

Abstract
Peripheral T-cell lymphomas (PTCLs) are uncommon, heterogeneous, and aggressive non-Hodgkin’s lymphomas. Despite progress in the last several years resulting in a deeper understanding of PTCL biology and pathogenesis, there is currently no accepted single standard of care for newly diagnosed patients, and for those with relapsed or refractory disease, prognosis is dismal. The National Cancer Institute convened a Clinical Trials Planning Meeting to advance the national clinical trial agenda in lymphoma. The objective was to identify unmet needs specific to five major lymphoma subtypes and develop strategies to address them. This consensus statement reviews recent advances in the molecular and genetic characterization of PTCL that may inform novel treatments, proposes strategies to test novel therapies in the relapsed setting with the hopes of rapid advancement into frontline trials, and underscores the need for the identification and development of active and biologically rational therapies to cure PTCL at higher rates, with iterative biomarker evaluation.
Recent Advances in Disease Characterization and Management
Epidemiology and Burden of Disease on Population
Peripheral T-cell lymphomas (PTCLs) are a heterogeneous, uncommon, and often aggressive group of non-Hodgkin’s lymphomas (NHL) representing approximately 10% to 15% of all new NHL diagnoses (1). The most recent iteration of the World Health Organization (WHO) classification describes over 20 distinct subtypes of PTCL (2). However, most incident cases are represented by the four most common subtypes: PTCL not otherwise specified (NOS), 34%; angioimmunoblastic T-cell lymphoma (AITL), 14%; anaplastic large cell lymphoma, anaplastic lymphoma kinase–negative (ALK- ALCL), 15%; and anaplastic large cell lymphoma, ALK + (ALK+ ALCL), 9% (3,4). Compared with B cell NHL, PTCLs carry inferior outcomes. Despite generally favorable response rates to chemotherapy, remissions are often not durable, and as such the natural history of PTCL is characterized by relapses and, in some cases, refractory disease. For that reason, upfront hematopoietic stem cell transplantation (HSCT) is often offered in first remission for patients who are fit and have chemosensitive disease. While phase II studies suggest a benefit of this approach, it is unclear whether the superior outcomes after HSCT are due to patient selection as opposed to a true benefit of HSCT (5,6). Unfortunately, many patients are either not candidates for transplantation or, conversely, are unable to attain a response adequate enough to proceed to transplantation at the time of relapse. For these patients, hopes of long-term disease control are very limited (Figure 1), with progression-free survival (PFS) of 5.5 months (7). In this setting, optimal therapeutic approaches remain undefined and represent an unmet clinical need.
Figure 1.
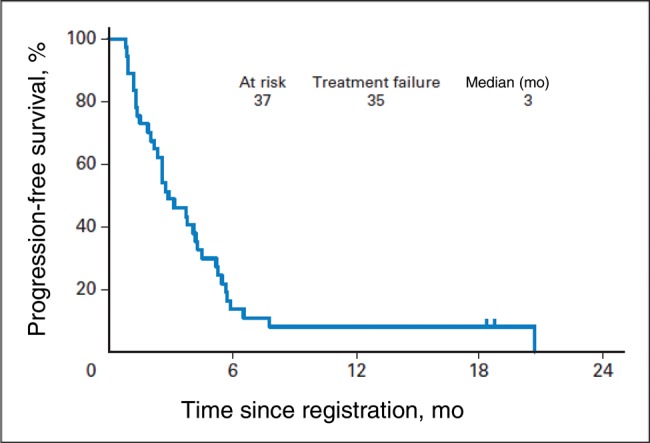
In the relapsed setting, outcomes for peripheral T-cell lymphoma (PTCL) are dismal. Noted here is a representative example from a recently completed Southwest Oncology Group trial evaluating alisertib in relapsed/refractory PTCL (33). This figure is being published with permission from the original authors and copyright holder.
Despite progress in the last several years resulting in a deeper understanding of PTCL, at present there is no accepted single standard of care for newly diagnosed patients. The most commonly used regimens have been derived from treatment of aggressive B-cell NHL, such as cyclophosphamide, doxorubicin, vincristine, prednisone (CHOP); cyclophosphamide, doxorubicin, vincristine, etoposide, prednisone (CHOEP); or infusional etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin (EPOCH). Each appears to provide a similar overall response rate (ORR) of approximately 70% to 80%, with a complete response (CR) rate of between 30% and 50% (5,6,8,9), and a five-year overall survival (OS) rate between 25% and 70%. When evaluated based on histology, however, outcomes are poorest for AITCL and PTCL-NOS, and slightly better for ALCL (4,8,10–12).
The National Clinical Trials Network (NCTN) has contributed to the treatment landscape of PTCL with several recently completed studies, although, with a few noted exceptions, therapeutic strategies for PTCL have been largely unexplored by the NCTN. The Eastern Cooperative Oncology Group (ECOG) studied the addition of humanized antivascular endothelial growth factor antibody bevacizumab in combination with CHOP in the E2404 study, given a role for angiogenesis in PTCL (13). This phase II study enrolled 46 patients, of whom 39 were evaluable for response. While high overall response rates were observed with bevacizumab-CHOP, the regimen failed to yield durable remissions and was associated with clinically significant and unacceptable grade 3 and 4 toxicities including febrile neutropenia, congestive heart failure, venous thrombosis, and gastrointestinal hemorrhage/perforation. At one year, progression-free survival (PFS) was 44%, with a median OS of 22 months after three years of follow-up (13).
