

Abstract
Peeling skin syndromes form a large and heterogeneous group of inherited disorders characterized by superficial detachment of the epidermal cornified cell layers, often associated with inflammatory features. Here we report on a consanguineous family featuring noninflammatory peeling of the skin exacerbated by exposure to heat and mechanical stress. Whole exome sequencing revealed a homozygous nonsense mutation in FLG2, encoding filaggrin 2, which cosegregated with the disease phenotype in the family. The mutation was found to result in decreased FLG2 RNA levels as well as almost total absence of filaggrin 2 in the patient epidermis. Filaggrin 2 was found to be expressed throughout the cornified cell layers and to colocalize with corneodesmosin that plays a crucial role in maintaining cell-cell adhesion in this region of the epidermis. The absence of filaggrin 2 in the patient skin was associated with markedly decreased corneodesmosin expression, which may contribute to the peeling phenotype displayed by the patients. Accordingly, using the dispase dissociation assay, we showed that FLG2 downregulation interferes with keratinocyte cell-cell adhesion. Of particular interest, this effect was aggravated by temperature elevation, consistent with the clinical phenotype. Restoration of corneodesmosin levels by ectopic expression rescued cell-cell adhesion. Taken together, the present data suggest that filaggrin 2 is essential for normal cell-cell adhesion in the cornified cell layers.
INTRODUCTION
Peeling skin syndrome (PSS) refers to a complex group of autosomal recessive disorders of cornification featuring superficial detachment of the epidermal cornified cell layers with no mucosal fragility (Kose et al., 2012; Levy and Goldsmith, 1982).
Two major clinical types of PSS have been reported: localized (acral) and generalized PSSs. Generalized PSS is further subdivided into a noninflammatory (type A) form and an inflammatory (type B) form. This latter type also features itching, atopic diathesis, allergic reactions, and failure to thrive (Kose et al., 2012).
These three clinical groups of PSS correspond to five genetic subtypes of the disease. Localized or acral PSS (PSS2; Online Mendelian Inheritance in Man, OMIM. Johns Hopkins University, Baltimore, MD. MIM Number: 609796: http://www.ncbi.nlm.nih.gov/omim/) can be caused by mutations in two genes: TGM5 (Cassidy et al., 2005), encoding transglutaminase 5, which catalyzes the formation of γ-glu-tamyl-ε-lysine isopeptide bonds between epidermal differentiation-associated proteins, and CSTA (Blaydon et al., 2011; Krunic et al., 2013) (PSS4; Online Mendelian Inheritance in Man, OMIM. Johns Hopkins University, Baltimore, MD. MIM Number: 607936: http://www.ncbi.nlm.nih.gov/omim/), encoding cystatin A, a cysteine protease inhibitor. Type B generalized PSS results from mutations in CDSN (PSS1; Online Mendelian Inheritance in Man, OMIM. Johns Hopkins University, Baltimore, MD. MIM Number: 270300: http://www.ncbi.nlm.nih.gov/omim/), encoding corneodesmosin (CDSN) (Oji et al., 2010), a component of the desmosomal plaques in the upper epidermal layers. CDSN is incorporated into desmosomes in the cornified layers (Garrod and Chidgey, 2008) and has been shown to play a critical role in cell-cell adhesion in both humans and animal models (Ishida-Yamamoto and Igawa, 2015; Matsumoto et al., 2008). Additional conditions that also feature inflammatory generalized peeling include Netherton syndrome (Online Mendelian Inheritance in Man, OMIM. Johns Hopkins University, Baltimore, MD. MIM Number: 256500: http://www.ncbi.nlm.nih.gov/omim/), also associated with CDSN deficiency, and severe skin dermatitis, multiple allergies, and metabolic wasting (SAM; Online Mendelian Inheritance in Man, OMIM. Johns Hopkins University, Baltimore, MD. MIM Number: 615508: http://www.ncbi.nlm.nih.gov/omim/) syndrome (Samuelov and Sprecher, 2014). Type A generalized PSS encompasses two major phenotypes: PSS type 5 (Online Mendelian Inheritance in Man, OMIM. Johns Hopkins University, Baltimore, MD. MIM Number: 617115: http://www.ncbi.nlm.nih.gov/omim/), which features late-onset peeling over the hands, feet, and knees as well as palmoplantar keratoderma and is caused by mutations in the SERPINB8 gene, which encodes a serine protease inhibitor (Pigors et al., 2016); and PSS type 3 (Online Mendelian Inheritance in Man, OMIM. Johns Hopkins University, Baltimore, MD. MIM Number: 616265: http://www.ncbi.nlm.nih.gov/omim/), which manifests with superficial and generalized peeling without pruritus or any associated signs (Cabral et al., 2012).
PSS3 was found in one family to be caused by a mutation in CHST8 encoding a carbohydrate sulfotransferase, N-ace-tylgalactosamine-4-O-sulfotransferase 1 (Cabral et al., 2012) (although this mutation may represent a neutral polymorphism; Fiete et al., 2017). Two recent studies identified the same homozygous variant in FLG2 encoding filaggrin 2, which cosegregated with PSS3 (Alfares et al., 2017; Bolling et al., 2018). The functional significance of this variant has not been studied.
