

Abstract
Background and Purpose
Microglial phenotype and phagocytic activity are deregulated in Parkinson's disease (PD). PPARγ agonists are neuroprotective in experimental PD, but their role in regulating microglial phenotype and phagocytosis has been poorly investigated. We addressed it by using the PPARγ agonist MDG548.
Experimental Approach
Murine microglial cell line MMGT12 was stimulated with LPS and/or MDG548, and their effect on phagocytosis of fluorescent microspheres or necrotic neurons was investigated by flow cytometry. Cytokines and markers of microglia phenotype, such as mannose receptor C type 1; MRC1), Ym1 and CD68 were measured by elisa and fluorescent immunohistochemistry. Levels of Beclin‐1, which plays a role in microglial phagocytosis, were measured by Western blotting. In the in vivo MPTP–probenecid (MPTPp) model of PD in mice, MDG548 was tested on motor impairment, nigral neurodegeneration, microglial activation and phenotype.
Key Results
In LPS‐stimulated microglia, MDG548 increased phagocytosis of both latex beads and necrotic cells, up‐regulated the expression of MRC1, CD68 and to a lesser extent IL‐10, while blocking the LPS‐induced increase of TNF‐α and iNOS. MDG548 also induced Beclin‐1. Chronic MPTPp treatment in mice down‐regulated MRC1 and TGF‐β and up‐regulated TNF‐α and IL‐1β immunoreactivity in activated CD11b‐positive microglia, causing the death of nigral dopaminergic neurons. MDG548 arrested MPTPp‐induced cell death, enhanced MRC1 and restored cytokine levels.
Conclusions and Implications
This study adds a novel mechanism for PPARγ‐mediated neuroprotection in PD and suggests that increasing phagocytic activity and anti‐inflammatory markers may represent an effective disease‐modifying approach.
Abbreviations
- CD
cluster of differentiation
- iNOS
inducible NOS
- IR
immunoreactivity
- MRC1
mannose receptor C type 1
- MPTP
1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine
- PD
Parkinson's disease
- PFA
paraformaldehyde
- SNc
substantia nigra pars compacta
- TZDs
thiazolidinediones
- Ym1
β‐N‐acetylhexosaminidase/chitinase‐like 3
Introduction
A chronic and deregulated glial response contributes to the progression and spreading of neurodegeneration in Parkinson's disease (PD) (Joers et al., 2017). Specifically, an imbalance in pro‐inflammatory/anti‐inflammatory microglia is regarded as a main pathological contributor to chronic neuroinflammation and neurodegeneration in PD and targeting cytokine production by microglia has been suggested as a promising disease‐modifying strategy in PD (Sanchez‐Guajardo et al., 2010; Tang and Le, 2016). Moreover, sporadic studies have reported an abnormal phagocytosis by microglia in PD, although whether it is up‐regulated or down‐regulated in the pathology is still a matter of debate (Salman et al., 1999; Gardai et al., 2013). Measurement of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=837 has been proposed as a marker of phagocytosis, and morphological analysis of microglia pointed to an increased phagocytic activity in PD (Barcia et al., 2013; Croisier et al., 2005; Depboylu et al., 2012; Doorn et al., 2014; Sanchez‐Guajardo et al., 2010; Theodore et al., 2008), while functional studies with latex beads indicate a defect in the phagocytic function of microglia or other immune cells (Salman et al., 1999; Gardai et al., 2013). Phagocytosis is a beneficial event, which has been traditionally associated with an anti‐inflammatory profile of microglia, whereas this may not be the case in PD, where microglia may acquire mixed pro‐inflammatory and anti‐inflammatory phenotypes. Therefore, whether phagocytosis is a beneficial or detrimental event in PD is still debated, and how it is modified by neuroprotective drugs has not been investigated (Nolan et al., 2013; Peña‐Altamira et al., 2015; Plaza‐Zabala et al., 2017).
A finely regulated production of pro‐inflammatory and anti‐inflammatory cytokines, growth factors and toxic molecules, such as free radicals and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2509, shapes microglia phenotype into either pro‐inflammatory or anti‐inflammatory. Moreover, a number of membrane markers characterize microglial phenotype and function, among which mannose receptor C type 1 (MRC1) and β‐N‐acetylhexosaminidase/chitinase‐like 3 (Ym1) have been associated with anti‐inflammatory phenotypes (Subramaniam and Federoff, 2017). MRC1 is a pattern recognition membrane receptor involved in innate and adaptive immunity, specifically in microglial phagocytosis. Expression of this receptor is controlled by cytokines, being down‐regulated by pro‐inflammatory and up‐regulated by anti‐inflammatory cytokines (Zimmer et al., 2003). Inside the cell, CD68 is mainly located on lysosomes, belonging to the family of lysosomal glycoproteins, and its rapid recirculation from endosomes and lysosomes to the plasma membrane may improve the function of the phagocytic machinery, although the role and involvement of this protein in phagocytosis has been recently questioned and remains to be determined (Song et al., 2011; Chistiakov et al., 2017). Increased CD68 expression has been related to dopamine neuron degeneration in PD (Croisier et al., 2005; Sanchez‐Guajardo et al., 2010). Finally, Beclin‐1 is an autophagy‐regulating protein recently shown to play a critical role in microglial phagocytosis of β‐amyloid (Lucin et al., 2013). Beclin‐1 expression is reduced in microglia of Alzheimer's disease patients, and lack or reduced levels of Beclin‐1 impair microglia phagocytosis in a model of Alzheimer's disease (Lucin et al., 2013). Moreover, Beclin‐1 levels can be induced by the exposure to phagocytosis‐promoting factors in macrophages (Wu et al., 2016).
The nuclear receptor http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=595 is a promising target for a number of neurodegenerative diseases with a relevant neuroinflammatory component, including PD (Nolan et al., 2013; Skerrett et al., 2014). The thiazolidinediones (TZDs) http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2694 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1056, and the more recently characterized PPARγ agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9888 effectively prevent or arrest nigrostriatal degeneration in vivo in the Parkinsonian rodent and monkey models and counteract the neuroinflammatory response in terms of inflammatory cytokine production (Swanson et al., 2011; Carta, 2013; Pisanu et al., 2014; Lecca et al., 2015). MDG548 was identified as a PPARγ agonist with a thiobarbituric acid structure, displaying a twofold higher PPARγ affinity and increased BBB permeability, compared with the TZDs (Nevin et al., 2012). PPARγ function has been extensively characterized in peripheral immune cells, where it mediates many immunomodulatory effects in macrophages, monocytes and granulocytes (Reddy, 2008). Hence, PPARγ acts as a master regulator of macrophage phenotype in physiological as well as pathological conditions, by driving the acquisition of an anti‐inflammatory, or alternative activation state (Bouhlel et al., 2007; Croasdell et al., 2015; Glass and Saijo, 2010; Odegaard and Chawla, 2011; Penas et al., 2013; Varga et al., 2011). Moreover, PPARγ governs the promotion of phagocytosis and tissue repair by macrophages (Nagy et al., 2012). Whether similar roles are fulfilled by PPARγ expressed in microglia in neurological disorders has recently started to be elucidated in vitro and in vivo models (Ajmone‐Cat et al., 2012; Pisanu et al., 2014; Savage et al., 2015; Zhao et al., 2015; Flores et al., 2016; Pinto et al., 2016). In PD, studies suggest that regulation of genes involved in the inflammatory response may represent one mechanism of PPARγ‐mediated neuroprotection, but modulation of microglia phagocytosis has not been investigated.
The specific aim of the present study was to investigate the effect of the PPARγ agonist MDG548 on phagocytosis and microglia phenotype in PD models, together with its disease‐modifying potential in vivo. Given the complexity of in vivo systems, and the broad distribution of PPARγ in different cell types, including immune cells, astrocytes and neurons, assessing the direct effects of PPARγ agonists on a selective cell type in vivo is questionable. For this reason, we first tested MDG548 on unstimulated and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5019‐stimulated microglia in an in vitro system made of pure MMGT12 microglial cells that are comparable with primary microglial cells with respect to several microglial markers (Briers et al., 1994; Michelucci et al., 2009; Heurtaux et al., 2010). In this model, we measured the phagocytosis of fluorescent latex beads or necrotic dopaminergic neuroblastoma cells and cell debris, the levels of CD68 and Beclin‐1 and the production of MRC1, pro‐inflammatory and anti‐inflammatory cytokines and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1250 (iNOS). Thereafter, we tested MDG548 in an in vivo model of PD obtained by the chronic administration of MPTP plus http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4357 (MPTPp), which reproduces the progressive nature of dopaminergic degeneration and inflammatory responses of PD. In this model, we investigated the effect of MDG548 on dopaminergic cell loss and motor impairment and on MRC1 levels and cytokine production by microglia.
Methods
MMGT12 microglial cells and treatments
The murine microglial cell line MMGT12 was used and was a generous gift from T. Heurtaux and A. Michelucci (Luxembourg). Cells were cultured in DF culture medium comprising DMEM/F12 (1:1, vol/vol), supplemented with 10% FBS without antibiotics, grown in humidified atmosphere of 5% CO2 at 37°C, harvested and seeded twice a week. For elisa and immunocytochemistry, MMGT12 cells were seeded into 12‐well plates previously filled with 18 mm coverslips. Cell density was 15 × 103 per well at the time of seeding to ensure 70% confluence on coverslips after 3 days in vitro (DIV), when most cells presented a common morphological appearance. Three days after plating, cells were treated with LPS or MDG548 or LPS plus MDG548 added either at the same time (concurrent, c) or after 2 h (sequential, s) according to Table 1.
Table 1.
Treatments of MMGT12 cells for in vitro experiments
| Treatment | LPS (μg·mL−1) | MDG548 (μM) |
|---|---|---|
| Vehicle | – | – |
| LPS | 1 | – |
| MDG548 | – | 10 |
| MDG548 | – | 50 |
| LPS + MDG548s | 1 | 10 |
| LPS + MDG548s | 1 | 50 |
| LPS + MDG548c | 1 | 10 |
| LPS + MDG548c | 1 | 50 |
MMGT12 cells were treated with LPS or MDG548 or LPS plus MDG548 added either at the same time (concurrent, c) or after 2 h (sequential, s).
