

Abstract
Proteomic characterization of blood plasma is of central importance to clinical proteomics and particularly to biomarker discovery studies. The vast dynamic range and high complexity of the plasma proteome have, however, proven to be serious challenges and have often led to unacceptable tradeoffs between depth of coverage and sample throughput. We present an optimized sample-processing pipeline for analysis of the human plasma proteome that provides greatly increased depth of detection, improved quantitative precision and much higher sample analysis throughput as compared with prior methods. The process includes abundant protein depletion, isobaric labeling at the peptide level for multiplexed relative quantification and ultra-high-performance liquid chromatography coupled to accurate-mass, high-resolution tandem mass spectrometry analysis of peptides fractionated off-line by basic pH reversed-phase (bRP) chromatography. The overall reproducibility of the process, including immunoaffinity depletion, is high, with a process replicate coefficient of variation (CV) of <12%. Using isobaric tags for relative and absolute quantitation (iTRAQ) 4-plex, >4,500 proteins are detected and quantified per patient sample on average, with two or more peptides per protein and starting from as little as 200 μl of plasma. The approach can be multiplexed up to 10-plex using tandem mass tags (TMT) reagents, further increasing throughput, albeit with some decrease in the number of proteins quantified. In addition, we provide a rapid protocol for analysis of nonfractionated depleted plasma samples analyzed in 10-plex. This provides ~600 quantified proteins for each of the ten samples in ~5 h of instrument time.
INTRODUCTION
Blood is estimated to contain well over 10,000 distinct proteins (not counting proteoforms that are post-translationally modified), with concentrations spanning a dynamic range of 10–12 orders of magnitude1,2. As it directly communicates with nearly all organs and tissues in the body and is one of the most easily accessible bodily fluids, blood and its liquid derivatives are thought to hold singular promise as a source of potential biomarkers. Indeed, the vast majority of existing clinical laboratory tests measure proteins, small molecules and other analytes in plasma or serum derived from whole blood. Despite decades of attempts to mine this biomarker resource, however, global-discovery plasma proteomics has met with limited success, in large measure because of low numbers of identified proteins (i.e., poor depth of detection). Individual studies of the plasma proteome have typically detected fewer than 1,000 proteins with high confidence3,4. As part of the Peptide Atlas project, Farrah and colleagues developed a high-confidence list of 1,929 plasma proteins by re-analyzing 91 high-quality data sets using a common database search and analysis platform and a protein false discovery rate (FDR) of 1%5. Recent analysis of additional data sets allowed further expansion to more than 3,500 plasma proteins; however, only ~2,500 of these proteins were confidently identified with at least two peptides, the ~1,000 remaining having been identified using only a single peptide and therefore being of lower confidence6. By contrast, current expert proteomic analyses of cells and tissues routinely yield >10,000 distinct proteins identified with two or more peptides per protein7–9. This large disparity in depth of detection is believed to be because of both the greater range in abundance of proteins in blood as compared with other tissues and the presence of a handful of large, highly abundant blood proteins that, after enzymatic digestion, yield vast numbers of abundant peptides that interfere with detection of peptides derived from less-abundant plasma proteins10.
To increase the depth of detection in plasma, methods have been developed to remove the most highly abundant proteins and to partition the remaining proteins or peptides into fractions with reduced complexity and dynamic range before analysis by liquid chromatography–tandem mass spectrometry (LC–MS/MS). Abundant protein depletion is now commonly done by immunoaffinity, with single depletion columns capable of removing the 6–20 dominant plasma proteins11–13. Considerably deeper (two to fivefold, depending on specific methods used) coverage of the plasma proteome is possible with more intensive depletion. For example, the IgY14–SuperMix tandem column system (Sigma) depletes ~60 highly to moderately abundant proteins in addition to the 14 most highly abundant proteins that are removed by the IgY14 column, enabling detection of proteins in the low nanogram/milliliter range12,13.
Abundant protein depletion has been most often used together with off-line protein- or peptide-level fractionation before LC–MS/MS analysis. Speicher and co-workers investigated multiple post-depletion plasma fractionation strategies—including SDS-PAGE for protein-level fractionation and isoelectric focusing, and bRP for peptide-level fractionation14—and showed that increasing the number of fractions by any method improves the sensitivity of detection in plasma, with high-pH reversed-phase (RP) fractionation yielding the highest number of low-abundant proteins. Combining fractions from the early, middle and late portions of the high-pH RP gradient further increased the depth of detection and chromatographic resolution by better equalizing the amount of peptide in each fraction and more uniformly distributing peptide retention times in the acidic-pH RP chromatographic separation that is done online to the MS15,16.
Although fractionation at the peptide level greatly increases the depth of analysis, a major drawback has been the significant increase in the amount of time it takes to analyze a single sample. This unfortunate tradeoff between depth and throughput has meant that studies in plasma proteomics that include relatively large numbers of samples (many tens to >100) typically have shallow depth of coverage (e.g., Cominetti et al.17 and Dayon et al.18), whereas studies emphasizing depth of coverage typically have small sample numbers (e.g., refs. 19–21). Our prior label-free workflow for biomarker candidate discovery in acute myocardial injury highlights this challenge of plasma proteomics19. After immunoaffinity depletion of the 14 most abundant proteins, peptides from each patient sample time point were partitioned using strong cation exchange chromatography (SCX) into 80 fractions, with each patient sample requiring 160 h of MS analysis time. Although the depth of analysis was good by contemporary standards (average of 900 proteins per sample using a minimum of two peptides for identification), the prohibitive analysis time severely constrained the number of samples that could be analyzed. Importantly, small sample numbers combined with the relative quantitative imprecision of a label-free workflow meant that only fold-changes >5× could be confidently considered differential (see Addona et al.19 for details).
To simultaneously achieve more precise relative quantification and higher analysis throughput in biomarker discovery studies, chemical labeling using commercially available iTRAQ and TMT isobaric reagents22–33 has begun to be used for plasma analysis. After the depletion of six abundant proteins, Cole et al. performed 8-plex iTRAQ labeling and SCX separation into 24 fractions, resulting in the quantification of ~900 proteins across 500 patient plasma samples spanning 72 iTRAQ experiments34. In the largest patient study yet reported, Cominetti et al. applied an automated plasma processing workflow that included depletion of 14 abundant proteins, 6-plex TMT peptide labeling and online LC–MS/MS analysis without fractionation to >1,000 patient samples17,18. Although each sample required only 3 h of instrument time, this study identified only 365 proteins in total across all samples. In an inter-laboratory study organized by the Association of Biomolecular Resource Facilities, centrally processed plasma underwent IgY14–SuperMix depletion, iTRAQ 4-plex labeling and SCX peptide separation into 30 fractions before distribution to participating laboratories for analysis35. Between 1,200 and 1,700 proteins were identified in each of the laboratories using current-generation high-performance LC and MS instruments.
Development of the protocol
Although a wide range of methods have been explored for plasma processing and analysis, an optimal approach has yet to be defined. In developing a processing and analysis protocol, our goals were to achieve very deep and reproducible coverage of the plasma proteome with precise relative quantification. This would allow sensitive detection of differential protein abundance between samples while retaining sufficient throughput to develop reasonable statistics. To this end, we have evaluated and optimized methods for abundant protein depletion, off-line peptide fractionation, online peptide separation and MS analysis36–38. The integrated approach that we developed incorporates IgY14–SuperMix immunoaffinity depletion, chemical labeling of peptides with isobaric mass tags (using either iTRAQ or TMT), peptide-level fractionation using basic RP with a concatenated fraction-pooling strategy and LC–MS/MS analysis using long (>20 cm) columns packed with sub-2-μm beads coupled to current, high-performance hybrid mass spectrometers such as the Thermo Q-Exactive Plus (Fig. 1). To demonstrate the effectiveness of the workflow, we analyzed a time-course series of plasma samples using iTRAQ 4-plex labeling. The samples were obtained from patients undergoing therapeutic septal ablation (planned myocardial infarction (PMI)) for the treatment of hypertrophic obstructive cardiomyopathy38. An average of 4,600 proteins were confidently detected and quantified in each of 16 patient samples with a peptide FDR of < 1.5%. This is the largest number of proteins detected in a single study of the plasma proteome to date, and the deep coverage combined with the use of isobaric labeling enabled confident identification of >300 novel candidate biomarkers of acute myocardial injury—six times more than what was achieved in the prior label-free study19.
Figure 1.
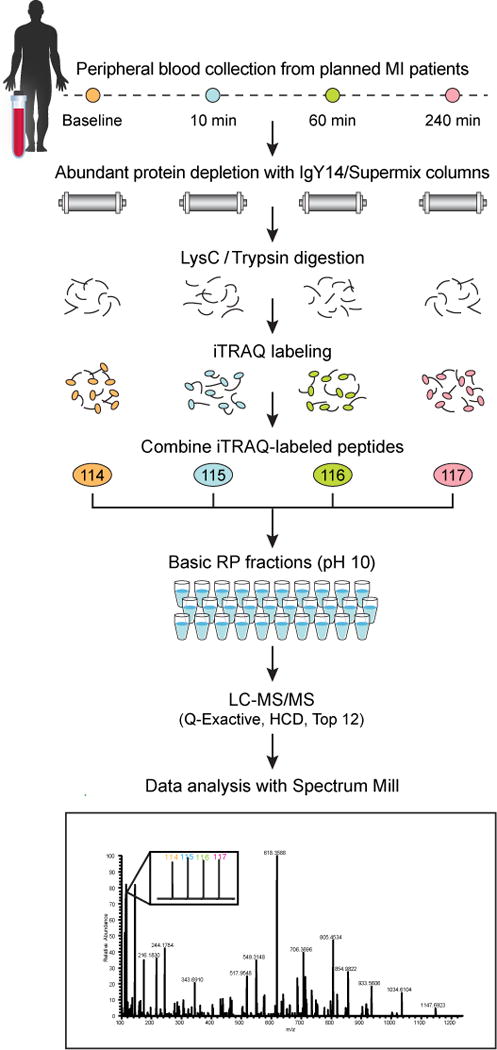
Overview of multiplexed workflow for discovery proteomics in plasma. This figure shows application of the workflow to a specific study in which iTRAQ 4-plex reagent was used to process and analyze four time points of planned myocardial infarction (PMI) patient samples. However, as shown in the protocol, other depletion and labeling strategies can be used to accommodate the goals of a given study. HCD, higher energy collisional dissociation. Image adapted with permission from Keshishian et al.38, American Society for Biochemistry and Molecular Biology.
The protocol described here provides complete step-by-step instructions for achieving deep, quantitative analysis of the plasma proteome by mass spectrometry, with multiplexing of samples for improved sensitivity, decreased preanalytical variability and higher throughput. Measures of overall process reproducibility are provided, as are methods for assessing reproducibility of the depletion step. Substantial details about iTRAQ/TMT-specific issues of data analysis are included. Although Spectrum Mill parameters and metrics are used as examples, the underlying concepts are described generally so that they can be applied when using other software packages that can handle iTRAQ/TMT data.
The protocol for deep, global analysis of plasma is illustrated using both 4-plex and 10-plex isobaric labeling strategies. A modified version of the protocol that enables, in principle, the analysis of up to 50 patient samples per day per instrument to a depth of ~600 proteins is also provided. All the methods described take advantage of the greater confidence in database searching provided by data collected at high resolution and accurate mass for both the intact precursor peptides and their fragment ions.