Here we show in an additional family with PSS3 that loss of expression of filaggrin 2 underlies PSS3 and destabilizes epidermal cell-cell adhesion.
RESULTS
Clinical features
We studied two siblings born to consanguineous parents of Arab Moslem origin who displayed noninflammatory type A PSS since birth. All other family members lacked cutaneous manifestations although atopic diathesis was seen among first-degree cousins of the affected children. Peeling, mostly involved the limbs, was occasionally seen on the trunk with sparing of the face and palmoplantar skin. It was most prominent during the summer time, after exposure to elevated ambient temperature, and was also triggered by minor trauma to the skin (Figure 1a). Skin lesions resolved leaving residual discolored skin (Figure 1b). Of note, the disease manifestations were much more prominent during childhood (Supplementary Figure S1 online) and improved with age. Skin biopsy revealed subcorneal separation with little or no dermal inflammatory infiltrate (Figure 1c). Electron microscopy demonstrated intraepidermal separation within the lower corneal layers (Figure 1d) and a reduction in the number of keratohyalin granules, most of which were deformed and irregularly shaped (Supplementary Figure S2 online).
Figure 1. Clinical and pathological features.

Individual II-8 displays at age 24: (a) superficial peeling of the skin after minor trauma of the left arm, (b) resolving with crust formation and hyperpigmentation. (c) A skin biopsy obtained from the arm area in individual II-8 demonstrates subcorneal (asterisk) blister formation (hematoxylin and eosin; scale bar = 50 μm). Note lack of inflammatory infiltrate. (d) Intracorneal detachment is demonstrated by electron microscopy (scale bar = 0.5 μm).
MUTATIONAL ANALYSIS
After having excluded by direct sequencing pathogenic mutations in the coding sequences of CDSN, CHST8, and CSTA, which have been associated with various forms of PSS (Cabral et al., 2012; Krunic et al., 2013; Oji et al., 2010), DNA samples extracted from individuals I-3 and II-8 (Figure 2b) were subjected to whole exome sequencing. A hitherto unreported homozygous transversion in FLG2, c.1065T>A (p.Y355*), was identified in affected individual II-8. The mutation was validated using direct sequencing (Figure 2a) and a PCR-restriction fragment length polymorphism assay (Figure 2b). The mutation was found to cosegregate with the disease phenotype in the family: the two patients (individuals II-7 and II-8) were found to carry the mutation in an homozygous state, whereas all other family members were found to carry the mutation in a heterozygous state or to be homozygous for the wild-type allele except for healthy individual II-9 who carries mutation c.1065T>A in a homozygous state in her peripheral blood leukocytes as a result of the fact that she received a bone marrow transplantation from patient II-7 as a treatment for thalassemia major.
Figure 2. Mutation analysis.

(a) Direct sequencing of FLG2 revealed a homozygous T>A transversion (arrow) at position c.1065 of the cDNA sequence in the two patients (left panel). The wild-type sequence (WT/WT) is given for comparison (right panel). (b) A PCR-restriction fragment length polymorphism assay (lower panel) was used to confirm cosegregation of the mutation with the disease phenotype throughout the family (upper panel; black symbols denote affected individuals; a gray symbol denotes an individual with no skin disease who received a bone marrow transplant from her affected sister).
The mutation was excluded using the PCR-restriction fragment length polymorphism assay from a cohort of 151 population-matched nonaffected individuals (not shown). Further confirming its pathogenicity, we could not identify the mutation in available public exome databases including gnomAD, ESP, 1000 Genomes, Ensembl, UCSC, and HGMD totaling more than 130,000 individual FLG2 sequences.
Consequences of mutation c.1065T>A in FLG2
Mutation c.1065T>A is predicted to result in the generation of a premature termination codon, which in turn is likely to lead to nonsense-mediated mRNA decay. To ascertain this possibility, we used quantitative real-time PCR to assess the effect of the mutation at the RNA level. We found out that FLG2 RNA levels were reduced by more than 80% in the skin of the patients as compared with normal skin (Figure 3a). Accordingly, filaggrin 2 protein expression was dramatically reduced in the stratum granulosum and absent in the stratum corneum in patients’ skin as compared with normal skin (Figure 3b).
Figure 3. FLG2 RNA and protein level in the epidermis.

(a) Real-time PCR analysis was used to assess FLG2 RNA expression in cDNA derived from keratinocytes from two healthy individuals (Control) and patient II-7 (Patient). Results are expressed as percentage of RNA expression relative to expression in control samples ± standard error of the mean. (b) Skin biopsies from a healthy individual (Supplementary Table S5 online; upper panel) and from patient II-8 (inferior panel) were stained with an anti-filaggrin 2-specific antibody (scale bar =50 μm). (c) Filaggrin 2 and corneodesmosin (CDSN) partially colocalize in the lower cornified cell layers (scale bars = 20 μm). (d) Filaggrin 2 and CDSN colocalize in skin section (left panel) and in keratinocytes culture (right panel) by confocal microscopy (three-dimensional image analysis; scale bars = 10 μm). (e) Proximity ligation assay was performed using antibodies directed against CDSN and filaggrin 2 and biopsies from healthy human skin (upper-left panel) or from three-dimensional keratinocyte cultures transfected with control siRNA (upper-right panel, siCTL); FLG2-specific siRNA (lower-left panel, siFLG2); or CDSN-specific siRNA (lower-right panel, siCDSN). Colocalization produces a red fluorescent spot (scale bar = 50 μm).