Phagocytosis of latex microspheres
The whole experiment was repeated three times (each on a different day), with an n = 3 of independent samples (3 separate plates) in each experimental group. We planned to perform three separate experiments with three independent samples for each experimental group since a higher n would have implied longer and inhomogeneous processing time, compromising the results. Upon verification of the homogeneity of the data, results from three experiments were collected, and statistical analysis was performed with a final n of nine samples for each experimental group.
MMGT12 cells were seeded on 24‐well plates (5 × 104 cells per well) and incubated in DF culture medium in absence or presence of drugs, according to Table 1. Control wells were treated with the corresponding vehicles. Twenty‐four hours later, medium was temporarily removed. Cells were washed in PBS, trypsinized in 0.05% trypsin for 3 min and re‐plated in the same wells, using the same medium for 1 h. Fluoresbrite Carboxy YG 6.0 μm Microspheres (cat#18141; Polysciences Inc., Warrington, PA, USA) were resuspended in PBS with 5.5 mM glucose, 1.5 mM magnesium and 1 mM calcium and pre‐opsonized by addition of 50% FBS and incubation for 30 min in 5% CO2 at 37°C. The 12 × 106 pre‐opsonized microspheres were resuspended in 12 mL of DF culture medium without FBS and distributed on cells (about 10 beads per cell) and incubated for 2 h. Cells were washed with PBS, trypsinized for 5 min and collected with 0.5 mL of culture medium in conical tubes (for flow cytometry), centrifuged at 290× g for 5 min, PBS washed and centrifuged again for 3 min. Cell pellet was suspended in 0.3 mL PBS containing 1% FBS. Cells were acquired in the green channel (502 nm) by FACSCanto II (BD Biosciences, Erenbodgem, Belgium). The percentage of green cells was determined on the single‐cell population by BD FACSDiva software.
SH‐SY5Y cells phagocytosis assay
For the technical reasons reported above, this experiment was carried out twice (each on a different day) with three independent samples (3 plates) each time, and thus, the final n of independent samples was 6 for each experimental group. Confluent dopaminergic neuroblastoma SH‐SY5Y cells (kind gift of Professor Corasaniti, UMG Catanzaro, Italy) were washed twice with PBS and stained for 40 min with 5 μM carboxyfluorescein diacetate succinimidyl ester according to the manufacturer's protocol (V12883, Molecular Probes; Thermo Fisher, Monza, Italy). The cells were collected, as the staining procedure caused cell detachment, washed twice in PBS and then plated 1:2 in RPMI containing 10% FBS. After 2 h, 6‐hydroxy‐dopamine (50 μM) was added for 50 h to induce cell death (99.9% necrosis). Then the MMGT12 cells (5 × 104cells per dish) were plated on 3.5 cm dishes containing sterile glass coverslips. Next day, they were treated with MDG548 and/or LPS according to Table 1, for 24 h.Five hours before the end of the experiment, the cultures were overlaid with necrotic SH‐SY5Y cells (2 × 105 cells per dish) resuspended in 100 μL PBS to give an expected cell ratio of 1:1. After 5 h, the medium was removed, and cultures were washed five times with ice‐cold PBS to remove the SH‐SY5Y cells. Coverslips were removed and placed briefly in the pre‐warmed fresh medium, before placing them one by one on glass slides for imaging with LEICA DMI 4000 B fluorescent microscope, equipped with LEICA DFC 350 FX camera. The rest of the cells were detached with trypsin–EDTA (0.05%) (Gibco; Life Technologies, USA), harvested with regular medium and transferred into conical flow cytometry tubes, processed and analysed with BD Canto II flow cytometer, as described for the phagocytosis of latex beads.
Cytokine quantification by elisa assay
MMGT12 cells were seeded on 24‐well plates (5 × 105 cells per well), in DF culture medium containing 5% FBS and then treated with LPS, MDG548 or LPS plus MDG548 added either at the same time (concurrent, c) or after 2 h (sequential, s) according to Table 1. After 24 h, media were collected and clarified by 400× g centrifugation for 10 min; supernatant samples (n = 5 for each experimental group) were collected in new tubes and frozen at −80°C until analysis. Mouse http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5074 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974 (https://www.thermofisher.com/us/en/home.html, Waltham, MA, USA), Ym1 (EIAab Science, Wuhan, China), MRC1/CD206 (Cloud‐Clone Corp., Katy, TX, USA) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4975 (Sigma‐Aldrich Corp., St. Louis, MO, USA) were assessed by sandwich elisa according to the manufacturer's instructions. Absorbance at 450 nm for all cytokines was measured with a microplate reader, model 680 (Bio‐Rad, Hercules, CA, USA). A standard curve was prepared by plotting absorbance value of the standard cytokine versus the corresponding concentration (pg·mL−1). The range of assay for cytokines was 15.6–1000 pg·mL−1 for TNF‐α, 45.0–5000 pg·mL−1 for IL‐10, 78.0–5000 pg·mL−1 for Ym1 and 15.6–1000 pg·mL−1 for MRC1.
Western blotting analysis
MMGT12 cells were seeded on six‐well plates at density 4 × 104 cells per well. After 3 days, cells were treated according to Table 1. Twenty‐four hours later, cells were lysed and processed for SDS‐PAGE and Western blotting analysis according to a previously published procedure (Janda et al., 2015). The experiment was repeated five times, and the samples were loaded on at least two different gels each time, and whenever possible, the mean OD signal was analysed and calculated for each experiment. Rabbit anti‐Beclin (MBL, cat#PD017, 1:2000) and rabbit anti‐GAPDH (cat#sc87752, 1:500; Santa Cruz Biotech. Inc, Santa Cruz, Dallas, TX, USA) were used as primary antibodies. HRP‐conjugated goat anti‐rabbit (cat#31460, 1:5000; Thermo Scientific) was used as a secondary antibody. Blots were developed with the enhanced chemiluminescence method, using ImmunoStar TM (cat# 170–5070; Bio‐Rad Lab. Inc.) and digitally acquired using a ChemiDoc XRS imaging system (Bio‐Rad Lab. Inc.).
In vivo model of PD in mice
All animal care and experimental procedures complied with the Directive 63/2010/EU on the protection of animals used for scientific purposes and were approved by the Ethics Committee of the University of Cagliari. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Three‐month‐old male C57BL/6J mice were purchased from Charles River (Calco, Italy) and randomized into six experimental groups (total n = 30). Animals were housed five per cage, in plastic cages with conventional bedding, in the animal facility of the Department of Biomedical Sciences, under controlled environmental conditions, with a 12:12 h light/dark cycle, 23–24°C, food and water ad libitum and the proper enrichment.
The control group of mice (n = 5) received saline as a vehicle. The remaining mice were randomly allocated into additional groups (n = 5 per group) to receive daily vehicle (30% DMSO, 10% PEG in water), plus 3 (MPTP3), 7 (MPTP7) and 10 (MPTP10) doses of MPTP (25 mg·kg−1 i.p.), plus probenecid (100 mg·kg−1 i.p.) (MPTPp). This MPTPp treatment was given twice a week up to 5 weeks. Group MPTP + MDG548 received 10 doses of MPTPp over 5 weeks (twice a week), plus daily MDG548 (2 mg·kg−1 i.p., 15 min before MPTPp injection) starting on the 24th day of MPTPp treatment and until the end of the experiment. The group MPTP + MDG548 + GW9662 received MPTPp and MDG548 as above, plus the PPARγ antagonist GW9662 (5 mg·kg−1 i.p.) 15 min before MDG548 injection. Three days after discontinuation of treatments, mice were anaesthetized with fentanyl (0.06 mg·kg−1 s.c.) and transcardially perfused with 4% paraformaldehyde (PFA) in 0.1 M PBS. Brains were removed, post‐fixed for 2 h and stored in 0.1% sodium azide in PBS until immunohistochemical processing. In a separate experiment, 15 mice (n = 5 per group: vehicle, MPTP10 and MPTP + MDG548) were anaesthetized and immediately killed without PFA perfusion, and their brains were used for RT‐PCR experiments.
Immunocytochemistry and immunohistochemistry
Cells plated as described in MMGT12 microglial cells and treatments section were incubated in a medium containing 5% FBS plus factors added according to Table 1. After 24 h, exposure media were saved for elisa assay, and cells were washed with PBS and fixed with 4% PFA/PBS for 15 min at room temperature under gentle agitation. Fixative solution was replaced with PBS, and cells were processed for immunocytochemistry. For iNOS, cells were incubated with iNOS antibody (iNOS monoclonal antibody 1:400; Santa Cruz Biotech. Inc); for CD68/PPARγ staining, cells were incubated with CD68 antibody (anti‐CD68 polyclonal antibody 1:200; Abbiotec, San Diego, CA, USA) plus PPARγ antibody (anti‐PPARγ mouse monoclonal antibody 1:50; Santa Cruz Biotech. Inc). After PBS washing, cells were incubated with a secondary antibody for CD68 [Alexa594‐conjugated AffiniPure Donkey Anti‐Rabbit IgG (H + L), Jackson ImmunoResearch], while three‐step detection was used to increase the signal of PPARγ by biotin‐conjugated IgG [IgG (H + L) Biotin‐Goat anti‐mouse (1:200) and streptavidin–fluorescein (1:200)] (Vector Labs. Ltd, Peterborough, UK). All five brains from each experimental group were used for immunohistochemistry. Coronal sections from mouse substantia nigra pars compacta (SNc) (40 μm thick) were vibratome cut and incubated with primary antibodies against tyrosine hydroxylase (TH; monoclonal anti‐TH 1:1000; Sigma‐Aldrich), http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=760#2452 (monoclonal anti‐CD11b 1:1000; Serotec, Oxford, UK), TNF‐α (TNF‐α polyclonal rabbit anti‐TNF‐α 1:800; Abbiotec), IL‐1β (IL‐1β polyclonal rabbit anti‐IL‐1β 1:200; Abbiotec), MRC1 (polyclonal anti‐MRC1 1:500; Santa Cruz Biotech. Inc) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5060 (TGF‐β polyclonal rabbit anti TGF‐1β 1:200; Abbiotec) and the secondary antibodies, except for TH where the classic avidin–peroxidase complex (ABC; Vector Labs. Ltd) protocol was applied using 3,30‐diaminobenzidine (Sigma‐Aldrich) as a chromogen. A three‐step detection was used to increase signal of cytokines and MRC1 by biotin‐conjugated IgG plus streptavidin–fluorescein (1:200; Vector Labs. Ltd).
