducing substituents at the =NH position. Their lead optimization efforts led to the identification of the first sulfoximine clinical candidate suloxifen, which is an N‐alkylated diphenyl sulfoximine (Figure 4).10c, 25
Figure 4.
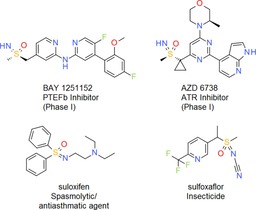
Examples of clinical and commercial sulfoximines: BAY 1251152, AZD 6738, suloxifen and sulfoxaflor.
Direct N‐alkylation of =NH sulfoximines is not a trivial task since the nucleophilicity of the nitrogen is dramatically reduced due to steric and electronic effects of the adjacent tetracoordinated sulfur.26 N‐Methylation of =NH sulfoximines has usually been achieved under Eschweiler–Clarke conditions or by the use of a strong methyl‐transfer agent.27 Introduction of more complex alkyl groups, however, remained difficult until Bolm and co‐workers introduced a new method employing alkyl bromides and KOH as a base in DMSO at room temperature.28
So far, there have been scant reports of the corresponding direct N‐alkylation of =NH sulfonimidamides, the first from Johnson and Lavergne.26, 29 They discovered that while Eschweiler–Clarke conditions resulted in degradation of the =NH sulfonimidamide starting materials, the use of primary alkyl bromides in combination with KH and the phase‐transfer catalyst Bu4NBr in 1,2‐dimethoxyethane gave the desired N‐alkylation products in good to excellent yields. In contrast, secondary bromides did not provide the N‐alkylated products under these conditions.
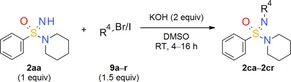
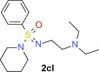
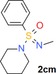
Along the lines of our concept to apply successful reaction conditions from our experience with =NH sulfoximines in drug discovery to =NH sulfonimidamides, we investigated the N‐alkylation of model compound 2 aa using alkyl bromides in the presence of KOH in DMSO. In the first reaction, tertiary =NH sulfonimidamide 2 aa (100 mg, 0.45 mmol, 1 equiv) was treated with bromoethane (9 a, 1.5 equiv) in DMSO in the presence of KOH (2 equiv) for 4 hours at room temperature, to give the desired N‐ethyl derivative 2 ca in 99 % isolated yield (Table 2). After this initial success, the substrate scope of this new process was explored using alkyl bromides 9 b–l. In contrast to Johnson and Lavergne's method,26 reaction of the secondary alkyl bromide 2‐bromopropane (9 b) also gave the desired product 2 cb, albeit in a significantly reduced yield of 38 %. Reaction of 2 aa with a variety of more complex alkyl bromides 9 c–k gave the corresponding N‐alkylation products, usually in good to moderate yields. Similar to the reported results of the N‐alkylation of sulfoximines,28 the use of N‐(2‐bromoethyl)‐N,N‐diethylamine (9 l) resulted in a low 13 % yield. However, the corresponding product 2 cl can be considered as a partially saturated sulfonimidamide analogue of the potent spasmolytic and antiasthmatic agent suloxifen (Figure 4). Since methyl bromide is a gas, we employed methyl iodide for the desired N‐methylation reaction of 2 aa to isolate the methylated product 2 cm in 59 % yield. Benzyl bromide (9 n) reacted with 2 aa in a similar yield of 57 % (2 cn), and product 2 co was isolated from the reaction with heterocyclic 3‐(bromomethyl)‐5‐methylisoxazole (9 o) in good yield. Moreover, the reactions of allyl bromide (9 p) and propargylic bromides 9 q,r also gave the corresponding products 2 cp–2 cr in good yields.
Table 2.
Exploration of the substrate scope of the N‐alkylation of tertiary =NH sulfonimidamide 2 aa: Variation of alkyl halide 9.
| ||
|---|---|---|
| Alkyl halide (R4Br/I) | Isolated yield [%] | |
| 9 a: bromoethane | 2 ca: 99 | |
| 9 b: 2‐bromopropane | 2 cb: 38 | |
| 9 c: (bromomethyl)cyclobutane | 2 cc: 61 | |
| 9 d: (bromomethyl)cyclopentane | 2 cd: 29 | |
| 9 e: (bromomethyl)cyclohexane | 2 ce: 71 | |
| 9 f: 4‐(bromomethyl)tetrahydro‐2H‐pyran | 2 cf: 60 | |
| 9 g: |
|
2 cg: 35 |
| 9 h: |
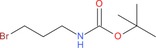
|
2 ch: 50 |
| 9 i: |
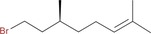
|
2 ci: 46 |
| 9 j: 1‐bromo‐2‐methoxyethane | 2 cj: 80 | |
| 9 k: ethyl bromoacetate | 2 ck: 40 | |
| 9 l: |
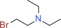
|
2 cl: 13 |
| 9 m: methyl iodide | 2 cm: 59 | |
| 9 n: benzyl bromide | 2 cn: 57 | |
| 9 o: |
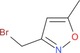
|
2 co: 62 |
| 9 p: allyl bromide | 2 cp: 72 | |
| 9 q: propargyl bromide | 2 cq: 63 | |
| 9 r: 1‐bromobut‐2‐yne | 2 cr: 71 | |
Bolm and co‐workers recently achieved the first N‐trifluoromethylation of =NH sulfoximines.30 This radical process relies on the use of TMSCF3 as a trifluoromethylating agent and catalytic amounts of Ag2CO3 and 1,10‐phenanthroline under an oxygen atmosphere. The introduction of trifluoromethyl substituents is now a well‐accepted strategy in medicinal chemistry, since it can significantly alter the properties of the resulting compounds, for example with respect to lipophilicity, metabolic stability and conformational behavior.31 Since N‐trifluoromethylation of =NH sulfonimidamides has not been reported, we were intrigued to apply the above conditions to model compound 2 aa. To our delight, the desired product 2 cs was formed and could be isolated in 44 % yield (Scheme 2) without further optimization.
Scheme 2.
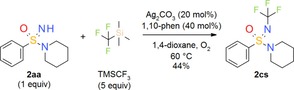
Synthesis of N‐trifluoromethylated tertiary sulfonimidamide 2 cs.