We then investigated the effect of filaggrin 2 deficiency on the expression of a number of biologically relevant proteins in the skin of the patients. CDSN has been shown to play a vital role in cell-cell adhesion within the cornified cell layers (Matsumoto et al., 2008; Oji et al., 2010). CDSN expression was found to be decreased by immunofluorescence in the epidermis of our patients in contrast with normal expression of other proliferation, epidermal differentiation, or cell-cell adhesion proteins such as E-cadherin, filaggrin, loricrin, keratin 10, and keratin 14 (Figure 4).
Figure 4. Protein expression in patient skin.
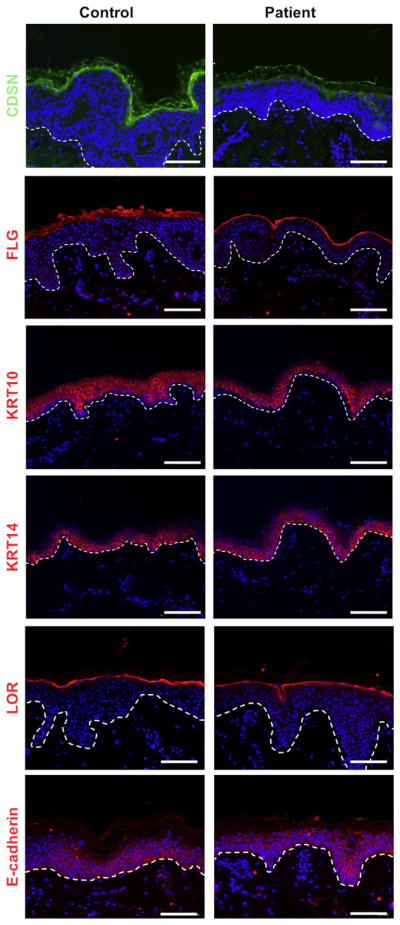
Skin biopsies obtained from individual II-8 were stained for corneodesmosin (CDSN), filaggrin (FLG), keratin 10 (KRT10), keratin 14 (KRT14), loricrin (LOR), and E-cadherin expression (dermoepidermal junction is marked with a dotted line; scale bars = 100 μm, blue staining, DAPI).
Further supporting the pathogenicity of the mutation, three-dimensional skin equivalents generated from the patient’s keratinocytes show abnormal differentiation (Supplementary Figure S3 online). Epidermal thickness and more specifically live keratinocyte layers were thinner in three-dimensional skin equivalents generated from the patient’s keratinocytes as compared with three-dimensional skin equivalents generated from normal keratinocytes, as previously shown with FLG2-knocked-down three-dimensional skin equivalents (Pendaries et al., 2015). Moreover, both filaggrin 2 and CDSN expression levels were reduced in three-dimensional skin equivalents, in agreement with their pattern of expression in patients’ skin (Supplementary Figure S3).
Functional consequences of filaggrin 2 deficiency
The above results as well as other recent data (Alfares et al., 2017; Bolling et al., 2018) support the possibility that filaggrin 2 deficiency is the proximal cause of type A PSS that manifests with heat-induced superficial peeling (Levy and Goldsmith, 1982). To ascertain directly the effect of filaggrin 2 deficiency on CDSN, which is downregulated in the skin of the patients, as well as on the expression of other adhesion proteins, we stained primary keratinocytes silenced for FLG2 for CDSN, desmoglein 1, and E-cadherin. Silencing FLG2 in vitro led to a reduction in the expression of CDSN (also confirmed by western blot analysis; Supplementary Figure S4 online) but did not affect desmoglein 1 or E-cadherin expression (Supplementary Figure S5 online). Immunohistochemistry of patient and control skin biopsies demonstrated that desmoglein 1 and desmocollin 1 were also present in the patient epidermis, albeit somewhat disorganized and with elevated cytoplasmic staining compared with the control (Supplementary Figure S6 online).
Filaggrin 2 and CDSN have been shown to be expressed in the stratum granulosum and stratum corneum (Telem et al., 2012; Wu et al., 2009). Microscopical analysis of normal skin biopsies and cultured keratinocytes revealed colocalization of filaggrin 2 and CDSN in both cases (Figure 3c and d, Supplementary Figure S7 Supplementary). The confocal microscopy results were further supported using the proximity ligation assay (PLA) of normal human tissue and 3D keratinocyte cultures, indicating that filaggrin 2 and CDSN are within close proximity (within 40–100 nm) (Figure 3e).
