

Abstract
Cisplatin-complexed gold nanoparticles (PtII-AuNP) provide a promising strategy for chemo-radiation based anticancer drugs. Effective design of such platforms necessitates reliable assessment of surface engineering on a quantitative basis, and its influence on drug payload, stability and release. In this paper, polyethylene glycol (PEG) stabilized PtII-AuNP was synthesized as a model anti-tumor drug platform, where PtII is attached via a carboxyl terminated dendron ligand. Surface modification by PEG and its influence on drug loading, colloidal stability and drug release were assessed. Complexation with PtII significantly degrades colloidal stability of the conjugate; however, PEGylation provides substantial improvement of stability in conjunction with an insignificant trade-off in drug loading capacity compared with the non-PEGylated control (< 20 % decrease in loading capacity). In this context, the effect of varying PEG concentration and molar mass was investigated. On a quantitative basis, the extent of PEGylation was characterized and its influence on dispersion stability and drug load was examined using electrospray differential mobility analysis (ES-DMA) hyphenated with inductively coupled plasma mass spectrometry (ICP-MS) and compared with attenuated total reflectance - FTIR. Using ES-DMA-ICP-MS, AuNP conjugates were size-classified based on their electrical mobility, while PtII loading was simultaneously quantified by determination of Pt mass. Colloidal stability was quantitatively evaluated in biologically relevant media. Finally, the pH-dependent PtII release performance was evaluated. We observed 9 % and 16 % PtII release at drug loadings of 0.5 PtII/nm2 and 1.9 PtII/nm2, respectively. The relative molar mass of PEG had no significant influence on PtII uptake or release performance, while PEGylation substantially improved the colloidal stability of the conjugate. Notably, the PtII release over 10 days (examined at 0.5 PtII/nm2 drug loading) remained constant for non-PEGylated, 1K-PEGylated and 5K-PEGylated conjugates.
TOC
Introduction
Since its discovery as an antitumor agent, cisplatin or cis-diamminedichloroplatinum(II) (abbreviated herein as PtII), has been applied in the treatment of various solid tumors (e.g., ovarian and testicular cancer, germ cell tumors, and non-small-cell carcinoma) with a cure rate as high as 90 % for testicular cancer. [1–6] When entering tumor cells, the active PtII drug reacts with DNA to form adducts, inhibits its replication and triggers apoptosis (programmed cell death). The synergistic combination of PtII and ionizing radiation induces additional damage to DNA, and degrades cell viability through formation of toxic Pt intermediates and inhibition of the DNA repair process [4].
However, clinical use of PtII is limited by severe side effects and poor patient compliance. [7] Indiscriminate uptake of the drug into both normal and cancerous tissue, coupled with rapid clearance and excretion through the kidney, limits the achievable dose delivered to tumor cells.[8] Despite these issues, cisplatin continues to serve as a first-line chemotherapy against specific malignancies and has been described as the “gold standard” treatment for cervical cancer. [9, 10]
In this context, nanoparticle (NP)-based drug delivery systems are a promising approach to improve targeting and enable high delivered doses, while minimizing the toxic side-effects to healthy tissue.[11, 12] Use of NP delivery platforms may enhance the accumulation of the drug in tumors, minimizing either kidney excretion or uptake by healthy cells, via the well-documented enhanced permeation and retention (EPR) effect.[13, 14] In other words, by attaching the drug to NPs, the physicochemical properties of the resulting complex are changed, leading to altered pharmacokinetics and biodistribution profiles. NPs can also be engineered through interfacial chemistry to serve multiple functions in addition to drug delivery (e.g., active and passive targeting, avoidance of the reticuloendothelial system, reduced interaction with plasma proteins, imaging of localized drug delivery. [15–19]
For instance, AuNPs have been used as a platform for drug delivery, due in part to their biocompatibility, facile synthesis and functionalization, and physico-chemical stability.[20–22] Cytotoxicity in both lung epithelial and colon cancer cell lines, and an unusual ability to penetrate the cell nucleus, have been demonstrated when the chemotherapy drug oxaliplatin (another member of the class of Pt-containing anti-tumor drugs) is complexed with AuNPs.[23] Additionally, AuNPs complexed with Pt-based therapeutics may provide an enhanced radiation therapeutic effect.[24–26] Lee et al. demonstrated Auger electron emissions from PtII-AuNP complexes triggered by external ionizing radiation, suggesting its potential as an X-ray absorbing adjuvant agent for chemo-radiation cancer therapy. [26]
Although the PtII-complexed AuNP vector provides a promising strategy for the use of targeted drug delivery in cancer treatment, one challenge is to engineer this platform on a quantitative basis to achieve optimal therapeutic performance.[27] In terms of efficacy, both drug loading and the stability of the drug complex (i.e., colloidal stability and the stability of active drug under different conditions) are of central importance. An optimal design should afford rational control facilitated by an understanding of the underlying surface properties in order to achieve the desired outcome. To support a quality by design approach, it is also necessary to have measurement tools required to accurately assess the critical physical and chemical quality attributes of the nano-drug complex, as well to quantify drug uptake and release. This paper describes and demonstrates such a strategy based principally on coupling of an aerosol-based ion mobility technique with plasma-based mass spectrometry. As we will show, this strategy provides critical information and insight regarding the efficacy of surface engineering in the tested system.
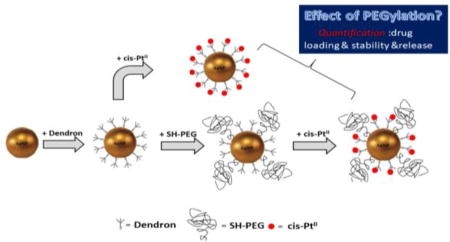
In the current study, we chose as our test bed a functional dendron structure, designated SH-G1-COOH (inset in Figure 1), which in our previous study exhibited the maximum available surface binding sites, but also the poorest colloidal stability. [27] The AuNP surface was modified with polyethyleneglycol (PEG) for the purpose of improving colloidal stability through additional steric hindrance. In addition, PEGylation has been shown to enhance biocompatibility and delay clearance, [28–30] though quantifying these effects was beyond the scope of this study. The present work investigates the effect of surface PEGylation on colloidal stability, drug loading and release performance using the test bed formulation.
Figure 1.

Cartoon depiction of conjugation and complexation steps in processing of AuNP-(dendron), AuNP-(dendron)-(SH-PEG) and PtII complexed products.