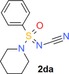
N‐Cyanation of =NH sulfoximines is another interesting design option in the life sciences. The resulting =N−CN sulfoximine group is, for instance, present in the only marketed sulfoximine so far, the insecticide sulfoxaflor (Figure 4).32 In the past, we and others have relied on the reaction of =NH sulfoximines with cyanogen bromide to prepare =N−CN sulfoximines.33 There is also one report by Gnamm and co‐workers of the successful application of this method for the preparation of an =N−CN sulfonimidamide derivative.34 However, given the high toxicity of the required cyanogen bromide, a new, environmentally benign procedure caught our attention. Thus, Cheng and co‐workers recently reported the direct copper‐catalyzed N‐cyanation of =NH sulfoximines using AIBN as a safe cyanide source.35 Given the successful N‐trifluoromethylation of =NH sulfonimidamide 2 aa under radical conditions (Scheme 2), we elected to apply the new radical N‐cyanation process of sulfoximines to =NH sulfonimidamide model compound 2 aa and at once isolated the corresponding product 2 da in 43 % yield (Scheme 3).
Scheme 3.
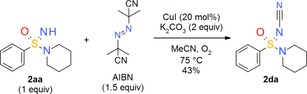
Synthesis of N‐cyanated tertiary sulfonimidamide 2 da.
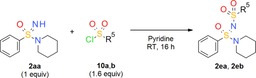
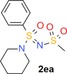
N‐Sulfonylated sulfoximines can be prepared by the reaction of =NH sulfoximines with sulfonyl chlorides.36 There are various reported N‐(sulfonyl)sulfonimidamides,37 but none obtained by the direct reaction of =NH sulfonimidamides with sulfonyl chlorides. Nevertheless, the reaction of model compound 2 aa with sulfonyl chlorides 10 a,b in pyridine at room temperature gave the desired coupling products in good yields (Table 3).
Table 3.
N‐Sulfonylation of tertiary =NH sulfonimidamide 2 aa with sulfonyl chlorides 10 a,b.
| |
|---|---|
| Sulfonyl chloride (R5SO2Cl) | Isolated yield [%] |
| 10 a: methanesulfonyl chloride | 2 ea: 62 |
| 10 b: p‐toluenesulfonyl chloride | 2 eb: 58 |
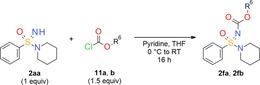
The carbamate group is a key structural motif in many approved drugs.38 Sulfoximines and sulfonimidamides also offer the opportunity to form carbamate‐type products by employing the =NH position. The first N‐(sulfoxylidene)carbamate was described in 197025a and products of this type are usually synthesized by the reaction of =NH sulfoximines with chloroformates.39 Carbamates based on sulfonimidamides were already described in 196340 but, to the best of our knowledge, there is only one literature example of a direct coupling of an =NH sulfonimidamide with a chloroformate.39b To gain further insight into this straightforward method, =NH sulfonimidamide model compound 2 aa was reacted with chloroformates 11 a,b in pyridine to give the coupling products in good yields (Table 4).
Table 4.
N‐Alkoxycarbonylation of tertiary =NH sulfonimidamide 2 aa with chloroformates 11 a,b (carbamate formation).
| |
|---|---|
| Chloroformate [R6OC(O)Cl] | Isolated yield [%] |
| 11 a: phenyl chloroformate | 2 fa: 60 |
| 11 b: ethyl chloroformate | 2 fb: 69 |
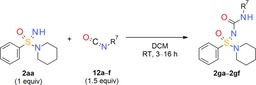
Like the carbamate group, the urea moiety is a very important pharmacophore in the life sciences.41 The formation of N‐(sulfoxylidene)urea derivatives via reaction of the =NH group of sulfoximines with isocyanates was described as early as 1965.42 We have also successfully applied this reaction in a variety of lead optimization programs.43 In comparison, very few sulfonimidamide‐based ureas have been described as yet.44 As far as we are aware, none of these products was obtained by the direct reaction of an =NH sulfonimidamide with an isocyanate. However, the reaction of =NH sulfonimidamide model compound 2 aa with isocyanates 12 a–f in dichloromethane at room temperature gave the desired coupling products in good yields (Table 5).
Table 5.
N‐Aminocarbonylation of tertiary =NH sulfonimidamide 2 aa with isocyanates 12 a–f (urea formation).
| ||
|---|---|---|
| Isocyanate (R7N=C=O) | Isolated yield [%] | |
| 12 a: n‐propyl isocyanate | 2 ga: 65 | |
| 12 b: phenyl isocyanate | 2 gb: 79 | |
| 12 c: 4‐nitrophenyl isocyanate | 2 gc: 82 | |
| 12 d: 2,5‐dimethoxyphenyl isocyanate | 2 gd: 75 | |
| 12 e: 4‐methoxyphenyl isocyanate | 2 ge: 82 | |
| 12 f: |
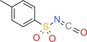
|
2 gf: 49 |
In a recent, preliminary assessment of the medicinal chemistry properties of sulfonimidamides, the behavior of =NH sulfonimidamide 2 aa was compared to the matched sulfonamide analogue 1 aa in selected in vitro assays.12, 45 The hydrolytic stability of both compounds at different pH values was investigated, along with the metabolic stability in liver microsomes (human, rat and mouse) and also rat hepatocytes in vitro. Furthermore, the Caco2 permeability and logD values were determined. In vitro, model compound 2 aa did not reveal any intrinsic flaw of the sulfonimidamide group with respect to its application in the life sciences.
To gain further knowledge of the properties of the neglected sulfonimidamide group, selected N‐functionalized sulfonimidamides were evaluated in the same in vitro panel (Table 6 contains selected examples; see the Supporting Information for additional examples). Similar to =NH sulfonimidamide 2 aa, the tested N‐functionalized sulfonimidamides revealed very high hydrolytic stabilities after 24 hours with stirring at pH 1, 7 and 10. The only exception was N‐trifluoromethylated sulfonimidamide 2 cs that revealed signs of hydrolysis at pH 1 and 10.
Table 6.
Comparison of the in vitro properties of sulfonamide 1 aa and the analogous =NH sulfonimidamide 2 aa with a structural variety of N‐functionalized sulfonimidamides.