Although the current protocol focuses on the analysis of plasma, it can be adapted for the analysis of other biofluids, including cyst fluid, urine and cerebrospinal fluid, which also have populations of abundant proteins that overlap in identity with those found in plasma. Variations in total protein content and precise abundant protein composition in these fluids can be addressed with appropriate protocol modifications, including those in regard to intensity of depletion and extent of fractionation.
Current limitations and future directions
Using the workflow described here, it is possible to routinely detect and quantify >4,500 proteins per sample in plasma. Although this far exceeds what has been attained with other established workflows, it still represents at best 50% of the plasma proteome, and further advances, especially in detection of low-abundance proteins, are desirable. Improvements in both sample preparation strategies and MS instrumentation will move us closer to this goal. Although many such developments are on the near horizon, some are ready for immediate adoption. Among these are the use of small interior diameter (i.d.), ultra-high-performance LC columns for off-line basic pH reverse-phase fractionation and coupling of high-field asymmetric waveform ion mobility spectrometry (FAIMS)39 to state-of-the-art MS instrumentation, both of which will help augment sensitivity for plasma analysis.
Experimental design
Starting material
The volume of starting plasma required is dependent on the depletion strategy, the intended plex level for labeling and the total protein amount required for off-line fractionation. The yield of protein from IgY14-depleted plasma is a minimum of ~3.5%, whereas IgY14–SuperMix tandem depletion yields a minimum of ~0.5%. Typically, 200 μl of plasma is required for IgY14–SuperMix depletion, but it is feasible to start with less (Table 1). This also has the advantage that smaller-capacity, lower-cost, higher-throughput depletion columns may be used. The study detailed in the protocol analyzed plasma from patients undergoing a therapeutic, PMI for hypertrophic cardiomyopathy. In this nonsurgical procedure, alcohol is injected via a catheter into the heart (alcohol ablation) to cause selective destruction of the hypertrophied portion of the left side of the interventricular septum37,38. In this study, we depleted 400 μl of plasma for each of the four time points (pre-alcohol ablation, and 10, 60 and 240 min post ablation) per patient using IgY14–SuperMix (Fig. 1). This large volume was used because we wanted several additional depleted samples (per time point per patient) for future analyses and the sample was not limiting.
TABLE 1.
Plasma volume requirement for labeling after IgY14-only and IgY14–SuperMix depletions.
| Protein amount for labeling post-depletion (μg) | IgY14 only
|
IgY14-SuperMix
|
||
|---|---|---|---|---|
| Min. nondepleted plasma volume required (μL)a | Min. plasma volume to deplete (μL)b | Min. nondepleted plasma volume required (μL)a | Min. plasma volume to deplete (μL)b | |
| 10 | 4.1 | 6.1 | 28.6 | 42.9 |
| 30 | 12.2 | 18.4 | 85.7 | 128.6 |
| 50 | 20.4 | 30.6 | 142.9 | 214.3 |
| 100 | 40.8 | 61.2 | 285.7 | 428.6 |
Volume requirement is based on the 3.5 and 0.5% minimum recovery after IgY14-only and IgY14–SuperMix depletions, respectively. A plasma concentration of 70 mg/ml was used for the calculations.
Minimum required volume is increased by 50% to account for losses during sample processing.
Protein amounts shown in Table 1 are based on a typical protein content of 70 mg/ml in plasma. It is important to check the plasma protein concentration upon which vendors base nominal column capacity and to adjust injection volumes accordingly if samples have substantially higher concentration than that reference plasma. Stated nominal capacities should not be exceeded. A lower bound for input protein has not been established, but we have achieved effective depletion with as little as 35% of nominal column capacity. The stated life span of the depletion columns can also vary, but our experience (and those of others13) indicates that the IgY14 and IgY14–SuperMix columns will continue to deplete with good reproducibility over ~100 injections. To ensure this consistency for large studies (>100 samples), it is important to obtain an appropriate number of IgY14–SuperMix columns belonging to the same lot to avoid possible performance differences because of specific populations of antibodies.
MATERIALS
REAGENTS
Human plasma (BioreclamationIVT, cat. no. HMPLEDTA2) ! CAUTION Informed consent must be in place when using patient plasma. Human plasma and other human biofluids should be handled in a Biosafety Level 2 (or higher) environment and with strict attention to Biosafety Level 2 procedures. Refer to the Centers for Disease Control and Prevention website (http://www.cdc.gov/biosafety/publications/bmbl5/BMBL5_sect_IV.pdf) for additional information.
Bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, cat. no. 23225)
10× dilution buffer for depletion: 100 mM Tris–HCl, 1.5 M NaCl, pH 7.4 (Sigma-Aldrich, cat. no. S4199) ▲ CRITICAL Preparation and use of vendor-specific reagents are recommended.
10× stripping buffer for depletion: 1 M glycine, pH 2.5 (Sigma-Aldrich, cat. no. S4324)
10× neutralization buffer for depletion: 1 M Tris–HCl, pH 8.0 (Sigma-Aldrich, cat. no. S4449)
N-Octyl-B-D-glucopyranoside, anagrade (ODG; Anatrace, cat. no. 0311)
Sodium azide (Sigma-Aldrich, cat. no. S2002) ! CAUTION Sodium azide is a health hazard level 4 compound, with flammability level 1 and reactivity level 3. Avoid contact with skin and eyes, and avoid inhalation. Store it separately at room temperature (RT; 25 °C) for up to 6 months based on Material Safety Data Sheet (MSDS) recommendations.
HPLC-grade water (J.T. Baker, cat. no. 4218-03)
Ammonium bicarbonate (Sigma-Aldrich, cat. no. A6141)
! CAUTION Ammonium bicarbonate is a health hazard level 2 compound. Avoid contact with skin and eyes, and avoid inhalation.
Urea (Sigma-Aldrich, cat. no. U0631) ! CAUTION Urea is a health hazard level 2 compound. Avoid contact with skin and eyes, and avoid and inhalation.
DTT (Thermo Fisher Scientific, cat. no. 20291) ! CAUTION DTT is a health hazard level 2 compound. Avoid contact with skin and eyes, and avoid inhalation.
Iodoacetamide (Sigma-Aldrich, cat. no. A3221) ! CAUTION IAA is a health hazard level 3 compound, with reactivity level 1. Avoid contact with skin and eyes, and avoid inhalation.
Lysyl endopeptidase (Lys-C; Wako, cat. no. 129-02541)
Sequencing-grade modified trypsin (Promega, cat. no. V511X)
Formic acid (FA; Sigma-Aldrich, cat. no. 56302) ! CAUTION FA is a health hazard level 4 compound, with flammability level 3. Avoid contact with skin and eyes, and avoid inhalation. Use it in a well-ventilated area per MSDS recommendations. Store at RT for up to 6 months.
Ammonium hydroxide solution, 28% (wt/vol) (NH4OH; Sigma-Aldrich, cat. no. 338818) ! CAUTION NH4OH is a health hazard level 4 compound. Avoid contact with skin and eyes, and avoid inhalation.
mColorpHast pH Test Strips, 5–10 range (VWR, cat. no. EM1.09533.0001)
Acetonitrile (MeCN; J.T. Baker, cat. no. 9829-03) ! CAUTION Acetonitrile is a health hazard level 4 compound, with flammability level 2. Avoid contact with skin and eyes, and avoid inhalation. Store/use it in a well-ventilated area per MSDS recommendations. Store at RT for up to 6 months. ▲ CRITICAL LC/MS-grade-quality MeCN is required.
Methanol (MeOH; Fluka, cat. no. 34966) ! CAUTION Methanol is a health hazard level 1 compound, with flammability level 3 and instability/reactivity level 0. Avoid contact with skin and eyes, and avoid inhalation. Store/use it in a well-ventilated area per MSDS recommendations. Store at RT for up to 6 months.
Synthetic peptide standards. We use an in-house set of seven synthetic peptides, but commercial peptide standards such as custom synthetic peptides from New England Peptide (Pierce peptide retention time calibration mixture; Thermo Fisher Scientific, cat. no. 88320) can also be used.
iTRAQ 4-plex reagent (AB Sciex, cat. no. 4466096)
TMT-6 reagent (Thermo Fisher Scientific, cat. no. 90068)
TMT-10 reagent (Thermo Fisher Scientific, cat. no. 90046)
Triethylammonium bicarbonate (TEAB; Sigma-Aldrich, cat. no. T7408-100ML)
HEPES (Sigma-Aldrich, cat. no. H4034)
50% hydroxylamine solution (Sigma-Aldrich, cat. no. 467804) ! CAUTION 50% (vol/vol) hydroxylamine solution is a health hazard level 2 compound. Avoid contact with skin or eyes, and avoid inhalation.
ReproSil-Pur, 120 Å, C18-AQ, 1.9-μm resin (Dr. Maisch, cat. no. r119.ag)
EQUIPMENT
Off-line HPLC system for plasma depletion and bRP separation: Agilent 1100 or 1200 series pump equipped with degasser, autosampler, UV detector, column-switching module and fraction collector. We use an Agilent 1200 system for plasma depletion and an Agilent 1100 system for bRP separation.
Seppro IgY14 LC20 column (Sigma-Aldrich, cat. no. SEP000-1KT). This column will deplete the 14 most abundant proteins effectively for up to 300 μl of plasma at a 70 mg/ml concentration; however, for depletion of 400 μl of plasma, as described in this protocol, we used a custom 25-ml IgY14 column. The same HPLC method can be used for both columns with slight adjustment of fraction collection start time. ▲ CRITICAL Store the column at 2–8°C.
Seppro SuperMix LC10 column (Sigma-Aldrich, cat. no. SEP000-1KT) This column is not intended for stand-alone use but only for tandem depletion after IgY14 depletion. Its capacity is matched to the flow-through of the IgY14 LC20 column. ▲ CRITICAL Store the column at 2–8°C.
PEEK sample loop, 2 ml, 0.76 mm i.d. (Agilent, cat. no. 0101-1234)
0.01- inch i.d., 1/16-inch outer diameter (o.d.) PEEK tubing (Upchurch, cat. no. 1531)
1-ml autosampler vial, for bRP (National Scientific, cat. no. C4010-14)
1-ml autosampler cap, for bRP (National Scientific, cat. no. C4010-55A)
0.22-μm cellulose acetate spin filters (Agilent, cat. no. 5185-5990)
3,000 MWCO concentrators, 15-ml capacity (Millipore, cat. no. UFC900324)
Sorvall ST 8 small benchtop centrifuge (Thermo Fisher, cat. no. 75007203) configured with TX-150 swinging bucket rotor (cat. no. 75005701) with 8 × 50-ml sealed tube adaptor
96-well (2 ml) collection plate (Whatman, cat. no. 7701-5200)
96-well microtiter plate (BD Falcon, cat. no. 353261, or equivalent)
Extraction manifold (Waters, cat. no. WAT200606).