An electrospray differential mobility analyzer hyphenated with inductively coupled plasma mass spectrometry (ES-DMA-ICP-MS) or a condensation particle counter (CPC), as described in a previous publication, [31] was used in the present study on a quantitative basis to systematically evaluate: (1) the relationship between loading of PtII and colloidal stability in relevant (proxy) test media such as phosphate buffered saline (PBS) and Dulbecco's modified Eagle’s medium (DMEM); (2) the effect of PEGylation (packing density and relative molar mass) on colloidal stability and PtII loading capacity; and (3) pH-dependent release performance of PtII from the AuNP-dendron delivery platform and the consequent influence of PEGylation. Additionally, attenuated total reflectance - FT infrared (ATR-FTIR) spectroscopy was employed to assess PEG bonding onto the AuNPs.
Experimental
Materials
Citrate stabilized gold colloid (10 nm nominal particle diameter) was obtained from Ted Pella, Inc. (Redding, CA) and produced by BBI Solutions (Cardiff, UK).* Thiolated polyethylene glycol (SH-PEG, 1, 5 and 10 kDa) was purchased from Nanocs Inc. (New York, NY). Cis-diamminedichloroplatinum (II) (trade name Platinol; formula cis-PtCl2(NH3)2) and silver nitrate (AgNO3, 99 %) were purchased from Sigma-Aldrich (St. Louis, MO). Sodium hydroxide (NaOH, pellets, 99 %) was obtained from Mallincrodt Chemicals (Phillipsburg, NJ). Phosphate buffered saline (PBS10×) and Dulbecco’s modified Eagle’s medium (DMEM) were obtained from HyClone (Logan, UT). The composition of pH 5 buffer used for release experiment contains 137 mmol/L of NaCl, 2.7 mmol/L KCl, 3.48 mmol/L of HAc and 8.32 mmol/L NaOAc to better match the electrolyte content of PBS.
All chemicals were used as received without further purification. Deionized (DI) water (18.2 MΩ·cm) was provided by an Aqua Solutions (Jasper, GA) biological grade water purification system.
Methods
PtII Preparation
The activated Pt(II) moiety for complexation, cis-[PtII(NH3)2(H2O)2](NO3)2, was prepared by modification of a procedure reported in the literature, as follows:[32] An aqueous solution of AgNO3 (16.9 mg, 0.1 mmol) in water (2.5 mL) was added drop-wise to cisplatin (15 mg, 0.05 mmol) in DI water (2.5 mL) and stirred in the dark for 1 h at 50 °C. A white precipitate (AgCl) was removed by filtration and the filtrate of the activated cisplatin (with Cl− displaced by the more labile NO3−) was used for complexation with surface modified AuNPs diluted as needed (in this work, PtII always refers to the activated form of cisplatin). The dendron was synthesized using a previously described method.[33] Among the different structural forms of dendrons, we selected SH-G1-COOH because it yielded the highest loading capacity for cisplatin (PtII). G1 expresses a branched structure analogous to a 1st generation dendrimer terminated with three COOH groups. SH-G1-COOH has a wedge-shaped branching structure as shown in Figure 1. The COOH groups provide complexing sites for PtII, and the thiol group provides covalent bonding to the AuNP surface. To activate (deprotonate) the carboxylate group, the dendron (5 mmol/L) was dissolved in NaOH (15 mmol/L) to obtain the maximum carboxylate population.
Conjugation of AuNP-(dendron) and AuNP-(dendron)-(SH-PEG)
As illustrated in Figure 1 the initial AuNP-(dendron) conjugate and the modified conjugate AuNP-(dendron)-(SH-PEG) were used for comparison. Carboxylate terminated dendron (5 mmol/L) was added drop-wise to a AuNP suspension (57.6 µg/mL) in a 1-to-10 volume ratio, and stirred for 5 h at room temperature. The resulting product (designated as AuNP-(dendron)) was purified by centrifugal filtration using a 100 kDa regenerated cellulose membrane, at 4000 rpm for 5 mins.
A series of AuNP-(dendron)-(SH-PEG) samples were prepared by drop-wise addition of 1 part by volume of an aqueous solution containing (1, 5 or 10) kDa SH-PEG at concentrations of (0.1, 0.5, 1.0, and 5.0) mmol/L, to 11 parts by volume of purified AuNP-dendron conjugate suspension with stirring at room temperature for 5 h. The resulting products were designated as AuNP-(dendron)-(SH-PEG)-(50-1), AuNP-(dendron)-(SH-PEG)-(10-1), AuNP-(dendron)-(SH-PEG)-(5-1), and AuNP-(dendron)-(SH-PEG)-(1-1), where X-1 represents the molar ratio (X:1) of dendron to SH-PEG for the reaction. The designation “xK” appended to the product name is shorthand for “x kDa” relative molar mass of SH-PEG. The full naming scheme is, for example, AuNP-(dendron)-(SH-PEG5K)-(50-1), for the 5 kDa PEGylated dendron ligated AuNPs with a 50:1 molar ratio of dendron to SH-PEG in solution. For convenience, 5K is shorthand for SH-PEG5K in all the figures, and similarly for 1K and 10K.
After reaction, the mixture was again purified by centrifugal filtration, at 4000 rpm for 5 min. As a control, citrate-stabilized AuNPs were conjugated with SH-PEG, and labeled as AuNP-(SH-PEG)-(5mM) where 5 mmol/L SH-PEG solution was added drop-wise to AuNP suspensions in a 1 to 11 volume ratio (final SH-PEG concentration in solution was 0.42 mmol/L)
Complexation with cisplatin
A solution of PtII was added drop-wise to either AuNP-dendron suspension or PEGylated AuNP-(dendron) suspension (in a volumetric ratio of 1 to 10), and was then mildly shaken using a vortex mixer (VWR, Radnor, PA) at 400 rpm in the dark for 2 h at room temperature. The final concentration of PtII for the reaction ranged from 0.6 µmol/L to 34 µmol/L (of Pt to Au mass ratio 1:448 to 1:8). The AuNP-containing suspension was modified to yield a pH of roughly 7 by addition of NaOH and then followed by mixing with PtII. After complexation, any free (uncomplexed) PtII was removed by gently spinning down the NPs in a microfuge at 14500 rpm and replacing the supernatant with DI water. Actual spinning time may vary from 30 mins to more than 1 h depending the stability and volume of the sample. The cleaning step was examined and optimized to ensure that free PtII would not influence the PtII loading measurement. (Supplemental Information (SI) Figure S1(a))
The effect of reaction time on PtII complexation was also examined (SI Figure S1(b)), based on which a 2 h reaction time was deemed sufficient, as no significant increase in PtII loading was observed over longer reaction times.