Stereological counting of TH immunoreactivity
All immunohistochemical reactions were analysed by an operator blinded to experimental groups and different from the experimenter that performed the behavioural tests and histology. http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1243‐immunoreactive and Nissl‐stained neurons were counted bilaterally in the SNc as previously described (Lecca et al., 2015). A dedicated software was used (Stereologer; System Planning and Analysis, Inc., Alexandria, VA, USA) linked to a motorized stage on the BX‐60 Olympus light microscope (Olympus Optical, GmbH, Hamburg, Germany). The total number of TH‐stained or Nissl‐stained cells was estimated by means of Optical Fractionator method, which combines the optical dissector with the fractionator sampling scheme, giving a direct estimation of the number of three‐dimensional objects unbiased by shape, size and orientation (Mouton et al., 2002). A systematic random sampling of cells within the area of interest was achieved by ‘Stereologer’ program. Equidistant counting frames (frame area = 50 μm2) were obtained. Sampling fraction was delimited at low power, and cells were sampled with a ×40 oil immersion objective through a defined depth with a 2 μm guard zone. The coefficient of error for each estimation and animal ranged from 0.05 to 0.1.
CD11b analysis
CD11b‐positive microglia were identified at ×100/1.25 oil magnification. For each animal, six fields of SNc were captured from both the left and the right SNc and analysed. For each microglial cell, the body and primary processes were outlined. The total area occupied by CD11b immunoreactivity (IR) and the mean area per cell was measured using NIH software ImageJ 1.47v (NIH, Bethesda, MD, USA).
Confocal analysis
Quantitative analysis for CD68 colocalization with PPARγ in MMGT12 cells, TNF‐α, IL‐1β, MRC1 and IL‐10 colocalization with CD11b in vivo was performed using a Leica 4D confocal laser scanning microscope, equipped with an argon–krypton laser (Carta et al., 2011). Images were digitized 24 h after the immunofluorescence procedure. Surface rendering, maximum intensity, colocalization and simulated fluorescence process algorithms were used (ImageJ 1.48q and Imaris 7.0). Volume of colocalized elements was determined as follows: for each dataset (40–60 images), a colocalization channel was automatically composed by Imaris 7.3. In the resulting stacks, four regions of interest (x = 40 μm; y = 40 μm; and z = 10 μm) were randomly chosen, and volume of the elements of interest was calculated, summed and expressed as volume μm−3 (n = 200).
RT‐PCR analysis of gene expression
The midbrain was dissected, and RNA was isolated using RNeasy Plus Universal Mini Kit (Qiagen, Ambion Inc., Austin, TX, USA) according to the manufacturer's instructions. MIQE guidelines were followed for RT‐PCR analysis. RNA (2 μg) was reverse transcribed and real‐time PCR for Tnf, and Mrc1 was performed in a 7300 Real‐Time PCR System (Life Technologies, Monza, Italy). The amplification mixture contained 20 ng of cDNA, 10 μL 2× TaqManGene Expression PCR Master mix (Life Technologies, Monza, Italy) and 1 μL of specific 20× TaqMan Gene Expression Assay (Mm00485148‐m1 for Mrc1/CD206 and Mm00443258‐m1/Tnf) (Pisanu et al., 2014). Samples were analysed in triplicate with β‐actin as housekeeping control (Pisanu et al., 2014). Relative gene expression was calculated according to the 2‐ΔΔCT method using the vehicle‐treated tissue as control.
Beam traverse test
This test was used to assess motor performance of mice and to investigate whether MDG548 rescued MPTP‐induced motor impairment (Schintu et al., 2009a). The beam was constructed as described (Schintu et al., 2009a). Mice were trained for two consecutive days to traverse the beam. On the test day, a grid (1 cm2) of corresponding width was placed 1 cm above the beam, and mice were videotaped while traversing it for a total of five trials. Limb slips were considered errors, and total errors were calculated by an operator blinded to experimental groups (Schintu et al., 2009b).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). Variance inhomogeneity was assessed by Bartlett's test for equal variances in all data sets and for all experimental designs. Where variance was homogenous, data were statistically compared by one‐way ANOVA followed by Tukey's HSD or Fisher's post hoc test when P < 0.05, with the software Statistica. When variances differed significantly (P < 0.05) with Bartlett's test, we applied a non‐parametric test (Kruskal‐Wallis) followed by Dunn's multiple comparison test. Level of significance was set at P < 0.05.
Materials
MPTP‐HCl (Sigma‐Aldrich, Milan, Italy) was dissolved in saline; probenecid (Sigma‐Aldrich) was dissolved in 5% NaHCO3; MDG548 (Specs ID number: AN‐698/15136006; 5‐[4‐(benzyloxy)‐3‐chlorobenzylidene]‐2‐thioxodihydropyrimidine‐4,6(1H,5H)‐dione; mol. weight: 372.83) was solubilized in 30% DMSO, 10% polyethylene glycol (PEG) and water. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3442 (Sigma‐Aldrich) was solubilized in 5% DMSO and water. For in vitro studies, MDG548 was dissolved in DMSO, while LPS (1 μg·mL−1; Sigma‐Aldrich) was dissolved in water. DMSO was added to vehicle and LPS‐treated wells instead of MDG548. The final concentration of DMSO in culture medium was 0.1% for all experimental groups.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c).
Results
MDG548 stimulated phagocytosis in MMGT12 microglia
We investigated the effect of MDG548 on phagocytosis in a functional beads‐based assay and neuronal cells‐based assay, in murine microglial cells MMGT12. We exposed the cells to the drug in the presence and absence of LPS for 24 h and then assessed their ability to engulf fluorescent latex microspheres (Figure 1A) or necrotic SH‐SY5Y and cell debris (Figure 1C), by co‐incubation and subsequent flow cytometry analysis. Control cells showed a basal level of phagocytosis ranging from 0.5% in the SH‐SY5Y (Figure 1D) assay to 6% in the beads assay (Figure 1B), which was unaffected by LPS alone (Figure 1A, B). Phagocytosis was significantly increased by MDG548 alone, at both tested doses, as compared with controls. Moreover, MDG548 added 2 h after adding LPS within 24 h incubation time (LPS + MDG10s) further increased the percentage of phagocytotic cells, while MDG548 applied at the same time with LPS was less effective in driving microglia phagocytosis (Figure 1B, D), as shown in Figure 2.
Figure 1.
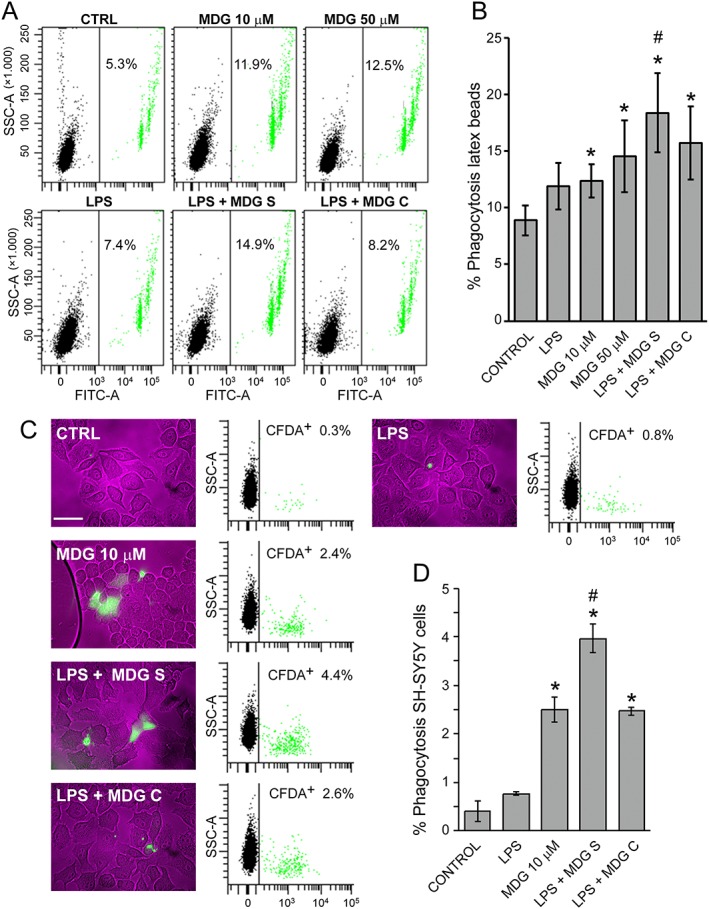
MDG548 increased phagocytosis of fluorescent latex microspheres and necrotic SH‐SY5Y cells. (A) Representative flow cytometry dot plots [green fluorescence vs. side scatter (SSC‐A)] of MMGT12 cells treated for 24 h as indicated: CTRL, vehicle control; MDG, MDG548 10 or 50 μM; LPS, 1 μg·mL−1 LPS; ‘LPS + MDG10 S’, LPS in association with MDG548 (10 μM), added 2 h after LPS or ‘LPS + MDG10 C’ added at the same time. After treatment, the cells were incubated with green fluorescent latex beads for 2 h. Note that cells engulfed one up to four beads within 2 h assay (1 bead cells and 4 bead cells are the most left and a small fourth green population, respectively). (B) The graph shows the means of nine independent samples ± SEM. (C) Representative phase contrast/green fluorescence pictures and flow cytometry dot plots (CFDA fluorescence vs. SSC‐A) of MMGT12 cells after a phagocytosis assay with necrotic dopaminergic SH‐SY5Y cells, stained with CFDA (green). The microglia were treated as indicated above (50 μM MDG548 was omitted), but necrotic green SH‐SY5Y cells (1:1 ratio) were added for 5 h, then carefully removed before the analysis of MMGT12 cells by flow cytometry and phase contrast and fluorescence microscopy analysis (white bar, 50 μm). (D) The graph shows the mean % of green MMGT cells analysed by flow cytometry (after engulfing green SH‐SY5Y cells) from six independent samples for each experimental group ± SEM. Data were analysed with ANOVA followed by Tukey's post hoc test and (for the latex beads assay) with the Kruskal‐Wallis test followed by Dunn's multiple comparison test. ^ P < 0.05, significantly different from control cells; * P < 0.05, significantly different from LPS alone‐treated cells; or # P < 0.05, significantly different from MDG548 only or LPS + MDG10c‐treated cells.