Eppendorf Thermomixer (VWR, cat. no. 89428-662)
Column for bRP separation: Zorbax 300 Extend, 2.1-mm i.d. × 250-mm length (Agilent, custom order)
Solid-phase extraction cartridge for sample desalting: Oasis HLB 1-cc Vac Cartridge, 30 mg of sorbent per cartridge, 37–55 μm particle size (Waters, cat. no. WAT036790)
Solid-phase extraction disk for stage tips: Empore C18 extraction disk (Supelco, cat. no. 66883-U)
Stainless-steel blunt-end needle, 16 gauge (Cadence Science, cat. no. 7938)
Adaptor for stage-tipping (Glygen, cat. no. CEN.24)
PicoFrit column, 360-μm o.d. × 75-μm i.d., 10-μm i.d. tip, 50-cm length (New Objective, cat. no. PF360-75-10-N-5)
300-μl autosampler vial, for LC–MS (Waters, cat. no. 186002639)
300-μl autosampler cap, for LC–MS (Waters, cat. no. 186000305)
LC system for online LC–MS analysis (Thermo Fisher Scientific, Proxeon Easy-nLC, model no. 1000) ▲ CRITICAL We use a Proxeon Easy-nLC 1000 conditions. However, any LC system that can deliver nanoflow rates and can operate up to a pressure of 1,000 bar can be used for peptide separation.
20-cm nanospray column heater (Phoenix S&T, cat. no. PST-CH-20U)
Column heater controller (Phoenix S&T, cat. no. PST-CHC)
Mass spectrometer (Thermo Fisher Scientific, model no. Q Exactive Plus)
Spectrum Mill MS Proteomics Workbench (Agilent Technologies)
Vacuum manifold (Waters)
REAGENT SETUP
Solutions for IgY14 and SuperMix depletion of human plasma
Dilute 10× stocks of all the buffers (dilution, stripping and neutralization) tenfold with HPLC-grade water. Buffer usage is ~148 ml of 1× dilution buffer per depletion, ~170 ml of 1× stripping buffer per depletion and 96 ml of 1× neutralization buffer per depletion. Prepare corresponding total volumes based on the number of depletions, adding an extra 10%.
Solution for conditioning concentrator filters
Prepare a 0.1% (wt/vol) ODG solution by dissolving 0.08 g of ODG in 80 ml of 1× dilution buffer.
Plasma digestion buffer
Prepare a 50 mM ammonium bicarbonate solution at pH 8.0 by adding 3.953 g of ammonium bicarbonate to 1 L of water. Measure and make sure that the pH is 8.0.
Solid-phase extraction desalting solvents
Solvent A, for equilibration and washing the cartridge, is 0.1% (vol/vol) FA, and solvent B, for elution, is 0.1% (vol/vol) FA in 80% (vol/vol) acetonitrile. Solid-phase extraction (SPE) desalting solvents can be made in advance and stored at RT; they are stable for up to several weeks.
Stage-tip desalting solvents
Solvent A, for equilibration and washing, is 0.1% (vol/vol) FA.
Solvent B, for elution, is 0.1% (vol/vol) FA in 50% (vol/vol) acetonitrile.
Stage-tip desalting solvents can be made in advance and stored at RT; they are stable for up to several weeks.
bRP solvents
Stock solution is 200 mM ammonium formate (NH4HCO2); to make 1 liter, slowly add 27.8 ml of 28% (wt/vol) ammonium hydroxide to 500 ml of HPLC-grade water, then add ~45 ml of 10% (vol/vol) FA to bring the pH to 10; bring the final volume to 1 liter with HPLC-grade water.
bRP solvent A is 20 mM ammonium formate, pH 10, in 2% (vol/vol) acetonitrile.
bRP solvent B is 20 mM ammonium formate, pH 10, in 90% (vol/vol) acetonitrile.
bRP solvents are stable at RT for up to several days.
UPLC–MS/MS solvents
Solvent A is 0.1% (vol/vol) FA in 3% (vol/vol) acetonitrile and solvent B is 0.1% (vol/vol) FA in 90% (vol/vol) acetonitrile. UPLC solvents are stable for up to a month at RT.
EQUIPMENT SETUP
Plasma depletion setup on the Agilent 1200 system: tandem IgY14–SuperMix depletion
To accommodate tandem column depletion, specific plumbing changes, modified methodologies and the use of a column-switching valve are required. Each are described below in detail. In addition, a simplified setup is also described for IgY14-only depletion.
Plumbing changes specific to the Agilent 1200 system are shown in Supplementary Figure 1. Briefly, to increase the sample loop volume, replace the needle seat extension tubing with a 2-ml PEEK sample loop. Replace the following tubing: line 1 (from the purge valve/pump to port 1 on the injection valve), line 2 (from port 6 of the injection valve to top of the IgY14 depletion column), line 3 (from the bottom of the IgY14 column to port 4 of the column-switching valve), line 4 (port 5 of the column-switching valve to the top of the SuperMix column), line 5 (from port 3 of the column-switching valve to the low dead volume T connector, which is also connected to the bottom of the SuperMix column) and line 6 (from the T connector to the detector) with 0.01-inch i.d., 1/16-inch o.d. PEEK tubing. ▲ CRITICAL It is important to note that, although our work was accomplished on an Agilent 1200 system, this procedure is universal to any high-pressure LC system. Loop capacity is ideal for 2-ml injection volumes, as neat plasma (maximum amount of 400 μl) must be diluted fivefold in 1× dilution buffer for optimal column performance. In addition, the inclusion of a 6-port column-switching capability between the tandem columns is required. Any HPLC system that can accommodate these requirements and handle 500 p.s.i. (34 bar) column backpressure is suitable to accomplish this workflow.
Injector-specific modifications to the Agilent 1200 system
Because of limitations in loop sizes >900 μl on the Agilent system, the incorporation of the 2-ml needle seat extension is required to accommodate larger injection volumes. During method editing, replace the standard injector parameters in the ‘Use injector’ program with those shown in the table below. In this scheme, a coordinated injection protocol delivers three separate sample volumes from three separate, specifically located sample vials (vial positions 1 (750 μl), 3 (750 μl) and 5 (500 μl)) for a total of 2 ml injected per sample. Set the draw speed to 100 μl/min, eject speed to 100 μl/min and draw position to 1 mm in the ‘general parameters’ section.
|
| |
| No. | Command |
|
| |
| 1 | DRAW 750 μl from sample (vial position 1) |
| 2 | EJECT 750 μl into seat |
| 3 | DRAW 750 μl from sample +2 (vial position 3) |
| 4 | EJECT 750 μl into seat |
| 5 | DRAW 500 μl from sample +4 (vial position 5) |
| 6 | INJECT |
|
| |
▲ CRITICAL This protocol is written for a 400-μl neat plasma sample. If less material is to be depleted, the volumes and/or number of vials to be injected from can be modified. For example, if 200 μl of neat plasma is to be depleted, the final diluted plasma volume would be 1 ml, which could be injected from only two vials as 500-μl individual injections.
Incorporation of column switching
In the ‘column thermostat’ module of the method builder within the ChemStation vendor software, construct a timetable for column switching as shown in table below:
|
| ||
| Line | Time (min) | Column |
|
| ||
| 1 | 0 | Column 1 (IgY14) |
| 2 | 55.01 | Column 2 (SuperMix) |
| 3 | 86.01 | Column 1 (IgY14) |
|
| ||
Customized fractionation method setup for tandem IgY14–SuperMix depletion
Finally, fractionation parameters are set in the method as described in the table below:
|
| |||
| Line | Time (min) | Trigger mode | No. of fractions |
|
| |||
| 1 | 40.5 | Time-based | 15 |
| 2 | 55.5 | Off | |
| 3 | 68 | Time-based | 17 |
| 4 | 85 | Off | |
| 5 | 90 | Time-based | 9 |
| 6 | 99 | Off | |
|
| |||
The goal is to use timed fractionation to collect the three eluted peaks: the flow-through (IgY14 and SuperMix depleted material), followed by the IgY14-bound material and, later, the SuperMix bound fraction. Figure 2 shows a typical UV trace at 280 nm.
Figure 2.
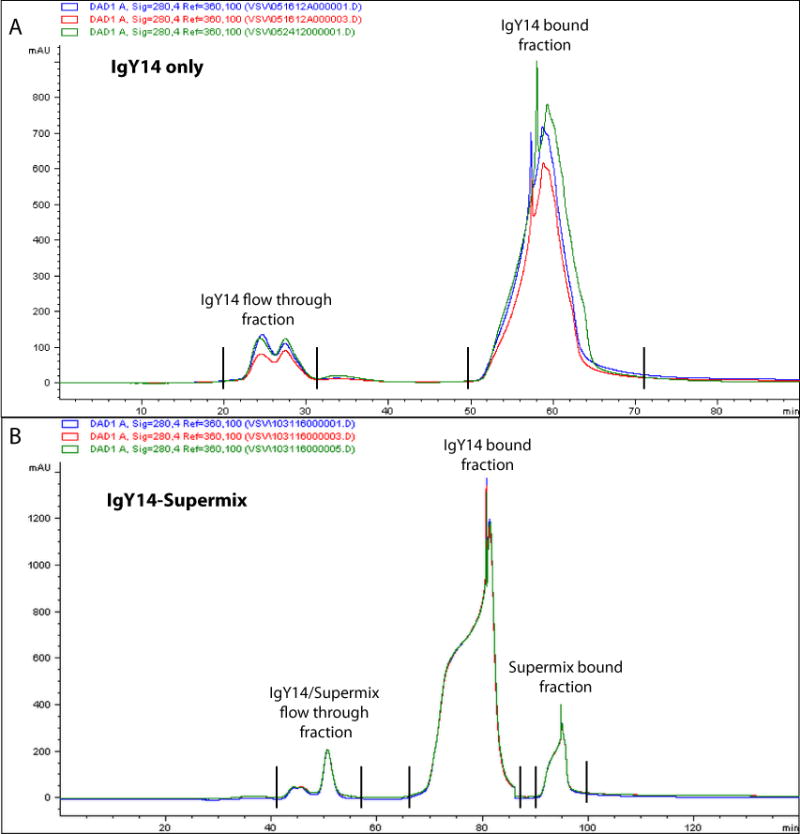
Chromatogram of plasma after immunoaffinity depletion. (a,b) Plasma chromatograms after IgY14-only (a) or tandem IgY14–SuperMix (b) depletion. Overlaid traces showing elution profiles of three reference plasma samples monitored at a 280-nm wavelength over the course of each depletion run. mAU, milli absorbance units.
Plasma depletion setup on the Agilent 1200 system: IgY14-only depletion
If IgY14-only depletion is desired, a simplified protocol can be constructed. Specific gradients for both tandem IgY14–SuperMix and IgY14 only are shown below. Each protocol uses the same regeneration method and the same customized injector protocol described above. Column switching is not used for IgY14-only depletion. However, the fractionation module is modified, as shown in the table below:
|
| |||
| Line | Time (min) | Trigger mode | No. of fractions |
|
| |||
| 1 | 22 | Time-based | 15 |
| 2 | 35 | Off | |
| 3 | 52 | Time-based | 17 |
| 4 | 69 | Off | |
|
| |||
Preparing the system for depletion
Once plumbing and method modifications are complete, purge all the solvent lines and then the entire system with water. Individually purge A1 with 1× dilution buffer, B2 with 1× stripping buffer and B1 with 1× neutralization buffer, each for 5 min at 5 ml/min. Run the dilution buffer for 15–30 min at 2 ml/min with no column. Connect the IgY14 and SuperMix columns and run the dilution buffer at 0.5 ml/min. Ramp up the flow rate to 2.0 ml/min and flush for another 30 min. If you are installing the column for the first time, run a blank injection before depleting samples. ▲ CRITICAL Depletion columns are stored at 4 °C indefinitely. They must reach RT before depletion. This can be achieved during the column flush. Do not exceed 34 bar of back-pressure on the column. When depletion is complete, flush the column(s) with 1× dilution buffer containing 0.02% sodium azide for 30 min at 2 ml/min and store the columns at 4 °C. ▲ CRITICAL Antibody-based columns require specific handling and care. It is prudent to periodically check system and column performance by depleting a reference plasma sample (e.g., from a commercial source such as BioreclamationIVT). We typically run a reference sample as the first depletion when the column has been stored for longer than 1 month. This strategy also allows for troubleshooting of typical LC issues, including leaks, clogged lines and any errors in methodologies and proper fractionation before applying the methods to actual patient samples.