To directly assess the possibility that filaggrin 2 may be required for efficient epidermal cell-cell adhesion, we used the dispase-based dissociation assay. In this system, keratinocytes are grown to confluence, epidermal sheets are then separated using dispase, and the resilience of the resulting epidermal sheets to mechanical stress is evaluated as previously described (Vodo et al., 2016). Keratinocytes grown under standard conditions at 37 °C and silenced for FLG2 exhibited a more than 50% decrease in cell-cell adhesion compared with keratinocytes transfected with control small interference RNA (siRNA). Moreover, when the same experiment was performed at 40 °C, FLG2-downregulated keratinocytes exhibited a more than 80% decrease in cell-cell adhesion, as compared with keratinocytes transfected with control siRNA (Figure 5a). These results indicate that filaggrin 2 may regulate cell-cell adhesion in a temperature-sensitive fashion, which is of relevance to the fact that PSS3 is exacerbated by high ambient temperature. We hypothesized that if the effect of FLG2 deficiency on CDSN expression is causally related to the disease phenotype, over-expression of CDSN should attenuate the deleterious effect of FLG2 downregulation on cell-cell adhesion. To address this possibility, we restored CDSN expression by introducing an ectopic expression construct (Supplementary Figure S8 online) and assessed the extent to which cell-cell adhesion was rescued in the dispase assay. We found that exogenous CDSN expression in normal keratinocytes silenced for FLG2 or of patient-derived keratinocytes markedly attenuated FLG2 deficiency-associated decrease in cell-cell adhesion (Figure 5b and c).
Figure 5. Downregulation of FLG2 compromises epidermal cell-cell adhesion and corneodesmosin (CDSN) overexpression rescues the phenotype.

(a) Keratinocytes were transfected with FLG2-specific small interference RNA (siRNA) or with control siRNA and then maintained at 37 °C or at 40 °C. (b) Keratinocytes were cotransfected with either FLG2-specific siRNA or with control siRNA together with either an empty vector (EV) or a CDSN expression vector. (c) Patient keratinocytes were transfected with the EV or CDSN expression vector. In all experiments described above, epidermal sheets were then released from the tissue plates with dispase and subjected to mechanical stress (upper panel); the resulting fragments were counted (lower panel). Results are expressed as number of fragments ± standard error of the mean and represent the mean of at least two independent experiments (two-sided t-test; *P < 0.05, **P < 0.01, n.s. = nonsignificant).
DISCUSSION
Filaggrin 2, a 248-kDa member of the S100 fused-type family of proteins (Kypriotou et al., 2012), consists of an N-terminal S100/EF hand region followed by two arrays of multiple tandem repeats termed A- and B-repeats. The A-repeat shares homology with hornerin, whereas the B-repeat is reminiscent of filaggrin (Wu et al., 2009). Filaggrin 2 has been suggested to play a role in the maintenance of the epidermal barrier given its partial similarity to filaggrin (Wu et al., 2009), given the facts that its expression is affected in atopic dermatitis (Trzeciak et al., 2017) and it is rich in histidine and glutamine residues (Wu et al., 2009), sources of the natural moisturizing factors urocanic acid and pyrrolidone carboxylic acid, respectively (Hon et al., 2013). In addition, polymorphisms in the FLG2 gene have been found to be associated with the propensity to develop atopic dermatitis (Margolis et al., 2014).
In contrast with this body of circumstantial evidence, our and recent other data (Alfares et al., 2017; Bolling et al., 2018) suggest that filaggrin 2 also, and possibly mainly, contributes to cell-cell adhesion in the cornified layers rather than to epidermal barrier integrity only. In this regard, it is of interest to note that no permeability defect was found in skin equivalents downregulated for filaggrin 2 (Pendaries et al., 2015). Supporting different roles for filaggrin and filaggrin 2, the two molecules were found to be differentially regulated when the epidermal barrier was challenged (Hansmann et al., 2012). In addition, although first-degree cousins of affected individuals (Figure 2b) showed atopic features, these were absent in the patients themselves who displayed normal IgE levels. The discordance between atopic features and peeling skin within a single family argues against a direct role of filaggrin 2 in epidermal barrier function.
How filaggrin 2 promotes cell-cell adhesion in the upper epidermis remains to be fully elucidated. Although desmoglein 1 appeared disorganized in vivo, the lack of histological features typical of desmoglein 1 deficiency in the patients, the normal expression and organization of desmoglein 1 in vitro, and of E-cadherin in vivo and in vitro suggests that the adhesion defect evident in the patients cannot be explained simply by changes in cadherin function (although a role of desmoglein 1 abnormal organization in the pathogenesis of PSS3 cannot be formally excluded). In contrast, filaggrin 2 deficiency was found to result in decreased expression of CDSN (Figure 4, Supplementary Figures S3–S5). If the physiological role of filaggrin 2 involves maintenance of cell-cell adhesion, the effect of filaggrin 2 deficiency on CDSN expression may thus explain the fact that its clinical consequences were mainly confined to the upper epidermal layers. Indeed, CDSN stabilizes specialized desmosomal junctions within the cornified layers only (Oji et al., 2010).