In the Southwest Oncology Group 0350 study, the PEGS regimen, consisting of cisplatin, etoposide, gemcitabine, and solumedrol, was studied in an attempt to abrogate drug resistance through the selection of drugs not effluxed by the ATP-dependent P-glycoprotein efflux pump (14). In this study, Mahadevan and colleagues reported their experience with 34 patients with PTCL. The combination resulted in a disappointing two-year PFS of 12% (95% confidence interval [CI] = 0.1% to 31%) and a two-year OS of 31% (95% CI = 8% to 54%). Subsequently, many other treatment combinations are being actively explored in PTCL, with the goal of developing biologically based rational therapeutic strategies.
Areas of Recent Progress in Biological Understanding of Disease
Recent advances in the molecular and genetic characterization of T-cell lymphomas have helped to delineate differences and similarities between the various subtypes. In the most recent World Health Organization (WHO) Classification of Lymphoid Neoplasms, multiple changes were made to the classification of both nodal and extranodal TCLs. Several recurrent mutations have been identified in small subsets of patients. These genetic alterations are being grouped mechanistically and inform novel treatments. As such, much of the additional genomic information available is being included in the new WHO classification (15).
In PTCL NOS, a targeted deep-sequencing approach in 28 patients revealed epigenetic deregulation as the most frequently mutated category, followed by constitutive T-cell receptor (TCR) activation as the most prominent genetic mechanisms in PTCL pathogenesis (16). Translocation t(5;9)(q33;q32) resulting in the inducible T-cell kinase-spleen tyrosine kinase (ITK-SYK) fusion gene and leading to constitutive activation of the T-cell receptor was found in less than 10% of PTCL NOS (17). Similarly, the recurrent translocation t(6:14) in PTCL juxtaposes the interferon regulatory factor 4 (IRF4) and the TCR alpha genes, resulting in overexpression of IRF4, driving MYC expression and contributing to oncogenesis (18). Expression of the T-cell transcription factor GATA-binding protein 3 (GATA 3) has been found to be elevated in a subset of PTCLs and regulates the production of T helper cells and cytokine production, which are associated with poor outcomes (19). These findings have been recapitulated in genome-wide expression profiling, suggesting the presence of two distinct molecular subgroups of PTCL NOS: one characterized by increased GATA3 expression with poor prognosis and the other by increased expression of t-box 21 (TBX21) transcription factors and corresponding targets (20).
The enzyme isocitrate dehydrogenase 2 (IDH2) is a component of the Krebs cycle that normally catalyzes the conversion of isocitrate to alpha-ketoglutarate, an intermediate for several enzymes involved in epigenetic regulation and adaptation to hypoxia. Neomorphic mutations in IDH2 result in production of 2-hydroxyglutarate, an oncometabolite that represses histone 3 lysine and DNA 5mC demethylation. In AITL, this represents the third most common genetic lesion identified, after the Tet methylcytosine dioxygenase 2 gene (TET2) and the Ras homolog gene family, member A (RHOA) mutations (21,22). TET2 mutations were seen with a high frequency in AITL and PTCL NOS with a T follicular helper cell phenotype. Moreover, epigenetic alterations and inactivating DNA (cytosine-5-)-methyltransferase 3 alpha (DNMT3A) mutations are seen in many cases of AITL, often in association with the TET2 mutations (23,24). Similarly, regulators of histone methylation were mutated in 25% of AITL cases, including mixed lineage leukemia/lymphoma (MLL2), lysine demethylase 6a (KDM6A), and MLL (20). To date, the presence or absence of these mutations suggests that TET2 mutations may be associated with poor prognosis (24); however, the mutations in epigenetic regulatory genes appear to be particularly frequent in AITL. This supports a potential role for targeting the epigenome in T-cell lymphomas (20,21,23,25) using DNA methyl-transferase inhibitor +/− histone deacetylase (HDAC) inhibitors or IDH2 inhibitors (eg, AG-221, NCT02273739).
In ALK+ ALCL, the ALK protein is constitutively expressed after fusing to partner genes through chromosomal translocations, most frequently a (2;5)(p23;q35) translocation fusing the ALK gene with NPM on chromosome 5, although other translocations have been reported (26). The ALK chimeras activate Signal transducer and activator of transcription factor 3 (STAT3), deregulation of which is required for maintenance of the neoplastic phenotype in ALK+ ALCL. A systematic characterization of the genetic alterations driving ALCLs was recently undertaken using sequencing strategies. Activating mutations of Janus kinase 1 (JAK1) and STAT3 genes were found in 20% of ALK- ALCLs, 38% of which displayed double lesions (27). Through next-generation sequencing (NGS), recurrent translocations of t(6;7) were recently identified in ALK-ALCLs, leading to a balanced translocation disrupting the dual-specificity phosphatase 22 (DUSP22) gene on chromosome 6, and joining to the fragile site, aphidicolin type gene (FRA) site on chromosome 7. This was associated with upregulation of microRNA 29 (28). DUSP22 is a dual-specificity phosphatase that inhibits T-cell antigen receptor signaling in reactive T-cells through mitogen-activated protein kinase (MAPK) and extracellular signal–regulated kinase (ERK2) inactivation. More recently, the presence of DUSP22 rearrangement was shown to be associated with a favorable outcome in ALK- ALCL (29). NGS studies have also identified recurrent mutations in p53 and related genes (p63, cyclin dependent kinase [CDKN2A], WW domain containing oxidoreductase [WWOX], ankryn repeat domain 11 [ANKRD11]) in PTCL; in particular, p63 rearrangements were observed in nearly 6% of patients with PTCL and are associated with inferior survival (30).