Cisplatin release
Uncomplexed PtII was first removed from samples using two rounds of cleaning via centrifugation in a microfuge and supernatant replacement by DI water. PtII release studies were carried out by ICP-MS measurement of free PtII at time points of (0, 2 and 10) day, after separating free (released) PtII from AuNP-bound PtII using centrifugal filtration (3 kDa regenerated cellulose membrane). The total load of PtII on AuNPs was obtained by ICP-MS measurement of digested cleaned PtII-AuNP conjugates. The percentage of PtII release at a specific time is defined as the ratio of free PtII to that of the total PtII. Two buffers (PBS with a pH of 7.4 and pH 5 buffer) were used for release studies.
Measurements
ES-DMA-ICP-MS (CPC)
The schematic of the instrumentation as shown in Figure 2, is composed of four major components: an ES device (Model 3480, TSI Inc., Shoreview, MN, (a)–(b)) for aerosolizing nanoparticle suspensions, a DMA (Model 3085, TSI Inc., (c)) for size classification based on electrical mobility, a gas-exchange device (GED (d)) to exchange air (typical carrier gas for ES) to ICP-MS compatible argon, and an ICP-MS (7700× and 7900×, Agilent Technologies, Santa Clara, CA (e)) for element-specific mass quantification, or a CPC (Model 3776, TSI Inc.) for number concentration measurement (not shown in Figure 2). This configuration enables simultaneous size-resolved elemental-composition or particle concentration measurement. ES-DMA was operated with a sheath flow of 10 L/min (argon) and an aerosol flow of 1 L/min (air). Aerosol flow out of the DMA is interfaced to the ICP-MS through the GED directly into the plasma (similar to a GC-MS connection). Conductive silicone tubing (0.31” ID × 0.5” OD, TSI Inc., Shoreview, MN, USA) is used to couple between devices (where ID and OD stands for inner diameter and outer diameter, respectively). To make a direct measurement of particle counts as a function of size the output of the DMA is coupled to the CPC (inlet flow of 1.5 L /min) to obtain a number-weighted particle size distribution. A detailed description of the ES-DMA setup can be found in the SI and elsewhere.[31] The ICP-MS was operated in time resolved acquisition mode (TRA), with a dwell time of 0.5 s, to monitor signal intensity (IPt(cps) and IAu(cps)) at a fixed DMA voltage. The DMA was then stepped to another voltage representing a specific mobility size so that an elemental characterization as a function of particle size could be obtained. The PtII loading on a AuNP is defined as the number of PtII molecules per unit surface area of a single particle. The ratio can be measured quantitatively by ICP-MS as: [27]
| (1) |
where IPt(cps) and IAu(cps) represents the signal intensity of 195Pt and 197Au detected by ICP-MS in counts per second (cps); SAuNP and VAuNP are the surface area and volume of a single AuNP (in this case, these parameters are obtained from the DMA diameter dp and assuming spherical geometry); Mm,Au and ρAu are the molar mass and density of Au, and Nav is Avogadro’s number; αAu-Pt is a calibration factor taking into account the relative sensitivity of ICP-MS for 197Au and 195Pt (see SI Figure S2 (a)–(b)).
Figure 2.

Schematic of hyphenated ES-DMA-ICP-MS. (a) ES. (b) 210Po neutralizer chamber. (c) DMA. (d) GED to exchange air with argon for ICP-MS compatibility. (e) ICP-MS.
ATR-FTIR
ATR-FTIR spectra were collected on a Thermo Scientific Nicolet iS50 (Thermo Fisher Scientific, Waltham, MA) equipped with Pike horizontal ATR (HATR) ZnSe 45° crystal (Pike Technologies, Madison, WI). Three samples, AuNP-(dendron), AuNP-(dendron)-(PEG5K)-(5-1) and AuNP-PEG5K, were purified by centrifugal filtration, dropcast onto the crystal, and dried prior to examination. Spectra presented were collected over 250 scans at 2.0 cm−1 resolution.
Results and Discussion
Molecular conjugation of dendron and SH-PEG on AuNPs
SH-PEG with three molar masses (1 kDa, 5 kDa and 10 kDa) were examined. Figure 3(a) shows the corresponding particle size distributions (PSDs) measured for AuNPs, their dendron conjugates and the product after reaction with SH-PEG5K. The PSDs for the 1 kDa and 10 kDa SH-PEG conjugation to AuNP-dendron are shown in SI Figure S3(a)–(b). The corresponding hydrodynamic size measured by DLS is provided in SI Table S1. For clarity, the number densities shown in Figure 3(a) are normalized to unity. As shown in Figure 3(a), the citrate-stabilized AuNPs (after centrifugal cleaning) have a size distribution centered at 11.4 nm prior to conjugation. Upon conjugation with the dendron, the peak size increases to 12.4 nm; the difference representing the dried dendron corona. After subsequent reaction with increasing concentrations of SH-PEG5K, from (0.1 to 5) mmol/L, the peak size continually increases from 14.8 nm to 15.6 nm; this increase is indicative of SH-PEG5K uptake onto the AuNP surface. The width of the size distribution, as determined from the peak-width at half-maximum, does not significantly change with conjugation (from 1.7 nm for citrate stabilized AuNPs to 1.8 nm for AuNP-(dendron)-(SH-PEG5K)), implying that conjugation does not alter the effective PSD shape or width (i.e., it does not induce significant agglomeration).
Figure 3.

Surface conjugation of SH-PEG and dendron on AuNPs. (a) Normalized number-weighted PSDs measured by ES-DMA-CPC. (b) Molecular binding of dendron and SH-PEG5K on AuNP characterized by ATR-FTIR. A 5-1 ratio of dendron to SH-PEG5K was used as a representative example for the ATR-FTIR study. Baseline subtraction was performed for each spectrum (absorption at wavenumber 1800 cm−1 was chosen as zero) and followed by normalization to the highest peak intensity. Dashed lines mark the characteristic vibrational modes from dendron (COO−, C=O) and SH-PEG5K (C-O).
The peak size in Figure 3(a) reaches a plateau value of 15.6 nm with increasing concentration of SH-PEG, yet is clearly smaller than the peak size (20.8 nm) obtained for AuNP-(SH-PEG5K) formed in the absence of the dendron. This clearly shows that the dendron partially inhibits adsorption of SH-PEG5K. Similar results were obtained for the 1 kDa and 10 kDa PEG. Note that the small DMA peaks situated below 10 nm are attributed to the nonvolatile remnants generated by the electrospray,[34] and in our case these remnants are negligibly small due to the previously described cleaning steps.