It is unclear how filaggrin 2 deficiency leads to down-regulation of CDSN. Evidence for their colocalization (Figure 3c–e) suggests that it is possible that the two proteins interact. Alternatively, filaggrin 2 deficiency was shown to cause an increase in pH in the upper stratum corneum (Pendaries et al, 2015). An increase in pH activates serine proteases such as kallikreins in the outer epidermis and, as a consequence, leads to increased degradation of corneodesmosome components such as CDSN (Elias and Wakefield, 2014). Moreover, we observed reduced CDSN protein levels at 40 °C in control cells (Supplementary Figure S5). It is of interest that high temperature decreases CDSN expression whereas CDSN expression is induced by cold-dry exposure (Boutrand et al., 2017). This phenomenon may also possibly be related to the fact that elevated temperature has been previously reported to be associated with destabilization of cell-cell adhesion (Patel et al., 1981). The superficial peeling seen in patients carrying loss-of-function mutations in FLG2 may therefore in part be due to keratinocyte inability to mobilize a sufficient amount of CDSN to the membrane. The fact that CDSN was able to rescue cell-cell adhesion in FLG2-deficient keratinocytes (Figure 5b and c) supports this possibility.
However, although filaggrin 2 deficiency was associated with decreased expression of CDSN in the cornified layers, heterozygous carriers of null mutations in CDSN are typically asymptomatic (Israeli et al., 2011; Oji et al., 2010), suggesting that abnormal expression of CDSN contributes, but cannot explain alone, abnormal cell-cell adhesion in filaggrin 2-deficient epidermis. The ability of CDSN to rescue the adhesion defect induced by filaggrin 2 deficiency (Figure 5) may possibly be accounted for by the fact that CDSN adhesive properties could compensate for the absence of other components of the desmosome such as desmoglein 1 (Jonca et al., 2011).
Other elements unrelated to the corneodesmosome may also explain the role of filaggrin 2 in the maintenance of cell-cell adhesion in the upper epidermal layers. Degradation products of filaggrin 2 have been shown to carry various functions including photoprotection (Pendaries et al., 2015), antimicrobial activity (Hansmann et al., 2015), and skin acidification, which in turn is well known to affect intra-epidermal cell-cell adhesion and desquamation (Matsui and Amagai, 2015).
In summary, the present data substantiate the notion that filaggrin 2 is essential for normal cell-cell adhesion in the cornified cell layers.
MATERIALS AND METHODS
Patients
All affected and healthy family members or their legal guardian provided written and informed consent according to a protocol approved by our institutional review board and by Israel National Committee for Genetic Studies in adherence with the Helsinki principles.
DNA extraction
Genomic DNA was extracted from peripheral blood leukocytes using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany) or from the OG-500 saliva collection kit (DNA Genotek Inc, Ottawa, Canada) according to the manufacturer’s instructions.
Exome sequencing
Details regarding exome sequencing can be found in Supplementary Materials and Methods and Supplementary Table S1 online.
Mutation analysis
Details regarding mutation analysis can be found in Supplementary Materials and Methods and Supplementary Tables S2 and S3.
PCR-restriction fragment length polymorphism
Details regarding restriction fragment length polymorphism can be found in Supplementary Materials and Methods.
Quantitative real-time PCR
Details regarding quantitative real-time PCR can be found in Supplementary Materials and Methods and Supplementary Table S4 online.
Immunohistochemistry and immunofluorescence studies
Details regarding immunohistochemistry and immunofluorescence can be found in Supplementary Materials and Methods and Supplementary Figure S9 online.
Electron microscopy
Skin biopsies were fixed in half-strength Karnowski’s fixative and 1% osmium tetroxide. Tissue blocks were dehydrated with ethanol, stained en bloc using 1% uranyl acetate in 50% ethanol, and embedded in Epon resin (TAAB, Aldermaston, UK). Ultrathin sections were stained with 1.5% uranyl acetate in methanol and Reynold’s lead citrate and were examined with a JEM-1010 transmission electron microscope (Jeol, Tokyo, Japan).
Cell cultures and reagents
Primary keratinocytes and fibroblasts were isolated from adult skin obtained from plastic surgery specimens or from shave biopsies after having received written informed consent from the donors according to a protocol reviewed and approved by our institutional review board as previously described (Samuelov et al., 2013). Primary keratinocytes were maintained in Keratinocytes Growth Medium (Lonza, Walkersville, MD). Fibroblasts were cultured in DMEM supplemented with 20% fetal calf serum (Biological Industries Israel, Beit-Haemek, Israel).
For FLG2 silencing, human keratinocytes were cultured in six-well plates at 37 °C in 5% CO2 in a humidified incubator. To downregulate FLG2 expression, we used human FLG2 siRNAs (Santa Cruz; sc-88656). As control siRNA, we used Stealth RNAi Negative Control Duplex (Invitrogen, Carlsbad, CA). Twenty-five pmol of siRNAs were transfected into keratinocytes using Lipofectamine RNAiMax (Invitrogen) or with Lipofectamine 2000 (Invitrogen) when cotransfected with pcDNA3.1 or pcDNA3.1 CDSN expression vectors (see below). The transfection medium was replaced after 6 hours with high calcium (1.6 mM)-containing keratinocyte growth medium.