Aurora A (AA) is a serine/threonine mitotic kinase implicated in oncogenesis through its role in cell division via regulation of chromatid separation. Overexpression of Aurora A kinase (AAK) has been observed in PTCL and is associated with a poorer prognosis (31). In 100 lymphoma samples across the spectrum of PTCL, AAK expression was highest in PTCL-NOS and ALCL and was detected in 68% of cases, including 100% of PTCL NOS (32). In early studies, Alisertib, an oral, selective, competitive, reversible inhibitor of AAK showed an ORR in four out of eight patients with relapsed PTCL (31). Larger studies have shown the ORR to be 30% (95% CI = 9% to 61%) in a phase II study (33) and 33% in a randomized phase III study when tested against other comparators (pralatrexate, romidepsin, gemcitabine) (34), which is consistent with many other available agents in T-cell lymphoma.
This emerging understanding of mutations, targets, and new drugs provides increased rationale for selecting agents and combinations for investigations into novel treatment of PTCLs based on their mechanisms of action.
Key Clinical Knowledge Gaps and Unmet Clinical Needs
PTCL is a rare disease and as such the cumulative incidence of many recently discovered mutations is quite low. Consequently, targeting these infrequent mutations in uncommonly occurring disease subtypes is a daunting task. Rather, an approach that targets overarching mechanisms in pathogenesis such as T-cell receptor signaling, epigenetic deregulation, and pathways such as JAK/STAT, phosphatidylinositol 3 (Pi3) kinase, or mammalian target of rapamycin (mTOR) activation may be a more feasible approach that could also apply to greater numbers of patients with various PTCL subtypes and thus provide broader benefit.
The areas most in need of further development in TCL include the identification and development of active and biologically rational therapies to cure PTCL at high rates. These novel agents and/or strategies would be initiated in the relapsed setting where the overall goals would be to generally improve overall response rates and durability. These discoveries would then be translated into the upfront setting with an aim toward improving CR rates and, if achieved, translating higher CR rates into improved PFS and OS. We recognize that given the current state of therapies, high cure rates are a longer-term goal and will likely be achieved in a step-wise fashion that will be based on other short- and intermediate-term goals. We aim to identify more active therapies and regimens for patients by developing novel platforms in efficiently conducted, and likely sequential, clinical trials to improve upon and/or replace current standard therapies. We also propose to increase our understanding of TCL with an emphasis on inclusion of correlative science in these clinical trials in order to identify biomarkers of response and resistance and thereby better select therapies for individual patients.
Current Landscape of Ongoing Clinical Trials
The key to optimizing therapy for patients with PTCL will involve the incorporation of novel agents into well-designed clinical studies to ultimately provide a precision approach to care. The past several years heralded a shift in PTCL, when after decades without meaningful progress in the treatment of relapsed or refractory disease, four drugs were approved worldwide for the treatment of recurrent PTCL. Antifolate pralatrexate and histone deacetylase inhibitor romidepsin have shown statistically significant efficacy in relapsed and refractory PTCL and were approved by the US Food and Drug Administration (FDA) in 2009 and 2011, respectively. Approval of romidepsin for PTCL was based on a pivotal phase II study of patients with relapsed or refractory PTCL (n = 131) that demonstrated an objective response rate of 25%, including 15% with CR; responses lasted a median of greater than two years. Brentuximab vedotin, a CD30-targeted immunoconjugate, demonstrated a high response rate of 85% in a phase II study of 58 patients with CD30+ ALCL who relapsed after prior therapy. Consequently, brentuximab vedotin was approved by the FDA and the European Medicines Agency in 2011 and 2012, respectively, for recurrent CD30+ ALCL. Most recently, the novel pan-histone deacetylase inhibitor belinostat was approved by the FDA in 2014. Belinostat has demonstrated meaningful efficacy and a favorable toxicity profile in a single-arm phase II trial of 129 patients with relapsed/refractory PTCL, with a response rate consistent with rates of other current agents at 28.5% and median duration of response of 13.6 months (95% CI = 4.5 to 29.4) (35). These conclusive results led to an accelerated approval by the FDA.
Approval of these novel agents has led to attempts to improve upon the frequently used CHOP backbone of treatment for PTCL (Table 1). As new agents have shown activity in the relapsed setting, several large international randomized studies have been recently completed, are ongoing, or have been planned comparing the addition of a new agent to the standard CHOP regimen. These studies include the ACT 1 (alemtuzumab + CHOP vs CHOP); NCT00646854, ECHELON 2 (brentuximab vedotin + CHP vs CHOP; NCT01777152); Ro-CHOP (romidepsin + CHOP vs CHOP; NCT01796002); and a planned study of Belinostat + CHOP vs Pralatrexate + CHOP vs CHOP.
Table 1.