ATR-FTIR spectra, shown in Figure 3(b), were recorded for AuNP-(dendron) and AuNP-(SH-PEG5K). The ATR-FTIR spectrum of the target species AuNP-(dendron)-(SH-PEG5K) was then compared with that of both AuNP-(dendron) and AuNP-(SH-PEG5K) in order to monitor the presence and relative quantity of each ligand. Each spectrum was normalized to the highest peak intensity for clarity. As shown in Figure 3(b), the AuNP-(dendron) exhibits a distinct peak at 1570 cm−1 designated as the anti-symmetric stretching for COO− from the carboxyl group of the dendron, and similarly the absorbance at 1410 cm−1 corresponds to the symmetric stretch for COO−. The small peak at 1710 cm−1 is assigned to C=O stretching (again associated with the dendron).[35] On the other hand, for AuNP-(SH-PEG5K), the peak at 1110 cm−1 is characteristic of C-O stretching in the SH-PEG5K structure.[36] Comparing the measured absorbance for AuNP-(dendron)-(SH-PEG5K), where SH-PEG5K was added post-dendron conjugation, the characteristic peaks for both dendron and SH-PEG5K appear, confirming the presence of both ligands. The physical measurement of PSDs using DMA combined with the ATR-FTIR spectroscopic results confirm that SH-PEG5K binds to the AuNP-(dendron) conjugate, and suggests that this reaction occurs without substantial displacement of the bound dendron. It should be emphasized that SH-PEG is a potential competitive binding ligand capable of displacing other ligands, but in this case the dendron-Au bond appears sufficiently stable to resist displacement and block some potential adsorption sites for SH-PEG.
To better understand the adsorption, and to examine dendron binding and possible steric hindrance due to SH-PEG, we quantify the extent of SH-PEG adsorption on the dendron conjugated AuNP using a refined two-layer geometric model. Prior work has demonstrated the capacity to quantify ligand surface packing density based on careful PSD measurements using ES-DMA.[37] The general concept of this analysis is that by assuming a random coil configuration for the linear SH-PEG molecule, the surface coverage of SH-PEG is obtained from the increase in cross-sectional area obtained via DMA. In the present study, we refine the core-shell model used previously to account for two ligand layers, as the contributions to particle size from either ligand cannot be neglected (Figure 4(a)).
Figure 4.

(a) Schematic illustration of the two-layer core-shell model used to quantify SH-PEG packing density used for Eq. (2) (b) Adsorption isotherm for SH-PEG on AuNP-dendron conjugates. (c) Citrate AuNP conjugated with SH-PEG 1K, 5K and 10K, at SH-PEG concentration of 0.42 mmol/L during reaction. Packing density is given in molecules/nm2. Error bars represent one standard deviation for at least four replicate measurements. In order to include ligand stability over the time scale of experiments, four replicate measurements were performed within a time scale of ≈10 days.
In the two-layer model, the net contribution of SH-PEG to the increase of cross-sectional area was obtained by subtracting the cross-section increase due to the dendron from the total increase. The cross-sectional area of the SH-PEG corona is a function of particle diameter, and therefore the packing density (σ in unit of molecules/nm2) of SH-PEG can be correlated with changes in particle diameter. [34,38] :
| (2) |
where <x2>0.5 is the random walk radius, the calculated value of which is 3.7 nm [38]for SH-PEG5K under a dried aerosol condition. The random walk radius for 1K and 10K could be calculated similarly using equations from previous work (i.e., 1.7 nm and 5.2 nm, respectively). [38]The surface packing density of SH-PEG was examined as a function of molar mass and SH-PEG concentration.
Figure 4(b) shows the adsorption isotherm derived for the surface packing density of SH-PEG onto AuNP-dendron conjugates versus PEG relative molar mass. With the increase of SH-PEG concentration, the packing density of SH-PEG increases and then plateaus. The plateau surface density, when dendron is pre-attached to the surface, is 0.003 molecules/nm2, 0.008 molecules/nm2 and 0.003 molecules/nm2, for SH-PEG1K, SH-PEG5K and SH-PEG10K, respectively (Conversion to molecules per AuNP in SI, Table S2). The saturated packing density of SH-PEG was compared with the surface coverage of the dendron molecule. Using the same conjugation method, Cho et al. [33] reported a packing density for dendron (i.e., SH-G1-COOH) by x-ray photoelectron spectroscopy (XPS) of 0.76 relative to mercaptoundecanoic acid (MUA). Ivanov et al. [39] reported that the absolute packing density of MUA on a roughly 12 nm diameter AuNP surface was 4.97 molecules/nm2 determined by XPS. Therefore, the estimated packing density for the dendron was ≈ 3.8 molecules/nm2. In comparison, the saturated molecular packing density of SH-PEG accounted for ≤ 0.2 % relative to the dendrons, (0.08 %, 0.2 % and 0.08 % for SH-PEG1K, SH-PEG5K and SH-PEG10K, respectively, in ratio to dendron). The surface is therefore covered primarily by dendrons, with a relatively small presence of SH-PEG on a number basis. The saturated molecular packing density by number is independent of PEG relative molar mass in this case. This independence indicates the limiting factor for PEG adsorption is the amount of dendron present. As shown below, a very different result is obtained when SH-PEG is conjugated directly to the citrate-stabilized AuNPs without the dendron corona present. By comparison, in terms of molar mass, the saturated packing density of PEG-1K, 5K, and 10K is 0.2 %, 3 % and 2.3 %, respectively, relative to that of dendron.
For comparison, 0.42 mmol/L of SH-PEG was added to citrate-stabilized AuNPs. At this concentration, the surface packing of SH-PEG reached saturation levels on the AuNP-dendron surface at ≤ 0.008 molecules/nm2, as indicated by the adsorption isotherms in Figure 4(b). By contrast, the resulting SH-PEG packing density on citrate-stabilized AuNPs, as shown in Figure 4(c), is ≈ 0.12 molecules/nm2, 0.08 molecules/nm2 and 0.02 molecules/nm2, for SH-PEG1K, SH-PEG5K and SH-PEG10K, respectively. The latter packing densities exceed those for AuNP-(dendron)-(SH-PEG1K, 5K, and 10K) conjugates by an order of magnitude. The sharp decrease in packing density with respect to an increase of SH-PEG molar mass on citrate-stabilized AuNPs is attributed to intermolecular steric hindrance between the SH-PEG molecules when more closely packed on the surface. Note that the higher relative molar mass of SH-PEG10K yields a higher occupied area per molecule on the AuNP surface, and therefore a lower molecular packing density.
We may conclude for this part of the study that the principal limiting factor for uptake of SH-PEG by the AuNP-dendron conjugate is the availability of bonding sites or open surface (due to pre-attached dendron) and apparently not a result of steric hindrance, which would increase with molar mass of SH-PEG.