Nanoflow C18 column
The nanospray column for online UPLC–MS/MS analysis is self-packed into a 75-μm i.d. PicoFrit column with ReproSil-Pur resin (120 Å, C18-AQ, 1.9 μm) to a length of 20–24 cm using a pressure bomb set to 950 p.s.i.
Chromatography gradients
bRP chromatography blank gradient: timetable summarizing the gradient used for conditioning the column before injection of the sample at a flow rate of 200 μl/min (Step 57).
|
| |
| Time interval (min) | Gradient (%B) |
|
| |
| 0 | 0 |
| 20 | 100 |
| 22 | 100 |
| 23 | 0 |
| 25 | 0 |
| 45 | 100 |
| 47 | 100 |
| 48 | 0 |
| 90 | 0 |
|
| |
bRP chromatography iTRAQ-labeled sample gradient: timetable summarizing the gradient used for fractionation of the iTRAQ-labeled sample at a flow rate of 200 μl/min (Step 59).
|
| |
| Time interval (min) | Gradient (%B) |
|
| |
| 0 | 0 |
| 5 | 0 |
| 13 | 15 |
| 46 | 28.5 |
| 51.5 | 34 |
| 64.5 | 60 |
| 90 | 60 |
|
| |
bRP chromatography TMT-labeled sample gradient: timetable summarizing the gradient used for fractionation of the TMT-labeled sample at a flow rate of 200 μl/min (Step 59).
|
| |
| Time interval (min) | Gradient (%B) |
|
| |
| 0 | 0 |
| 1 | 0 |
| 7 | 8 |
| 9 | 20 |
| 49 | 40 |
| 59 | 60 |
| 90 | 60 |
|
| |
UPLC–MS RP chromatography gradient: timetable summarizing the gradient for LC–MS/MS analysis of basic RP fractions at a flow rate of 200 nl/min (Step 66).
|
| |
| Time interval (min) | Gradient (%B) |
|
| |
| 0 | 0 |
| 2 | 6 |
| 152 | 35 |
| 160 | 60 |
| 163 | 90 |
| 173 | 90 |
| 174 | 50 |
| 184 | 50 |
|
| |
Gradient for unfractionated LC–MS/MS analysis: timetable summarizing the gradient for LC–MS/MS analysis of unfractionated sample at a flow rate of 200 nl/min (Step 72).
|
| |
| Time interval (min) | Gradient (%B) |
|
| |
| 0 | 0 |
| 2 | 6 |
| 242 | 20 |
| 272 | 35 |
| 280 | 60 |
| 283 | 90 |
| 293 | 90 |
| 294 | 50 |
| 304 | 50 |
|
| |
Q Exactive Plus HCD–MS/MS method
MS parameters used for the LC–MS/MS analysis of fractionated and unfractionated samples (Steps 66 and 72) are shown below.
|
| |
| Method parameter | Value |
|
| |
| Polarity | Positive |
| Full MS | |
| Microscans | 1 |
| Resolution | 70,000 |
| Automatic gain control (AGC) target | 3 × 106 ion counts |
| Maximum ion time | 5 ms |
| Scan range | 300–1,800 m/z |
| Data-dependent MS2 (dd-MS2) | |
| Microscans | 1 |
| Resolution | 17,500 (iTRAQ, TMT- 6); 35,000 (TMT-10) |
| AGC target | 5 × 104 ion counts |
| Maximum ion time | 120 ms |
| Loop count | 12 |
| Isolation window | 1.6 m/z |
| Isolation offset | 0.3 m/z |
| Fixed first mass | 100 m/z |
| Normalized collision energy | 27 (iTRAQ), 29 (TMT) |
| Data-dependent (dd) settings | |
| Underfill ratio | 5% |
| Charge exclusion | Unassigned, 1, 7 |
| Peptide match | Preferred |
| Exclude isotopes | On |
| Dynamic exclusion | 20 s |
|
| |
▲ CRITICAL 17,500 resolution is used for MS2 in the experiments in which an iTRAQ or TMT-6 labeling strategy is used, as it provides enough resolution for resolving reporter ions. However, to resolve C and N series reporter ions for the TMT-10 labeling strategy, 35,000 resolution is used for MS2. ▲ CRITICAL We optimized collision energy for our instrument and used normalized collision energy (NCE) values of 27 and 29 for the iTRAQ and TMT tags, respectively. To achieve optimal fragmentation of iTRAQ- or TMT-labeled peptides, collision energy should be optimized on the instrument used for analyses of those samples.
PROCEDURE
Plasma sample preparation for abundant protein depletion ● TIMING 30 min + overnight conditioning of concentrators
-
1|
Prepare Millipore Amicon concentrators a day before the depleted samples are ready. Condition them by adding 2 ml of 0.1% (wt/vol) ODG solution to the concentrator and let them stand at RT overnight.
▲ CRITICAL STEP Passivation and preconditioning of concentrator membrane material is considered prudent in order to achieve minimal nonspecific peptide binding and subsequent loss.
-
2|
Remove a 3- to 5-μl unprocessed plasma pre-depletion sample. This sample can be stored at 2–8 °C for 24 h for measuring the pre-depletion protein concentration using a BCA assay at a later time point (Step 18).
-
3|
Prepare dilution of plasma by adding 400 μl of plasma to 1,700 μl of 1× dilution buffer (Reagent Setup). An excess volume of 100 μl is included to account for sample volume losses during the spin filter step. The final effective dilution is slightly more diluted than the recommended fivefold dilution.
-
4|
Add 700 μl of diluted plasma to three separate 0.22-μm cellulose acetate spin filter tubes. Spin at 16,000g at 4 °C for 10 min.
-
5|
Transfer the combined flow-through from all spin filters and re-distribute into three separate 1-ml autosampler vials.
▲ CRITICAL STEP Some volume may be lost in the spin filters. Ensure that the final volume of diluted plasma in the three 1-ml autosampler vials is 780 μl, 780 μl and 530 μl (30 μl more than each of the three sample pickups; see Equipment Setup) for the user-defined injector sequence. If not, add 1× dilution buffer to compensate.
Plasma depletion of highly abundant proteins ● TIMING 4 h
-
6|
Highly abundant plasma proteins can be depleted either by using an IgY14 column (option A) or by using IgY14 and SuperMix tandem columns (option B).
-
Plasma depletion using only an IgY14 column ● TIMING 3 h per sample
- For depletion of plasma using only the IgY14 column, use the step elution timetable and flow-rate settings outlined in the table below. Measure the UV absorbance at 280 nm. Depleted plasma elutes as a single peak from ~22 min to 35 min into 12–15 fractions, each containing 2 ml (Fig 2a).
Time interval (min) Dilution buffer (%) Stripping buffer (%) Flow rate (ml/min)
0 100 0 0.5 22 100 0 0.5 22.01 100 0 2 40 100 0 2 40.01 0 100 2 90 0 100 2
-
Plasma depletion using IgY14–SuperMix tandem columns ● TIMING 4 h per sample
▲ CRITICAL For large-scale studies, obtain the appropriate number of IgY14–SuperMix columns to ensure that the same lot of antibodies is being used for all of the samples.
- For depletion of plasma using both IgY14 and SuperMix columns, use the step elution timetable and flow-rate settings outlined in the table below. Measure the UV absorbance at 280 nm.
Time interval (min) Dilution buffer (%) Stripping buffer (%) Flow rate (ml/min)
0 100 0 0.5 40 100 0 0.5 40.01 100 0 2 55 100 0 2 55.01 0 100 2 140 0 100 2
Inject the sample. The depleted plasma elutes as a single peak from ~37 min to 52 min into 12–15 fractions, each containing 2 ml (Fig. 2b). Fractions of the SuperMix-bound material must be collected only if the SuperMix column is being used as a form of abundance-based fractionation rather than for depletion.
-
-
7|After each depletion step, regardless of depletion strategy, neutralize the acidic stripping conditions and re-equilibrate the column using the timetable outlined below before the next plasma sample is run.
Time interval (min) Neutralization buffer (%) Dilution buffer (%) Flow rate (ml/min)
0 100 0 2 48 100 0 2 48.01 0 100 2 97 0 100 2
? TROUBLESHOOTING
Concentration and buffer exchange of flow-through fractions after depletion ● TIMING 5 h per set of 4–8 samples, depending on centrifuge rotor configuration
-
8|
Spin the 0.1% (wt/vol) ODG solution for ~30 min at 4,000g at 4 °C, and dispose of both filtrate and retentate as waste. Leave the membrane moist until ready for the addition of the samples.
-
9|
Choose the fractions that contain the peak representing the depleted portion (typically distributed within 12 fractions). While selecting fractions, keep in mind that the total capacity of a single Amicon concentrator is no more than 15 ml. In this example, a sequence of two consecutive concentration steps, each with 12 ml, efficiently achieves this.
-
10|
Pipette the first six selected fractions into a single concentrator (12 ml). Keep the collection plate refrigerated during all spins to reduce any possible sample degradation.
-
11|
Centrifuge at 4,000g for 45 min at 4 °C. The sample retentate should be ~250 μl. Collect the filtrate and freeze it at −80 °C by placing it in the −80 °C freezer. This sample can be further analyzed (i.e., metabolite and/or small-molecule profiling) at a later time point.
-
12|
Add the next six selected fractions onto the retentate of each concentrator (12 ml).
-
13|
Repeat Step 11.
-
14|
In case more than 12 fractions are selected, repeat Steps 10 and 11 for the other fractions.
-
15|
Perform a buffer exchange by adding 12 ml of the desired buffer (i.e., 50 mM ammonium bicarbonate with or without urea) to each concentrator. Centrifuge at 4,000g for 120 min at 4 °C.
▲ CRITICAL STEP Typically, 50 mM ammonium bicarbonate is used without urea. However, if digestion will immediately follow this step, urea can be added for a final concentration of 6 M.
-
16|
Transfer the retentate to a 1.5-ml tube. The sample volume should be ~250 μl. Rinse each concentrator by adding ~100–150 μl of additional exchange buffer and centrifuge at 4,000g for an additional 10 min at 4 °C. The goal is to rinse the membrane in order to optimize sample recovery while using minimal volume. We observe that most of the rinse volume is retained during this spin, probably because of the membrane condition at that stage.
-
17|
Transfer the sample retentate (~100 μl) to the previous sample retentate. The total volume should be ~ 300–350 μl. Add exchange buffer to achieve a final volume of 400 μl—the original plasma starting volume. If starting with another volume for plasma depletion (e.g., 200 μl or 300 μl), then after buffer exchange adjust the volume to be the same as the starting volume.
-
18|
Perform a BCA assay to determine the protein concentration in both the depleted and the previously saved nondepleted plasma sample from Step 2. Dilute the nondepleted plasma sample 1:100 in 50 mM ammonium bicarbonate. IgY14–SuperMix-depleted samples can be assayed as a 1:2 dilution. For comparison, if IgY14 only is used for depletion, the proper dilution for post-depletion BCA would be 1:5. The final concentrations for IgY14–SuperMix-depleted samples are usually ~0.5–2% of the original 70 mg/ml of unprocessed plasma (0.35–1.4 mg/ml), whereas for IgY14 only the yield ranges from 3.5 to 5% (2.45–3.5 mg/ml).