Current landscape of key ongoing and recently completed phase 3 trials*
| Clinical study | Setting | Therapies being studied | Results |
|---|---|---|---|
| ACT 1 (NCT00646854) | Newly diagnosed, frontline | Alemtuzumab + CHOP vs CHOP (CHOP given every 2 wks) | Primary end point: event-free survival; recruitment complete |
| ECHELON 2 (NCT01777152) | Newly diagnosed, frontline | Brentuximab + CHP vs CHOP | Primary end point: progression-free survival; recruitment ongoing |
| Ro-CHOP (NCT01796002) | Newly diagnosed, frontline | Romidepsin + CHOP vs CHOP | Primary end point: progression-free survival; recruitment ongoing |
| Lumiere (NCT01482962) | Relapsed and refractory | Alisertib vs pralatrexate vs romidepsin vs gemcitabine | Overall response rate: alisertib 33%, pralatrexate 40% (95% CI = 0.34 to 1.23); pralatrexate 40%; romidepsin 59%; gemcitabine 35%; median PFS in months: 3.7, 3.4, 8, and 1.9, respectively |
CHOP = cyclophosphamide, doxorubicin, oncovin, and prednisone; CI = confidence interval; NCT = National Clinical Trial; PFS = progression-free survival; Ro = romidepsin.
Other strategies include the use of HSCT as a common consolidation strategy in the frontline setting for TCL. Prospective phase II studies and retrospective series have shown long-term PFS and OS of 41% to 51% and 44% to 48%, respectively, on an intent-to-treat basis, which compares favorably with the historical and prospective data with CHOP or CHOP-like regimens without consolidation (5,6). While many centers in the United States have adopted consolidation of first remission for HSCT, there is no randomized study to confirm a benefit over chemotherapy alone, and there are no ongoing studies evaluating this question.
In the setting of relapsed disease, advances have been made in the therapeutic armamentarium for PTCL. However, the identification of new therapies to date has been largely empiric. Recent efforts at targeted sequencing have identified several recurrent mutations (listed above) that are promising for the development of new therapies for TCL. However, several of these targets are present in only small subsets of patients with PTCL, presenting a major challenge for study accrual.
As there are four FDA-approved agents for relapsed PTCL—belinostat, brentuximab vedotin (ALCL only), pralatrexate, and romidepsin—combining novel agents is a feasible strategy for improving current therapies. With the exception of brentuximab vedotin for ALCL, the ORR in the relapsed or refractory setting is less than 30% and median PFS is less than five months in the pivotal studies of each of the newer agents (36). Often, in phase I or small phase II signal-finding studies, multiple agents are currently under evaluation as single agents or components of combination therapies for PTCL. These include the FDA-approved agents that are currently being studied as part of novel combinations with each other or with other frequently used agents such as bortezomib, carfilzomib, lenalidomide, alemtuzumab, 5-azacitidine, gemcitabine, bendamustine, and others. Investigational agents such as PI3 kinase inhibitors, alisertib, mogamulizumab, immune checkpoint inhibitors, and others are also being explored alone or in combination.
Mogamulizumab is a humanized IgG1 monoclonal antibody against chemokine receptor 4 (CCR4). Expression of CCR4 in nodal TCL and cutaneous TCL, as well as in patients with adult T-cell lymphoma/leukemia, makes mogamulizumab an appealing targeted agent (37). A multicenter phase II study in patients with relapsed PTCL as well as cutaneous TCL demonstrated a promising ORR of 35% (95% CI = 20% to 53%), with a 14% CR. Median PFS was low, at three months (95% CI = 1.6 to 4.9 months) (38). A phase III study of mogamulizumab vs vorinostat in CTCL is ongoing (NCT01728805).
The immunomodulatory drug lenalidomide was studied in a multicenter phase II study of patients with relapsed and refractory TCL (39). Fifty-four patients received 25 mg of lenalidomide for 21 of 28 days for up to 24 months. While the ORR was similar to those of other agents in relapsed TCL at 22% (12 of 54 patients, 95% CI = 12.0 to 35.6), those with AITL did best, with an ORR of 31% and a CR in 15% of patients. Similarly, a Canadian phase II study tested the efficacy of lenalidomide in 40 patients with untreated and relapsed and refractory PTCL (40). The ORR was 29%, with an 8% CR rate in the entire group, but among those previously treated the median OS was 12 months. Unfortunately, the duration of response was low, at five months. To evaluate the role of lenalidomide doublet and triplet combinations, other studies are ongoing, in combination with romidepsin (NCT01742793) and carfilzomib (NCT02341014).
Recommended High-Priority Clinical Trial Questions
Trial Structure Including Current Standards of Care
The committee believes that, given the current inadequacies in standard therapy for TCL, simultaneous studies should be conducted so patients progressing on one therapy would have immediate access to other novel approaches. This can best be achieved by studying patients with relapsed or refractory disease. A sequential study design could begin with a series of novel mechanisms based combinations with the ultimate aim of developing a regimen to replace CHOP in the upfront setting.
One approach could include a randomized phase II trial to select the most promising combinations with the highest CR or ORR rate. Selection of these combinations should be based on preclinical rationale and preliminary efficacy demonstrated in phase I/II trials and pilot studies of novel doublets or triplets. Moreover, approaches that target mechanisms of pathogenesis may be a useful paradigm that can be applied to various PTCL subtypes to optimize outcome. Novel doublets have recently been studied or are currently being studied in early trials including romidepsin + lenalidomide, which showed a 60% ORR in relapsed PTCL (41). Other combinations being investigated include romidepsin and pralatrexate, being evaluated currently in an ongoing phase I study (NCT01947140). The combination shows efficacy in vitro, suggesting the potential for synergism (42). Similarly, the combination of alisertib and romidepsin is being evaluated in a phase I study based on preclinical data supporting efficacy of the combination (43). Other possibilities would include targeting epigenetic modifications using a doublet of a histone deacetylase inhibitor and hypomethylating agent, as is already ongoing with 5-azacitidine + romidepsin (NCT01998035). The most promising of these doublets could be compared with select combinations in a subsequent randomized phase II trial. Once a preferred backbone is established, third agents could be added in parallel phase I studies, followed by randomized phase II studies of the new combinations.