Organotypic cell cultures
Details regarding organotypic cell cultures can be found in Supplementary Materials and Methods.
Proximity ligation assay
To perform transient knockdown with siRNA, human primary keratinocytes were electroporated using the Amaxa Nucleofector System (Lonza) according to the manufacturer’s instructions. Keratinocytes were counted and resuspended in Ingenio Electroporation Solution (Mirus) and siRNA (final concentration 20 μM) and then electroporated using program X-001. Nontargeting siRNA (negative control) (Dharmacon), siRNA against human CDSN (M-011238-01, Dharmacon) with targeting sequences: 5′-GGACAA AGCUCUUC CUCUU-3′ and 5′-CCAGGGACCUUGGCUAAGA-3′, 5′-CAACUCU UACC GCGGAAUA-3′, 5′-UGACCUACAGUAAGGGUAA-3′, and siRNA against human filaggrin 2 (sc-88656, Santa Cruz Biotechnology), with targeting sequences: 5′-GAUCU GGAUCAAACAG UU-3′, 5′-GGUAGCCAGUCUUGUAGUA-3′, and 5′-GCAUGAG UCUA CAUCAAGU-3′, were used for transient knockdown in keratinocytes.
For stratified, three-dimensional keratinocyte cultures, human primary keratinocytes were expanded and grown at an air-medium interface according to published protocols (Simpson et al., 2010). These lifted cultures were grown for 6 days, at which time they were lysed in Urea Sample Buffer (8 M deionized urea; 1% SDS; 10% glycerol; 60 mM Tris, pH 6.8; 0.1% pyronin-Y; and 5% β-mercaptoethanol) for biochemical analysis or fixed in 10% neutral-buffered formalin, and embedded in paraffin.
Reagents used to conduct PLA in situ were purchased from Sigma-Aldrich and used according to the manufacturer’s protocol (Sigma-Aldrich, MO). Cells were rinsed and fixed with 4% paraformaldehyde and permeabilized with 0.2% triton X-100. After incubation with a primary antibody overnight at 4 °C (sheep anti-human CDSN [R&D Systems] and rabbit anti-human filaggrin 2 [Novus Biologicals]), samples were incubated with PLA secondary antibodies conjugated to DNA oligonucleotides for 60 minutes at 37°C. Samples were then subjected to a 30-minute incubation at37 °C for ligation of nucleotides, followed by a 100-minute incubation at 37 °C for rolling circle polymerization, resulting in the production of fluorescent dots (shown in red) if the antigens targeted by secondary antibodies were in close proximity (40–100 nm). Note that the donkey-anti-goat secondary antibody used to recognize the sheep anti-CDSN antibody was labeled using the In Situ Probemaker kit, whereas FLG2 was recognized by a rabbit-anti-FLG2 bound by a commercially available anti-rabbit PLA probe (Sigma-Aldrich). Fluorescent spots appearing at the site of CDSN/FLG2 proximity were detected by wide-field microscopy using the Axioimager and the Apotome2 (Zeiss) used with the ×20 objective and an maximum intensity projection of the Z-sections was used.
Dispase-based dissociation assay
Human keratinocytes were grown to confluence in triplicates on six-well plates at 37 °C in 5% CO2 in a humidified incubator. After 5 days, keratinocytes at 90–95% confluency were transfectedwith FLG2-specific siRNA (sc-88656) or control siRNA-Stealth RNAi Negative Control Duplex (Invitrogen). Efficacy of siRNA-mediated FLG2 down-regulation was quantified using real-time PCR (Supplementary Figure S10 online). In the rescue experiments, pcDNA3.1 (Abnova, Walnut, CA) or pcDNA3.1 CDSN expression vector (GenScript, Piscataway, NJ) were cotransfected with FLG2-specific siRNA or control siRNA-Stealth RNAi Negative Control Duplex.
The transfection medium was replaced after 6 hours with Keratinocytes Growth Medium high Ca+2 (1.6 mM). Forty-eight hours after transfection, keratinocyte cultures were washed twice with phosphate buffered saline, incubated in 2 ml of dispase II (2.4 units/ml, Roche Diagnostics, Basel, Switzerland) at 37 °C for 35 minutes and detached from the plate as monolayers. Cell sheets were carefully transferred to a 15 ml tube containing 5 ml phosphate buffered saline and subjected to mechanical stress that consisted of five inversions. The number of fragments was counted by two independent evaluators. Of note, the treatment of cells with dispase alone had no effect on filaggrin 2 expression (Supplementary Figure S11 online).
Western blotting
Details regarding western blotting can be found in Supplementary Materials and Methods.