■ PAUSE POINT Samples can be frozen and stored at −80 °C for several weeks if the final buffer does not contain urea. Samples or solutions are frozen by placement into −80 °C freezer. Thawing of previously frozen samples is performed gently on ice.
Plasma sample preparation for enzymatic digestion ● TIMING 1.5 h
▲ CRITICAL We prepared the entire 400 μl of depleted plasma for digestion, but this step can be done with as little as 50 μl of depleted plasma. Note that it is important at this stage to have enough total protein per sample for chemical labeling and subsequent processing.
-
19|
Denature the plasma samples by adding dry urea to a final concentration of 6 M.
-
20|
Reduce disulfide bonds by adding DTT to a final concentration of 20 mM. Incubate the sample at 37 °C for 30 min.
-
21|
Alkylate reduced cysteine residues by adding iodoacetamide to a final concentration of 50 mM. Incubate at RT in the dark for 30 min.
Enzymatic digestion ● TIMING 15–18 h
-
22|
Reduce the urea concentration to 2 M by diluting the sample 1:3 with 50 mM ammonium bicarbonate.
-
23|
Add Lys-C at an enzyme/substrate ratio of 1:50 and incubate at 30 °C with shaking on a table-top shaker set at 850 r.p.m. for 2 h.
▲ CRITICAL STEP Before incubation, check the pH with pH indicator strips to make sure that the pH is 8.5. Adjust as needed with the addition of 5–10 μl of 1 M Tris buffer.
-
24|
Further dilute the urea concentration to <1 M with 50 mM ammonium bicarbonate.
-
25|
Add trypsin at an enzyme/substrate ratio of 1:50 and incubate at 37 °C with shaking on a table-top shaker set at 850 r.p.m. overnight (15−18 h).
▲ CRITICAL STEP Again, before incubation, check the pH with pH indicator strips to make sure that the pH is ~8. Adjust as needed with the addition of 1 M Tris buffer.
-
26|
Quench the digestion with FA to a final concentration of 1%.
Desalting of digested plasma sample by SPE ● TIMING 1 h per set of four samples + >3 h drying time
▲ CRITICAL We use Oasis HLB 1 cc (30 mg) SPE cartridges (Waters) for desalting 90–300 μg of plasma. The size of the cartridge should be adjusted when desalting less or more plasma sample. Waters recommends a sample capacity of 0.03–1% of total cartridge sorbent. Desalting is achieved using a Waters extraction manifold.
-
27|
Wash the cartridge three times with 500 μl of SPE solvent B (see Reagent Setup for SPE desalting solvents).
▲ CRITICAL STEP These volumes are designed to efficiently bind and elute the specified amount of protein content for a cartridge of this size. Use of smaller- or larger-capacity cartridges would require smaller or larger volumes. Check with the vendor for recommendations.
-
28|
Equilibrate the cartridge four times with 500 μl of SPE solvent A (see Reagent Setup for SPE desalting solvents).
-
29|
Load the digested sample and adjust the vacuum to ensure that the flow rate is very slow. We estimate our flow rate at ~1 drop in 2–3 s.
-
30|
Wash four times with 750 μl of solvent A.
-
31|
Elute three times with 500 μl of solvent B into single 1.5-ml Eppendorf tubes.
-
32|
Freeze the sample by placing it in a −80 °C freezer, and completely dry it via vacuum centrifugation.
■ PAUSE POINT Samples can be frozen and stored at −80 °C for several weeks.
Sample preparation for chemical labeling ● TIMING 45 min
-
33|
Reconstitute the dried digested plasma in 0.1% (vol/vol) FA in 3% (vol/vol) acetonitrile.
▲ CRITICAL STEP The reconstitution volume can be the same as the original unprocessed plasma volume.
-
34|
Take an aliquot for BCA to determine the concentration of digested plasma (BCA is performed on a 1:10 diluted digest in triplicate).
-
35|
On the basis of the BCA results, make 100-μg aliquots of the digested sample, freeze them and completely dry them via vacuum centrifugation.
■ PAUSE POINT Samples can be frozen by placing them in a −80 °C freezer and can be stored at −80 °C for several weeks.
Labeling of the digested, depleted plasma samples
-
36|
The digested plasma samples can be labeled using iTRAQ 4-plex reagent (option A) or using TMT-6 or TMT-10 reagent (option B). Selection of the labeling strategy is based on the study goal and the number of samples included in the multiplex experiment. iTRAQ 4 provides somewhat larger numbers of confident protein identifications, whereas TMT reagents allow higher multiplexing and throughput (see Anticipated Results below).
-
iTRAQ labeling of digested, depleted plasma samples ● TIMING 1.5 h
▲ CRITICAL Labeling of plasma samples requires 2× more iTRAQ reagent than is typically used for peptide labeling in cell line or tissue extracts. On occasion, to achieve >95% labeling efficiency we have had to use additional reagent after label incorporation testing.
Keep the vials containing iTRAQ reagents on ice.
Vortex each reagent tube briefly and spin down on a benchtop centrifuge at RT for 15–20 s at 2,000g.
Combine the contents of two vials of the same iTRAQ reagent into one vial per label. Vortex well and spin down on a benchtop centrifuge at RT for 15–20 s at 2,000g.
-
Reconstitute dried 100-μg aliquots of digested plasma in 30 μl of 1 M TEAB.
▲ CRITICAL STEP For plasma labeling, we follow the vendor’s recommendation to use a 1 M TEAB solution instead of the dissolution buffer supplied in the iTRAQ kit, for better control of the reaction pH.
-
Add 50 μl of 100% ethanol per aliquot of reagent used for labeling.
▲ CRITICAL STEP The amount of ethanol added must be adjusted to the amount of iTRAQ reagent; e.g., two vials of iTRAQ reagent require the addition of 100 μl of ethanol to the plasma sample.
Transfer the contents of the appropriate iTRAQ label solution to the corresponding plasma sample.
Mix well and incubate at RT for 1 h with shaking on a table-top shaker set at 850 r.p.m.
Remove aliquots from each sample for the label incorporation and mixing quality control (QC) tests: take 3 μl per sample for the label incorporation test, and 2 μl per sample to be mixed with the other samples for the mixing test. Desalt these QC samples using stage tips (see below).
-
Freeze the samples at −80 °C until the QC samples are analyzed, typically for 1–2 d.
▲ CRITICAL STEP Do not quench the labeling reaction until assessment of the label incorporation test results.
▲ CRITICAL STEP The presence of ethanol in label reaction volumes can suppress complete freezing of samples. It is recommended to perform QC analysis as quickly as possible in order to quench and further process the sample.
-
TMT labeling of digested, depleted plasma samples ● TIMING 1.5 h
Reconstitute the peptides in 100 μl of dissolution buffer (50 mM HEPES) per 100 μg of digested plasma and vortex well to mix.
Reconstitute each TMT reagent (0.8 mg contained in each vial labels 100 μg of sample) in 41 μl of anhydrous acetonitrile. Keep on ice for 5 min.
Vortex the reagent tubes briefly and spin down on a benchtop centrifuge at RT for 15–20 s at 2,000g.
Add each reagent to the corresponding aliquot of digested plasma, mix well and incubate at RT for 1 h with shaking on a table-top shaker set at 850 r.p.m.
After 1 h, remove aliquots from each sample for the label incorporation and mixing QC tests: take 3 μl per sample for the label incorporation test and 2 μl per sample to be mixed with the other samples for the mixing test. Desalt these QC samples using stage tips (see below).
-
Freeze the samples at −80 °C until the QC samples are analyzed.
▲ CRITICAL STEP Do not quench the labeling reaction until assessment of the label incorporation test results.
-
Stage-tip desalting ● TIMING 1 h
-
37|
Prepare C18 stage tips by using Empore C18 extraction disks, as described by Rappsilber et al.40. Pack two plugs of C18 material into each stage tip (100-μl pipette tips) for a total binding capacity of ~25 μg total. Create extraction disks using a 16-gauge blunt-end metal needle to hole-punch the ~1-mm disks. We recommend using adaptors to the hold stage-tip pipette tip at the orifice of each 2-ml microcentrifuge tube.
-
38|
Condition the stage tips with 100 μl of methanol. Centrifuge at 3,000g for 3 min at RT and discard the liquid from the collection vial. Repeat this step two times. All subsequent centrifugation steps for stage-tip desalting are for the same duration at the same speed and at RT.
-
39|
Wash the stage tips with 100 μl of 50% (vol/vol) acetonitrile in 0.1% (vol/vol) FA (stage-tip solvent B); repeat the centrifugation step. Discard the liquid.
-
40|
Equilibrate the stage tips twice with 100 μl of 0.1% (vol/vol) FA (stage-tip solvent A). Repeat the centrifugation step. Discard the liquid.
-
41|
Reconstitute the samples in 100 μl of 0.1% (vol/vol) FA in 3% (vol/vol) acetonitrile and add the samples to the stage tip.
-
42|
Centrifuge the samples. Save the flow-through at 4 °C.
▲ CRITICAL STEP We typically save the flow-through of samples in case there is a catastrophic failure of the constructed stage tip. Recovery of the sample is not optimal but can be achieved. Once normal analysis of the sample is confirmed, the flow-through can be discarded.
-
43|
Wash the samples twice with 100 μl of solvent A. Repeat the centrifugation step. Discard the liquid.
-
44|
Replace the collection vial with a new 1.5-ml screw cap vial for the collection of eluate.
-
45|
Elute the sample with 60 μl of solvent B. Repeat the centrifugation step.
-
46|
Transfer the eluate to an HPLC vial, freeze it at −80 °C and completely dry it by vacuum centrifugation.
-
47|
Reconstitute the dried sample in 10 μl of 5% (vol/vol) FA in 3% (vol/vol) acetonitrile and analyze by LC–MS/MS to check the label incorporation (see below).
▲ CRITICAL STEP As it can take a considerable amount of instrument time to analyze each of the individual label incorporation test samples, particularly for TMT-10, one can consider analyzing the mixing test sample for both labeling efficiency and mixing consistency, and only analyze the individual label incorporation test samples for troubleshooting purposes if the labeling efficiency is suspect in the mixing test sample.
Data analysis and assessment of labeling efficiency ● TIMING 1 h
-
48|
In order to evaluate the completeness of free-amine labeling, configure the database searches to allow for both peptide N termini and lysine side chains to be present in either labeled or unlabeled form. With Spectrum Mill, accounting for partial labeling is accomplished with a fixed and mixed modifications search cycle strategy that runs a search four consecutive times with different sets of modifications in each round and then produces a single integrated output. The four cycles are as follows: (i) all unmodified, (ii) both peptide N termini and lysines labeled, (iii) only lysines labeled and (iv) only peptide N termini labeled. As the primary amine groups of lysine side chains (pKa ~10) are more reactive than peptide N-terminal amines (pKa ~7.5), incomplete labeling tends to be observed predominantly in the form of unlabeled peptide N termini. Furthermore, as long as a peptide includes at least one label, reporter ion quantification is viable. After running a four-cycle partial-labeling database search configuration with a suitable (1%) peptide spectral match (PSM)-level FDR threshold on a label-check aliquot, labeling efficiency percentage metrics are calculated in the Spectrum Mill Quality Metrics module: full label % = (100 × fully labeled PSMs)/total PSMs and label % = (100 × 1 or more labeled PSMs)/total PSMs.