End Points for Clinical Trials in PTCL
The preferred end point for considering when to move a regimen to front line is CR, preferably in conjunction with a predictive biomarker (Figure 2). Defining a response rate in the relapsed setting that warrants consideration as a frontline regimen is arbitrary and based on many other factors such as the toxicity of the regimen and durability of responses. However, given that the goal of a frontline therapy is cure, we believe that a CR of greater than 30% in the relapse setting suggests robust enough activity to be considered.
Figure 2.

Proposed strategy for developing the next generation of clinical trials in T-cell lymphoma (TCL). A) Relapsed disease with representative examples. B) Frontline setting with representative examples. C) Clinical trials in TCL that include a putative biomarker (as an example, CD30). TCL = T-cell lymphoma.
Biomarkers in PTCL
Biomarker discovery should be an important component of the tier 1 studies (Figure 2). This will increase understanding of the mechanisms of response and resistance and support simultaneous tier 2 studies involving targeted agents, to which patients progressing on the tier 1 studies could be assigned on the basis of biological characteristics. Notable biomarkers relevant to TCL are listed in Table 2. While most current treatment choices derive from studies in which patients with a broad spectrum of TCL subtypes were treated nonselectively, two agents with targets that are more clearly defined are the CD30 targeted antibody drug conjugates brentuximab vedotin and crizotinib, an oral small-molecule tyrosine kinase inhibitor of ALK and several other kinases. As understanding of PTCL increases, there is an opportunity to better refine therapeutic decisions for patients and identify new agents. Recent studies with brentuximab vedotin have provided into disease settings other than anaplastic large cell lymphoma in which it may have the greatest activity and which groups of patients are most likely to benefit from these studies. A recent multi-institution collaborative project reported activity of brentuximab across a wide range of CD30 expression levels in mycosis fungoides and Sezary syndrome (44). We are also gaining an early understanding of targeted therapies such as histone deacetylase inhibitors and PI3 kinase inhibitors in PTCL. Recent mutation analyses in a large series of patients with T-cell lymphoma have also identified rare but recurrent druggable targets, such as Nuclear factor-κB (NF-kB) via proteasome inhibitors, IDH2 mutations, and PI3kinase/AKT (20).
Table 2.
Peripheral T-cell lymphoma biomarkers*
| Recommendations | Standard of care | Integral | Integrated | Exploratory | Validated | Identifies high-/low-risk groups | Identifies therapeutic target | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Histopathology | ||||||||||
| Complete immunophenotypic panel by flow cytometry and/or immunohistochemistry for CD3, CD4, CD8, CD30, etc. | Yes | Yes | Yes | No | Yes | Yes | No | |||
| Disease-specific phenotype: ALK, CD2, CD5, CD7, CD56, EBV (EBER ISH), PD1, granzyme, perforin, TIA1, BetaF1, TCR-GM1 | Yes | Yes | Yes | No | Yes | Yes | No | |||
| Molecular/genetic | ||||||||||
| Targeted mutational panel for known PTCL mutations (eg, TET2, IDH2, DNMT3A, FYN, RHOA, CD28, Stat5b in gamma-delta subtype) | No | No | Yes | Yes | No | Yes | Yes | |||
| GEP for diagnostic signatures (GATA3, TBX1, gamma-delta) | No | No | No | Yes | No | Yes | Maybe | |||
ALK = anaplastic lymphoma kinase; Beta F1 = T-cell beta chain antigen receptor; CD = complement of differentiation; DNMT3A = DNA (cytosine-5-)-methyltransferase 3 alpha; EBER ISH = Epstein Barr virus in situ hybridization; EBV = Epstein Barr virus; FYN = tyrosine kinase Fyn; GATA3 = T-cell-specific transcription factor binds to the sequence G-A-T-A; GEP = gene expression profiling; IDH2 = isocitrate dehydrogenase gene; PD1 = programmed death 1; RHOA = Ras homolog gene family, member A, a GTPase protein of the Rho family; Stat5b = signal transducer and activator of transcription 5B; TBX1 = T box 1 gene; TCR-GM1 = T-cell receptor glycosphingolipid; TET2 = Tet methylcytosine dioxygenase 2 gene; TIA1 = T-cell intracytoplasmic antigen.
Promising Novel Agents/Pathways for Study
Targeting the Epigenome
HDAC inhibitors appear to have activity across a broad range of PTCLs, with three commonly used and FDA-approved agents in this class: romidepsin, vorinostat, and belinostat (45–47). While most of these agents have a broad range of activity in PTCL, there is some insight into which patients are most likely to respond. For belinostat, ORR in AITL was 46%, compared with only 15% for ALK- ALCL, and many of the most durable responses to romidepsin are seen in those with AITL (45). Genomic profiling in PTCL has led to the identification of recurrent mutations in epigenetic regulator genes in both PTCL-NOS and AITL, which can help tailor treatment strategies in future trials.