| Compound | Recovery [%][a] (pH) |
F
max
(h/r/m)‐LMs [%][b] |
F
max
rHep [%][b] |
P
app A‐B [nm s−1][c] |
Efflux ratio[c] |
logD
pH 7.5[d] |
|---|---|---|---|---|---|---|
|
100 (1) 100 (7) 100 (10) |
69 (h) 10 (r) 26 (m) |
4.3 | 393 | 0.64 | 2.6 |
|
100 (1) 100 (7) 100 (10) |
97 (h) 30 (r) 79 (m) |
14 | 378 | 0.59 | 1.9 |
|
100 (1) 100 (7) 100 (10) |
100 (h) 3.4 (r) 8.2 (m) |
3.3 | 164 | 1.5 | 1.9 |
|
100 (1) 100 (7) 100 (10) |
100 (h) 33 (r) 83 (m) |
20 | 363 | 0.67 | 2.0 |
|
100 (1) 100 (7) 100 (10) |
87 (h) 11 (r) 47 (m) |
7.8 | 256 | 0.73 | 2.2 |
|
100 (1) 100 (7) 100 (10) |
84 (h) 18 (r) 47 (m) |
19 | 404 | 0.57 | 2.4 |
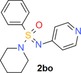
|
100 (1) 100 (7) 100 (10) |
82 (h) 18 (r) 67 (m) |
26 | 400 | 0.61 | 2.6 |
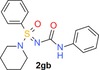
|
100 (1) 100 (7) 100 (10) |
65 (h) 26 (r) 28 (m) |
11 | 404 | 0.57 | 3.0 |
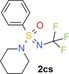
|
56 (1) 100 (7) 91 (10) |
19 (h) 10 (r) 9.3 (m) |
6.8 | 192 | 0.70 | 3.7 |
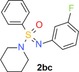
|
100 (1) 100 (7) 100 (10) |
8.3 (h) 0.5 (r) 16 (m) |
2.1 | 252 | 0.45 | 4.3 |
[a] Hydrolytic stability measured as recovery of test compound after 24 hours with stirring at pH 1 (HCl buffer), pH 7 (phosphate‐buffered saline) and pH 10 (sodium borate buffer).48 [b] Predicted hepatic metabolic first pass given as the maximum oral bioavailability F max based on a metabolic stability assay using (i) pooled human liver microsomes (hLMs), (ii) pooled rat liver microsomes (rLMs), (iii) pooled mouse liver microsomes (mLMs) and (iv) freshly harvested rat hepatocytes (rHep).49 [c] P app A‐B (apical to basolateral) and efflux ratio (ER) data were generated in a bidirectionally performed Caco2 permeability assay in a 24‐well format; ER was calculated as P app B‐A/P app A‐B.49 [d] Determined by reversed‐phase HPLC.50
As expected, introduction of a substituent at the =NH position significantly influences the lipophilicity of the resulting compounds. Depending on the nature of the substituent, logD values in the range of 1.9 (2 cl) up to 4.3 (2 bc) were recorded. Introduction of a methyl group at the =NH position, for instance, resulted in an increased logD value from 1.9 (2 aa) to 2.4 (2 cm), whereas the introduction of a trifluoromethyl group raised the logD value to 3.7 (2 cs).
All compounds were tested in a Caco2 screening assay and revealed high permeability coefficients (P app A–B) and no evidence of drug efflux. This behavior can be attributed to the low molecular weight and the rather lipophilic nature of these small and fragment‐like compounds (see logD values at pH 7.5, Table 6).
In vitro pharmacokinetic studies in liver microsomes of human, rat and mouse origin, and rat hepatocytes, with selected N‐functionalized sulfonimidamides revealed a clear species dependence of the predicted metabolic stabilities, given as the maximum oral bioavailability F max 46 (Table 6 and Supporting Information). From the studies with rat hepatocytes, low metabolic stabilities (F max<30 %) were observed for all tested sulfonimidamides as well as sulfonamide 1 aa. Similar instabilities were also observed with rat liver microsomes, which is well in line with the unrestricted membrane permeability observed in Caco2 cells and hints at a major involvement of Phase I metabolism for these compounds in rats. Based on liver microsomes from humans, however, the metabolic stabilities cover the whole range, from high (F max>70 %; e.g., 2 cl, 2 ea) to low (F max<30 %; e.g., 2 bc). This range of metabolic stabilities in human liver microsomes correlates nicely with the corresponding logD values of the test compounds (see the Supporting Information), which is in line with the general trend for many chemical series in the life sciences.47 However, it is noteworthy that the main sites of metabolism and the involved metabolic enzymes were not determined in these in vitro studies.
Conclusions
Although overlooked in the life sciences for a long time, the sulfonimidamide functional group has recently been the subject of a growing interest as a versatile pharmacophore. Based on the premise that the =NH position of tertiary sulfonimidamides should show a similar reactivity as the =NH position of sulfoximines, we have successfully applied various reported reaction conditions for the N‐functionalization of sulfoximines to the tertiary =NH sulfonimidamide model compound 2 aa. Using this methodology, we have synthesized an unprecedented set of structurally diverse sulfonimidamides (47 compounds). The described N‐functionalization reactions include arylation, alkylation, trifluoromethylation, cyanation, sulfonylation, alkoxycarbonylation (carbamate formation) and aminocarbonylation (urea formation). Generally, only isolated examples of these transformations have been reported previously, and we have utilized many of the described reactions for the first time for the N‐functionalization of =NH sulfonimidamides. In vitro studies of selected, structurally diverse N‐functionalized sulfonimidamides from our set of compounds have not revealed any intrinsic flaw of the sulfonimidamide group with respect to its application as a versatile pharmacophore in drug discovery. The combination of our recently reported one‐pot synthesis of tertiary =NH sulfonimidamides by NH transfer to simple sulfinamides and the subsequent N‐functionalization of the newly formed =NH position employing the now‐outlined set of reactions offers rapid access to highly complex and structurally diverse molecules, addressing novel chemical space with properties suitable for application in the life sciences.
Experimental Section
N‐Arylation of sulfonimidamide 2 aa: General procedure A
In a dry MW vial flushed with Ar, sulfonimidamide 2 aa (100 mg, 0.45 mmol, 1.1 equiv) and an aryl bromide (0.40 mmol, 1.0 equiv) were dissolved in toluene (6.8 mL). The mixture was then degassed for 10 min and Pd(OAc)2 (4.5 mg, 0.02 mmol, 5 mol %), Xantphos (23 mg, 0.04 mmol, 10 mol %) and Cs2CO3 (200 mg, 0.60 mmol, 1.5 equiv) were added at RT. The reaction mixture was heated to 100 °C and stirred overnight. Once the starting material had been consumed (monitored by TLC), the mixture was cooled, diluted with methyl tert‐butyl ether, filtered through a pad of Celite under reduced pressure and washed with methyl tert‐butyl ether. Solvent was removed under reduced pressure and the crude product was purified by flash column chromatography.
1. ‐(N,S‐Diphenylsulfonimidoyl)piperidine (2 ba)
Prepared according to general procedure A, from bromobenzene; crude purified by flash column chromatography (KP‐Sil, 0–30 % EtOAc in PE) to give 2 ba as a white solid (104 mg, 86 %): m.p. 118–120 °C; 1H NMR (400 MHz, CDCl3): δ=8.01–7.96 (m, 2 H), 7.62–7.52 (m, 3 H), 7.27 (d, J=6.1 Hz, 2 H), 7.25 (d, J=2.4 Hz, 2 H), 6.98 (tt, J=5.70, 2.91 Hz, 1 H), 3.10–2.99 (m, 4 H), 1.58–1.49 (m, 4 H), 1.37 ppm (quin, J=6.08 Hz, 2 H); 13C NMR (101 MHz, CDCl3): δ=143.9, 136.6, 132.3, 129.9, 128.9, 127.9, 123.9, 121.8, 47.6, 25.4, 23.7 ppm; IR (KBr): =3074, 2939, 2849, 1585, 1485, 1306, 1219, 931, 781, 694 cm−1; HRMS (ESI‐TOF) m/z [M+H]+ calcd for C17H21N2OS: 301.1375, found: 301.1378.