Figure 2.
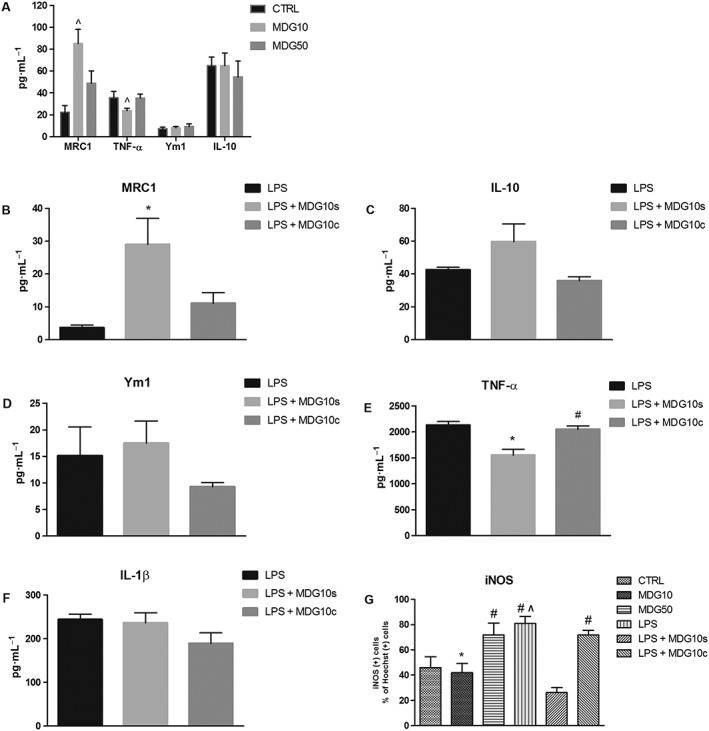
MDG548 modified the production of TNF‐α, MRC1 and iNOS in MMGT12 cells. (A) MDG548 effect at the doses of 10 and 50 μM (MDG10 and MDG50, respectively). (B‐F) Effect of LPS alone or in association with MDG548 10 μM, added 2 h after LPS (LPS + MDG10s) or at the same time point (LPS + MDG10c). TNF‐α, IL‐1β, IL‐10, MRC1 and Ym1 were measured by elisa. (G) Effect of MDG548, LPS alone or in association with MDG548 on iNOS expression. iNOS was measured by immunohistochemistry. ^ P < 0.05, significantly different from control cells; * P < 0.05, significantly different from LPS alone‐treated cells; # P < 0.05, significantly different from LPS + MDG10s‐treated cells.
MDG548 altered microglia phenotype in LPS‐stimulated MMGT12 microglia
We investigated the effect of MDG548 on pro‐inflammatory and anti‐inflammatory markers in MMGT12 cells. MDG548 displayed a dose‐dependent and opposite effect on MRC1 and TNF‐α levels. elisa assay showed that 10 μM, but not 50 μM, of MDG548 significantly increased MRC1 levels and decreased TNF‐α levels in unstimulated cells, as compared with vehicle (control) (Figure 2A). Ym1 and IL‐10 were unaffected by MDG548, while IL‐1β levels were in the low physiological range and therefore undetectable (data not shown). When cells were stimulated with LPS, MRC1 and IL‐10 decreased below vehicle levels, while the secretion of TNF‐α and IL‐1β markedly increased above vehicle (compare absolute values of control in Figure 2A with Figure 2B, C, E, F). Again, Ym1 was unaffected (Figure 2D). Finally, when MDG548 10 μM was added 2 h after LPS (LPS + MDG10s), it reversed the LPS‐induced decrease of MRC1 to control levels and attenuated the LPS‐induced increase of TNF‐α (Figure 2B, E). Moreover, IL‐10 showed a tendency to increase after LPS + MDG10s (Figure 2C), while IL‐1β and Ym1 were unaffected (Figure 2D, F). The same changes were not observed when MDG548 was applied at the same time as LPS. We also measured the iNOS IR in cells and found that LPS significantly increased the number of iNOS positive cells as compared with vehicle, while LPS + MDG10s totally reversed such effect (Figure 2G).
MDG548 increased the expression of CD68 and Beclin‐1
In order to further investigate MDG548 effect on phagocytic activity, we analysed the expression of CD68, a scavenger receptor suggested to be involved in macrophage and microglial phagocytosis, by evaluating immunoreactivity for CD68 (CD68IR) in MMGT12 cells treated as in Table 1 (Figure 3). A preliminary experiment assessing the expression of PPARγ in MMGT12 cells showed constitutive expression in all cells. We therefore analysed CD68 IR colocalization in PPARγ‐stained cells. LPS and MDG548 alone did not change CD68 IR, while MDG548 (10 μM) added after LPS strongly enhanced CD68 IR (Figure 3A). Again, the effect was limited to the experiment where MDG548 was administered 2 h after LPS, as adding MDG548 concomitantly with LPS did not affect CD68 levels.
Figure 3.
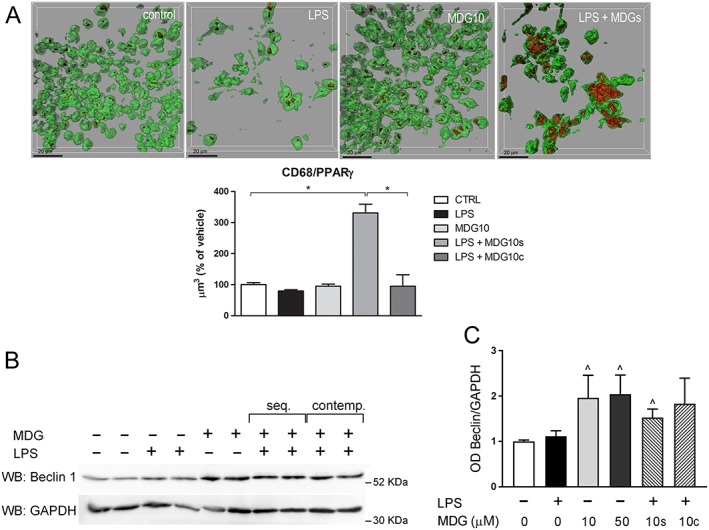
MDG548 increased the expression of phagocytosis marker CD68 andautophagy/phagocytosis regulator Beclin‐1. (A) Cells were treated for 24 h with LPS or with LPS in association with MDG548 10 μM, added 2 h after LPS (LPS + MDG10s) or at the same time point (LPS + MDG10c) and analysed for immunofluorescent co‐staining of CD68 (red) and PPARγ (green). n = 5 for each experimental group, *P < 0.05, significantly different as indicated. (B, C) After treatments described in (A), cells were lysed, and total cell lysates were analysed by Western blotting. Five independent experiments were performed. (B) Representative immunoblot for Beclin‐1 showing two independent samples for each experimental group. (C) Densitometric analysis of Beclin‐1 normalized to respective GAPDH signal (loading control). Mean ± SEM from n = 5 independent experiments. Data were analysed with the Kruskal‐Wallis test followed by Dunn's multiple comparison test; ^ P < 0.05, significantly different from control cells.
In search of a possible molecular mechanism of PPARγ‐mediated effect on phagocytosis, we also investigated the expression of Beclin‐1/BECN1. Beclin‐1 plays an important role in both autophagy and phagocytosis in different cell types, including microglia (Lucin et al., 2013). MMGT12 cells were treated as described in Table 1, and we assessed Beclin‐1 levels by Western blotting. We observed a marked increase of Beclin‐1 expression in response to MDG548, no matter if it was applied alone or together with LPS. This increase was statistically significant for all MDG548‐treated samples, except for LPS + MDG10c treatment (Figure 3C, D). This indicated that Beclin‐1 expression was regulated by PPARγ and may play a role in MDG548‐induced phagocytosis.
MDG548 arrested nigral degeneration and motor impairment in MPTPp‐treated mice
Next, we addressed the ability of MDG548 to arrest the MPTP‐induced nigral degeneration and motor impairment in the MPTPp mouse model of PD, which is a widely used and validated model of progressive PD (Schintu et al., 2009b). In accordance with previous reports (Schintu et al., 2009b), mice chronically treated with MPTPp showed progressive neurodegeneration in the SNc depending on the number of MPTPp injections (3, 7 or 10 respectively in groups MPTP3, MPTP7 and MPTP10), reaching a 40% reduction of TH+ cells after 10 doses of MPTPp, compared with vehicle‐treated mice (Figure 4A, D). The daily administration of MDG548 starting on the 24th day of MPTPp treatment (group MPTP + MDG548) arrested the loss of dopaminergic TH‐positive cells (Figure 4A, D), while the simultaneous administration of the PPARγ antagonist GW9662 (group MPTP + MDG548 + GW9662) attenuated the effect of MDG548, suggesting a PPARγ‐mediated mechanism of neuroprotection (Figure 4A, D). The analysis of Nissl staining confirmed the MPTPp‐induced cell loss and the protection by MDG548 (number of stained cells for vehicle: 16 238.4 ± 517.74; MPTP10: 8586.00 ± 884.27; and MPTP + MDG548: 13 508.00 ± 499.01). Moreover, the chronic administration of MPTPp caused a progressive motor impairment, as revealed by the increase of stepping errors measured by the beam walking test, as previously reported (Figure 4E) (Schintu et al., 2009b). Daily MDG548 administration starting from the 24th day of MPTPp treatment fully reversed MPTPp‐induced motor impairment, providing functional validation of neuroprotection and suggesting a disease‐modifying effect of PPARγ stimulation. Consistent with these findings, the effects of MDG548 were blocked by co‐administration of PPARγ antagonist GW9662 (Figure 4E).