Targeting ALK
Crizotinib is an example of therapy for PTCL based upon an understood mechanism and defined target. As in PTCL, ALK harbors an activating mutation in lung cancer and crizotinib is now commonly used in the treatment of the 3% to 5% of patients with non–small cell lung cancers with a rearrangement of the ALK gene (48). Although the presence of mutated ALK in ALCL is a favorable prognostic marker, relapses following chemotherapy occur, with poor outcomes (3). A small study of crizotinib in ALCL demonstrated promising response rates, suggesting a role for ALK inhibition in TCL. Several other ALK inhibitors are being actively developed.
Targeting CD30
The antibody drug conjugate brentuximab vedotin is an anti-CD30 monoclonal antibody fused via a protease-cleavable linker to the antitubulin agent monomethyl auristatin E. CD30 is a transmembrane receptor of the tumor necrosis factor (TNF) superfamily that is often expressed by activated lymphocytes and in some lymphoid malignancies, including ALCL, with more variable expression in other T-cell lymphomas. A phase II study of 58 patients with relapsed/refractory systemic ALCL showed a promising 86% ORR (95% CI = 74.6% to 93.9%), with a 57% CR rate (95% CI = 43.2% to 69.8%) (49). A phase II study in PTCLs other than ALCL with any degree of CD30 expression had an ORR of 41% (50). New insights into alternate mechanisms of action and predictors of response may allow extension of the use of brentuximab vedotin to a wider population of patients with PTCL.
Aurora A Kinase Inhibition
Inhibition of aurora A kinase has previously been shown to cause cell cycle arrest and apoptosis in preclinical studies (31). The oral, reversible inhibitor of AAK alisertib has shown promising response rates. A single-arm intergroup phase II study of 37 patients with relapsed and refractory PTCL as well as transformed mycoses fungoides demonstrated an ORR of 30% (95% CI = 9% to 61%) (33). Because of these findings, a randomized phase III clinical trial comparing alisertib with standard options of pralatrexate, romidepsin, or gemcitabine in subjects with relapsed PTCL has recently completed enrollment, with no discernible difference in response rates among the agents (34).
Phosphatidylinositol-3-kinase (PI3K) inhibition
The PI3K family consists of several serine/threonine and lipid kinases. These enzymes and their downstream targets, primarily Akt, play a major role in multiple cellular processes, including growth, differentiation, metabolism, survival, and cellular proliferation (51). Aberrant activation of the PI3K/AKT/mTOR pathway in lymphoma results from several alterations, including mutation or gene amplification, leading to increased pathway activity (52). A subset of patients with PTCL have been treated with the PI3 kinase delta and gamma inhibitor ipi-145 (duvelisib), with an ORR of 42%, including responses in 53% of those with PTCL (53). Further studies of PI3 kinase inhibition as part of a combinatorial strategy for T-cell lymphoma are in development.
Targeting FYN Tyrosine Kinase
The FYN tyrosine kinase is one of the predominant SRC family kinases expressed in T lymphocytes (54) and has an integral role in T-cell activation upon T-cell receptor stimulation. Recently mutations in FYN tyrosine kinase were noted in a small number (3%, 4/137) of TCLs (PTCL NOS and AITL). While the mutation is quite rare, dasatinib, a multikinase inhibitor that blocks ABL1 and SRC kinases, was shown to inhibit the activity of FYN mutant proteins and impair the growth of transformed cells expressing mutated FYN (54). Based on this preclinical data, SRC kinase inhibition with dasatinib may represent a targeted therapy in the rare PTCL cases with activating mutations in the FYN kinase gene.
JAK/STAT inhibition
JAK/STAT signaling is aberrantly activated in lymphoma by multiple mechanisms, including inappropriate autocrine and paracrine cytokine stimulation (55) as well as activating mutations (56). Preclinical studies involving the JAK3 inhibitor CP-690550 demonstrated suppression of the dissemination of NK/T-cell tumor cells in the bone marrow, spleen, and blood (57). An oral JAK 3 inhibitor, tofacitinib, is currently approved for the treatment of rheumatoid arthritis (58), and trials of other JAK3 inhibitors are in development. Similarly, the oral JAK inhibitor ruxolitinib is being studied in relapsed B-cell NHL and PTCL (NCT01431209).
Conclusions and Recommendations
The continuous discovery of mutations, targets, and new drugs provides a rationale for selecting treatments for PTCL based on mechanism of action and tumor biology. CHOP + X approaches are already ongoing, and no other novel regimens have sufficient preliminary data to warrant being studied in untreated patients. A randomized study evaluating the role of ASCT in the first-line setting should be critically assessed, but the committee has concerns about feasibility. Exploratory studies assessing biomarker development and/or MRD as companions to standard of care in the upfront setting are recommended. In the relapsed setting, sequential studies to develop optimal novel combinations (doublets and triplets) with prospective evaluation of biomarkers for discovery and to triage patients for phase I and II studies of targeted agents are recommended.
Funding
Partial support was from the Office of the Director, Coordinating Center for Clinical Trials, National Cancer Institute/National Institutes of Health.
Notes
The funders had no role in the writing of the review or decision to submit it for publication.
This manuscript resulted from a Clinical Trials Planning Meeting in Lymphoma, November 21–22, 2014.