Binding of cisplatin to surface engineered AuNPs
In the next phase of this investigation, we evaluate the effects of PtII complexation on conjugated AuNPs and, conversely, the effects of surface engineering on PtII complexation. Figure 5(a) shows the element-mass weighted particle size distribution for PtII complexed AuNPs obtained by ES-DMA-ICP-MS. Before analysis, free PtII was removed from the PtII-AuNP-(dendron)- (with or without SH-PEG) suspension by centrifugation. In this case we can simultaneously track signal intensity for 197Au and 195Pt (IPt(cps) and IAu(cps)) as a function of the mobility diameter, dp (determined solely by the voltage applied to the DMA). Note that the signal intensity for 197Au is significantly higher than that for195Pt, however the two curves are clearly superimposed and indicate sufficient signal-to-noise for characterizing size classified particles. These results also confirm that Pt is associated with the AuNPs and not free in solution. From this result, the average PtII loading on AuNP-(dendron) conjugates can be determined using equation (1).
Figure 5.

Quantification of PtII loading by ES-DMA-ICP-MS. (a) Representative elemental mass-based particle size distributions for AuNP-(dendron)-(SH-PEG5K) at CPtII = 11.4 µmol/L. Blue: 197Au trace; black: 195Pt trace, (b) Loading of PtII on AuNP-dendron vs the concentration of PtII, (c) Loading of PtII vs the packing density of SH-PEG5K on AuNP at CPtII = 11.4 µmol/L (dashed line is only a guide), (d) PtII loading vs SH-PEG molar mass (1kDa, 5kDa and 10kDa), at around saturation packing density (i.e. 5 to 1 dendron to SH-PEG) at CPtII = 11.4 µmol/L. Error bars for σPtII represent one standard deviation about the mean value derived from Pt/Au across a range of particle sizes in the mass-based PSD for one measurement in the same sub-sample. Uncertainties for same subsample (but with measurement replicates) and between different samples are based on relative standard deviation and averaged < 9 %, demonstrating good precision and sample-to-sample reproducibility (for details see SI Figure S4).
Exploiting the capacity to quantify PtII loading on AuNPs, we examine the effect of PtII solution concentration. As shown in Figure 5(b), the loading of PtII (σPtII) increased gradually with respect to increased PtII concentration during complex formation, and plateaued at ≈ 3 PtII molecules/nm2, indicating that PtII has reached a saturation level under the conditions studied (PtII loading in molecules/nm2 as well as in molecules/AuNP were summarized in SI, Table S2). Since surface coverage of the dendron is ≈ 3.8 molecules/nm2 and each dendron molecule contains three carboxylate groups, this yields ≈ 11 PtII/nm2 of available complexing sites. The saturated PtII loading of ≈ 3 PtII/nm2 suggests formation of some bidentate complexes.[26]
Next, we address the quantitative effect of SH-PEG surface packing (σSH-PEG and relative molar mass of SH-PEG) on PtII loading. Figure 5(c) shows how the PtII loading changes with respect to the SH-PEG5K packing density (σSH-PEG), while fixing the available PtII complexation sites. In this case, PtII loading is relatively insensitive to the packing density SH-PEG5K – ranging from (0 to 0.008) molecules/nm2 – on AuNP-(dendron)-(SH-PEG5K). With PEGylation, the loading capacity decreased by less than 20 %. Considering that SH-PEG5K binding density on the AuNP-(dendron) surface amounts to less than 0.2 % by number of the total dendron present, the available binding sites (COO−) for AuNP-dendron and AuNP-(dendron)-(SH-PEG5K) should be roughly equivalent. Similarly, there is a very mild effect of SH-PEG molar mass (1K, 5K and 10K in this case) on PtII loading (see Figure 5(d)). For samples susceptible to agglomerate formation during the centrifugal cleaning process, σPtII was reported from agglomerates since σPtII was previously demonstrated to be independent of agglomeration states.[27]
In combination with the quantification of dendron and SH-PEG obtained in Figure 4 and Figure 5, we conclude that attachment of SH-PEG does not significantly degrade the loading capacity for the active drug. This insensitivity is likely due to the low PEG adsorption density observed. Additionally, the smaller, more mobile PtII is able to access available carboxyl sites that are essentially shadowed, but not blocked, by the larger hydrophilic PEG that extends out from the surface. Similarly, the molar mass effect on PtII uptake is weak, since the number of binding sites per PEG remains constant, and larger polymers simply extend further out from the surface where PtII binding is not effected. In fact, there is a slight increase observed in PtII loading with increasing PEG molar mass. What remains to be determined is the effect of SH-PEG on colloidal stability of the PtII complexed conjugate.
Colloidal stability (by ES-DMA-ICP-MS)
Previous work has shown that complexation of PtII itself leads to colloidal destabilization of the conjugate due to electrostatic interactions between the anionic carboxylates and the positively charged PtII.[27] The principal motivation for PEGylation is to counteract that effect. Here we investigate the efficacy of PEGylation to preserve stability in biologically relevant media. ES-DMA has the capacity to quantify nanoparticle agglomeration kinetics.[40] The high sensitivity of ICP-MS as a detector enables sensitivity for low-order AuNP agglomerates at very low concentrations and should also be useful in tracking agglomeration in complex media (in this case, DMEM and PBS solution), as the element-specific detection enables direct identification of the agglomeration component (whereas CPC is nonspecific with respect to detection).
The raw spectrum obtained by ES-DMA-ICP-MS is a mobility size-resolved intensity (197Au) distribution (as shown in SI Figure S5). Each distinguishable peak is assigned to an oligomer state based on mobility size:[41] monomer, dimer, trimer, tetramer, etc. The ICP-MS intensity IAu,n for oligomer n is related to the oligomer mass through an appropriate calibration factor. For a single oligomer, the mass of n-mer (an oligomer consisting of n primary particles) is n times the mass of a primary AuNP. Therefore, the number of oligomer particles is related to the 197Au intensity as follows:
| (3) |
where Ψ represents a function that includes the sensitivity factor for 197Au, αAu, and the density and volume of a primary size AuNP, ρAu and VAu (i.e., Ψ = f (αAu, ρAu, VAu)). Nn represents the number of nanoparticle agglomerates containing n primary particles. It should be noted that for a given set of experimental conditions, Ψ is a constant and not a function of the oligomer state.
In order to investigate the colloidal stability of nanoparticles, we utilize the degree of agglomeration (DA). The degree of agglomeration is defined here as follows: [40]
| (4) |
Combining with equation (4), DA is expressed as:
| (5) |
Therefore, DA represents the average number of particles per agglomerate and will increase from unity with respect to time for an unstable system. Considering the resolving power of ES-DMA-ICP-MS to distinguish oligomers, the present analysis is limited to tetramers, and therefore the maximum DA in this study is four.