Supplementary Material
Acknowledgments
We are grateful to the patients and their family for their participation to this study. We would like to thank J. Segre, National Human Genome Research Institute, for the gift of the rabbit antikeratin 5 and keratin 10 antibodies. The study was supported by the Eli and Ruth Ram foundation (ES), the National Institutes of Health Grants R01 AR041836 and R37 AR043380 (KJG), and by the Edmond J. Safra Center for Bioinformatics at Tel Aviv University (TR).
Abbreviations
- CDSN
corneodesmosin
- PSS
peeling skin syndrome
- siRNA
small interference RNA
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
Supplementary material is linked to the online version of the paper at www.jidonline.org, and at https://doi.org/10.1016/j.jid.2018.04.032.
References
- Alfares A, Al-Khenaizan S, Al Mutairi F. Peeling skin syndrome associated with novel variant in FLG2 gene. Am J Med Genet A. 2017;173:3201–4. doi: 10.1002/ajmg.a.38468. [DOI] [PubMed] [Google Scholar]
- Blaydon DC, Nitoiu D, Eckl KM, Cabral RM, Bland P, Hausser I, et al. Mutations in CSTA, encoding Cystatin A, underlie exfoliative ichthyosis and reveal a role for this protease inhibitor in cell-cell adhesion. Am J Hum Genet. 2011;89:564–71. doi: 10.1016/j.ajhg.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolling MC, Jan SZ, Pasmooij AMG, Lemmink HH, Franke LH, Yenamandra VK, et al. Generalized ichthyotic peeling skin syndrome due to FLG2 mutations [e-pub ahead of print] J Invest Dermatol. 2018 doi: 10.1016/j.jid.2018.01.038. [DOI] [PubMed]
- Boutrand LB, Thepot A, Muther C, Boher A, Robic J, Guere C, et al. Repeated short climatic change affects the epidermal differentiation program and leads to matrix remodeling in a human organotypic skin model. Clin Cosmet Investig Dermatol. 2017;10:43–50. doi: 10.2147/CCID.S120800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral RM, Kurban M, Wajid M, Shimomura Y, Petukhova L, Christiano AM. Whole-exome sequencing in a single proband reveals a mutation in the CHST8 gene in autosomal recessive peeling skin syndrome. Genomics. 2012;99:202–8. doi: 10.1016/j.ygeno.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy AJ, van Steensel MA, Steijlen PM, van Geel M, van der Velden J, Morley SM, et al. A homozygous missense mutation in TGM5 abolishes epidermal transglutaminase 5 activity and causes acral peeling skin syndrome. Am J Hum Genet. 2005;77:909–17. doi: 10.1086/497707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias PM, Wakefield JS. Mechanisms of abnormal lamellar body secretion and the dysfunctional skin barrier in patients with atopic dermatitis. J Allergy Clin Immunol. 2014;134:781–91e1. doi: 10.1016/j.jaci.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiete D, Mi Y, Beranek M, Baenziger NL, Baenziger JU. The glycan-specific sulfotransferase (R77W)GalNAc-4-ST1 putatively responsible for peeling skin syndrome has normal properties consistent with a simple sequence polymorphisim. Glycobiology. 2017;27:450–6. doi: 10.1093/glycob/cwx018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta. 2008;1778:572–87. doi: 10.1016/j.bbamem.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Hansmann B, Ahrens K, Wu Z, Proksch E, Meyer-Hoffert U, Schroder JM. Murine filaggrin-2 is involved in epithelial barrier function and down-regulated in metabolically induced skin barrier dysfunction. Exp Dermatol. 2012;21:271–6. doi: 10.1111/j.1600-0625.2012.01449.x. [DOI] [PubMed] [Google Scholar]
- Hansmann B, Schroder JM, Gerstel U. Skin-derived C-terminal filaggrin-2 fragments are Pseudomonas aeruginosa-directed antimicrobials targeting bacterial replication. PLoS Pathog. 2015;11:e1005159. doi: 10.1371/journal.ppat.1005159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon KL, Leung AK, Barankin B. Barrier repair therapy in atopic dermatitis: an overview. Am J Clin Dermatol. 2013;14:389–99. doi: 10.1007/s40257-013-0033-9. [DOI] [PubMed] [Google Scholar]
- Ishida-Yamamoto A, Igawa S. The biology and regulation of corneodesmosomes. Cell Tissue Res. 2015;360:477–82. doi: 10.1007/s00441-014-2037-z. [DOI] [PubMed] [Google Scholar]
- Israeli S, Zamir H, Sarig O, Bergman R, Sprecher E. Inflammatory peeling skin syndrome caused by a mutation in CDSN encoding corneodesmosin. J Invest Dermatol. 2011;131:779–81. doi: 10.1038/jid.2010.363. [DOI] [PubMed] [Google Scholar]
- Jonca N, Leclerc EA, Caubet C, Simon M, Guerrin M, Serre G. Corneodesmosomes and corneodesmosin: from the stratum corneum cohesion to the pathophysiology of genodermatoses. Eur J Dermatol. 2011;21(Suppl 2):35–42. doi: 10.1684/ejd.2011.1264. [DOI] [PubMed] [Google Scholar]