References
- 1. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. The Non-Hodgkin's Lymphoma Classification Project. Blood. 1997;89(11):3909–3918. [PubMed] [Google Scholar]
- 2. Swerdlow SHN, Jaffe E, Pileri S, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed Lyon, France: International Agency for Research on Cancer; 2008. [Google Scholar]
- 3. Ellin F, Landstrom J, Jerkeman M, et al. Real-world data on prognostic factors and treatment in peripheral T-cell lymphomas: A study from the Swedish Lymphoma Registry. Blood. 2014;124(10):1570–1577. [DOI] [PubMed] [Google Scholar]
- 4. Vose J, Armitage J, Weisenburger D, International TCLP. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–4130. [DOI] [PubMed] [Google Scholar]
- 5. Reimer P, Rudiger T, Geissinger E, et al. Autologous stem-cell transplantation as first-line therapy in peripheral T-cell lymphomas: Results of a prospective multicenter study. J Clin Oncol. 2009;27(1):106–113. [DOI] [PubMed] [Google Scholar]
- 6. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093–3099. [DOI] [PubMed] [Google Scholar]
- 7. Mak V, Hamm J, Chhanabhai M, et al. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: Spectrum of disease and rare long-term survivors. J Clin Oncol. 2013;31(16):1970–1976. [DOI] [PubMed] [Google Scholar]
- 8. Simon A, Peoch M, Casassus P, et al. Upfront VIP-reinforced-ABVD (VIP-rABVD) is not superior to CHOP/21 in newly diagnosed peripheral T cell lymphoma. Results of the randomized phase III trial GOELAMS-LTP95. British J Haematol. 2010;151(2):159–166. [DOI] [PubMed] [Google Scholar]
- 9. Escalon MP, Liu NS, Yang Y, et al. Prognostic factors and treatment of patients with T-cell non-Hodgkin lymphoma: The M. D. Anderson Cancer Center experience. Cancer. 2005;103(10):2091–2098. [DOI] [PubMed] [Google Scholar]
- 10. Schmitz N, Trumper L, Ziepert M, et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma: An analysis of patients with T-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood. 2010;116(18):3418–3425. [DOI] [PubMed] [Google Scholar]
- 11. Savage KJ, Chhanabhai M, Gascoyne RD, Connors JM. Characterization of peripheral T-cell lymphomas in a single North American institution by the WHO classification. Annals Oncol. 2004;15(10):1467–1475. [DOI] [PubMed] [Google Scholar]
- 12. Smith SM, Burns LJ, van Besien K, et al. Hematopoietic cell transplantation for systemic mature T-cell non-Hodgkin lymphoma. J Clin Oncol 2013;31(25):3100–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ganjoo K, Hong F, Horning SJ, et al. Bevacizumab and cyclosphosphamide, doxorubicin, vincristine and prednisone in combination for patients with peripheral T-cell or natural killer cell neoplasms: An Eastern Cooperative Oncology Group study (E2404). Leuk Lymphoma. 2014;55(4):768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mahadevan D, Unger JM, Spier CM, et al. Phase 2 trial of combined cisplatin, etoposide, gemcitabine, and methylprednisolone (PEGS) in peripheral T-cell non-Hodgkin lymphoma: Southwest Oncology Group Study S0350. Cancer. 2013;119(2):371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Swerdlow SH, Campo E, Pileri SA. et al. The 2016 Revision of the World Health Organization (WHO) Classification of Lymphoid Neoplasms. Lyon, France: International Agency for Research on Cancer; 2016. [Google Scholar]
- 16. Schatz JH, Horwitz SM, Teruya-Feldstein J, et al. Targeted mutational profiling of peripheral T-cell lymphoma not otherwise specified highlights new mechanisms in a heterogeneous pathogenesis. Leukemia. 2015;29(1):237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dierks C, Adrian F, Fisch P, et al. The ITK-SYK fusion oncogene induces a T-cell lymphoproliferative disease in mice mimicking human disease. Cancer Res. 2010;70(15):6193–6204. [DOI] [PubMed] [Google Scholar]
- 18. Boddicker RL, Kip NS, Xing X, et al. The oncogenic transcription factor IRF4 is regulated by a novel CD30/NF-kappaB positive feedback loop in peripheral T-cell lymphoma. Blood. 2015;125(20):3118–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang T, Feldman AL, Wada DA, et al. GATA-3 expression identifies a high-risk subset of PTCL, NOS with distinct molecular and clinical features. Blood. 2014;123(19):3007–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iqbal J, Wright G, Wang C, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123(19):2915–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cairns RA, Iqbal J, Lemonnier F, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119(8):1901–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Odejide O, Weigert O, Lane AA, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123(9):1293–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lemonnier F, Couronne L, Parrens M, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120(7):1466–1469. [DOI] [PubMed] [Google Scholar]
- 25. Piccaluga PP, Agostinelli C, Califano A, et al. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J Clin Invest. 2007;117(3):823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263(5151):1281–1284. [DOI] [PubMed] [Google Scholar]
- 27. Crescenzo R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27(4):516–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feldman AL, Dogan A, Smith DI, et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood. 2011;117(3):915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parrilla Castellar ER, Jaffe ES, Said JW, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014;124(9):1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vasmatzis G, Johnson SH, Knudson RA, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood. 2012;120(11):2280–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yakushijin Y, Hamada M, Yasukawa M. The expression of the aurora-A gene and its significance with tumorgenesis in non-Hodgkin's lymphoma. Leuk Lymphoma. 2004;45(9):1741–1746. [DOI] [PubMed] [Google Scholar]
- 32. Friedberg JW, Mahadevan D, Cebula E, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol. 2014;32(1):44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barr PM, Li H, Spier C, et al. Phase II Intergroup Trial of alisertib in relapsed and refractory peripheral T-cell lymphoma and transformed mycosis fungoides: SWOG 1108. J Clin Oncol. 2015;33(21):2399–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Connor OA, Ozcan M, Jacobsen E, et al. First multicenter, randomized phase 3 study in patients (Pts) with relapsed/refractory (R/R) peripheral T-cell lymphoma (PTCL): Alisertib (MLN8237) versus investigator's choice (Lumiere trial; NCT01482962). Paper presented at the 57th Meeting of the American Society for Hematology; December 2015; Orlando, FL.