Figure 4.
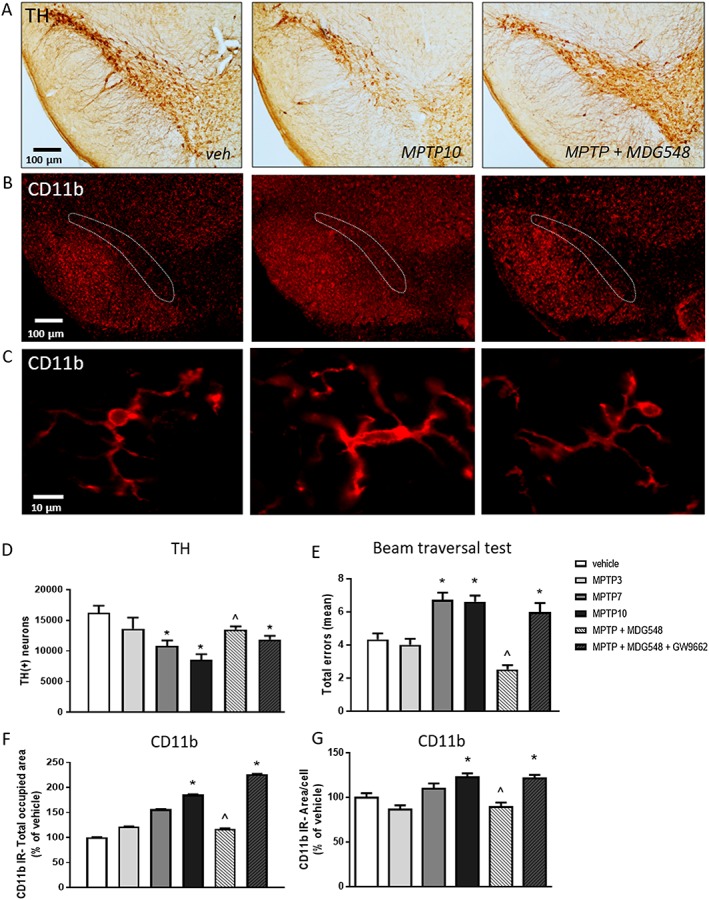
MDG548 arrested MPTPp‐induced motor deficits and nigral neurodegeneration in mice. Mice received from 3 up to 10 doses of MPTPp over 5 weeks (MPTP3, MPTP7 and MPTP10; n = 5 for each experimental group). MDG548 (2 mg·kg−1 i.p.) was added daily to MPTP10 group starting from the 24th day of MPTPp treatment (MPTP + MDG548; n = 5). (A) Representative images of the TH‐labelled substantia nigra from mice treated with vehicle (veh), MPTP10 and MPTP + MDG548. (B, C) Representative images of CD11b immunostaining in the substantia nigra from mice receiving the same treatment than in (A). In all experimental groups (n = 5 for each experimental group), (D) the total number of TH‐positive cells was measured by stereological counting; (E) motor impairment was measured by the beam walking test and expressed as total step errors; (F, G) CD11b IR was measured in the SNc as the total area occupied by CD11b‐positive cells and as the mean of CD11b‐positive area per cell. *P < 0.05 significantly different from veh‐treated mice; ^ P < 0.05 significantly different from MPTP10. See Methods for details.
MDG548 attenuated MPTPp‐induced microgliosis in the SNc
The chronic MPTPp treatment induced a gradual increase of microgliosis, as indicated by the increase of the total area occupied by CD11b IR in the analysed region (Figure 4B, F). Moreover, cells displayed changes in cell morphology after the MPTPp treatment, as indicated by measurement of the mean area, obtained by outlining the body and primary processes for each cell (Figure 4C, G). Morphology switched from a shape with a small body and highly ramified processes to an amoeboid form, in accordance with previous studies (Schintu et al., 2009b). MDG548 attenuated the changes induced by MPTPp in cell proliferation as well as in cell morphology, while GW9662 counteracted the effect of MDG548 (Figures 4B and 5F, G).
Figure 5.
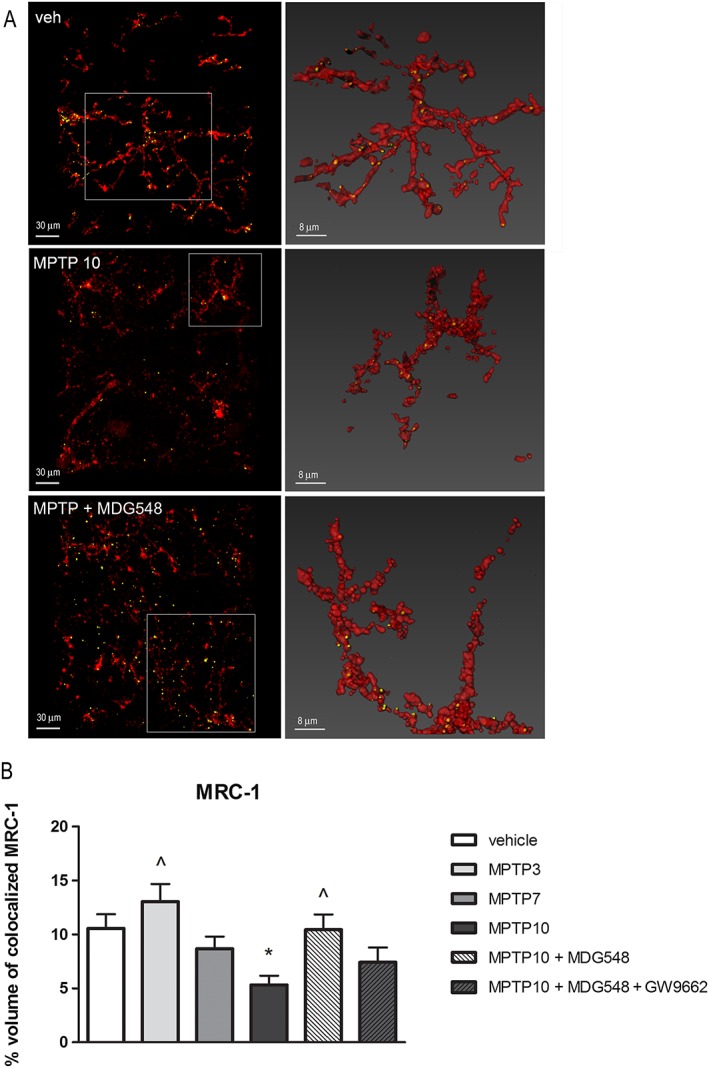
MDG548 restored the MRC1 IR in microglia of MPTPp‐treated mice. Representative confocal images showing MRC1 (yellow) in CD11b (red)‐positive cells in the SNc from mice chronically treated with vehicle (veh), MPTP10 or MPTP + MDG548. The percentage of colocalization of MRC1 in CD11b‐positive cells was calculated in all the six experimental groups (n = 5 for each experimental group). *P < 0.05 significantly different from veh; ^ P < 0.05, significantly different from MPTP10.
MDG548 reversed the MPTPp‐induced changes in MRC1 and cytokines in microglia
We performed confocal analysis of MRC1 IR and cytokines colocalized with CD11b in the SNc. The analysis showed that chronic MPTPp decreased MRC1 and TGF‐β IR in CD11b‐positive cells of the SNc, while increased TNF‐α and IL‐1β IR (Figures 5, 6, 7). The co‐administration of MDG548 with MPTPp enhanced the levels of MRC1 and TGF‐β colocalized with CD11b, restoring them to physiological values, and reduced levels of TNF‐α and IL‐1β IR (Figures 5, 6, 7), while GW9662 attenuated these effects of MDG548. Analysis of Mrc1 mRNA showed a decrease after MPTP10 treatment, compared with vehicle, which was restored by MPTP + MDG548 (Table 2). In addition, expression of Tnf mRNA did not change after MPTP10 or MPTP + MDG548, suggesting that the chronic exposure to MPTPp might induce a self‐repression of TNF‐α expression (Table 2).
Figure 6.