▲ CRITICAL STEP We typically apply a threshold of 95% labeling. If the threshold is not met, then the samples will be re-labeled (see Troubleshooting section for relabeling procedure) before proceeding with further peptide-level fractionation. In our experience, TMT labeling is more robust and less likely to require re-labeling than iTRAQ labeling.
-
49|
Analyze the resulting mass spectrometry data from the mixing test sample to verify whether the total protein amount from each sample is the same. The Spectrum Mill Quality Metrics module does this by summing the reporter ion intensity (after applying isotopic correction) for each channel across all the confidently identified PSMs in the data set. Then, for each sample, the mixing percentages are calculated using the most abundant reporter ion channel sum as a common denominator, and each of the other channels as a numerator.
▲ CRITICAL STEP Unless particular samples are available in limited quantity, we require each sample to have a reporter ion intensity sum that is >50% of the most abundant sample.
? TROUBLESHOOTING
Quenching of labeling reactions ● TIMING 20 min+ >2 h drying time
-
50|
Once QC analysis is complete and shows satisfactory labeling (>95% labeling efficiency) and a 1:1:1:1 ratio in the mixing test, thaw the samples by placing them on ice.
-
51|
To quench the iTRAQ reaction, add 1 M Tris, pH 8, to each reagent–plasma reaction vial to a final concentration of 100 mM. Let it react for 15 min at RT with shaking at 850 r.p.m.
-
52|
To quench the TMT labeling reaction, add 8 μl of 5% hydroxylamine and let it react at RT for 15 min with shaking at 850 r.p.m.
-
53|
Mix all the labeled samples together and dry by vacuum centrifugation.
■ PAUSE POINT Samples can be frozen and stored at −80 °C for 1–2 d if needed.
-
54|
Reconstitute the sample in 500 μl of 0.1% (vol/vol) FA in 3% (vol/vol) acetonitrile for desalting.
Desalting of labeled plasma sample by SPE ● TIMING 1 h per sample + >3 h drying time
-
55|
Desalt the labeled plasma sample by following Steps 27−32.
(Optional) Basic pH reversed-phase fractionation ● TIMING 2.5 h per separation + >2 h drying time
▲ CRITICAL Perform the steps below only if fractionation of the sample for deep plasma analysis is desired. If you are analyzing the sample without fractionation with single LC–MS/MS, proceed to Step 62. We use 2.1 mm × 250 mm columns for bRP fractionation, which are suitable for 100–400 μg of total loading capacity; if you are working with lower amounts of total protein, the gradients are transferrable to 1 mm × 250 mm microscale separations run at 50 μl/min for up to a 50-μg total load.
-
56|
Perform a QC of the HPLC system before fractionation. We typically accomplish this by injecting peptide standards in triplicate before running the actual sample. For this scale of bRP, 50 pmol of a mixture of seven in-house synthetic peptide standards is injected and evaluated for retention time reproducibility, signal intensity and peak resolution (Fig. 3a). Selection of synthetic peptides should be based on their availability, as well as chromatographic behavior such as retention times spanning the duration of the gradient. For example, tryptic peptides of 10–15 aa in length with good solubility in aqueous buffer and consistent elution behavior on RP C18 are ideal. We choose peptides that are either from a species different from that of our sample of interest or that are isotopically labeled with heavy amino acids to reduce any contamination in subsequent runs on the column. In addition, column backpressure is informative for system performance. For this scale and configuration, backpressure is typically in the 90–110 bar range.
Figure 3.
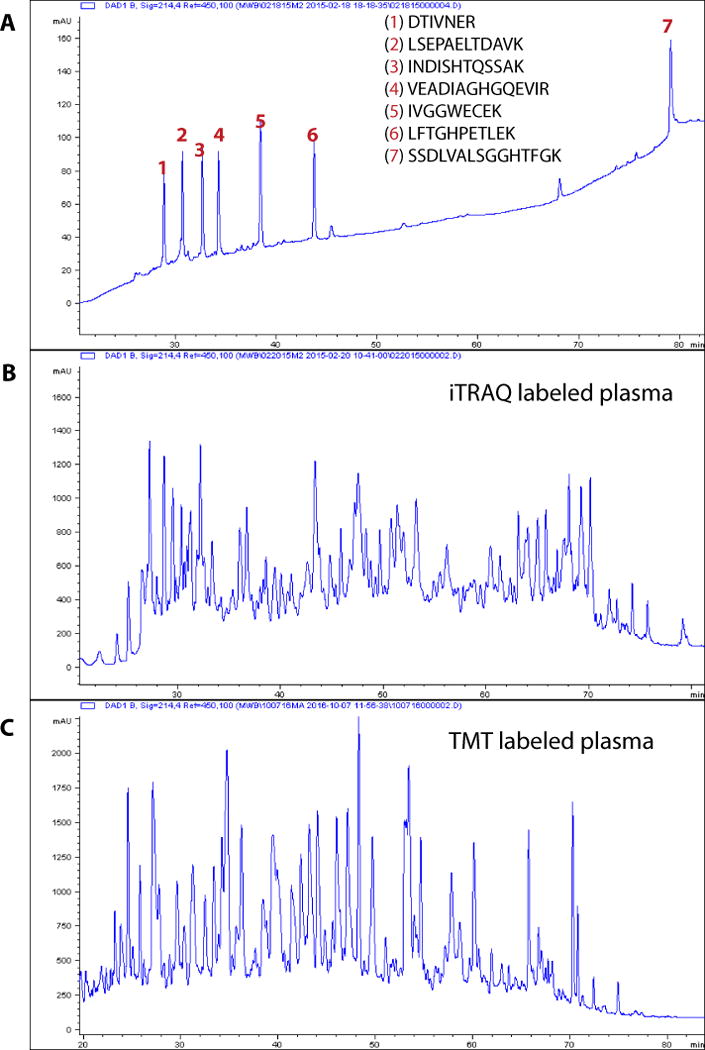
QC of bRP chromatography and representative bRP chromatograms of iTRAQ- and TMT-labeled plasma peptides. (a–c) Basic pH reversed-phase chromatograms of a standard peptide mixture (a) and plasma (b,c). Mixture consisting of seven peptides is used as a QC of the HPLC system. Sequences of peptides and their elution profiles monitored at 214 nm are shown in a. Typical elution profiles of depleted, digested and iTRAQ- or TMT-labeled plasma monitored at 214 nm are shown in b and c, respectively.
? TROUBLESHOOTING
-
57|
Prepare the LC system for sample injection. Single sample separation consists of a sequence of two methods: blank gradient followed by the sample gradient. Once the QC of peptide standards indicates that the system is running properly, run a blank gradient to condition the column for the sample (see Equipment Setup). This is achieved by injecting 10 μl of bRP solvent A (refer to Reagent Setup for bRP solvents) from an HPLC vial with the gradient outlined below as blank gradient.
-
58|
Upon completion of the blank run, reconstitute the sample in 540 μl of bRP solvent A. (see Reagent Setup for bRP solvents.) Vortex for 30 s. Centrifuge for 10 min at 10,000g at 4 °C to remove any material that did not go into solution. Transfer 530 μl to the HPLC vial. Inject 500 μl.
▲ CRITICAL STEP Extra volume is needed to avoid injecting air bubbles into the system during the sample injection.
-
59|
For separation of iTRAQ- and TMT-labeled samples, use the corresponding gradient and flow-rate settings outlined in the tables in the Equipment Setup. For an example chromatogram, see Figure 3b,c. In either bRP separation, 88 total fractions are collected into a Whatman 2-ml 96-well plate at a flow rate of 200 μl/min. For the iTRAQ-labeled sample, collection begins at 4 min, resulting in two fractions collected for the first 16 min (1.6 ml per well), followed by 84 fractions collected for 54 min (128.6 μl per well), followed by the final 10 min collected into two wells (1 ml per well). For the TMT-labeled sample, collection begins at 6 min, with the first 18 min collected into two wells (1.8 ml per well), followed by 52 min collected into 84 fractions (123.8 μl per well), followed by the final 14 min collected into two wells (1.4 ml per well).
-
60|
After separation, pool the bRP fractions (as described below) to generate 30 final fractions. The plate layout describes the recombination of wells, in which A1 is combined with H3 as fraction 1, A2 is combined with H4 as fraction 2, A3, C7 and E11 are combined as fraction 3 and so on (see the table below). Recombined pools for fractions 3 through 30 can be placed directly into HPLC vials (maximum volume is 400 μl) for reduced handling, although other vials with larger maximum volumes are commercially available.
▲ CRITICAL STEP Concatenation can be achieved into even smaller subsets depending on the number of final fractions desired. Our method reduces 84 individual wells to 28 final fractions (three wells with disparate hydrophobicity) plus 2 fractions from early and late portions of the chromatogram that are sparser in peptide content. For example, one could also recombine the 84 fractions into 12 repooled fractions by combining seven evenly distributed collected wells.
|
| ||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
|
| ||||||||||||
| A | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| B | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| C | 25 | 26 | 27 | 28 | 29 | 30 | 3 | 4 | 5 | 6 | 7 | 8 |
| D | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 |
| E | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | 3 | 4 |
| F | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
| G | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 |
| H | 29 | 30 | 1 | 2 | – | – | – | – | – | – | – | – |
|
| ||||||||||||
-
61|
Vacuum-centrifuge the pooled fractions to dryness.
■ PAUSE POINT The samples can be frozen by placing them in a −80 °C freezer, and they can be stored at −80 °C until ready for LC–MS/MS analysis.
UPLC–MS/MS for deep proteome analysis with 30 fractions ● TIMING 5 d per multiplexed sample
-
62|
Reconstitute each fraction in 20 μl of 5% (vol/vol) FA in 3% (vol/vol) acetonitrile solution, vortex for 2 min and spin down in a benchtop centrifuge at RT for 30 s at 2,000g.
-
63|
Set up the nanospray column packed to a 20-cm length (see Equipment Setup) with the column heater set to 50 °C.
-
64|
Set up the UPLC system with UPLC solvents A and B (see Reagent Setup for UPLC solvents).
-
65|
Before injection of the samples, run a QC test of the LC and MS performance by injecting a standard peptide mixture.
-
66|
Inject 2 μl of each fraction for LC–MS/MS analysis. LC and MS parameters used specific to the UPLC and Q Exactive Plus MS systems are outlined in the Equipment Setup.
▲ CRITICAL STEP Run a peptide QC sample after every 6 fractions to clean the system and check the mass accuracy.
-
67|
Freeze the remaining sample for fractions by placing them in a −80 °C freezer space.
UPLC–MS/MS for single (unfractionated) LC–MS/MS analysis of depleted, digested plasma sample ● TIMING 5 h per sample
-
68|
Reconstitute the sample at 1 μg/μl in 5% (vol/vol) FA in 3% (vol/vol) acetonitrile solution, vortex for 2 min and spin down in a benchtop centrifuge at RT for 30 s at 2,000g.
-
69|
Set up the nanospray column packed to a 50-cm length (see Equipment Setup) with the column heater set to 50 °C.
-
70|
Set up the UPLC system with UPLC solvents A and B (see Reagent Setup for UPLC solvents).
-
71|
Before injection of the samples, run a QC test of the LC and MS performance by injecting a standard peptide mixture.