Figure 6(a) quantifies the effect of added PtII on DA versus reaction time, t, for various PtII concentrations. At low PtII concentrations (CPtII = 1.1 µmol/L and 5.7 µmol/L) and for reaction times as long as 12.5 days, DA remains close to unity, demonstrating that the monomer specie is dominant in solution and the conjugate is colloidally stable before mixing with PtII; therefore, DA=1 is assumed for t=0 (0 h stands for the point immediately after adding PtII). With an increase in PtII concentration to 11.4 µmol/L, a gradual increase in agglomeration with respect to time was observed. At PtII concentrations as high as 34.1 µmol/L, DA approaches ≈ 3.0 in less than 4 h, indicating a substantial increase in agglomeration kinetics. We attribute the reduced stability at higher cisplatin loadings to an increase in neutralization of carboxylate groups resulting in weaker charge stabilization. Steric interactions due to the dendron corona are apparently insufficient to provide stability at high PtII loading.
Figure 6.

Quantifying colloidal stability using ES-DMA-ICP-MS. Initial mixing of cisplatin with AuNP conjugates occurs at 0 h, with the x-axis showing time after mixing or reaction time. Before mixing with PtII, samples are stable and therefore DA=1 was assumed at t=0. (a) Effect of PtII on colloidal stability. (b) Effect of PEGylation on stability in different media. The parameter DA was calculated from mass distribution measurements using DMA-ICP-MS. A relative standard deviation of 7 % is estimated based on the mean standard deviation performed at each size step. [27]
Figure 6(b) characterizes the effect of PEGylation on colloidal stability at a fixed PtII concentration (CPtII = 11.4 µmol/L). We examined PEG with three relative molar masses (1 kDa, 5 kDa and 10 kDa). Since no significant influence of SH-PEG packing density on PtII loading was observed, we chose to investigate the highest SH-PEG packing density for each relative molar mass. In comparison to the PtII-Au-dendron complexes, where DA increased gradually to roughly 3 within one day, the PEGylated Au-(dendron)-(SH-PEG1K), Au-(dendron)-(SH-PEG5K) and Au-(dendron)-(SH-PEG10K) remained close to unity for up to 24 d. This result unequivocally confirms that PEGylation imparts stabilization to otherwise unstable highly loaded PtII complexed AuNPs. The effect was independent of SH-PEG relative molar mass. Similar colloidal stability has been reported for AuNPs where PtII was conjugated indirectly through a linker molecule.[42]While UV and DLS were used to assess stability in this previous work, in the present study DMA-ICP-MS shows a clear advantage for quantifying the degree of agglomeration.
Colloidal stability in biologically relevant media (PBS and DMEM) was also examined. The AuNP-dendron complexed with PtII precipitated immediately (visually evident) when in contact with either PBS or DMEM buffer. However PEGylated samples exhibited visibly improved stability with a DA less than ≈ 1.5 over a period of two days. Since electrostatic induced stability is generally screened at biological saline levels, the enhanced dispersion stability can be attributed to steric interactions provided by the hydrophilic PEG chains attached to the surface. Although SH-PEG uses less than 2 % of dendron surface coverage, it is sufficient to provide obvious enhancement of dispersion stability for the PtII-AuNP platform, either in water or more complex biological media, with an insignificant trade-off in drug loading.
Cisplatin Release Performance
Nanoparticles were incubated at pH 5 mimicking the acidic pH of the endolysosomal compartment of a tumor, and it is close to the pKa for organic carboxylic acid groups (around pH 4.5). PBS was selected as a control to mimic pH conditions during blood circulation (pH 7.4), where PtII release should be minimal. In PBS (Figure (a)), the PtII-AuNP platforms examined in this study showed essentially no release of PtII, while at pH 5 (Figure 7 (b)), a clear preferential release relative to PBS was observed.
Figure 7.

PtII release under (a) PBS and (2) pH 5 (acetate buffer). The platform studied included AuNP-dendron, AuNP-(dendron)-(SH-PEG1K) and AuNP-(dendron)-(SH-PEG5K) at two different PtII loading concentrations (low PtII : CPtII = 1.1 µmol/L and high PtII: CPtII = 11.4 µmol/L), which are labeled as Pt (H) (PtII at high concentration) and Pt (L) (PtII at low concentration). The PtII loading was measured 1.9 PtII/nm2 for 5K-Pt (H), and 0.5 PtII/nm2 for dendon-Pt (L). 0.5PtII/nm2 was estimated for 1K-Pt (L) as well as for 5K-Pt (L) since no significant change in PtII loading was expected with PEGylation (Figure 5(c)). Error bars represent one standard deviation of two replicate experiments.
As an example, at day 10, AuNP-(dendron)-(SH-PEG5K)-high PtII exhibited a 10-fold increase in PtII release at pH 5 compared with PBS. Under acidic conditions, a release burst occurred within 2 d, followed by a slow release between (2 and 10) d. Notably, PEG relative molar mass (1 kDa and 5 kDa) showed no significant influence on PtII release performance. Overall, release performance is limited, but by increasing PtII loading (high PtII compared with the average of the three low concentrations shown in Figure 7), the release percentage increases approximately 2-fold (9 % to 16 %); this increased loading would not be achievable without PEG modification of the surface. The absolute amount of PtII release was about 6× greater for high PtII loading compared with that of low PtII loading. It should be noted that in the published literature, the plateau percentage of PtII released ranges between 10 % and 100 % for carboxylated ligands (on a time scale < 10 d).[26, 43–46] The present results clearly fall within this range, but toward the lower end. The stability of the dicarboxylate complex with PtII [46] provides an opportunity to surface engineer further improvements in the release performance in conjunction with the demonstrated improvements in stability due to PEG; however, this was beyond the scope of the present study.
Summary and Conclusions
In this study, we built upon previous efforts that established the capacity for a novel hyphenated technique, ES-DMA-ICP-MS, to examine, on a quantitative basis, the effects of surface engineering on drug loading and colloidal stability for a model nanoscale anti-tumor drug delivery platform. The model platform consisted of cisplatin (PtII) complexed to a carboxylate-terminated dendron tethered covalently by thiols to the surface of nominally 10 nm diameter AuNPs. In the present study, we extended this foundational work to analyze a more complex system containing multiple ligands (SH-PEG and thiolated carboxyl-terminated dendron). We quantified the effects of SH-PEG molar mass and concentration on PtII surface loading, and the impact of the dendron ligand on PEG uptake by the conjugate. We also examined the capacity for PEG adsorption to improve the colloidal stability of the PtII-loaded nano-conjugate, its primary role in this system, and the effect of adsorbed PEG on pH-dependent in vitro release of PtII. We show that SH-PEG uptake is inhibited by the dendron ligand if the ligand is pre-attached to the AuNP surface, but that once bound to the surface PEG does not significantly impact PtII loading capacity or release performance, while it does substantially improve colloidal stability both in water and in biologically relevant media such as PBS and DMEM. The decrease in PtII loading capacity upon PEGylation was observed to be less than 20 %. The 10 day release of PtII (examined at 0.5 PtII/nm2 drug loading) was 0.9 % regardless of PEGylation status. At higher drug loading (1.9 PtII/nm2), 16 % PtII release was reported. An optimal balance between drug loading and conjugate stability under biologically relevant conditions can thus be achieved. This study shows that by employing a strategy that combines separation with analysis and includes quantitative and orthogonal measurements, one can monitor and control properties that determine the efficacy, and, by extension, the safety, of nano-based drug delivery platforms. The lack of such quantitative measurements is one factor limiting the clinical application of these novel materials.