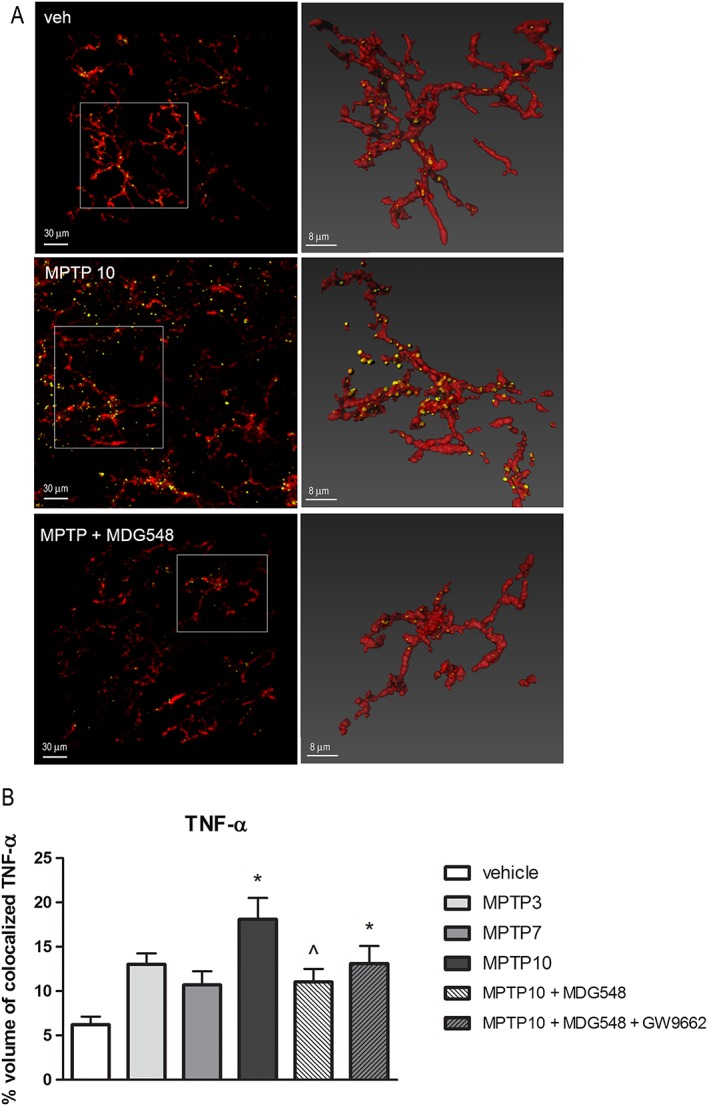
MDG548 decreased the TNF‐α IR in microglia of MPTPp‐treated mice. Representative confocal images showing TNF‐α (yellow) in CD11b (red)‐positive cells in the SNc from mice chronically treated with vehicle (veh), MPTP10 or MPTP + MDG548. The percentage of colocalization of TNF‐α in CD11b‐positive cells was calculated in all the six experimental groups (n = 5 for each experimental group). *P < 0.05, significantly different from veh; ^ P < 0.05, significantly different from MPTP10.
Figure 7.
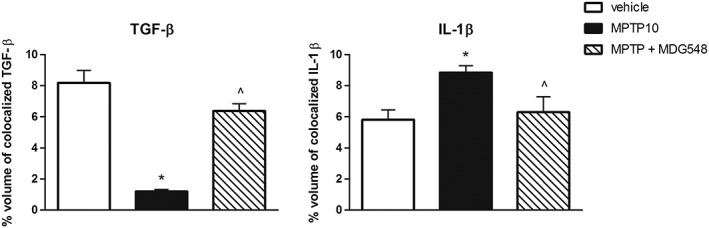
MDG548 restored TGF‐β and IL‐1β IR in microglia of MPTPp‐treated mice. The percentage of colocalization of TGF‐β and IL‐1β in CD11b‐positive cells was calculated in the most representative experimental groups vehicle, MPTP10 and MPTP + MDG548 (n = 5 for each experimental group). *P < 0.05, significantly different from vehicle; ^ P < 0.05, significantly different from MPTP10.
Table 2.
Analysis of Mrc1 and Tnf mRNA expression in the mouse midbrain
| MRC1 | TNF | |
|---|---|---|
| V | 0.84 ± 0.13 | 1.22 ± 0.16 |
| MPTP10 | 0.31 ± 0.02* | 0.75 ± 0.07 |
| MPTP + MDG548 | 0.61 ± 0.08 | 1.03 ± 0.21 |
Expression of mRNA for MRC1 and TNF as measured by RT‐PCR in homogenates of midbrain tissue, 24 h after completion of chronic treatment with vehicle (V), 10 doses of MPTPp in association with daily vehicle (MPTP10) or with daily MDG548 (MPTP + MDG548).
P < 0.05, significantly different from vehicle by Dunn's multiple comparison test.
Discussion
The neuroprotective and disease‐modifying potential of PPARγ agonists in PD and in other neurodegenerative pathologies has been widely reported, though the underlying mechanism is still debated (Mandrekar‐Colucci and Landreth, 2011; Carta, 2013). Here, we show that the PPARγ agonist MDG548 stimulated microglia phagocytic activity and modulated the profile of cytokines expression leading an alternative activation state in microglia, while suppressing inflammatory responses. These effects were observed in vitro in cultures of the microglial cell line MMGT12 as well as in vivo in a model of progressive PD, where MDG548 also displayed a disease‐modifying activity, arresting the nigral dopaminergic degeneration. Several studies have demonstrated the pivotal role of PPARγ in peripheral macrophage/monocytes polarization and have shown that PPARγ is required for maturation of alternatively activated macrophages in physiological as well as pathological conditions (Odegaard and Chawla, 2011). Our results demonstrate that PPARγ exerted a similar function in microglia in an experimental model of PD and suggest that promoting phagocytosis together with the production of anti‐inflammatory cytokines and suppression of pro‐inflammatory molecules may underlie PPARγ‐mediated neuroprotection in PD. Our results are compatible with earlier studies showing that PPARγ stimulation boosted the M2‐like microglia phenotype and accelerated functional recovery in rodent models of cerebral haemorrhage or stroke (Zhao et al., 2015; Flores et al., 2016) and promoted phagocytosis in a model of Alzheimer's disease (Yamanaka et al., 2012; Savage et al., 2015). The role of microglial phagocytosis in PD, whether beneficial or detrimental, is debated. Although some studies have proposed an increased phagocytic activity in microglia (Croisier et al., 2005; Theodore et al., 2008; Sanchez‐Guajardo et al., 2010; Doorn et al., 2014; Fourgeaud et al., 2016), others have reported a defective phagocytosis in this cell type or in other immune cells (Salman et al., 1999; Gardai et al., 2013). Moreover, an increasing number of studies indicate that microglia contribute to the progression of neurodegeneration in PD, by acquiring an unremitting pro‐inflammatory phenotype, with chronic production of pro‐inflammatory cytokines, iNOS, ROS and reactive nitrogen species (Boka et al., 1994; Joers et al., 2017; López González et al., 2016; Mogi et al., 2007; Pisanu et al., 2014; Sawada et al., 2006). The present study points to a reduced phagocytic activity by microglia in the MPTPp progressive model of PD, by showing a decreased MRC1 IR in these cells, which was associated with increased production of inflammatory cytokines.
To specifically address PPARγ stimulation in relation to microglial function, we first investigated the direct effect of MDG548 on cultured MMGT12 microglia, where the expression of PPARγ was first confirmed immunohistochemically, as shown in Figure 3. Although limitations of the in vitro model used here must be taken into consideration, these results provided evidence for the direct effect of PPARγ stimulation on microglial cells, which cannot be determined in vivo, where the diffuse PPARγ expression in different cell types does not allow the determination of their function in a selected cell population. We measured the microglia phagocytic activity by two functional assays, which measured engulfment of latex beads or necrotic cells. Thereafter, we assessed MDG548 effect in a classical inflammatory model such as LPS stimulation, by analysing both typical markers of pro‐inflammatory profile, such as TNF‐α, IL‐1β and iNOS, as well as molecules typically expressed by alternatively activated macrophages, such as IL‐10, MRC1 and Ym1 (Croasdell et al., 2015). In addition, we measured the expression of scavenger receptor CD68. Finally, we addressed a possible modulation of Beclin‐1, a recently identified regulator of phagocytosis (Lucin et al., 2013).
We observed that PPARγ stimulation with MDG548 induced a significant induction of phagocytosis of necrotic cells in untreated microglia and slightly increased the phagocytosis of latex beads. This was associated with a decrease of TNF‐α release and a robust increase of MRC1 and Beclin‐1 expression, suggesting that PPARγ stimulation itself induced a phagocytic microglia profile while lessening the production of inflammatory mediators. In particular, cytokine production was down‐regulated by 10 μM MDG548 but not 50 μM, which instead significantly increased iNOS IR. In a previous study, we found that 10 μM MDG548 was neuroprotective, while 50 μM was cytotoxic to neurons, suggesting that additional intracellular pathways may be activated by such higher concentrations (Lecca et al., 2015). Interestingly, LPS‐stimulated MMGT12 microglia did not show any phagocytic activity or changes in Beclin‐1 expression, while displayed increased levels of TNF‐α, IL‐1β and iNOS with a great suppression of MRC1, suggesting that LPS induced a pro‐inflammatory phenotype with impaired phagocytic activity. When MDG548 was administered to LPS‐stimulated microglia, we observed an amplified effect on phagocytosis as compared with LPS alone‐treated cells and an increase of CD68. Previous studies have demonstrated the PPARγ‐dependent regulation of CD68 expression in peripheral macrophages (Odegaard et al., 2007). Moreover, MDG548 reversed the LPS‐induced changes in cytokines and iNOS production. Remarkably, MDG548 was ineffective when added concurrently with LPS in all the assays performed in the present study. Although the investigation of mechanisms behind this finding goes beyond the scope of the present study, it should be pointed out that MDG548 might affect two main mechanisms of PPARγ‐mediated control of microglia activation. PPARγ inhibits the expression of inflammatory genes via the interaction at multiple steps with the LPS‐activated NF‐κB pathway, including a direct protein–protein interaction with NF‐κB (Varga et al., 2011). This mechanism may be more efficient after LPS‐induced release of NF‐κB from NF‐κB–IκB inactive complex. Moreover, PPARγ stimulates M2 polarization in macrophages through a crosstalk with the transcription factor STAT6 in its active phosphorylated state (Szanto et al., 2010). Recent studies have demonstrated that NF‐κB activation also induces an intracellular pathway for resolution of inflammation and M2 polarization of macrophages (Porta et al., 2009; Tugal et al., 2013). Therefore, the previous activation of M1‐related programs by LPS in microglia may paradoxically potentiate the effect of a subsequently administered PPARγ agonist. Our finding is of particular relevance for PD, where disease‐modifying drugs are expected to change a pre‐existing inflammatory environment. MDG548 significantly induced Beclin‐1 levels; however, this effect was independent from LPS administration in contrast to the observations on phagocytosis and CD68 expression. As Beclin‐1 plays also a role in autophagy regulation and it is induced when autophagy is stimulated (Janda et al., 2015), Beclin‐1 up‐regulation by MDG548 may reflect a pro‐autophagic capacity of this compound. Such an additional pharmacological activity of MDG548 would be beneficial, considering that autophagy has become a potential therapeutic target in PD (Janda et al., 2012; Janda et al., 2015; Plaza‐Zabala et al., 2017).