N‐Alkylation of sulfonimidamide 2 aa: General procedure B28
In a dry MW vial flushed with Ar, sulfonimidamide 2 aa (100 mg, 0.45 mmol, 1.0 equiv) and KOH (50 mg, 0.89 mmol, 2.0 equiv) were stirred in DMSO (0.7 mL) at RT for 15 min. Then, an alkyl bromide/iodide (0.67 mmol, 1.5 equiv) was added and the mixture was stirred at RT for 4–16 h. Once the starting material had been consumed (monitored by TLC), H2O was added and the aqueous layer was extracted with DCM (3×10 mL). The combined organic phases were filtered through water‐repellent filter paper. Solvent was removed under reduced pressure and the crude product was purified by flash column chromatography or preparative HPLC.
2. ‐(N‐Ethyl‐S‐phenylsulfonimidoyl)piperidine (2 ca)
Prepared according to general procedure B, from bromoethane; crude purified by flash column chromatography (KP‐Sil, 0–50 % EtOAc in hexane) to give 2 ca as a colorless oil (111 mg, 99 %): 1H NMR (400 MHz, CDCl3): δ=7.66–7.63 (m, 2 H), 7.33–7.24 (m, 3 H), 3.10 (dq, J=12.3, 7.2 Hz, 1 H), 2.93 (dq, J=12.3, 7.2 Hz, 1 H), 2.71 (qt, J=11.7, 5.4 Hz, 4 H), 1.39 (quin, J=5.6 Hz, 4 H), 1.17 (tt, J=8.3, 4.7 Hz, 2 H), 1.06 ppm (t, J=7.2 Hz, 3 H); 13C NMR (101 MHz, CDCl3): δ=136.1, 131.8, 128.5, 127.7, 47.7, 36.8, 25.5, 23.7, 18.3 ppm; IR (KBr): =3063, 2934, 2853, 1254, 1153, 914 cm−1; HRMS (ESI‐TOF) m/z [M+H]+ calcd for C13H21N2OS: 253.1375, found: 253.1379.
N‐Trifluoromethylation of sulfonimidamide 2 aa[30] 1‐[S‐Phenyl‐N‐(trifluoromethyl)sulfonimidoyl]piperidine (2 cs)
In a MW vial charged with sulfonimidamide 2 aa (100 mg, 0.45 mmol, 1.0 equiv), TMSCF3 (4.44 mL, 2.22 mmol, 5.0 equiv; 0.5 m solution in THF), Ag2CO3 (24 mg, 0.09 mmol, 0.2 equiv), 1,10‐phenanthroline (32 mg, 0.18 mmol, 0.4 equiv) and 1,4‐dioxane (8.9 mL) were added. Then, a balloon charged with O2 was attached to the MW vial and the solution was degassed for 10 min. The mixture was stirred and heated in an oil bath at 60 °C for 16 h. Then, the solution was cooled, solvent removed in vacuo and the crude product purified by flash column chromatography (KP‐Sil, 0–50 % EtOAc in PE) to give 2 cs as a yellow oil (57 mg, 44 %): 1H NMR (400 MHz, CDCl3): δ=7.89–7.86 (m, 2 H), 7.65–7.60 (m, 1 H), 7.57–7.53 (m, 2 H), 3.11–3.00 (m, 4 H), 1.68–1.62 (m, 4 H), 1.48–1.42 ppm (m, 2 H); 13C NMR (101 MHz, CDCl3): δ=135.6, 133.3, 129.2, 127.5, 121.6 (q, J=255 Hz), 47.5, 25.3, 23.7 ppm; IR (neat): =3069, 2944, 2856, 1256, 1077, 921 cm−1; HRMS (ESI‐TOF) m/z [M+H]+ calcd for C12H16N2OF3S: 293.0930, found: 293.0935.
N‐Cyanation of sulfonimidamide 2 aa[35] N‐[Oxo(phenyl)(piperidin‐1‐yl)‐λ6‐sulfanylidene]cyanamide (2 da)
In a MW vial charged with sulfonimidamide 2 aa (100 mg, 0.45 mmol, 1.0 equiv), AIBN (108 mg, 0.66 mmol, 1.5 equiv), CuI (16 mg, 0.09 mmol, 0.2 equiv), K2CO3 (122 mg, 0.88 mmol, 2.0 equiv) and MeCN (6.7 mL) were added. Then, a balloon charged with O2 was attached to the MW vial and the solution was degassed for 10 min. The mixture was stirred and heated in an oil bath at 75 °C for 16 h. Then, the solution was cooled, filtered and the solids washed with MeCN. The liquid phase was collected, solvent removed in vacuo and the crude product purified by flash column chromatography (KP‐Sil, 0–100 % EtOAc in PE) to give 2 da as a brown oil (47 mg, 43 %): 1H NMR (400 MHz, CDCl3): δ=7.87–7.84 (m, 2 H), 7.72–7.67 (m, 1 H), 7.62–7.57 (m, 2 H), 3.14 (td, J=5.6, 2.2 Hz, 4 H), 1.77–1.63 (m, 4 H), 1.54–1.46 ppm (m, 2 H); 13C NMR (101 MHz, CDCl3): δ=134.4, 133.6, 129.7, 127.8, 111.2, 47.4, 25.1, 23.4 ppm; IR (neat): =3063, 2924, 2853, 2151, 1268, 1200, 924 cm−1; HRMS (ESI‐TOF) m/z [M+H]+ calcd for C12H16N3OS: 250.1009, found: 250.1011.
N‐Sulfonylation of sulfonimidamide 2 aa: General procedure C
Sulfonimidamide 2 aa (100 mg, 0.45 mmol, 1.0 equiv) was dissolved in pyridine (3.0 mL) under Ar atmosphere; then, the appropriate sulfonyl chloride (0.71 mmol, 1.6 equiv) was added. The reaction mixture was stirred at RT overnight; then, the reaction was quenched with aqueous NaHCO3 and diluted with EtOAc (3 mL). The mixture was transferred to a separating funnel, the aqueous layer was extracted with EtOAc (3×10 mL), and the combined organic phases were washed with brine (3×10 mL), then filtered through water‐repellent filter paper. Solvent was removed in vacuo and the crude product purified (when needed).