-
72|
Inject 2 μl of the sample for LC–MS/MS analysis. The LC gradient used for this analysis is outlined in the Equipment Setup. Use the same MS parameters on the Q Exactive Plus MS system as described in the Equipment Setup.
▲ CRITICAL STEP Run a peptide QC sample after every six samples to clean the system and check the mass accuracy.
-
73|
Freeze the remaining sample for fractions by placing it in a −80 °C freezer space.
Data analysis ● TIMING >1 d
-
74|
Analyze the data using the Spectrum Mill MS Proteomics Workbench. Other search engines can also be used for searching the data, as long as the program can handle iTRAQ/TMT data. Use the parameters from the table below for searching plasma data for discovery:
-
75|Analyze the final sample data set with a two-cycle fixed and mixed modifications search in Spectrum Mill. This option runs two consecutive searches with different sets of fixed modifications in each round and then produces a single integrated output. The two cycles allow for (i) both labeled peptide N termini and labeled lysines and (ii) only labeled lysines. Other search engines might allow similar strategies to be executed by configuring the label as a variable modification on peptide N termini and on lysines.
Parameter Value
Variable modification Fixed modification Oxidation (M) Acetyl (Protein N-term) Deamidation PyroGlu (Q) Carbamidomethyl (C) iTRAQ or TMT-10 Maximum missed cleavages 4 Maximum charge 5 Precursor mass tolerance (p.p.m.) 20 Product mass tolerance (p.p.m.) 20 Peptide FDR 0.01 Protein FDR 0
-
76|
Correct the reporter ion intensities for isotopic impurities before using the reporter ion signals in each MS/MS spectrum for quantitative calculations41.
▲ CRITICAL STEP Each lot of reagent obtained from its manufacturer is accompanied by a certificate of analysis (this is also available on the manufacturer’s website) that contains isotopic correction factors to be used by the quantification software. These correction factors are primarily used to account for naturally occurring levels of 13C at the unlabeled carbons in the mass-tag portion of the labeling reagents. Because very-high-purity sources of 15N and 13C are routinely available and used for the labeled positions in the reagent, nearly full incorporation is typical. The correction factors provided by the reagent manufacturer represent mass spectrometry measurements of the isotope profile for each lot of reagent and thus co-mingle the contributions of unlabeled carbon and source of heavy isotope. Because there are more unlabeled carbons in the TMT structure than the iTRAQ structure, attention to isotopic correction to achieve accurate quantification is more important for TMT than for iTRAQ.
▲ CRITICAL STEP In complex samples such as plasma, the typical precursor mass window used for MS/MS (i.e., 0.7–3.0 m/z) passes more ions than just the dominant peak in any given m/z window. If these additional ions have an isobaric mass-tag reporter as part of the structure, then fragmentation of these ions will produce mass-tag ions that add to the reporter ion series from the dominant peak29,30. As most peptides in a sample derive from proteins whose levels are not changing (or not changing significantly), the interfering signals from this background in every mass-tag channel compresses the observed ratios of regulated peptides/proteins in the samples. When combining reporter ion quantification from multiple PSMs to calculate the protein level, several software programs offer mechanisms to exclude PSMs that exhibit significant interference. A precursor ion purity filter is commonly used that determines the ratio of the intensity of the precursor ion and isotopes of the primary peptide identified in the MS/MS spectrum to the total intensity in the mass window isolated for precursor ion transmission27,42. Use of a precursor ion purity of 50–70% is recommended. Various software packages use different mechanisms for combining the PSM-level measurements to determine the protein level from which the constituent peptides are derived. Spectrum Mill takes the ratios at the PSM level, and then calculates the protein-level ratio as the median of all PSM ratios27. This strategy diminishes the overall impact of outliers, but otherwise gives each PSM an equal contribution to the protein level ratio. Another common strategy is to sum the intensity for each reporter ion channel from the multiple PSMs contributing to the protein before taking the ratios. This strategy seeks to weight the contribution of each PSM by the reporter ion signal strength, with the implicit assumption that low-abundance signals are less accurate. Other variants of these basic approaches involve combining multiple PSMs to the peptide level first before calculating the protein level. These strategies seek to limit the bias of individual peptides that are observed in multiple PSMs because of different precursor charge states or sample-handling modifications.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
TABLE 2.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 7 | Decrease in the total number of proteins identified | Issue with depletion or depletion column aging | Run a QC the columns by digesting and analyzing an aliquot of depleted reference plasma sample (e.g. from a commercial source such as BioreclamationIVT) by LC-MS/MS. An Increase in the total intensity of the most abundant proteins targeted for depletion such as albumin and immunoglobulins would indicate degrading columns. See Supplementary Figure 2. |
| 49 | The iTRAQ labeling efficiency is < 95% | Insufficient labeling reagent is used | If iTRAQ labeling efficiency is suboptimal, a second reaction with another aliquot of label can be done to improve it. To do that, thaw the original reaction mixture, add 50μL of 100% ethanol and transfer contents of one vial of iTRAQ label solution to reaction mixture. React for 1 h at RT. Repeat the QC analysis. |
| The TMT labeling efficiency is < 95% | Insufficient labeling reagent was used | If TMT labeling efficiency is suboptimal, a second reaction with another aliquot of label can be done to improve it. To do that, thaw the original reaction mixture, and transfer contents of one vial of TMT label solution to reaction mixture. React for 1 h at room temperature. Repeat the QC analysis. | |
| 56 | Loss of resolution and/or broadening peak width of the peptide standards on bRP column | Deteriorating RP column or issue with the HPLC system | Troubleshoot the HPLC system to make sure the pumps, injector and fraction collector are performing optimally. If performance of HPLC system is optimal, test another RP column. |
| Increased back pressure on bRP column | Deteriorating RP column or issue with the HPLC system | Troubleshoot the HPLC system to make sure the pumps, injector and fraction collector are performing optimally. If performance of HPLC system is optimal, test another RP column. |
● TIMING
Steps 1–5, plasma sample preparation: 30 min + overnight conditioning of concentrators
Steps 6 and 7, plasma sample depletion: 4 h
Steps 8–18, concentration and buffer exchange: 5 h per set of 4–8 samples
Steps 19–21, sample preparation for enzymatic digestion: 1.5 h
Steps 22–26, enzymatic digestion: 15–18 h
Steps 27–32, peptide desalting by SPE: 1 h per set of four samples + >3 h drying time
Steps 33–35, sample preparation for chemical labeling: 45 min
Step 36A, chemical labeling using iTRAQ reagent: 1.5 h
Step 36B, chemical labeling using TMT reagent: 1.5 h
Steps 37–47, stage-tip desalting: 1 h
Steps 48 and 49, data analysis and assessment of labeling efficiency: 1 h
Steps 50–54, quenching of labeling reaction: 20 min + >2 h drying time
Step 55, labeled peptide desalting by SPE: 1 h + >3 h drying time
Steps 56–61, bRP fractionation: 2.5 h + >2 h drying time
Steps 62–67, LC–MS/MS of 30 fractions: 5 d per multiplexed sample
Steps 68–73, single LC–MS/MS: 5 h per sample
Steps 74–76, data analysis: >1 d
ANTICIPATED RESULTS
The number of peptides and proteins quantified in the deep, global discovery experiments of depleted plasma using the iTRAQ-4 and TMT-10 versions of the workflow are displayed in Table 3, together with the numbers obtained for single-shot, long-column analysis of depleted plasma in 10-plex.
TABLE 3.
Number of peptides and proteins quantified in a typical discovery experiment in plasma using this workflow.
| Number of peptides quantified
|
Number of proteins quantified with two or more peptides
|
|||
|---|---|---|---|---|
| IgY14 only | IgY14-SuperMix | IgY14 only | IgY14-SuperMix | |
| Deep profiling with iTRAQ 4-plex labelinga | 22,100b | 35,240b | 2,570b | 4,631b |
| Deep profiling with TMT 10-plex labelinga | NA | 33,079c | NA | 3,175c |
| Single shot analysis with TMT 10-plex labelingd | 4,361 | 9,811 | 317 | 652 |
Fractionation of labeled peptides into 30 fractions by basic RP before LC–MS/MS analysis.
Average number of peptides and proteins quantified in three iTRAQ experiments in three different patient samples.
Number of peptides and proteins quantified in a single TMT10 experiment.
Direct analysis after labeling (no peptide-level fractionation) by LC–MS/MS using a 50-cm column and a 4.5-h gradient.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
Typical chromatograms obtained when performing IgY14-only or IgY14–SuperMix depletions are shown in Figure 2a,b, respectively. The first peak to elute in both depletion strategies is the ‘flow-through’, reflecting the depleted sample that must always be collected. With IgY14–SuperMix depletion, another two peaks are observed, representing very highly abundant proteins bound to the IgY14 column and moderately abundant proteins bound to the SuperMix column. Deterioration of the column over multiple injections within a given sample analysis campaign can be assessed by analyzing the change in the peak area ratio of the flow-through from the IgY14–SuperMix to the IgY14 bound of the reference plasma sample (Supplementary Fig. 2). The SuperMix column eluate can be collected and analyzed separately if desired, in which case the column is being used as a form of abundance-based fractionation rather than exclusively for depletion. For multiple samples from the same patient population, depletion chromatograms should overlay tightly.
Concerns have been expressed about potential variability from injection to injection in immunoaffinity depletion of plasma, which could result in false positives and false negatives in the resulting proteomic discovery experiments13,43. The reproducibility of our entire workflow, including depletion, digestion, isobaric labeling, bRP fractionation and LC–MS/MS, has a median CV of 9% and a CV of <12% for 75% of the data, well within the acceptable range for discovery work (Fig. 4).
Figure 4.
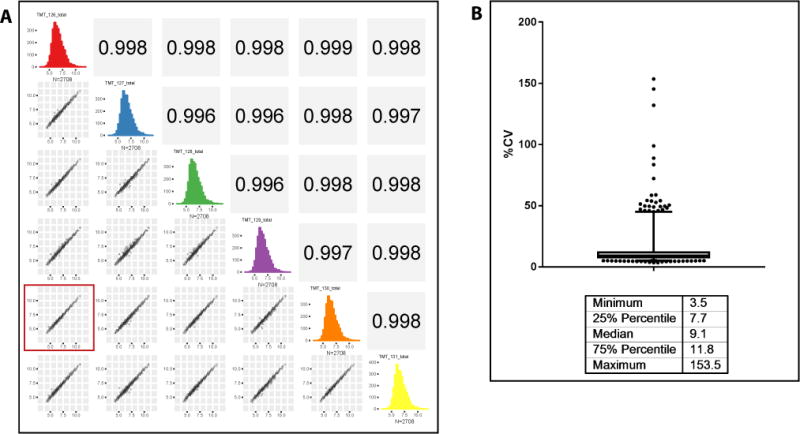
Reproducibility of the plasma processing workflow. Five different aliquots of a pooled plasma sample were depleted using the IgY14–SuperMix tandem depletion strategy and digested. Single 30-μl aliquots of each of the four depletions and two aliquots of the fifth depletion were labeled using TMT6 reagent. After assessing labeling efficiency, the samples were mixed and desalted. The multiplexed sample was fractionated by basic pH reversed-phase chromatography and all 30 fractions were analyzed on a Q Exactive Plus mass spectrometer using in-house packed 75-μm ID picofrit columns packed with 1.9-μm beads to 20-cm length. Data analysis was done using a Spectrum Mill MS Proteomics Workbench. Protein-level total intensity as reported by Spectrum Mill was used for assessing reproducibility. (a) Scatter plots showing correlation of all the replicates. The plot highlighted with the red square is for the two replicates of the same depletion, showing that the correlation is not any different from those for different depletions. (b) Bar and whisker plot of coefficients of variation (CV), showing their median and range.