In vitro findings supported the observations made in the in vivo PD model, where the PPARγ agonist administered in the late stages of the disease was able to switch the MPTPp‐induced phenotype of activated microglia and arrested the ongoing neurodegeneration. After chronic MPTPp treatment, microglia acquired a pro‐inflammatory profile, as indicated by the increases of TNF‐α and IL‐1β and the decreases of MRC1 and TGF‐β (Figure 8). MDG548 was administered in the late disease phase to investigate its disease‐modifying potential. Following PPARγ stimulation, we observed a switch in the profile and function of microglia, which acquired a phenotype with high levels of MRC1 and TGF‐β and low TNF‐α and IL‐1β, suggesting the acquisition of an ‘alternative’ activation state with increased phagocytic activity (Figure 8). The lack of modification of Tnf mRNA suggests that chronic exposure to MPTP might induce a feedback self‐repression of gene expression at the analysed time point related to protein overproduction, in line with previous findings (Pisanu et al., 2014). Our results also showed that MDG548 was neuroprotective as it arrested the MPTP‐induced loss of SNc neurons and rescued the motor impairment, confirming a PPARγ‐mediated protective effect in a PD model. Previous studies have reported TZD‐induced changes of microglial response in terms of cytokines and iNOS production, both in rodent and primate models of PD, as well as a neuroprotective action of these drugs and MDG548 itself in PD models (Kitamura et al., 1999; Cunard et al., 2002; Ji et al., 2010; Carta et al., 2011; Swanson et al., 2011; Pisanu et al., 2014; Lecca et al., 2015). Interestingly, MDG548 was effective at a dose fivefold lower than TZDs, due to higher receptor affinity and BBB permeability (Dehmer et al., 2004; Schintu et al., 2009a; Nevin et al., 2012). The use of TZDs has recently encountered several safety concerns, which largely limit their translation to the clinic in PD (Carta and Simuni, 2015; Simon et al., 2015). Particularly, as TZDs were originally developed for the treatment of type 2 diabetes, their pharmacokinetic features limit CNS entrance, requiring elevated dosage in neurological disorders.
Figure 8.
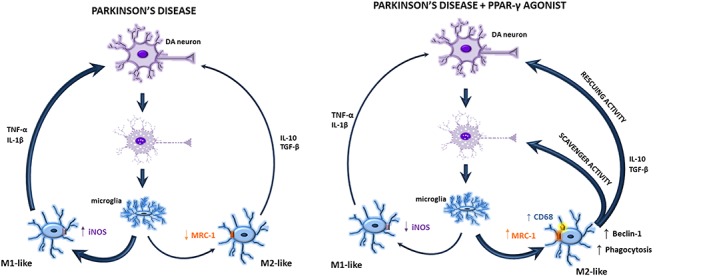
Diagram summarizing the observed effects and suggested mechanism of PPAR‐γ‐mediated neuroprotection in the MPTPp model of PD. Damaged dopaminergic (DA) neurons activate microglia to assume a M1‐like pro‐inflammatory phenotype with increased release of inflammatory molecules and defective phagocytosis. Administration of the PPAR‐γ agonist boosts phagocytosis and stimulates microglia to acquire an anti‐inflammatory phenotype, therefore resulting in both rescuing and scavenging activity.
In conclusion, numerous studies indicate that microglia are subjected to many plastic changes of their phenotype both in physiological and pathological conditions, and targeting the microglial phenotype is a recognized potential therapeutic target in neurological disorders. The present study suggests that boosting the phagocytic function of microglia and the acquisition of an anti‐inflammatory phenotype may represent a novel mechanism of PPARγ‐mediated neuroprotection (Figure 8). Despite the discouraging results recently provided by clinical trials in PD patients with the TZD pioglitazone, our study adds to an increasing literature providing the rationale to pursue clinical investigation of PPARγ in neurodegenerative diseases.
Author contributions
D.L. performed all the in vivo mice treatments, behavioural tests, analysis and statistics. E.J. and C.M. performed the phagocytosis in vitro and Western blot experiments with relative data analysis. G.M. and L.B. performed all the immunohistochemistry. A.D. developed and standardized the procedure to culture microglial cell line MMGT12. F.A., S.S. and B.B. performed all the elisa assays. M.A.C. performed the blinded stereological analysis. G.S. performed RT‐PCR experiments. S.S. performed all the confocal analysis of immunohistochemical reactions. A.R.C. conceived, supervised and coordinated the work, interpreted the data and mainly drafted the manuscript. All authors fulfilled the four ICJME authorship criteria, having contributed to experimental design, revised the work and approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was supported by the Fondazione Banco di Sardegna, grant number U629.2013/AI.553.MGB. We acknowledge Professor T. Heurtaux and A. Michelucci for kindly providing MMGT12 cells.
Lecca, D. , Janda, E. , Mulas, G. , Diana, A. , Martino, C. , Angius, F. , Spolitu, S. , Casu, M. A. , Simbula, G. , Boi, L. , Batetta, B. , Spiga, S. , and Carta, A. R. (2018) Boosting phagocytosis and anti‐inflammatory phenotype in microglia mediates neuroprotection by PPARγ agonist MDG548 in Parkinson's disease models. British Journal of Pharmacology, 175: 3298–3314. 10.1111/bph.14214.
References
- Ajmone‐Cat MA, Lavinia Salvatori M, De Simone R, Mancini M, Biagioni S, Bernardo A et al (2012). Docosahexaenoic acid modulates inflammatory and antineurogenic functions of activated microglial cells. J Neurosci Res 90: 575–587. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide To PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174 (Suppl 1): S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia C, Ros CM, Ros‐Bernal F, Gomez A, Annese V, Carrillo‐de Sauvage MA et al (2013). Persistent phagocytic characteristics of microglia in the substantia nigra of long‐term Parkinsonian macaques. J Neuroimmunol 261: 60–66. [DOI] [PubMed] [Google Scholar]
- Boka G, Anglade P, Wallach D, Javoy‐Agid F, Agid Y, Hirsch EC (1994). Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson's disease. Neurosci Lett 172: 151–154. [DOI] [PubMed] [Google Scholar]
- Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S et al (2007). PPARγ activation primes human monocytes into alternative M2 macrophages with anti‐inflammatory properties. Cell Metab 6: 137–143. [DOI] [PubMed] [Google Scholar]
- Briers TW, Desmaretz C, Vanmechelen E (1994). Generation and characterization of mouse microglial cell lines. J Neuroimmunol 52: 153–164. [DOI] [PubMed] [Google Scholar]
- Carta AR, Frau L, Pisanu A, Wardas J, Spiga S, Carboni E (2011). Rosiglitazone decreases peroxisome proliferator receptor‐γ levels in microglia and inhibits TNF‐α production: new evidences on neuroprotection in a progressive Parkinson's disease model. Neuroscience 194: 250–261. [DOI] [PubMed] [Google Scholar]
- Carta AR (2013). PPAR‐γ: therapeutic prospects in Parkinson's disease. Curr Drug Targets 14: 743–751. [DOI] [PubMed] [Google Scholar]
- Carta AR, Simuni T (2015). Thiazolidinediones under preclinical and early clinical development for the treatment of Parkinson's disease. Expert Opin Investig Drugs 24: 219–227. [DOI] [PubMed] [Google Scholar]
- Chistiakov DA, Killingsworth MC, Myasoedova VA, Orekhov AN, Bobryshev YV (2017). CD68/macrosialin: not just a histochemical marker. Lab Invest 97: 4–13. [DOI] [PubMed] [Google Scholar]
- Croasdell A, Duffney PF, Kim N, Lacy SH, Sime PJ, Phipps RP (2015). PPARγ and the innate immune system mediate the resolution of inflammation. PPAR Res 2015: Article ID 549691, 20 pages. 10.1155/2015/549691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croisier E, Moran LB, Dexter DT, Pearce RKB, Graeber MB (2005). Microglial inflammation in the Parkinsonian substantia nigra: relationship to α‐synuclein deposition. J Neuroinflammation 2: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunard R, Ricote M, DiCampli D, Archer DC, Kahn DA, Glass CK et al (2002). Regulation of cytokine expression by ligands of peroxisome proliferator activated receptors. J Immunol 168: 2795–2802. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Brit J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB (2004). Protection by pioglitazone in the MPTP model of Parkinson's disease correlates with IκBα induction and block of NFκB and iNOS activation. J Neurochem 88: 494–501. [DOI] [PubMed] [Google Scholar]
- Depboylu C, Stricker S, Ghobril JP, Oertel WH, Priller J, Höglinger GU (2012). Brain‐resident microglia predominate over infiltrating myeloid cells in activation, phagocytosis and interaction with T‐lymphocytes in the MPTP mouse model of Parkinson disease. Exp Neurol 238: 183–191. [DOI] [PubMed] [Google Scholar]
- Doorn, K.J. , Moors, T. , Drukarch, B. , van de Berg, W.D. j., Lucassen, P.J. , van Dam, A.M. , 2014. Microglial phenotypes and toll‐like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathol Commun 2: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores JJ, Klebe D, Rolland WB, Lekic T, Krafft PR, Zhang JH (2016). PPARγ‐induced upregulation of CD36 enhances hematoma resolution and attenuates long‐term neurological deficits after germinal matrix hemorrhage in neonatal rats. Neurobiol Dis 87: 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourgeaud L, Través PG, Tufail Y, Leal‐Bailey H, Lew ED, Burrola PG et al (2016). TAM receptors regulate multiple features of microglial physiology. Nature 532: 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardai SJ, Mao W, Schüle B, Babcock M, Schoebel S, Lorenzana C et al (2013). Elevated α‐synuclein impairs innate immune cell function and provides a potential peripheral biomarker for Parkinson's disease. PLoS One 8: e71634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K (2010). Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol 10: 365–376. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurtaux T, Michelucci A, Losciuto S, Gallotti C, Felten P, Dorban G et al (2010). Microglial activation depends on β‐amyloid conformation: role of the formylpeptide receptor 2. J Neurochem 114: 576–586. [DOI] [PubMed] [Google Scholar]