N‐[Oxo(phenyl)(piperidin‐1‐yl)‐λ6‐sulfanylidene]methanesulfonamide (2 ea)
Prepared according to general procedure C, from methanesulfonyl chloride; crude purified by preparative HPLC to give 2 ea as a white solid (83 mg, 62 %): m.p. 93–95 °C; 1H NMR (400 MHz, CDCl3): δ=7.92–7.90 (m, 2 H), 7.67–7.63 (m, 1 H), 7.59–7.54 (m, 2 H), 3.23 (ddd, J=11.4, 7.1, 4.0 Hz, 2 H), 3.18 (s, 3 H), 3.10 (ddd, J=11.5, 6.9, 3.9 Hz, 2 H), 1.74–1.60 (m, 4 H), 1.49 ppm (quin, J=5.9 Hz, 2 H); 13C NMR (101 MHz, CDCl3): δ=136.1, 133.7, 129.4, 127.6, 47.2, 45.0, 25.1, 23.5 ppm; IR (KBr): =2939, 2856, 1311, 1246, 1099, 918 cm−1; HRMS (ESI‐TOF) m/z [M+H]+ calcd for C12H19N2O3S2: 303.0837, found: 303.0838.
N‐Alkoxycarbonylation of sulfonimidamide 2 aa (carbamate formation): General procedure D51
To a solution of sulfonimidamide 2 aa (100 mg, 0.45 mmol, 1.0 equiv) and pyridine (54 μL, 0.67 mmol, 1.5 equiv) in THF (0.9 mL) under Ar atmosphere at 0 °C, the corresponding chloroformate (0.67 mmol, 1.5 equiv) was added. As soon as the chloroformate was in solution, a white precipitate formed. The temperature was allowed to rise to RT and the mixture was stirred overnight. The reaction was quenched with H2O and diluted with Et2O (3 mL). The mixture was transferred to a separating funnel, the aqueous layer was extracted with Et2O (3×10 mL), and the combined organic phases were washed with 1 m HCl (3×10 mL) and brine (3×10 mL), then filtered through water‐repellent filter paper. Solvent was removed in vacuo and the crude product purified (when needed).
Phenyl [oxo(phenyl)(piperidin‐1‐yl)‐λ6‐sulfanylidene]carbamate (2 fa)
Prepared according to general procedure D, from phenyl chloroformate; crude purified by flash column chromatography (KP‐Sil, 0–50 % EtOAc in PE) to give 2 fa as a white solid (90 mg, 60 %): m.p. 114–117 °C; 1H NMR (400 MHz, CDCl3): δ=7.94–7.91 (m, 2 H), 7.65–7.60 (m, 1 H), 7.58–7.53 (m, 2 H), 7.34–7.29 (m, 2 H), 7.17–7.11 (m, 3 H), 3.25–3.15 (m, 4 H), 1.66 (quin, J=5.6 Hz, 4 H), 1.53–1.47 ppm (m, 2 H); 13C NMR (101 MHz, CDCl3): δ=156.1, 151.8, 136.2, 133.4, 129.3, 129.2, 127.9, 125.3, 121.8, 46.7, 25.3, 23.7 ppm; IR (neat): =3068, 2945, 2856, 1687, 1274, 1248, 1194, 877 cm−1; HRMS (ESI‐TOF) m/z [M+H]+ calcd for C18H21N2O3S: 345.1267, found: 345.1275.
N‐Aminocarbonylation of sulfonimidamide 2 aa (urea formation): General procedure E52
To a solution of sulfonimidamide 2 aa (100 mg, 0.45 mmol, 1.0 equiv) in anhydrous DCM (0.9 mL), the corresponding isocyanate (0.66 mmol, 1.5 equiv) was added dropwise at RT under Ar atmosphere. The reaction mixture was stirred until a precipitate formed (3–16 h) and starting material had been consumed (TLC analysis). Et2O was added, and the precipitate was collected by filtration under reduced pressure and washed with Et2O. The solid was purified by flash column chromatography (when needed).
3. ‐[Oxo(phenyl)(piperidin‐1‐yl)‐λ6‐sulfanylidene]‐3‐propylurea (2 ga)
Prepared according to general procedure E, from n‐propyl isocyanate; crude purified by flash column chromatography (KP‐Sil, 0–100 % EtOAc in PE) to give 2 ga as a colorless oil (89 mg, 65 %): 1H NMR (400 MHz, CDCl3): δ=7.87 (dt, J=8.4, 1.3 Hz, 2 H), 7.59–7.55 (m, 1 H), 7.51 (ddd, J=8.3, 6.5, 1.2 Hz, 2 H), 5.16 (brs, 1 H), 3.17–3.06 (m, 6 H), 1.63 (quin, J=5.4 Hz, 4 H), 1.55–1.42 (m, 4 H), 0.92–0.88 ppm (m, 3 H); 13C NMR (101 MHz, CDCl3): δ=158.4, 137.1, 132.7, 129.0, 127.7, 46.9, 42.4, 25.4, 23.7, 23.3, 11.5 ppm; IR (neat): =3252, 2931, 2855, 1617, 1520, 1244, 931 cm−1; HRMS (ESI‐TOF) m/z [M+H]+ calcd for C15H24N3O2S: 310.1584, found: 310.1602.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank M. Bergmann and K. Greenfield for valuable support with the manuscript.
F. Izzo, M. Schäfer, P. Lienau, U. Ganzer, R. Stockman, U. Lücking, Chem. Eur. J. 2018, 24, 9295.
Contributor Information
Prof. Robert Stockman, Email: Robert.Stockman@nottingham.ac.uk.
Ulrich Lücking, Email: ulrich.luecking@bayer.com.
References
- 1.
- 1a. Dai H.-X., Stepan A. F., Plummer M. S., Zhang Y.-H., Yu J.-Q., J. Am. Chem. Soc. 2011, 133, 7222; For recent reviews, see for example: [DOI] [PubMed] [Google Scholar]
- 1b. Scott K. A., Njardarson J. T., Top. Curr. Chem. 2018, 376, 5; [DOI] [PubMed] [Google Scholar]
- 1c. Ajeet, Mishra A. K., Kumar A., Am. J. Pharmacol. Sci. 2015, 3, 18; [Google Scholar]
- 1d. Kołaczek A., Fusiarz I., Ławecka J., Branowska D., Chemik 2014, 68, 620; [Google Scholar]
- 1e. Shoaib Ahmad Shah S., Rivera G., Ashfaq M., Mini-Rev. Med. Chem. 2013, 13, 70. [PubMed] [Google Scholar]
- 2. Levchenko E. S., Derkach N. Y., Kirsanov A. V., Zh. Obshch. Khim. 1962, 32, 1208. [Google Scholar]
- 3. Chinthakindi P. K., Naicker T., Thota N., Govender T., Kruger H. G., Arvidsson P. I., Angew. Chem. Int. Ed. 2017, 56, 4100; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4160. [Google Scholar]
- 4.