- Kose O, Safali M, Koc E, Arca E, Acikgoz G, Ozmen I, et al. Peeling skin diseases: 21 cases from Turkey and a review of the literature. J Eur Acad Dermatol Venereol. 2012;26:844–8. doi: 10.1111/j.1468-3083.2011.04166.x. [DOI] [PubMed] [Google Scholar]
- Krunic AL, Stone KL, Simpson MA, McGrath JA. Acral peeling skin syndrome resulting from a homozygous nonsense mutation in the CSTA gene encoding cystatin A. Pediatr Dermatol. 2013;30:e87–8. doi: 10.1111/pde.12092. [DOI] [PubMed] [Google Scholar]
- Kypriotou M, Huber M, Hohl D. The human epidermal differentiation complex: cornified envelope precursors, S100 proteins and the “fused genes” family. Exp Dermatol. 2012;21:643–9. doi: 10.1111/j.1600-0625.2012.01472.x. [DOI] [PubMed] [Google Scholar]
- Levy SB, Goldsmith LA. The peeling skin syndrome. J Am Acad Dermatol. 1982;7:606–13. doi: 10.1016/s0190-9622(82)70140-9. [DOI] [PubMed] [Google Scholar]
- Margolis DJ, Gupta J, Apter AJ, Ganguly T, Hoffstad O, Papadopoulos M, et al. Filaggrin-2 variation is associated with more persistent atopic dermatitis in African American subjects. J Allergy Clin Immunol. 2014;133:784–9. doi: 10.1016/j.jaci.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Amagai M. Dissecting the formation, structure and barrier function of the stratum corneum. Int Immunol. 2015;27:269–80. doi: 10.1093/intimm/dxv013. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Zhou Y, Matsuo S, Nakanishi H, Hirose K, Oura H, et al. Targeted deletion of the murine corneodesmosin gene delineates its essential role in skin and hair physiology. Proc Natl Acad Sci USA. 2008;105:6720–4. doi: 10.1073/pnas.0709345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oji V, Eckl KM, Aufenvenne K, Natebus M, Tarinski T, Ackermann K, et al. Loss of corneodesmosin leads to severe skin barrier defect, pruritus, and atopy: unraveling the peeling skin disease. Am J Hum Genet. 2010;87:274–81. doi: 10.1016/j.ajhg.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H, Marcelo C, Voorhees JJ, Diaz LA. In vitro alterations of epidermal cell adhesion induced by temperature, substrate, and cations. J Invest Dermatol. 1981;76:474–9. doi: 10.1111/1523-1747.ep12521148. [DOI] [PubMed] [Google Scholar]
- Pendaries V, Le Lamer M, Cau L, Hansmann B, Malaisse J, Kezic S, et al. In a three-dimensional reconstructed human epidermis filaggrin-2 is essential for proper cornification. Cell Death Dis. 2015;6:e1656. doi: 10.1038/cddis.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigors M, Sarig O, Heinz L, Plagnol V, Fischer J, Mohamad J, et al. Loss-of-function mutations in SERPINB8 linked to exfoliative ichthyosis with impaired mechanical stability of intercellular adhesions. Am J Hum Genet. 2016;99:430–6. doi: 10.1016/j.ajhg.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, Isakov O, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. 2013;45:1244–8. doi: 10.1038/ng.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelov L, Sprecher E. Peeling off the genetics of atopic dermatitis-like congenital disorders. J Allergy Clin Immunol. 2014;134:808–15. doi: 10.1016/j.jaci.2014.07.061. [DOI] [PubMed] [Google Scholar]
- Simpson CL, Kojima S, Getsios S. RNA interference in keratinocytes and an organotypic model of human epidermis. Methods Mol Biol. 2010;585:127–46. doi: 10.1007/978-1-60761-380-0_10. [DOI] [PubMed] [Google Scholar]
- Telem DF, Israeli S, Sarig O, Sprecher E. Inflammatory peeling skin syndrome caused a novel mutation in CDSN. Arch Dermatol Res. 2012;304:251–5. doi: 10.1007/s00403-011-1195-z. [DOI] [PubMed] [Google Scholar]
- Trzeciak M, Sakowicz-Burkiewicz M, Wesserling M, Dobaczewska D, Glen J, Nowicki R, et al. Expression of cornified envelope proteins in skin and its relationship with atopic dermatitis phenotype. Acta Derm Venereol. 2017;97:36–41. doi: 10.2340/00015555-2482. [DOI] [PubMed] [Google Scholar]
- Vodo D, Sarig O, Geller S, Ben-Asher E, Olender T, Bochner R, et al. Identification of a functional risk variant for pemphigus vulgaris in the ST18 gene. PLoS Genet. 2016;12:e1006008. doi: 10.1371/journal.pgen.1006008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Hansmann B, Meyer-Hoffert U, Glaser R, Schroder JM. Molecular identification and expression analysis of filaggrin-2, a member of the S100 fused-type protein family. PLoS One. 2009;4:e5227. doi: 10.1371/journal.pone.0005227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.