Supplementary Material
Acknowledgments
The authors thank Dr. Suvajyoti Guha at FDA and Dr. Arnab Mukherjee at NIST, for their thorough reviews and comments on the manuscript. Work performed by JT was supported under the following financial assistance award - cooperative research agreement 70NANB13H021 from U.S. Department of Commerce, National Institute of Standards and Technology.
Footnotes
The identification of any commercial product or trade name does not imply endorsement or recommendation by the National Institute of Standards and Technology.
Author Contributions
J.Tan and T.J.Cho contributed equally to this work. V.A. Hackley and M.R. Zachariah contributed to the experimental design, interpretation and writing of the manuscript. D.-H. Tsai contributed to experimental design, manuscript modification. J. Liu contributed to results discussion and manuscript modification. J.M. Pettibone contributed to FTIR design and results interpretation. R. You contributed to aggregation simulation.
References
- 1.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–20. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 2.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nature Reviews Cancer. 2007;7(8):573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 3.Jung YW, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chemical Reviews. 2007;107(5):1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 4.Seiwert TY, Salama JK, Vokes EE. The concurrent chemoradiation paradigm - general principles. Nature Clinical Practice Oncology. 2007;4(2):86–100. doi: 10.1038/ncponc0714. [DOI] [PubMed] [Google Scholar]
- 5.Rose PG. New treatment paradigm for locally advanced cervical cancer? Nature Reviews Clinical Oncology. 2011;8(7):388–390. doi: 10.1038/nrclinonc.2011.85. [DOI] [PubMed] [Google Scholar]
- 6.Bosl GJ, Bajorin DF, Sheinfeld J. In: Cancer of the Testis. DeVita VTJ, Hellman S, Rosenberg SA, editors. Lippincott Williams & Wilkins; Philadelphia: 2001. [Google Scholar]
- 7.Barabas K, Milner R, Lurie D, Adin C. Cisplatin: a review of toxicities and therapeutic applications. Vet Comp Oncol. 2008;6(1):1–18. doi: 10.1111/j.1476-5829.2007.00142.x. [DOI] [PubMed] [Google Scholar]
- 8.Wheate NJ, Walker S, Craig GE, Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010;39(35):8113–27. doi: 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- 9.Boulikas T, Vougiouka M. Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs (review) Oncol Rep. 2004;11(3):559–95. [PubMed] [Google Scholar]
- 10.Johnstone TC, Suntharalingam K, Lippard SJ. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem Rev. 2016;116(5):3436–3486. doi: 10.1021/acs.chemrev.5b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–6. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 12.Hu CM, Zhang L. Therapeutic nanoparticles to combat cancer drug resistance. Curr Drug Metab. 2009;10(8):836–41. doi: 10.2174/138920009790274540. [DOI] [PubMed] [Google Scholar]
- 13.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka T, Shiramoto S, Miyashita M, Fujishima Y, Kaneo Y. Tumor targeting based on the effect of enhanced permeability and retention (EPR) and the mechanism of receptor-mediated endocytosis (RME) Int J Pharm. 2004;277(1–2):39–61. doi: 10.1016/j.ijpharm.2003.09.050. [DOI] [PubMed] [Google Scholar]
- 15.Paciotti GF, Kingston DGI, Tamarkin L. Colloidal gold nanoparticles: A novel nanoparticle platform for developing multifunctional tumor-targeted drug delivery vectors. Drug Development Research. 2006;67(1):47–54. [Google Scholar]
- 16.Bazak R, Houri M, El Achy S, Kamel S, Refaat T. Cancer active targeting by nanoparticles: a comprehensive review of literature. J Cancer Res Clin Oncol. 2015;141(5):769–84. doi: 10.1007/s00432-014-1767-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li SD, Huang L. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochim Biophys Acta. 2009;1788(10):2259–66. doi: 10.1016/j.bbamem.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bose T, Latawiec D, Mondal P, Mandal S. Overview of nano-drugs characteristics for clinical application: the journey from the entry to the exit point. J Nanopart Res. 2014;16(8):2527. [Google Scholar]
- 20.Ghosh P, Han G, De M, Kim CK, Rotello VM. Gold nanoparticles in delivery applications. Adv Drug Deliv Rev. 2008;60(11):1307–15. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 21.Connor EE, Mwamuka J, Gole A, Murphy CJ, Wyatt MD. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small. 2005;1(3):325–7. doi: 10.1002/smll.200400093. [DOI] [PubMed] [Google Scholar]
- 22.FRENS G. Controlled Nucleation for the Regulation of the Particle Size in Monodisperse Gold Suspensions. Nature. 1973;241(105):20–22. [Google Scholar]
- 23.Brown SD, Nativo P, Smith JA, Stirling D, Edwards PR, Venugopal B, Flint DJ, Plumb JA, Graham D, Wheate NJ. Gold Nanoparticles for the Improved Anticancer Drug Delivery of the Active Component of Oxaliplatin. J Am Chem Soc. 2010;132(13):4678–84. doi: 10.1021/ja908117a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter JD, Cheng NN, Qu YQ, Suarez GD, Guo T. Nanoscale energy deposition by x-ray absorbing nanostructures. Journal of Physical Chemistry B. 2007;111(40):11622–11625. doi: 10.1021/jp075253u. [DOI] [PubMed] [Google Scholar]
- 25.Pradhan AK, Nahar SN, Montenegro M, Yu Y, Zhang HL, Sur C, Mrozik M, Pitzer RM. Resonant X-ray enhancement of the Auger effect in high-Z atoms, molecules, and nanoparticles: potential biomedical applications. J Phys Chem A. 2009;113(45):12356–63. doi: 10.1021/jp904977z. [DOI] [PubMed] [Google Scholar]