- 4a. Chen Y., Gibson J., RSC Adv. 2015, 5, 4171; [Google Scholar]
- 4b. Nandi G. C., Arvidsson P., Adv. Synth. Catal. 2018, DOI: 10.1002/adsc.201800273. [DOI] [Google Scholar]
- 5. Steinkamp A.-D., Schmitt L., Chen X., Fietkau K., Heise R., Baron J. M., Bolm C., Skin Pharmacol. Physiol. 2016, 29, 281. [DOI] [PubMed] [Google Scholar]
- 6. Davies T. Q., Hall A., Willis M. C., Angew. Chem. Int. Ed. 2017, 56, 14937; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15133. [Google Scholar]
- 7. Chowdhury S., Dehnhardt C. M., Focken T., Grimwood M. E., Hemeon I. W., McKerrall S., Sutherlin D. (Genentech, Inc.), WO 2017172802, 2017.
- 8. Wen J., Cheng H., Dong S., Bolm C., Chem. Eur. J. 2016, 22, 5547. [DOI] [PubMed] [Google Scholar]
- 9. Richards-Taylor C. S., Martínez-Lamenca C., Leenaerts J. E., Trabanco A. A., Oehlrich D., J. Org. Chem. 2017, 82, 9898. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Johnson C. R., Aldrichimica Acta 1985, 18, 3; [Google Scholar]
- 10b. Reggelin M., Zur C., Synthesis 2000, 1; [Google Scholar]
- 10c. Lücking U., Angew. Chem. Int. Ed. 2013, 52, 9399; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9570; [Google Scholar]
- 10d. Frings M., Bolm C., Blum A., Gnamm C., Eur. J. Med. Chem. 2017, 126, 225; [DOI] [PubMed] [Google Scholar]
- 10e. Sirvent J. A., Lücking U., ChemMedChem 2017, 12, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Lücking U., Jautelat R., Krüger M., Brumby T., Lienau P., Schäfer M., Briem H., Schulze J., Hillisch A., Reichel A., Wengner A. M., Siemeister G., ChemMedChem 2013, 8, 1067; [DOI] [PubMed] [Google Scholar]
- 11b. Lücking U., Scholz A., Lienau P., Siemeister G., Kosemund D., Bohlmann R., Briem H., Terebesi I., Meyer K., Prelle K., Denner K., Bömer U., Schäfer M., Eis K., Valencia R., Ince S., von Nussbaum F., Mumberg D., Ziegelbauer K., Klebl B., Choidas A., Nussbaumer P., Baumann M., Schultz-Fademrecht C., Rühter G., Eickhoff J., Brands M., ChemMedChem 2017, 12, 1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Izzo F., Schäfer M., Stockman R., Lücking U., Chem. Eur. J. 2017, 23, 15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Zenzola M., Doran R., Degennaro L., Luisi R., Bull J., Angew. Chem. Int. Ed. 2016, 55, 7203; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7319; [Google Scholar]
- 13b.For recent developments concerning the use of PhI(OAc)2 in the synthesis of sulfoximines, see also: Tota A., Zenzola M., Chawner S. J., John-Campbell S. St., Carlucci C., Romanazzi G., Degennaro L., Bull J. A., Luisi R., Chem. Commun. 2017, 53, 348; [DOI] [PubMed] [Google Scholar]
- 13c. Lohier J.-F., Glachet T., Marzag H., Gaumont A.-C., Reboul V., Chem. Commun. 2017, 53, 2064; [DOI] [PubMed] [Google Scholar]
- 13d. Xie Y., Zhou B., Zhou S., Zhou S., Wei W., Liu J., Zhan Y., Cheng D., Chen M., Li Y., Wang B., Xue X.-S., Li Z., ChemistrySelect 2017, 2, 1620; [Google Scholar]
- 13e. Bull J. A., Degennaro L., Luisi R., Synlett 2017, 28, 2525. [Google Scholar]
- 14.
- 14a. Lücking U., Krüger M., Jautelat R., Siemeister G. (Schering Aktiengesellschaft), WO 2005037800, 2005;
- 14b. Lücking U., Siemeister G., Jautelat R. (Schering Aktiengesellschaft), WO 2006099974, 2006;
- 14c. Lücking U. (Schering Aktiengesellschaft), EP 1710246, 2006;
- 14d. Lücking U., Kettschau G., Briem H., Schwede W., Schäfer M., Thierauch K.-H., Husemann M. (Schering Aktiengesellschaft), WO 2006108695, 2006;
- 14e. Luecking U., Nguyen D., von Bonin A., von Ahsen O., Krueger M., Briem H., Kettschau G., Prien O., Mengel A., Krolikiewicz K., Boemer U., Bothe U., Hartung I. (Schering Aktiengesellschaft), WO 2007071455, 2007;
- 14f. Lücking U., Siemeister G., Bader B. (Schering Aktiengesellschaft), WO 2007079982, 2007;
- 14g. Luecking U., Siemeister G., Jautelat R. (Bayer Schering Pharma Aktiengesellschaft), WO 2008025556, 2008;
- 14h. Prien O., Eis K., Lücking U., Guenther J., Zopf D. (Bayer Schering Pharma Aktiengesellschaft), DE 102007024470, 2008;
- 14i. Hartung I., Bothe U., Kettschau G., Luecking U., Mengel A., Krueger M., Thierauch K.-H., Lienau P., Boemer U. (Bayer Schering Pharma Aktiengesellschaft), WO 2008155140, 2008;
- 14j. Nguyen D., Bonin A. Von, Haerter M., Schirok H., Mengel A., Ahsen O. Von (Bayer Schering Pharma Aktiengesellschaft), WO 2009089851, 2009;
- 14k. Härter M., Beck H., Ellinghaus P., Berhoerster K., Greschat S., Thierauch K.-H., Süssmeier F. (Bayer Schering Pharma Aktiengesellschaft), WO 2010054763, 2010;
- 14l. von Nussbaum F., Karthaus D., Anlauf S., Delbeck M., Li V. M.-J., Meibom D., Lustig K., Schneider D. (Bayer Schering Pharma Aktiengesellschaft), WO 2010115548, 2010;
- 14m. Schwede W., Klar U., Möller C., Rotgeri A., Bone W. (Bayer Schering Pharma Aktiengesellschaft), WO 2011009531, 2011;
- 14n. Lücking U., Cleve A., Haendler B., Faus H., Köhr S., Irlbacher H. (Bayer Schering Pharma Aktiengesellschaft), WO 2011029537, 2011;
- 14o. Lücking U., Bohlmann R., Scholz A., Siemeister G., Gnoth M. J., Boemer U., Kosemund D., Lienau P., Ruether G., Schulz-Fademrecht C. (Bayer Intellectual Property GmbH), WO 2012160034, 2012.