For the PMI patient samples, we selected 4-plex iTRAQ labeling of the peptides because the change in relative protein abundance across the 4-time-point perturbational course within each patient was the most important comparison to be made. To obtain maximal depth of proteome coverage, labeled peptides derived from a total of 400 μg of protein (100 μg protein from each sample per iTRAQ channel) were partitioned into 30 concatenated bRP fractions. Each of these fractions contained >10 μg—a considerable excess, as we inject only a 1–2 μg total protein equivalent on 20 cm × 75 μm columns packed with sub-2-μm beads for LC–MS/MS analysis. As little as 100 μl of starting plasma volume would provide equivalent analysis depth and a sufficient amount of sample (after all sample processing) for a replicate injection, if required (Table 1). Using the workflow described above, including IgY14–SuperMix depletion, an average of 4,600 proteins were confidently detected and quantified in each of 16 patient samples with peptide FDRs <1.5% (Supplementary Fig. 3). The anticipated number of confidently identified and quantified proteins reduces to an average of 2,500 with the less intensive IgY14-only depletion38.
Fractionating multiplexed samples by bRP chromatography reduces the complexity of the sample, therefore increasing the total number of identifications. The gradients for separation of iTRAQ-4 versus TMT-6- or TMT-10-labeled peptides are different owing to the different chemical properties of the tags and have been individually optimized so as to obtain as equal a distribution of peptide peaks across the entire gradient as possible (for example, see Fig. 3). The chromatograms of patient samples from the same study should look very similar. The corresponding gradient is used with synthetic peptides to assess performance of the system and column over time; peaks in this elution profile should also overlay (Fig. 3a).
Specific experiments may prioritize throughput over depth; our protocol can flexibly accommodate those requirements. In cases in which larger numbers of samples are being compared, or when channels are reserved for a reference sample to facilitate quantitative comparison between experiments8, alternative reagents allow up to ten samples to be multiplexed, albeit with decrements in the number of proteins confidently identified and quantified (Table 3)44. Eliminating off-line fractionation greatly increases sample analysis throughput, but decreases the depth of coverage by 5–8 fold (Table 3). By combining labeling with TMT-10 and eliminating off-line fractionation before LC–MS/MS, up to 600 proteins can be confidently identified and quantified (with two or more peptides/protein) by single-shot LC–MS/MS analysis using 50-cm columns with a 4.5-h gradient and packed with sub-2-μm beads (Supplementary Fig. 4).
Supplementary Material
Acknowledgments
We thank N. Udeshi for reading the manuscript and providing valuable feedback. This work was supported in part by grants from the National Institutes of Health: HHSN268201000033C and R01HL096738 from the National Heart, Lung, and Blood Institute (NHLBI; to S.A.C.) and grants U24CA160034 from the National Cancer Institute (NCI) Clinical Proteomics Tumor Analysis Consortium initiative and U01CA152990 from the NCI Early Detection Research Network program (to M.A.G.).
Footnotes
AUTHOR CONTRIBUTIONS
H.K., M.W.B., H.S., K.R.C., M.A.G. and S.A.C. developed the protocol. L.W., M.W.B. and H.S. optimized and ran QC checks on many aspects of the protocol. H.K., M.W.B., H.S., K.R.C., M.A.G. and S.A.C. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html. Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 2.Hortin GL, Jortani SA, Ritchie JC, Jr, Valdes R, Jr, Chan DW. Proteomics: a new diagnostic frontier. Clin Chem. 2006;52:1218–1222. doi: 10.1373/clinchem.2006.067280. [DOI] [PubMed] [Google Scholar]
- 3.Pernemalm M, Lehtio J. Mass spectrometry-based plasma proteomics: state of the art and future outlook. Expert Rev Proteomics. 2014;11:431–448. doi: 10.1586/14789450.2014.901157. [DOI] [PubMed] [Google Scholar]
- 4.States DJ, et al. Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study. Nat Biotechnol. 2006;24:333–338. doi: 10.1038/nbt1183. [DOI] [PubMed] [Google Scholar]
- 5.Farrah T, et al. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.006353. M110,006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrah T, et al. State of the human proteome in 2013 as viewed through PeptideAtlas: comparing the kidney, urine, and plasma proteomes for the biology- and disease-driven Human Proteome Project. J Proteome Res. 2014;13:60–75. doi: 10.1021/pr4010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geiger T, Wehner A, Schaab C, Cox J, Mann M. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.014050. M111,014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mertins P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mertins P, et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics. 2014;13:1690–1704. doi: 10.1074/mcp.M113.036392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hortin GL, Sviridov D, Anderson NL. High-abundance polypeptides of the human plasma proteome comprising the top 4 logs of polypeptide abundance. Clin Chem. 2008;54:1608–1616. doi: 10.1373/clinchem.2008.108175. [DOI] [PubMed] [Google Scholar]
- 11.Pieper R, et al. Multi-component immunoaffinity subtraction chromatography: an innovative step towards a comprehensive survey of the human plasma proteome. Proteomics. 2003;3:422–432. doi: 10.1002/pmic.200390057. [DOI] [PubMed] [Google Scholar]
- 12.Qian WJ, et al. Enhanced detection of low abundance human plasma proteins using a tandem IgY12-SuperMix immunoaffinity separation strategy. Mol Cell Proteomics. 2008;7:1963–1973. doi: 10.1074/mcp.M800008-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi T, et al. IgY14 and SuperMix immunoaffinity separations coupled with liquid chromatography-mass spectrometry for human plasma proteomics biomarker discovery. Methods. 2012;56:246–253. doi: 10.1016/j.ymeth.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao Z, Tang HY, Wang H, Liu Q, Speicher DW. Systematic comparison of fractionation methods for in-depth analysis of plasma proteomes. J Proteome Res. 2012;11:3090–3100. doi: 10.1021/pr201068b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song C, et al. Reversed-phase-reversed-phase liquid chromatography approach with high orthogonality for multidimensional separation of phosphopeptides. Anal Chem. 2010;82:53–56. doi: 10.1021/ac9023044. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 2011;11:2019–2026. doi: 10.1002/pmic.201000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cominetti O, et al. Proteomic biomarker discovery in 1000 human plasma samples with mass spectrometry. J Proteome Res. 2016;15:389–399. doi: 10.1021/acs.jproteome.5b00901. [DOI] [PubMed] [Google Scholar]
- 18.Dayon L, Nunez Galindo A, Corthesy J, Cominetti O, Kussmann M. Comprehensive and scalable highly automated MS-based proteomic workflow for clinical biomarker discovery in human plasma. J Proteome Res. 2014 doi: 10.1021/pr500635f. [DOI] [PubMed]
- 19.Addona TA, et al. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat Biotechnol. 2011;29:635–643. doi: 10.1038/nbt.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huttenhain R, et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 2012;4:142ra194. doi: 10.1126/scitranslmed.3003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whiteaker JR, et al. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat Biotechnol. 2011;29:625–634. doi: 10.1038/nbt.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bantscheff M, et al. Robust and sensitive iTRAQ quantification on an LTQ Orbitrap mass spectrometer. Mol Cell Proteomics. 2008;7:1702–1713. doi: 10.1074/mcp.M800029-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erickson BK, et al. Evaluating multiplexed quantitative phosphopeptide analysis on a hybrid quadrupole mass filter/linear ion trap/orbitrap mass spectrometer. Anal Chem. 2015;87:1241–1249. doi: 10.1021/ac503934f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gan CS, Chong PK, Pham TK, Wright PC. Technical, experimental, and biological variations in isobaric tags for relative and absolute quantitation (iTRAQ) J Proteome Res. 2007;6:821–827. doi: 10.1021/pr060474i. [DOI] [PubMed] [Google Scholar]
- 25.McAlister GC, et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem. 2012;84:7469–7478. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McAlister GC, et al. Analysis of the acidic proteome with negative electron-transfer dissociation mass spectrometry. Anal Chem. 2012;84:2875–2882. doi: 10.1021/ac203430u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mertins P, et al. iTRAQ labeling is superior to mTRAQ for quantitative global proteomics and phosphoproteomics. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.014423. M111,014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nilsson CL, et al. Quantitative phosphoproteomic analysis of the STAT3/IL-6/HIF1alpha signaling network: an initial study in GSC11 glioblastoma stem cells. J Proteome Res. 2010;9:430–443. doi: 10.1021/pr9007927. [DOI] [PubMed] [Google Scholar]
- 29.Ow SY, et al. iTRAQ underestimation in simple and complex mixtures: ‘The Good, the Bad and the Ugly’. J Proteome Res. 2009;8:5347–5355. doi: 10.1021/pr900634c. [DOI] [PubMed] [Google Scholar]
- 30.Rauniyar N, Yates JR., III Isobaric labeling-based relative quantification in shotgun proteomics. J Proteome Res. 2014;13:5293–5309. doi: 10.1021/pr500880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ross PL, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 32.Savitski MM, et al. Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. J Proteome Res. 2013;12:3586–3598. doi: 10.1021/pr400098r. [DOI] [PubMed] [Google Scholar]
- 33.Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cole RN, et al. The plasma proteome identifies expected and novel proteins correlated with micronutrient status in undernourished Nepalese children. J Nutr. 2013;143:1540–1548. doi: 10.3945/jn.113.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones KA, et al. Immunodepletion plasma proteomics by tripleTOF 5600 and Orbitrap elite/LTQ-Orbitrap Velos/Q exactive mass spectrometers. J Proteome Res. 2013;12:4351–4365. doi: 10.1021/pr400307u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burgess MW, Keshishian H, Mani DR, Gillette MA, Carr SA. Simplified and efficient quantification of low-abundance proteins at very high multiplex via targeted mass spectrometry. Mol Cell Proteomics. 2014;13:1137–1149. doi: 10.1074/mcp.M113.034660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keshishian H, Addona T, Burgess M, Kuhn E, Carr SA. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics. 2007;6:2212–2229. doi: 10.1074/mcp.M700354-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keshishian H, et al. Multiplexed, quantitative workflow for sensitive biomarker discovery in plasma yields novel candidates for early myocardial injury. Mol Cell Proteomics. 2015;14:2375–2393. doi: 10.1074/mcp.M114.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swearingen KE, Moritz RL. High-field asymmetric waveform ion mobility spectrometry for mass spectrometry-based proteomics. Expert Rev Proteomics. 2012;9:505–517. doi: 10.1586/epr.12.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 41.Shadforth IP, Dunkley TPJ, Lilley KS, Bessant C. i-Tracker: for quantitative proteomics using iTRAQ (TM) BMC Genomics. 2005;6:145. doi: 10.1186/1471-2164-6-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wenger CD, et al. Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat Methods. 2011;8:933–935. doi: 10.1038/nmeth.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel BB, et al. Assessment of two immunodepletion methods: off-target effects and variations in immunodepletion efficiency may confound plasma proteomics. J Proteome Res. 2012;11:5947–5958. doi: 10.1021/pr300686k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pichler P, et al. Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-plex compared to TMT 6-plex and iTRAQ 8-plex on LTQ Orbitrap. Anal Chem. 2010;82:6549–6558. doi: 10.1021/ac100890k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.