- 26.Lee SM, Tsai DH, Hackley VA, Brechbiel MW, Cook RF. Surface-engineered nanomaterials as X-ray absorbing adjuvant agents for Auger-mediated chemo-radiation. Nanoscale. 2013;5(12):5252–5256. doi: 10.1039/c3nr00333g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai DH, Cho TJ, Elzey SR, Gigault JC, Hackley VA. Quantitative analysis of dendron-conjugated cisplatin-complexed gold nanoparticles using scanning particle mobility mass spectrometry. Nanoscale. 2013;5(12):5390–5. doi: 10.1039/c3nr00543g. [DOI] [PubMed] [Google Scholar]
- 28.Ryan SM, Mantovani G, Wang XX, Haddleton DM, Brayden DJ. Advances in PEGylation of important biotech molecules: delivery aspects. Expert Opinion on Drug Delivery. 2008;5(4):371–383. doi: 10.1517/17425247.5.4.371. [DOI] [PubMed] [Google Scholar]
- 29.Klibanov AL, Maruyama K, Beckerleg AM, Torchilin VP, Huang L. Activity of amphipathic poly(ethylene glycol) 5000 to prolong the circulation time of liposomes depends on the liposome size and is unfavorable for immunoliposome binding to target. Biochim Biophys Acta. 1991;1062(2):142–8. doi: 10.1016/0005-2736(91)90385-l. [DOI] [PubMed] [Google Scholar]
- 30.Howard MD, Jay M, Dziubla TD, Lu X. PEGylation of Nanocarrier Drug Delivery Systems: State of the Art. Journal of Biomedical Nanotechnology. 2008;4(2):133–148. [Google Scholar]
- 31.Elzey S, Tsai DH, Yu LL, Winchester MR, Kelley ME, Hackley VA. Real-time size discrimination and elemental analysis of gold nanoparticles using ES-DMA coupled to ICP-MS. Anal Bioanal Chem. 2013;405(7):2279–88. doi: 10.1007/s00216-012-6617-z. [DOI] [PubMed] [Google Scholar]
- 32.Comenge J, Sotelo C, Romero F, Gallego O, Barnadas A, Parada TG, Dominguez F, Puntes VF. Detoxifying antitumoral drugs via nanoconjugation: the case of gold nanoparticles and cisplatin. PLoS One. 2012;7(10):e47562. doi: 10.1371/journal.pone.0047562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho TJ, Zangmeister RA, Maccuspie RI, Patri AK, Hackley VA. Newkome-type dendron stabilized gold nanoparticles: Synthesis, reactivity, and stability. Chemistry of materials : a publication of the American Chemical Society. 2011;23(10):2665–2676. doi: 10.1021/cm200591h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsai D-H, DelRio FW, Pettibone JM, Lin P-A, Tan J, Zachariah MR, Hackley VA. Temperature-Programmed Electrospray–Differential Mobility Analysis for Characterization of Ligated Nanoparticles in Complex Media. Langmuir. 2013;29(36):11267–11274. doi: 10.1021/la402311c. [DOI] [PubMed] [Google Scholar]
- 35.Petroski J, El-Sayed MA. FTIR study of the adsorption of the capping material to different platinum nanoparticle shapes. Journal of Physical Chemistry A. 2003;107(40):8371–8375. [Google Scholar]
- 36.Mansur HS, Orefice RL, Mansur AAP. Characterization of poly(vinyl alcohol)/poly(ethylene glycol) hydrogels and PVA-derived hybrids by small-angle X-ray scattering and FTIR spectroscopy. Polymer. 2004;45(21):7193–7202. [Google Scholar]
- 37.Pease LF, Tsai DH, Zangmeister RA, Zachariah MR, Tarlov MJ. Quantifying the surface coverage of conjugate molecules on functionalized nanoparticles. Journal of Physical Chemistry C. 2007;111(46):17155–17157. [Google Scholar]
- 38.Tsai DH, DelRio FW, MacCuspie RI, Cho TJ, Zachariah MR, Hackley VA. Competitive Adsorption of Thiolated Polyethylene Glycol and Mercaptopropionic Acid on Gold Nanoparticles Measured by Physical Characterization Methods. Langmuir. 2010;26(12):10325–10333. doi: 10.1021/la100484a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ivanov MR, Haes AJ. Anionic Functionalized Gold Nanoparticle Continuous Full Filling Separations: Importance of Sample Concentration. Analytical Chemistry. 2012;84(3):1320–1326. doi: 10.1021/ac2022376. [DOI] [PubMed] [Google Scholar]
- 40.Tsai DH, Pease LF, 3rd, Zangmeister RA, Tarlov MJ, Zachariah MR. Aggregation kinetics of colloidal particles measured by gas-phase differential mobility analysis. Langmuir. 2009;25(1):140–6. doi: 10.1021/la703164j. [DOI] [PubMed] [Google Scholar]
- 41.Tsai DH, Cho TJ, DelRio FW, Gorham JM, Zheng JW, Tan JJ, Zachariah MR, Hackley VA. Controlled Formation and Characterization of Dithiothreitol-Conjugated Gold Nanoparticle Clusters. Langmuir. 2014;30(12):3397–3405. doi: 10.1021/la500044y. [DOI] [PubMed] [Google Scholar]
- 42.Craig GE, Brown SD, Lamprou DA, Graham D, Wheate NJ. Cisplatin-Tethered Gold Nanoparticles That Exhibit Enhanced Reproducibility, Drug Loading, and Stability: a Step Closer to Pharmaceutical Approval? Inorganic Chemistry. 2012;51(6):3490–3497. doi: 10.1021/ic202197g. [DOI] [PubMed] [Google Scholar]
- 43.Ye L, Letchford K, Heller M, Liggins R, Guan D, Kizhakkedathu JN, Brooks DE, Jackson JK, Burt HM. Synthesis and characterization of carboxylic acid conjugated, hydrophobically derivatized, hyperbranched polyglycerols as nanoparticulate drug carriers for cisplatin. Biomacromolecules. 2011;12(1):145–55. doi: 10.1021/bm101080p. [DOI] [PubMed] [Google Scholar]
- 44.Kulhari H, Pooja D, Singh MK, Chauhan AS. Optimization of carboxylate-terminated poly(amidoamine) dendrimer-mediated cisplatin formulation. Drug Dev Ind Pharm. 2015;41(2):232–8. doi: 10.3109/03639045.2013.858735. [DOI] [PubMed] [Google Scholar]
- 45.Yan X, Gemeinhart RA. Cisplatin delivery from poly(acrylic acid-co-methyl methacrylate) microparticles. J Control Release. 2005;106(1–2):198–208. doi: 10.1016/j.jconrel.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 46.Paraskar AS, Soni S, Chin KT, Chaudhuri P, Muto KW, Berkowitz J, Handlogten MW, Alves NJ, Bilgicer B, Dinulescu DM, Mashelkar RA, Sengupta S. Harnessing structure-activity relationship to engineer a cisplatin nanoparticle for enhanced antitumor efficacy. PNAS. 2010;107:12435–12440. doi: 10.1073/pnas.1007026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.