- 15. Bolm C., Hildebrand J. P., Tetrahedron Lett. 1998, 39, 5731. [Google Scholar]
- 16.A recent search in Reaxys identified more than 100 documents describing N-arylation reactions of =NH sulfoximines.
- 17.
- 17a. Wortmann L., Lücking U., Lefranc J., Briem H., Koppitz M., Eis K., von Nussbaum F., Bader B., Wengner A. M., Siemeister G., Bone W., Lienau P., Grudzinska-Goebel J., Moosmayer D., Eberspächer U., Schick H. (Bayer Schering Pharma Aktiengesellschaft), WO 2016020320, 2016;
- 17b. Lücking U., Koppitz M., Lefranc J., Wortmann L., Wengner A. M., Siemeister G., Bömer U., Bader B., Lienau P., Schick H. (Bayer Schering Pharma Aktiengesellschaft), WO 2017121684, 2017.
- 18.Unpublished results.
- 19.
- 19a. Funes Maldonado M., Sehgelmeble F., Bjarnemark F., Svensson M., Ahman J., Arvidsson P. I., Tetrahedron 2012, 68, 7456; [Google Scholar]
- 19b.For an application of this method, see: Langkopf E., Blum A. (Boehringer Ingelheim International GmbH), WO 2016023789, 2016.
- 20.
- 20a. Battula S. R. K., Subbareddy G. V., Chakravarthy I. E., Tetrahedron Lett. 2014, 55, 517; [Google Scholar]
- 20b. Nandi G. C., Kota S. R., Govender T., Kruger H. G., Arvidsson P. I., Tetrahedron 2014, 70, 5428. [Google Scholar]
- 21. Siemeister G., Luecking U., Wengner A. M., Lienau P., Steinke W., Schatz C., Mumberg D., Ziegelbauer K., Mol. Cancer Ther. 2012, 11, 2265. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Scholz A., Oellerich T., Hussain A., Lindner S., Luecking U., Walter A. O., Ellinghaus P., Valencia R., von Nussbaum F., Mumberg D., Brands M., Ince S., Serve H., Ziegelbauer K., Cancer Res. 2016, 76, 3022; [Google Scholar]
- 22b. Scholz A., Luecking U., Siemeister G., Lienau P., Boemer U., Ellinghaus P., Walter A. O., Valencia R., Ince S., von Nussbaum F., Mumberg D., Brands M., Ziegelbauer K., Cancer Res. 2015, 75, DDT02-02; [Google Scholar]
- 22c. Luecking U., Scholz A., Lienau P., Siemeister G., Kosemund D., Bohlmann R., Eis K., Gnoth M., Terebesi I., Meyer K., Prelle K., Valencia R., Ince S., von Nussbaum F., Mumberg D., Ziegelbauer K., Klebl B., Choidas A., Nussbaumer P., Baumann M., Schultz-Fademrecht C., Ruehter G., Eickhoff J., Brands M., Cancer Res. 2015, 75, 2828. [Google Scholar]
- 23. Foote K. M., Lau A., Nissink J. W. M., Future Med. Chem. 2015, 7, 873. [DOI] [PubMed] [Google Scholar]
- 24.U. T. Luecking, A. Scholz, D. Kosemund, R. Bohlmann, H. Briem, P. Lienau, G. Siemeister, I. Terebesi, K. Meyer, K. Prelle, R. Valencia, S. Ince, F. von Nussbaum, D. Mumberg, K. Ziegelbauer, M. Brands, AACR Annual Meeting, Washington, DC, April, 2017, MS.CH01.01 #984.
- 25.
- 25a. Satzinger G., Stoss P., Arzneim.-Forsch. 1970, 20, 1214; [PubMed] [Google Scholar]
- 25b. Satzinger G., Drug News Perspect. 2001, 14, 197. [DOI] [PubMed] [Google Scholar]
- 26. Johnson C. R., Lavergne O. M., J. Org. Chem. 1993, 58, 1922. [Google Scholar]
- 27.
- 27a. Williams T. R., Booms R. E., Cram D. J., J. Am. Chem. Soc. 1971, 93, 7338; [Google Scholar]
- 27b. Schmidbaur H., Kammel G., Chem. Ber. 1971, 104, 3234; [Google Scholar]
- 27c. Johnson C. R., Schroeck C. W., Shanklin J. R., J. Am. Chem. Soc. 1973, 95, 7424. [Google Scholar]
- 28. Hendriks C. M. M., Bohmann R. A., Bohlem M., Bolm C., Adv. Synth. Catal. 2014, 356, 1847. [Google Scholar]
- 29.For more recent examples, see for example:
- 29a. Khan T. A., Scott J. D., Cumming J. N. (Merck Sharp & Dohme Corp.), WO 2014150340, 2014;
- 29b.Reference [6];
- 29c.For the indirect preparation of N-alkylated sulfonimidamides, see: Nandi G. C., Kota S. R., Naicker T., Govender T., Kruger H. G., Arvidsson P. I., Eur. J. Org. Chem. 2015, 2861; [Google Scholar]
- 29d. Nandi G. C., Kota S. R., Wakchaure P. B., Chinthakindi P. K., Govender T., Kruger H. G., Naicker T., Arvidsson P. I., RSC Adv. 2015, 5, 62084. [Google Scholar]
- 30. Teng F., Cheng J., Bolm C., Org. Lett. 2015, 17, 3166. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Bassetto M., Ferla S., Pertusati F., Future Med. Chem. 2015, 7, 527; [DOI] [PubMed] [Google Scholar]
- 31b.Reference [30], and references therein.
- 32.
- 32a. Sparks T. C., Watson G. B., Loso M. R., Geng C., Babcock J. M., Thomas J. D., Pestic. Biochem. Physiol. 2013, 107, 1; [DOI] [PubMed] [Google Scholar]
- 32b. Arndt K. E., Bland D. C., Irvine N. M., Powers S. L., Martin T. P., McConnell J. R., Podhorez D. E., Renga J. M., Ross R., Roth G. A., Scherzer B. D., Toyzan T. W., Org. Process Res. Dev. 2015, 19, 454. [Google Scholar]
- 33.See, for example:
- 33a. Sauer D. T., Shreeve J. M., Inorg. Chem. 1972, 11, 238; [Google Scholar]
- 33b. García Mancheño O., Bolm C., Org. Lett. 2007, 9, 2951; [DOI] [PubMed] [Google Scholar]
- 33c. Huang J. X., Zhu Y., Rogers R. B., Loso M. R., Hill R. L., Thomas J. D., Meade T., Gifford J. M., DeMark J. J., Benjamin B. M. (Dow Agrosciences LLC), US 20080108665, 2008;
- 33d. Lücking U., Cleve A., Haendler B., Faus H., Köhr S., Irlbacher H. (Bayer Schering Pharma Aktiengesellschaft), WO 2011029537, 2011.