- Janda E, Isidoro C, Carresi C, Mollace V (2012). Defective autophagy in Parkinson's disease: role of oxidative stress. Mol Neurobiol 46: 639–661. [DOI] [PubMed] [Google Scholar]
- Janda E, Lascala A, Carresi C, Parafati M, Aprigliano S, Russo V et al (2015). Parkinsonian toxin‐induced oxidative stress inhibits basal autophagy in astrocytes via NQO2/quinone oxidoreductase 2: implications for neuroprotection. Autophagy 11: 1063–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joers V, Tansey MG, Mulas G, Carta AR (2017). Microglial phenotypes in Parkinson's disease and animal models of the disease. Prog Neurobiol 155: 57–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Wang H, Zhang F, Li X, Xiang L, Aiguo S (2010). PPARγ agonist pioglitazone inhibits microglia inflammation by blocking p38 mitogen‐activated protein kinase signaling pathways. Inflamm Res 59: 921–929. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. J Pharmacol Pharmacother 1: 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Kakimura J, Matsuoka Y, Nomura Y, Gebicke‐Haerter PJ, Taniguchi T (1999). Activators of peroxisome proliferator‐activated receptor‐γ (PPARγ) inhibit inducible nitric oxide synthase expression but increase heme oxygenase‐1 expression in rat glial cells. Neurosci Lett 262: 129–132. [DOI] [PubMed] [Google Scholar]
- Lecca D, Nevin DK, Mulas G, Casu MA, Diana A, Rossi D et al (2015). Neuroprotective and anti‐inflammatory properties of a novel non‐thiazolidinedione PPARγ agonist in vitro and in MPTP‐treated mice. Neuroscience 302: 23–35. [DOI] [PubMed] [Google Scholar]
- López González I, Garcia‐Esparcia P, Llorens F, Ferrer I (2016). Genetic and transcriptomic profiles of inflammation in neurodegenerative diseases: Alzheimer, Parkinson, Creutzfeldt–Jakob and Tauopathies. Int J Mol Sci 17: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin KM, O'Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ et al (2013). Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron 79: 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar‐Colucci S, Landreth GE (2011). Nuclear receptors as therapeutic targets for Alzheimer's disease. Expert Opin Ther Targets 15: 1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P (2009). Characterization of the microglial phenotype under specific pro‐inflammatory and anti‐inflammatory conditions: effects of oligomeric and fibrillar amyloid‐β. J Neuroimmunol 210: 3–12. 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Mogi M, Kondo T, Mizuno Y, Nagatsu T (2007). p53 protein, interferon‐γ, and NF‐κB levels are elevated in the Parkinsonian brain. Neurosci Lett 414: 94–97. [DOI] [PubMed] [Google Scholar]
- Mouton PR, Gokhale AM, Ward NL, West MJ (2002). Stereological length estimation using spherical probes. J Microsc 206: 54–64. [DOI] [PubMed] [Google Scholar]
- Nagy L, Szanto A, Szatmari I, Széles L (2012). Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol Rev 92: 739–789. [DOI] [PubMed] [Google Scholar]
- Nevin DK, Peters MB, Carta G, Fayne D, Lloyd DG (2012). Integrated virtual screening for the identification of novel and selective peroxisome proliferator‐activated receptor (PPAR) scaffolds. J Med Chem 55: 4978–4989. [DOI] [PubMed] [Google Scholar]
- Nolan YM, Sullivan AM, Toulouse A (2013). Parkinson's disease in the nuclear age of neuroinflammation. Trends Mol Med 19: 187–196. [DOI] [PubMed] [Google Scholar]
- Odegaard JI, Chawla A (2011). Alternative macrophage activation and metabolism. Annu Rev Pathol 6: 275–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo‐Gonzalez RR, Goforth MH, Morel CR, Subramanian V et al (2007). Macrophage‐specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447: 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña‐Altamira E, Prati F, Massenzio F, Virgili M, Contestabile A, Bolognesi ML et al (2015). Changing paradigm to target microglia in neurodegenerative diseases: from anti‐inflammatory strategy to active immunomodulation. Expert Opin Ther Targets 15: 1–14. [DOI] [PubMed] [Google Scholar]
- Penas F, Mirkin GA, Hovsepian E, Cevey A, Caccuri R, Sales ME et al (2013). PPARγ ligand treatment inhibits cardiac inflammatory mediators induced by infection with different lethality strains of Trypanosoma cruzi . Biochim Biophys Acta 1832: 239–248. [DOI] [PubMed] [Google Scholar]
- Pinto M, Nissanka N, Peralta S, Brambilla R, Diaz F, Moraes CT (2016). Pioglitazone ameliorates the phenotype of a novel Parkinson's disease mouse model by reducing neuroinflammation. Mol Neurodegener 11: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisanu A, Lecca D, Mulas G, Wardas J, Simbula G, Spiga S et al (2014). Dynamic changes in pro‐ and anti‐inflammatory cytokines in microglia after PPAR‐γ agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson's disease. Neurobiol Dis 71: 280–291. [DOI] [PubMed] [Google Scholar]
- Plaza‐Zabala A, Sierra‐Torre V, Sierra A (2017). Autophagy and microglia: novel partners in neurodegeneration and aging. Int J Mol Sci 18 pii: E598. 10.3390/ijms18030598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D et al (2009). Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor κB. Proc Natl Acad Sci U S A 106: 14978–14983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy RC (2008). Immunomodulatory role of PPAR‐γ in alveolar macrophages. J Invest Med 56: 522–527. [DOI] [PubMed] [Google Scholar]
- Salman H, Bergman M, Djaldetti R, Bessler H, Djaldetti M (1999). Decreased phagocytic function in patients with Parkinson's disease. Biomed Pharmacother 53: 146–148. [DOI] [PubMed] [Google Scholar]
- Sanchez‐Guajardo V, Febbraro F, Kirik D, Romero‐Ramos M (2010). Microglia acquire distinct activation profiles depending on the degree of α‐synuclein neuropathology in a rAAV based model of Parkinson's disease. PLoS One 5: e8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage JC, Jay T, Goduni E, Quigley C, Mariani MM, Malm T et al (2015). Nuclear receptors license phagocytosis by Trem2+ myeloid cells in mouse models of Alzheimer's disease. J Neurosci 35: 6532–6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada M, Imamura K, Nagatsu T (2006). Role of cytokines in inflammatory process in Parkinson's disease. J Neural Transm Suppl 70: 373–381. [DOI] [PubMed] [Google Scholar]
- Schintu N, Frau L, Ibba M, Caboni P, Garau A, Carboni E et al (2009a). PPARγ‐mediated neuroprotection in a chronic mouse model of Parkinson's disease. Eur J Neurosci 29: 954–963. [DOI] [PubMed] [Google Scholar]
- Schintu N, Frau L, Ibba M, Garau A, Carboni E, Carta AR (2009b). Progressive dopaminergic degeneration in the chronic MPTPp mouse model of Parkinson's disease. Neurotox Res 16: 127–139. [DOI] [PubMed] [Google Scholar]
- Simon DK, Simuni T, Elm J, Clark‐Matott J, Graebner AK, Baker L et al (2015). Peripheral biomarkers of Parkinson's disease progression and pioglitazone effects. J Parkinsons Dis 5: 731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerrett R, Malm T, Landreth G (2014). Nuclear receptors in neurodegenerative diseases. Neurobiol Dis 72 (Pt A): 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Lee C, Schindler C (2011). Deletion of the murine scavenger receptor CD68. J Lipid Res 52: 1542–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam SR, Federoff HJ (2017). Targeting microglial activation states as a therapeutic avenue in Parkinson's disease. Front Aging Neurosci 9: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson, C.R. , Joers, V. , Bondarenko, V. , Brunner, K. , Simmons, H. a, Ziegler, T.E. , et al, 2011. The PPAR‐γ agonist pioglitazone modulates inflammation and induces neuroprotection in Parkinsonian monkeys. J Neuroinflammation 8: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A et al (2010). STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ‐regulated gene expression in macrophages and dendritic cells. Immunity 33: 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Le W (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol 53: 1181–1194. [DOI] [PubMed] [Google Scholar]
- Theodore S, Cao S, McLean PJ, Standaert DG (2008). Targeted overexpression of human α‐synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol 67: 1149–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugal D, Liao X, Jain MK (2013). Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol 33: 1135–1144. [DOI] [PubMed] [Google Scholar]
- Varga T, Czimmerer Z, Nagy L (2011). PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta 1812: 1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HM, Wang J, Zhang B, Fang L, Xu K, Liu RY (2016). CpG‐ODN promotes phagocytosis and autophagy through JNK/P38 signal pathway in Staphylococcus aureus‐stimulated macrophage. Life Sci 161: 51–59. [DOI] [PubMed] [Google Scholar]
- Zhao XR, Gonzales N, Aronowski J (2015). Pleiotropic role of PPARγ in intracerebral hemorrhage: an intricate system involving Nrf2, RXR, and NF‐κB. CNS Neurosci Ther 21: 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer H, Riese S, Régnier‐Vigouroux A (2003). Functional characterization of mannose receptor expressed by immunocompetent mouse microglia. Glia 42: 89–100. [DOI] [PubMed] [Google Scholar]
- Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT (2012). PPARγ/RXRα‐induced and CD36‐mediated microglial amyloid‐β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci 32: 17321–17331. [DOI] [PMC free article] [PubMed] [Google Scholar]