- 35. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol. 2015;33(23):2492–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coiffier B, Federico M, Caballero D, et al. Therapeutic options in relapsed or refractory peripheral T-cell lymphoma. Cancer Treat Rev. 2014;40(9):1080–1088. [DOI] [PubMed] [Google Scholar]
- 37. Ishida T, Inagaki H, Utsunomiya A, et al. CXC chemokine receptor 3 and CC chemokine receptor 4 expression in T-cell and NK-cell lymphomas with special reference to clinicopathological significance for peripheral T-cell lymphoma, unspecified. Clin Cancer Res. 2004;10(16):5494–5500. [DOI] [PubMed] [Google Scholar]
- 38. Ogura M, Ishida T, Hatake K, et al. Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J Clin Oncol. 2014; 32(11):1157–1163. [DOI] [PubMed] [Google Scholar]
- 39. Morschhauser F1, Fitoussi O, Haioun C, et al. A phase 2, multicentre, single-arm, open-label study to evaluate the safety and efficacy of single-agent lenalidomide (Revlimid) in subjects with relapsed or refractory peripheral T-cell non-Hodgkin lymphoma: The EXPECT trial. Eur J Cancer. 2013;49(13):2869–2876. [DOI] [PubMed] [Google Scholar]
- 40. Toumishey E, Prasad A, Dueck G, et al. Final report of a phase 2 clinical trial of lenalidomide monotherapy for patients with T-cell lymphoma. Cancer. 2015;121(5):716–723. [DOI] [PubMed] [Google Scholar]
- 41. Mehta-Shah N, Lunning MA, Ruan J. et al. A phase I/II trial of the combination of romidepsin and lenalidomide in patients with relapsed/refractory lymphoma and myeloma. Paper presented at the 13th International Conference on Malignant Lymphoma; June 2015; Lugano, Switzerland.
- 42. Jain S, Jirau-Serrano X, Zullo KM, et al. Preclinical pharmacologic evaluation of pralatrexate and romidepsin confirms potent synergy of the combination in a murine model of human T-cell lymphoma. Clin Cancer Res. 2015;21(9):2096–2106. [DOI] [PubMed] [Google Scholar]
- 43. Fanale MA, Hagemeister FB, Fayad L, et al. A phase I trial of alisertib plus romidepsin for relapsed/refractory aggressive B- and T-cell lymphomas. Paper presented at the 56th meeting of the American Society for Hematology; December 2014; San Francisco, CA.
- 44. Kim YH, Tavallaee M, Sundram U, et al. Phase II investigator-initiated study of brentuximab vedotin in mycosis fungoides and sezary syndrome with variable CD30 expression level: A multi-institution collaborative project. J Clin Oncol. 2015;33(32):3750–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631–636. [DOI] [PubMed] [Google Scholar]
- 46. Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–3115. [DOI] [PubMed] [Google Scholar]
- 47. O'Connor OA, Masszi TM, Savage KJ, et al. Belinostat, a novel pan-histone deacetylase inhibitor (HDACi), in relapsed or refractory peripheral T-cell lymphoma (R/R PTCL): Results from the BELIEF trial. J Clin Oncol. 2015;33(23):2492–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. [DOI] [PubMed] [Google Scholar]
- 49. Pro B, Advani RH, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J Clin Oncol. 2012;30(18):2190–2196. [DOI] [PubMed] [Google Scholar]
- 50. Horwitz SM, Advani RH, Bartlett NL, et al. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood. 2014;123(20):3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang CH, Mandelker D, Schmidt-Kittler O, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318(5857):1744–1748. [DOI] [PubMed] [Google Scholar]
- 52. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nature Rev Cancer. 2002;2(7):489–501. [DOI] [PubMed] [Google Scholar]
- 53. Horwitz S, Porcu P, Flinn I, et al. Duvelisib (IPI-145), a phosphoinositide-3-kinase-δ,γ inhibitor, shows activity in patients with relapsed/refractory T-cell lymphoma. Paper presented at the 56th Meeting of the American Society for Hematology; December 2014; San Francisco, CA.
- 54. Palomero T, Couronne L, Khiabanian H, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46(2):166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013;32(21):2601–2613. [DOI] [PubMed] [Google Scholar]
- 56. Kucuk C, Jiang B, Hu X, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat Commun. 2015;6:6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Koo GC, Tan SY, Tang T, et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012;2(7):591–597. [DOI] [PubMed] [Google Scholar]
- 58. Fleischmann R, Kremer J, Tanaka Y. et al. Efficacy and safety of tofacitinib in patients with active rheumatoid arthritis: Review of key phase II studies. Int J Rheum Dis. 2016; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]