- 34. Gnamm C., Jeanguenat A., Dutton A. C., Grimm C., Kloer D. P., Crossthwaite A. J., Bioorg. Med. Chem. Lett. 2012, 22, 3800. [DOI] [PubMed] [Google Scholar]
- 35. Teng F., Yu J.-T., Zhou Z., Chu H., Cheng J., J. Org. Chem. 2015, 80, 2822. [DOI] [PubMed] [Google Scholar]
- 36.See, for example:
- 36a. Cram D. J., Day J., Rayner D. R., Von Schriltz D. M., Duchamp D. J., Garwood D. C., J. Am. Chem. Soc. 1970, 92, 7369; [Google Scholar]
- 36b. Craig D., Geach N. J., Pearson C. J., Slawin A. M. Z., White A. J. P., Williams D. J., Tetrahedron 1995, 51, 6071; [Google Scholar]
- 36c. Craig D., Grellepois F., White A. J. P., J. Org. Chem. 2005, 70, 6827; [DOI] [PubMed] [Google Scholar]
- 36d. Subasinghe N. L., Ballentine S., Travins J. M., Khalil E. M., Ali F., Leonard K. A., Gushue J. M., Winter M. P., Hufnagel H., Cummings M. D. (Janssen Pharmaceutica N. V.), WO 2006101860, 2006;
- 36e. Lucking U., Siemeister G., Bader B. (Bayer Schering Pharma Aktiengesellschaft), US 20070191393, 2007.
- 37.See for example:
- 37a. Kresze G., Seyfried C., Trede A., Justus Liebigs Ann. Chem. 1968, 715, 223; [Google Scholar]
- 37b. Johnson C. R., Jonsson E. U., Bacon C. C., J. Org. Chem. 1979, 44, 2055; [Google Scholar]
- 37c. Levchenko E. S., Kozlov E. S., Zh. Obshch. Khim. 1963, 33, 3551; [Google Scholar]
- 37d. Buendia J., Grelier G., Darses B., Jarvis A. G., Taran F., Dauban P., Angew. Chem. Int. Ed. 2016, 55, 7530; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7656. [Google Scholar]
- 38. Ghosh A. K., Brindisi M., J. Med. Chem. 2015, 58, 2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.See, for example:
- 39a. Williams T. R., Cram D. J., J. Org. Chem. 1973, 38, 20; [Google Scholar]
- 39b. Jones M. R., Cram D. J., J. Am. Chem. Soc. 1974, 96, 2183; [Google Scholar]
- 39c.Reference [14a].
- 40. Levchenko E. S., Kozlov E. S., Kirsanov A. V., Zh. Obshch. Khim. 1963, 33, 565. [Google Scholar]
- 41.
- 41a. Garuti L., Roberti M., Bottegoni G., Ferraro M., Curr. Med. Chem. 2016, 23, 1528; [DOI] [PubMed] [Google Scholar]
- 41b. Sikka P., Sahu J. K., Mishra A. K., Hashim S. R., Med. Chem. 2015, 5, 479. [Google Scholar]
- 42. Wehr R., J. Chem. Soc. 1965, 3004. [Google Scholar]
- 43.See, for example:
- 43a.Refs. [14e] and [14o];
- 43b. Luecking U., Siemeister G., Jautelat R. (Bayer Schering Pharma Aktiengesellschaft), US 20080167330, 2008;
- 43c. Lücking U., Wasnaire P., Scholz A., Lienau P., Siemeister G., Stegmann C., Bömer U., Zheng K., Gao P., Chen G., Xi J. (Bayer Schering Pharma Aktiengesellschaft), WO 2015155197, 2015.
- 44.
- 44a. Toth J. E., Ray J., Deeter J., J. Org. Chem. 1993, 58, 3469; [Google Scholar]
- 44b. Toth J. E., Grindey G. B., Ehlhardt W. J., Ray J. E., Boder G. B., Bewley J. R., Klingerman K. K., Gates S. B., Rinzel S. M., Schultz R. M., Weir L. C., Worzalla J. F., J. Med. Chem. 1997, 40, 1018; [DOI] [PubMed] [Google Scholar]
- 44c. Uchida K., Kanai T., Homma M., Aratake S., Ishimori T. (Kyowa Hakko Kirin Co., Ltd.), EP 2927214, 2015;
- 44d. Uchida K., Kanai T., Homma M., Aratake S., Ishimori T. (Kyowa Hakko Kirin Co., Ltd.), WO 2014084330, 2014.
- 45.For the first assessment of the in vitro properties of sulfonimidamides and matched sulfonamides, see: Sehgelmeble F., Janson J., Ray C., Rosqvist S., Gustavsson S., Nilsson L. I., Minidis A., Holenz J., Rotticci D., Lundkvist J., Arvidsson P. I., ChemMedChem 2012, 7, 396. [DOI] [PubMed] [Google Scholar]
- 46. Benet L. Z., Zia-Amirhosseini P., Toxicol. Pathol. 1995, 23, 115. [DOI] [PubMed] [Google Scholar]
- 47. Smith D. A. in Computer-Assisted Lead Finding and Optimisation (Eds.: H. Van de Waterbeemd, B. Testa, G. Folkers), Wiley-VCH, Weinheim, 1997, pp. 265–276. [Google Scholar]
- 48.For the assay description, see: Kerns E. H., Di L., Drug-Like Properties: Concepts, Structure Design and Methods, Elsevier Inc./Academic Press, Burlington, 2008, pp. 353–356. [Google Scholar]
- 49.For the assay description, see reference [43c].
- 50.For the assay description, see: Minick D. J., Frenz J. H., Patrick M. A., Brent D. A., J. Med. Chem. 1988, 31, 1923. [DOI] [PubMed] [Google Scholar]
- 51. Virolleaud M.-A., Sridharan V., Mailhol D., Bonne D., Bressy C., Chouraqui G., Commeiras L., Coquerel Y., Rodriguez J., Tetrahedron 2009, 65, 9756. [Google Scholar]
- 52. Frings M., Thomé I., Bolm C., Beilstein J. Org. Chem. 2012, 8, 1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary