

Abstract
Current knowledge of the genetic architecture of key reproductive events across the female life course is largely based on association studies of European descent women. The relevance of known loci for age at menarche (AAM) and age at natural menopause (ANM) in diverse populations remains unclear. We investigated 32 AAM and 14 ANM previously-identified loci and sought to identify novel loci in a trans-ethnic array-wide study of 196,483 SNPs on the MetaboChip (Illumina, Inc.). A total of 45,364 women of diverse ancestries (African, Hispanic/Latina, Asian American and American Indian/Alaskan Native) in the Population Architecture using Genomics and Epidemiology (PAGE) Study were included in cross-sectional analyses of AAM and ANM. Within each study we conducted a linear regression of SNP associations with self-reported or medical record-derived AAM or ANM (in years), adjusting for birth year, population stratification, and center/region, as appropriate, and meta-analyzed results across studies using multiple meta-analytic techniques. For both AAM and ANM, we observed more directionally consistent associations with the previously reported risk alleles than expected by chance (p-valuesbinomial≤0.01). Eight densely genotyped reproductive loci generalized significantly to at least one non-European population. We identified one trans-ethnic array-wide SNP association with AAM and two significant associations with ANM, which have not been described previously. Additionally, we observed evidence of independent secondary signals at three of six AAM trans-ethnic loci. Our findings support the transferability of reproductive trait loci discovered in European women to women of other race/ethnicities and indicate the presence of additional trans-ethnic associations both at both novel and established loci. These findings suggest the benefit of including diverse populations in future studies of the genetic architecture of female growth and development.
Introduction
Age at menarche (AAM) and age at natural menopause (ANM) are important events in the reproductive lifespan of a woman. Menarche, the initiation of the female menstrual cycle, occurs at 12 years on average [1,2]. In the United States (US), mean AAM is lower for African and Mexican American women, and higher for non-Hispanic women of European descent [2,3]. Yet, epidemiologic data on the average AAM of Asian American, Native Hawaiian and American Indian/Alaskan Native women are generally lacking. An earlier age at menarche has been associated with early life obesity and risk for a variety of diseases including breast and endometrial cancer, diabetes, and coronary heart disease [4–6].
Menopause, the cessation of the menstrual cycle that signifies the end of the reproductive lifespan, occurs at 51 years on average, with the majority of women experiencing a natural onset of menopause (not surgically or drug-induced) sometime between ages 45–55 years [7]. Similar to AAM, race/ethnicity appears to be an independent predictor of ANM in the US, with African and Mexican American women having earlier ANM, as compared to non-Hispanic women of European and Japanese descent [8,9]. Epidemiologic investigations of ANM in other US racial/ethnic groups are still needed. Earlier ANM is influenced by smoking status and can confer increased risk for cardiovascular disease and osteoporosis later in life, while later ANM can increase the risk of hormone-related female cancers, such as breast and endometrial cancers [10,11].
For both AAM and ANM, population-level changes have been observed in the US over the last century, wherein the average AAM decreased [1] and the average ANM has increased [12]. These trends may reflect the population-level shifts in the race/ethnicities of females living currently in the US, secular trends in obesity or smoking prevalence, or other environmental conditions supportive of a longer average female reproductive lifespan.
Given the racial/ethnic differences in AAM and ANM in the US, there remains significant interest in identifying the genetic factors that influence the timing of these reproductive events in diverse populations. Numerous candidate gene and genome-wide association studies (GWAS) have been performed for AAM and ANM, and as a result, more than 360 and 40 loci have been associated with AAM and ANM, respectively [13–23]. Although the vast majority of these studies have included only women of European descent in their discovery and validation samples, more recent GWAS have begun to include women of African (up to ~18,000 women) and East Asian ancestry (up to ~16,000 women), but have not discovered any additional loci [24–28]. Recent generalizability studies have also begun to include these populations as well as Hispanic/Latina, Native Hawaiian, and American Indian/Alaskan Native descent women [29–31] to more fully describe the transferability and allele frequency heterogeneity of these established AAM and ANM loci, as well as to discover novel race/ethnic-specific loci.
Recently developed methods for trans-ethnic meta-analysis now allow researchers to combine several populations, while accounting for heterogeneity between racial/ethnic groups [32,33]. Previous genetic epidemiologic research indicates that trans-ethnic meta-analyses improve the power to discover variants of low and moderate effect sizes and may reveal allelic heterogeneity at known genetic loci [17,26,27]. Additionally, trans-ethnic approaches may help narrow the interval of interest around loci discovered in European-descent populations. The Population Architecture using Genomics and Epidemiology (PAGE) Study, a consortium of ancestrally diverse genetic studies from the US, is well-positioned to investigate the genetics of complex traits within a trans-ethnic context [34].
Herein, we sought to analyze the roughly 200,000 SNPs genotyped on the MetaboChip (Illumina, Inc., San Diego, CA, USA), a high-density genotyping array of primarily cardiometabolic loci [35], for association with reproductive milestones in the ancestrally diverse study participants of the PAGE Study [34]. Given the known overlap between the genetic underpinnings of AAM, and related cardiometabolic traits [22], the MetaboChip provides a densely genotyped resource to search for novel reproductive associations and broadly investigate the overlap of cardiometabolic and reproductive traits. Using race/ethnicity-stratified meta-analyses (20,398 African American, 15,856 Hispanic/Latina, 8,572 Asian American, and 538 American Indian/Alaskan Native women) and a trans-ethnic modified random-effects meta-analysis of up to 42,826 ancestrally diverse women, we sought to (i) establish how many index AAM and ANM SNPs, previously described in European-descent populations, also generalize to diverse racial/ethnic groups of women in the PAGE Study, (ii) their trans-ethnic localization, and (iii) to identify novel AAM or ANM associations on the MetaboChip.
Materials and methods
Study participants and phenotyping
The PAGE Study was designed to generalize and estimate common genetic effects across multiple ancestral populations [34]. Briefly, the first phase of the PAGE study was comprised of a coordinating center, four large study sites/consortia [Causal Variants Across the Life Course (CALiCo) Consortium, including the Atherosclerosis Risk in Communities (ARIC) Study, Coronary Artery Risk Development in Young Adults (CARDIA), the Hispanic Community Health Study/Study of Latinos (HCHS/SOL); Epidemiologic Architecture for Genes Linked to Environment (EAGLE)-accessing the Vanderbilt University Medical Center’s biorepository (BioVU); Multiethnic Cohort (MEC); the Women’s Health Initiative (WHI)], and additional collaborating studies [The Hypertension Genetic Epidemiology Network (HyperGEN) Study, the MEC-Slim Initiative in Genomic Medicine for the Americas Type 2 Diabetes Consortium (MEC-SIGMA), Multi-Ethnic Study of Atherosclerosis (MESA), and Mount Sinai School of Medicine BioBank (BioME)]. We provide a detailed description of each study included in this analysis in our S1 Text. The datasets generated as part of the PAGE study can be accessed through the dbGaP repository (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000356). All studies in this analysis obtained Institutional Review Board approval and written informed consent from all participants, with the exception of EAGLE BioVU, which obtained Institutional Review Board approval to follow an opt-out consent process, described in detail separately [36,37].
Self-reported AAM (onset of first menses) and ANM (cessation of regular menses) in years were collected by questionnaire or via medical record [38]. AAM and ANM were harmonized across the studies, as reported previously [30]. Detailed descriptions of the pseudo-continuous coding and outlier exclusions are provided in the S2 Text.
Genotyping and imputation
The custom Illumina, Inc. iSELECT array, MetaboChip, genotyped 196,483 autosomal SNPs including the high-density genotyping of 257 regions associated with cardiometabolic traits as of 2009 [35]. As described in S2 Text, for three studies MetaboChip SNPs were imputed [MEC SIGMA, BioME, WHI African Americans [39]]. Additionally, we excluded SNPs with low minor allele frequencies (MAF), <0.1%, or that had deviations from Hardy-Weinberg Equilibrium (HWE), p-value<1x10-6. Additional information on the specific implementation of HWE filtering and other SNP-level quality control procedures is provided in the S2 Text.
Forty-six index SNPs had been previously associated with either AAM or ANM (or if unavailable on the MetaboChip, a proxy SNP r2≥0.8 in 1000 Genomes CEU sample) and represented distinct genetic loci (r2<0.2) (S1 Table). These SNPs included all two of the known AAM and five of the known ANM loci as of when the MetaboChip was designed, including the two strongest and most widely-generalizable AAM and ANM signals to date (LIN28B and MCM8) [22,23]. Additionally, seven of the previously-associated SNPs were located within six densely-genotyped loci (S2 Table) that were associated with AAM or ANM after the initial design of the MetaboChip.
S2 Text provides additional information on the following person-level exclusions. Briefly, we identified and excluded individuals with high inbreeding coefficients, F > 0.15 [40], and either excluded one woman of each 1st degree relative pair [41], or modeled relatedness using generalized estimating equations [42] and linear mixed models [43]. We generated principal components using Eigensoft for each study [44,45] and excluded ancestral outliers [46]. We excluded samples with phenotype-genotype sex discordance, low person-level call rate (<95%), or excessive heterozygosity.
Only study- and race/ethnic-specific sample sizes of 50 women or more were carried forward to statistical analyses and summarized descriptively in S3 and S4 Tables. Collectively, the studies represent 20,398 African American, 15,856 Hispanic/Latina, 8,572 Asian American, and 538 American Indian/Alaskan Native women who provided informed consent (or in the case of BioVU, did not opt out from the approved research), and had complete information on genetics, reproductive phenotypes and covariates.
Statistical modeling and analyses
Within each study the MetaboChip SNP and reproductive trait associations were modeled under an additive genetic model and adjusted for birth year, principal components, or if applicable, also center and Type 2 Diabetes case/control status. Within each racial/ethnic group we then implemented fixed-effect inverse-variance weighted meta-analyses using METAL (version from 2011-03-25) [47], to estimate race/ethnic-specific effects for those SNPs that were informed by more than half of the maximum race/ethnic sample (n = 123,493–157,710). Additional information on our data visualization and post-hoc power analyses are provided in the S1 Text.
Array-wide significance for novel SNPs was defined as a Bonferroni p-value<2.5x10-7 to account for the total number of autosomal SNPs on the MetaboChip (n = 196,483). We concluded that the observed association was directionally consistent, if the trait-decreasing allele of the trans-ethnic analysis was the same as the trait-decreasing allele of previous report(s). Furthermore, using a binomial distribution we tested (p-values binomial <0.05) if we observed more directional consistency than we would expect by chance (i.e. assuming that 50% of all tests would be consistent by chance alone). Generalization of previously described reproductive loci at index SNPs (or their proxies) to our samples was declared if 1) our estimate was directionally consistent with the previous reports, and 2) the SNP association had a p-value<0.0016 for AAM or p-value<0.0036 for ANM, corresponding to a Bonferroni correction for the number of independent AAM (n = 32) and ANM (n = 14) loci tested. SNPs located within densely genotyped reproductive loci, which were not index SNPs or their proxies, were considered to be significant if their association was less than a p-value Bonferroni-corrected for the number of independent signals within the given locus (independent signals pruned to r2<0.2 in ARIC African Americans), resulting in p-value thresholds ranging from 9.0x10-5 to 2.8x10-4 (S4 Table). Within each densely genotyped locus, statistically significant race/ethnic-specific lead SNPs (i.e. those with the lowest p-values in the locus) were considered to be potentially independent of the index SNP and warranting of additional conditional analyses, if they were in moderate to low linkage disequilibrium, LD (r2<0.5 in 1000 Genomes CEU sample).
At each of the densely genotyped reproductive loci, publicly available reference samples from 1000 Genomes were utilized to estimate the number of SNPs (and their base pair locations) in the European (CEU), African (YRI), Hispanic/Latino (MXL, PUR, CLM), and Asian (JPT) reference populations that are in high LD (r2≥0.8) with the previously reported AAM and ANM index SNPs. The percentage reduction in the putative interval of interest was then calculated by contrasting the populations with the smallest and largest LD blocks associated with these index SNPs (S5 Table).
Modified random-effects meta-analysis
Trans-ethnic meta-analyses of AAM and ANM were conducted using a modified random-effects meta-analysis of study/race-ethnic-specific results, as implemented in Metasoft by Han and Eskin, which applies a likelihood ratio test to allow the existence of heterogeneity to be dependent on the hypothesis of association—either the alternative (random-effects) or the null hypotheses (a fixed null effect) [48]. We excluded American Indian/Alaskan Native women from the trans-ethnic meta-analyses, due to their relatively small sample size as compared to the combined trans-ethnic sample of the other racial/ethnic groups (1% for both AAM and ANM). For SNP-associations with more than half of the maximum trans-ethnic sample size for the specific trait, we estimated modified random-effects up to 22 AAM and 23 ANM study subsamples of African, Hispanic/Latina and Asian ancestry.
Secondary signal analysis
Next, we tested for the presence of statistically significant secondary signals using an approximate conditional method in Genome-wide Complex Trait Analysis (GCTA, version 64) [49,50] and using the same trans-ethnic reference samples as above to estimate trans-ethnic LD patterns. Adjusting for the significant lead trans-ethnic SNP at each locus, we contrasted the unconditional and approximate conditional p-values of the SNPs within the region. If an unconditional SNP association was suggestive (p-value<0.05) and not heterogeneous across race/ethnic groups in the trans-ethnic modified random-effect analysis, but became array-wide or Bonferroni-significant after adjusting for the lead SNP in the region, we concluded that this was evidence for a secondary signal in the region. This approach was repeated until no additional significant conditional SNP associations arose.
Results
The epidemiology of ages at menarche and natural menopause
Our final analytic samples were comprised of 44,367 and 17,100 women with AAM and ANM information from four broad racial/ethnic groups (Table 1). The biobank studies (EAGLE BioVU, BioME) and HCHS/SOL represented a wide range of ages (S3 and S4 Tables). The median age was lower and the median birth year more recent in the AAM samples, than in the ANM samples. In both the AAM and ANM analytic samples, the obesity prevalence at examination was the highest in African American women and lowest in the East Asian women (47 versus 10% weighted prevalence). MEC Native Hawaiian, Hispanic/Latina, and WHI American Indian/Alaskan Native women had intermediate obesity prevalence estimates (37–45%). In the ANM analysis samples, the prevalence of current cigarette smoking at examination was the highest in the Native Hawaiian (20%) and African American women and lowest in other Asian samples of women (14% versus 6% weighted prevalence). American Indian/Alaskan Native and Hispanic/Latina women had intermediate prevalence estimates of smoking (10–11%).
Table 1. Descriptive statistics for the age at menarche (AAM, n = 44,367) and natural menopause (ANM, n = 17,100) analytic samples.
| Study | Sample Size | Median Age at report (years) | Median Birth Year | Median* AAM or ANM (years) | Obesity (%) | ||
| MENARCHE | |||||||
| AfA | ARIC | 2,056 | 53 | 1935 | 13 | 48 | |
| EAGLE BioVU | 656 | 41 | 1972 | 12 | 51 | ||
| CARDIA | 990 | 25 | 1960 | 12 | 21 | ||
| MEC | 4,410 | 59 | 1933 | - | 39 | ||
| BioME** | 373 | 47 | 1966 | 12 | 48 | ||
| WHI** | 11,724 | 61 | 1935 | - | 51 | ||
| H/L | HCHS/SOL | 7,027 | 48 | 1962 | 13 | 45 | |
| MEC | 859 | 59 | 1934 | - | 32 | ||
| SIGMA-Diab** | 910 | 59 | 1935 | - | 49 | ||
| SIGMA-Cont** | 910 | 59 | 1934 | - | 17 | ||
| BioME** | 512 | 49 | 1964 | 12 | 45 | ||
| WHI | 5,129 | 59 | 1936 | - | 37 | ||
| AsA | MEC-Hawaiian | 1,364 | 54 | 1939 | - | 37 | |
| MEC-Japanese | 3,725 | 59 | 1935 | - | 10 | ||
| WHI | 3,184 | 63 | 1933 | - | 12 | ||
| AI/AN | WHI | 538 | 61 | 1935 | - | 45 | |
| Study | Sample Size | Median Age at report (years) | Median Birth Year | Median* AAM or ANM (years) | Obesity (%) | Current Smoking (%) | |
| NATURAL MENOPAUSE | |||||||
| AfA | ARIC | 569 | 56 | 1931 | 49 | 49 | 25 |
| CARDIA | 150 | 54 | 1956 | 49 | 63 | 18 | |
| HyperGen | 189 | 57 | 1940 | 47 | 63 | 20 | |
| MEC | 1,598 | 62 | 1930 | - | 38 | 16 | |
| MESA | 583 | 65 | 1936 | 49 | 52 | 17 | |
| WHI** | 4,209 | 62 | 1934 | 50 | 50 | 12 | |
| H/L | HCHS/SOL | 1,940 | 58 | 1952 | 49 | 49 | 16 |
| MEC | 416 | 60 | 1934 | - | 31 | 12 | |
| SIGMA-Diab** | 225 | 60 | 1933 | - | 31 | 9 | |
| SIGMA-Cont** | 61 | 61 | 1932 | - | 20 | 8 | |
| MESA | 509 | 63 | 1938 | 49 | 51 | 12 | |
| BioME** | 93 | 55 | 1958 | 50 | 55 | - | |
| WHI | 2,027 | 60 | 1936 | 50 | 36 | 6 | |
| AsA | MEC-Hawaiian | 567 | 57 | 1936 | - | 33 | 20 |
| MEC-Japanese | 1,822 | 61 | 1932 | - | 7 | 8 | |
| MESA | 299 | 65 | 1936 | 50 | 5 | 2 | |
| WHI | 1,659 | 63 | 1932 | 50 | 10 | 4 | |
| AI/AN | WHI | 184 | 61 | 1935 | 50 | 41 | 10 |
*Median estimates not available for MEC or WHI for AAM, or MEC for ANM, due to their categorical ascertainment of these traits (see S1 and S2 Text for more information on the pseudo-continuous recoding of these traits).
**Studies which include imputed MetaboChip SNP data.
Abbreviations: AfA = African American, AI/AN = American Indian/Alaskan Native, AsA = Asian American, Cont = Type 2 Diabetes Controls, Diab = Type 2 Diabetes Cases, H/L = Hispanic/Latina
Generalization of previously reported reproductive trait associations
In our trans-ethnic AAM analyses in women of African, Hispanic/Latina and Asian descent, we generalized the association at LIN28B with AAM at array-wide significance (S1 Fig). Even though genotyping in the region is sparse on the MetaboChip, the strongest SNP association in the region (rs7759938) was a previously published European descent index SNP [13,17] and was directionally consistent with the previously reported risk allele (T). This SNP association was significant in the African, Hispanic/Latina, and Asian American samples after adjusting for the number of independent loci tested with AAM, and directionally consistent in American Indian/Alaskan Native women (Table 2).
Table 2. Generalization of five previously described age at menarche and natural menopause loci to multiple race/ethnic groups or trans-ethnically.
| GWAS Ref* | SNP | GWAS Decreas-ing Allele* | Previous GWAS Effect (years)* | Chr | BP | Gene(s)* | Pop | Freq | Effect (years) | SE | P-value | P-value hetero-geneity | Sam-ple Size |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MENARCHE | |||||||||||||
| [20] | rs823114* | G | -0.03 | 1 | 205719532 | NUCKS1, RAB7L1 | AfA | 0.754 | -0.044 | 0.02 | 0.024 | 0.146 | 20199 |
| H/L | 0.589 | -0.064 | 0.021 | 0.002 | 0.098 | 15343 | |||||||
| AsA | 0.469 | -0.008 | 0.02 | 0.744 | 0.819 | 8266 | |||||||
| AI/AN | 0.568 | -0.100 | 0.11 | 0.382 | - | 538 | |||||||
| TE** | 0.636 | -0.042 | 0.02 | 1.36E-03 | - | 43808 | |||||||
| [13,17] | rs7759938 | T | -0.09 | 6 | 105378954 | LIN28B | AfA | 0.464 | -0.080 | 0.02 | 2.15E-06 | 0.501 | 20206 |
| H/L | 0.719 | -0.106 | 0.02 | 3.22E-06 | 0.671 | 15345 | |||||||
| AsA | 0.702 | -0.171 | 0.03 | 1.00E-10 | 0.733 | 8269 | |||||||
| AI/AN | 0.699 | -0.302 | 0.12 | 0.011 | - | 538 | |||||||
| TE** | 0.601 | -0.106 | 0.01 | 4.71E-18 | - | 43820 | |||||||
| [13,17] | rs2090409 | A | -0.1 | 9 | 108967088 | TMEM38B | AfA | - | - | - | - | - | - |
| H/L | 0.325 | -0.087 | 0.02 | 7.72E-05 | 0.728 | 15342 | |||||||
| AsA | 0.432 | -0.034 | 0.02 | 0.165 | 0.061 | 8249 | |||||||
| AI/AN | 0.324 | -0.043 | 0.11 | 0.700 | - | 538 | |||||||
| TE** | 0.358 | -0.060 | 0.02 | 1.35E-04 | - | 28222 | |||||||
| NATURAL MENOPAUSE | |||||||||||||
| [15,18] | rs11668309* | T | -0.49 | 19 | 55833460 | BRSK1, TMEM150B | AfA | 0.238 | -0.031 | 0.03 | 0.278 | 0.258 | 7294 |
| H/L | 0.343 | -0.2071 | 0.06 | 2.52E-04 | 0.892 | 5271 | |||||||
| AsA | 0.145 | -0.038 | 0.04 | 0.361 | 0.821 | 4348 | |||||||
| AI/AN | 0.376 | 0.175 | 0.50 | 0.729 | - | 185 | |||||||
| TE** | 0.241 | -0.065 | 0.02 | 0.011 | - | 16913 | |||||||
| [19] | rs236114 | C | -0.5 | 20 | 5935385 | MCM8 | AfA | 0.904 | 0.017 | 0.04 | 0.694 | 0.951 | 7108 |
| H/L | 0.832 | -0.215 | 0.07 | 2.99E-03 | 0.408 | 5270 | |||||||
| AsA | 0.945 | -0.100 | 0.10 | 0.303 | 0.722 | 4049 | |||||||
| AI/AN | 0.870 | -0.278 | 0.75 | 0.710 | - | 185 | |||||||
| TE** | 0.901 | -0.051 | 0.03 | 0.205 | - | 16427 | |||||||
*When the index SNP was not genotyped on the MetaboChip, the proxy SNP in tight linkage disequilibrium (r2> = 0.8 in 1000 Genomes pilot 1 CEU) with the lowest p-value in the African American sample was chose to represent the index signal. If more than one SNP represented the same locus (within 500kb of each other) on the MetaboChip, only the SNPs r2<0.2 in ARIC African Americans (or HCHS/SOL Hispanic/Latinos, when missing) were included in this table allowing preference for index SNPs, and in most cases SNPs from multiple citations. The decreasing (coded) allele and previous effect size for proxies were assigned assuming that the risk index SNP would have a similar allele frequency (either minor or major) and effect as the selected proxy SNP.
**Modified random-effects trans-ethnic meta-analysis across three racial/ethnic groups (African, Hispanic/Latina, and Asian Americans). American Indian/Alaskan Native samples were not included due to their relative small sample size.
Significant SNP-associations (p<0.05/32 menarche SNPs tested; p<0.05/14 menopause SNPs tested; all SNPs shown in S1 Table) shown in bold.
Nominally significant p-values (p<0.05) shown in italics.
All SNPs are oriented on positive strand and positions based on Build 37.
Abbreviations: AfA = African American, AI/AN = American Indian/Alaskan Native, AsA = Asian American, BP = Base pair, Chr = Chromosome, Freq = Frequency for coded decreasing allele, GWAS = Genome-wide association study, H/L = Hispanic/Latina, Pop = Racial/ethnic group or trans-ethnic analysis, TE = Trans-ethnic modified random effects, MA = Minor Allele, N = Sample Size, SE = Standard Error, SNP = Single nucleotide polymorphism.
In addition, we observed Bonferroni-significant evidence of generalization to diverse racial/ethnic groups at two other AAM loci. The index/proxy AAM SNPs at NUCKS1 and TMEM38B were most strongly associated in the Hispanic/Latina subsample, and were also significant in the trans-ethnic meta-analysis and directionally consistent with the previously reported risk allele in all race/ethnic groups (Table 2).
In our trans-ethnic ANM analyses, we observed evidence of diverse generalization at two ANM loci after accounting for the number of independent loci tested with ANM. The proxy SNP at BRSK1 and the selected index SNP at MCM8 were significantly associated in Hispanic/Latinas (trait-decreasing allele frequency, TDAF, 34% and 83%; p-value<3x10-3; Table 2). The BRSK1 and MCM8 associations were not significant in the trans-ethnic meta-analysis or directionally consistent with the trait-decreasing allele across all race/ethnic groups.
The first index SNP at MCM8, [rs236114 [19]] was in moderate LD (1000 Genomes AMR r2 = 0.36) with another SNP, rs16991615 [15,18,23], which was both Bonferroni significantly associated in Hispanic/Latinas (TDAF 94%, p-value = 7.14x10-6) in the trans-ethnic sample (TDAF 97%, p-value = 1.91x10-6; S2 Fig) and associated with ANM in a directionally-consistent manner across all race/ethnic groups. Yet within the 9 Hispanic/Latina studies this SNP association exhibited evidence of effect heterogeneity (p-valueheterogeneity = 3.43x10-4), which in S3 Fig appeared to be driven by ANM-increasing effects among the MEC and MEC SIGMA Type 2 Diabetes cases, which was inconsistent with the previously reported ANM reducing allele and with the observed direction of effect in the WHI Native American/American Indian subsample. Although this apparent effect heterogeneity could be due to chance, it is also possible it could reflect differences in relevant pre-menopausal environments/health statuses of these specific MEC subsamples (e.g. gene-environment interactions). Additionally, approximate conditional analyses revealed that the significant association between rs16991615 and ANM at MCM8 may be independent from rs236114 in our sample of Hispanic/Latinas (p-valueconditional = 6.2x10-4).
Next, across all 32 AAM and 14 ANM loci on the MetaboChip, we assessed the directional consistency between our race/ethnic-specific and trans-ethnic results and previously reported risk-associated alleles (S1 Table). The number of directionally-consistent SNP associations with AAM exceeded our expectation in all race/ethnic groups (p-valuesbinomial<0.01) and trans-ethnic results (p-valuesbinomial = 1.2x10-6), with the exception of American Indian/Native American women (p-valuebinomial = 0.11). For ANM the number of directionally consistent SNP-associations also exceeded our expectation based on chance in all race/ethnic (p-valuebinomial≤0.03) and trans-ethnic results (p-valuebinomial = 0.01), with the exception of African American women (p-valuebinomial = 0.18).
Generalization at densely genotyped reproductive trait loci
Three of the six densely genotyped ANM loci, SEC16B, BDNF and FTO, generalized to the trans-ethnic sample at a lead SNP that was in moderate LD with at least one previously reported index SNP for AAM and ANM (r2>0.2; Table 3 and Fig 1). At SEC16B, the lead Hispanic/Latina SNP (rs78368018-A; MAF 0.3%) was significant after Bonferroni correction and also in moderate LD with the index SNP (rs633715-C; trans-ethnic MAF 14.9%). However, the lead SNP had nominal evidence of effect heterogeneity across three studies of Hispanic/Latinas (p-valueheterogeneity = 0.03, Table 2) that was driven by a subsample of the MEC (S4 Fig). Patterns of LD at BDNF and FTO revealed that the AAM signal aligned more closely with the primary BMI signal than with other independent signals for BMI previously reported at these loci [51,52]. At four additional, albeit non-significant, densely genotyped loci, LD patterns revealed that our lead AAM or ANM SNPs were dependent on the previously reported index SNPs (S2 Table; S5 Fig).
Table 3. Three densely-genotyped MetaboChip loci with Bonferroni-significant associations with age at menarche across multiple race/ethnic groups or trans-ethnically.
| GWAS Ref | Gene | Chr | Start/Stop BP | P-value* | Index GWAS SNP (Position) | Pop | Top MetaboChip SNP | Position | MA | Freq | Effect (years) | SE | P-value | P-value hetero-geneity | Sam-ple Size |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [13] | SEC16B | 1 | 177753776/177936525 | 2.82E-04 | rs633715 (177852580) | AfA | rs75552107 | 177925677 | G | 0.158 | 0.127 | 0.04 | 6.85E-04 | 0.891 | 18599 |
| H/L | rs78368018 | 177777983 | A | 0.003 | -0.825 | 0.21 | 9.42E-05 | 0.038 | 12787 | ||||||
| AsA | rs7518576 | 177849963 | T | 0.459 | 0.081 | 0.03 | 1.19E-03 | 0.885 | 8265 | ||||||
| TE** | rs604388 | 177877979 | C | 0.427 | -0.060 | 0.01 | 3.39E-05 | - | 31349 | ||||||
| [20] | BDNF | 11 | 27452706/ 27749725 | 2.48E-04 | rs7103411 (27700125) | AfA | rs113940328 | 27530168 | C | 0.053 | 0.140 | 0.04 | 2.31E-04 | 0.510 | 20209 |
| H/L | rs11030104 | 27684517 | G | 0.173 | 0.076 | 0.03 | 5.04E-03 | 0.851 | 15346 | ||||||
| AsA | rs1491850 | 27749725 | C | 0.412 | 0.073 | 0.02 | 3.38E-03 | 0.933 | 8273 | ||||||
| TE** | rs4923463 | 27672500 | G | 0.177 | 0.068 | 0.02 | 3.54E-05 | - | 43824 | ||||||
| [13] | FTO | 16 | 53539509/ 54185787 | 8.98E-05 | rs9939609 (53820527) | AfA | rs76299885 | 53950231 | A | 0.136 | -0.119 | 0.04 | 3.48E-03 | 0.483 | 19545 |
| H/L | rs112372930 | 53898024 | A | 0.017 | 0.325 | 0.09 | 5.28E-04 | 0.112 | 13527 | ||||||
| AsA | rs150356630 | 53830590 | T | 0.075 | -0.198 | 0.05 | 1.43E-04 | 0.864 | 8273 | ||||||
| TE** | rs11642841 | 53845487 | A | 0.149 | -0.073 | 0.02 | 2.07E-05 | - | 43797 |
*Bonferroni correction for the number of SNPs r2<0.2 in region in MetaboChip data from the ARIC African Americans (n = 1419 males, n = 2332 females), using a 50-SNP window and shifting the window in each iteration by 5 SNPs.
**Strongest SNP marker in modified random-effects trans-ethnic meta-analysis across three race/ethnic groups (African, Hispanic/Latina, and Asian Americans).
Significant SNP-associations below specific Bonferroni p-values for a given locus are shown in bold.
Nominally significant heterogeneity p-values (p<0.05) shown in italics.
Trans-ethnic P-value represented a modified Han and Eskin p-value.
All SNPs are oriented on positive strand and positions based on Build 37.
Abbreviations: AfA = African American, AsA = Asian American, BP = Base pair, Chr = Chromosome, Freq = Frequency for coded decreasing allele, GWAS = Genome-wide association study, H/L = Hispanic/Latina, Pop = Racial/ethnic group or trans-ethnic analysis, TE = Trans-ethnic modified random effects, MA = Minor Allele, N = Sample Size, Ref = Reference, SE = Standard Error, SNP = Single nucleotide polymorphism.
Fig 1. Regional plots for age at menarche Bonferroni-significant loci at SEC16B (Panel A), BDNF (Panel B) and FTO (Panel C), showing previously published body mass index (BMI) primary and secondary SNP associations, using a modified random-effects trans-ethnic meta-analysis of more than 31,000 women.
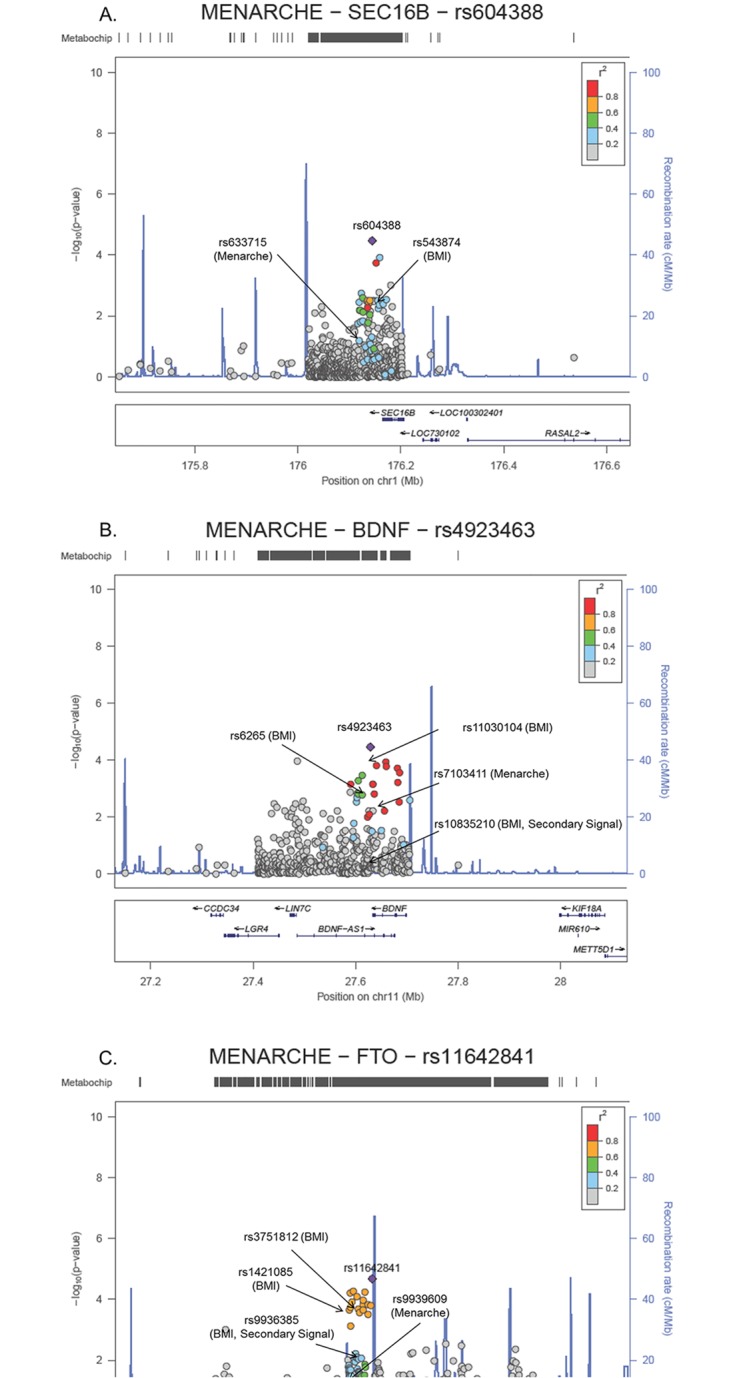
Lastly, we harnessed publicly available information on the LD blocks tagged by the index SNPs for AAM and ANM to inform narrowing of the putative interval around the loci that generalized to Hispanic/Latinas. Specifically, we found that the percent reduction in the base pair interval of interest (based on the location of SNPs in strong LD, r2≥0.8, with the index SNP of interest) was 77–96% across five AAM loci (SEC16B, TRIM66, BDNF, GPRC5B, FTO; S5 Table) or, in the case of TMEM18 this approach pointed to one other SNP (rs7559547). For the ANM signal at FNDC4, the percent reduction was less dramatic (28% reduction) in the base pair interval of interest. The largest LD blocks were found in the 1000 Genomes CEU, whereas the smallest LD blocks were noted in either YRI or AMR reference populations.
Trans-ethnic array-wide associations
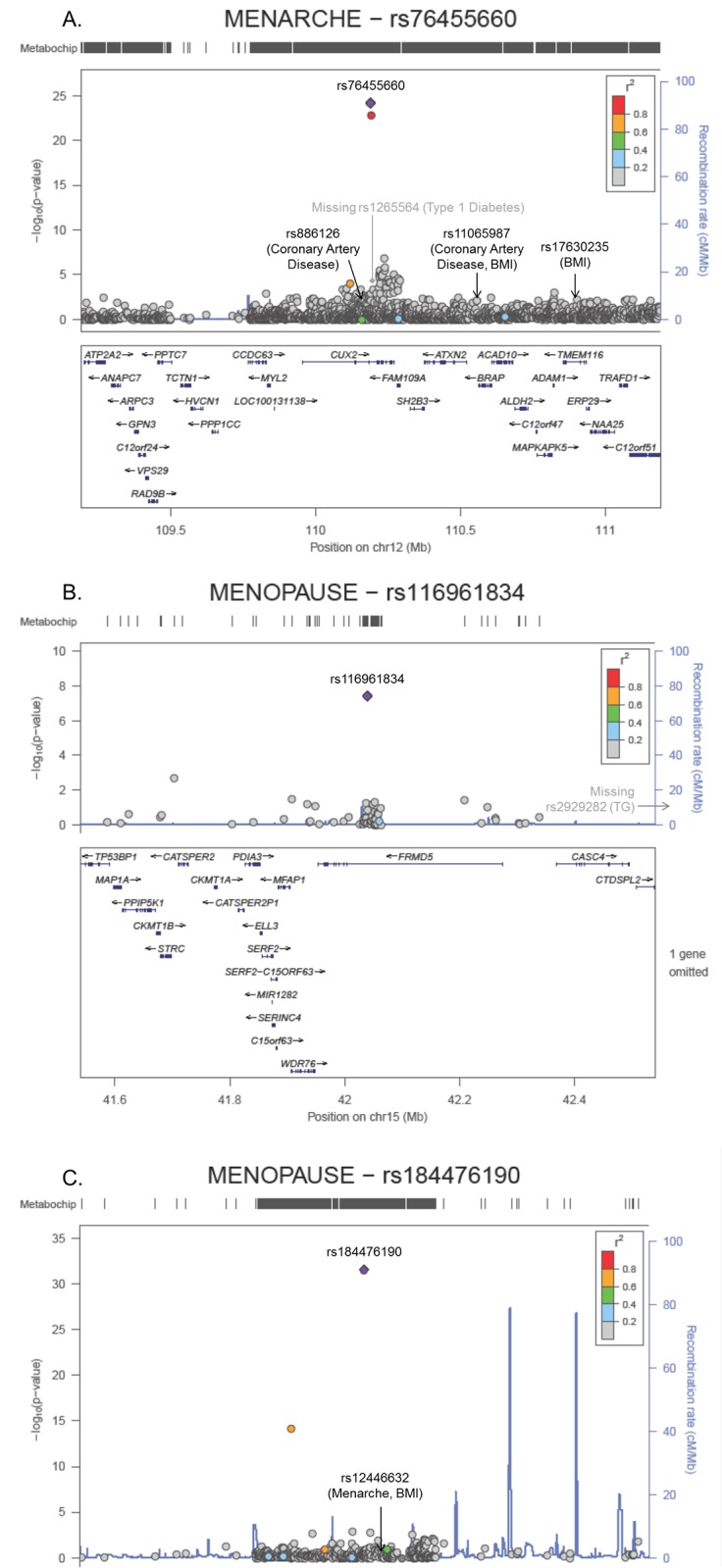
In our trans-ethnic meta-analyses, we observed evidence of array-wide (p-value<2.5x10-7) novel associations with AAM at CUX2, and with ANM at FRMD5 and GPRC5B. The lead SNPs at these three loci were all highly variable across studies but were on average low frequency SNPs (Table 4), and in weak LD with most SNPs in the region (r2<0.2; Fig 2A–2C). As shown in S6 Fig, the estimated effect for each novel SNP was strongest in Hispanic/Latinas than the other racial/ethnic groups, and in the case of GPRC5B showed evidence of heterogeneity among Hispanic/Latinas (Table 4). The lead SNPs were observed in predominantly one ancestral group, such as African (rs76455660 at CUX2, MAF = 1.7% 1000 Genomes AFR; rs184476190 at GPRC5B, MAF = 0.9% AFR) and Asian ancestries (rs116961834 at FRMD5, MAF = 6.6% in 1000 Genomes EAS), and several other race/ethnic samples were filtered out due to low frequency (MAF<0.1%), which yielded analytic sample sizes 61–75% of the total trans-ethnic sample for the given trait.
Table 4. Three loci with trans-ethnic array-wide significant modified random-effects associations* at novel age at menarche or natural menopause loci.
| SNP | Chr | BP | Gene | Decreasing Allele | Pop | Freq | Freq Range | Effect* (years) | SE | P-value | P-value Het or Tau2 | N |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MENARCHE | ||||||||||||
| rs76455660 | 12 | 111705293 | CUX2 | C | AfA | 0.837 | 0.806–0.991 | -0.106 | 0.04 | 7.06E-03 | 1E-01 | 19435 |
| H/L | 0.998 | 0.998–0.999 | -0.661 | 0.06 | 2.32E-26 | 4E-01 | 7255 | |||||
| TE | 0.955 | 0.806–0.999 | -0.108 | 0.12 | 6.29E-25 | 0.09 | 26690 | |||||
| NATURAL MENOPAUSE | ||||||||||||
| rs116961834 | 15 | 44252119 | FRMD5 | T | AfA | 0.015 | 0.002–0.038 | 0.138 | 0.25 | 5.77E-01 | 5E-01 | 4109 |
| H/L | 0.001 | 0.001–0.002 | -1.390 | 0.20 | 1.15E-11 | 6E-02 | 4273 | |||||
| AsA | 0.087 | 0.055–0.128 | -0.016 | 0.04 | 7.06E-01 | 7E-01 | 4348 | |||||
| TE | 0.045 | 0.001–0.128 | -0.095 | 0.14 | 3.74E-08 | 0.13 | 12730 | |||||
| rs184476190 | 16 | 19893802 | GPRC5B | C | AfA | 0.037 | 0.007–0.136 | -0.200 | 0.11 | 6.22E-02 | 4E-01 | 6715 |
| H/L | 0.002 | 0.002–0.003 | -3.081 | 0.27 | 2.72E-30 | 1E-07 | 4273 | |||||
| TE | 0.030 | 0.001–0.136 | -0.494 | 0.44 | 2.91E-32 | 1.61 | 10988 | |||||
*Strongest SNP marker in modified random-effects trans-ethnic meta-analysis informed by studies from up to three race/ethnic groups (African, Hispanic/Latina, and Asian American women). P-value represented a modified Han and Eskin p-value. Race/ethnic fixed-effect estimates and p-values of heterogeneity are shown for illustrative purposes, as the modified random-effects meta-analysis was run on studies separately.
Significant SNP-associations below specific Bonferroni p-values for a given locus are shown in bold.
All SNPs are oriented on positive strand and positions based on Build 37.
Abbreviations: AfA = African American, AsA = Asian American, BP = Base pair, Chr = Chromosome, Freq = Frequency for coded decreasing allele, Het = Heterogeneity p-value from race/ethnic specific fixed-effect meta-analysis, H/L = Hispanic/Latina, N = Sample Size, Pop = Population, SE = Standard Error, SNP = Single nucleotide polymorphism, TE = Trans-ethnic modified random effects.
Fig 2. Regional plots of the novel array-wide significant age at menarche (Panel A: CUX2) and natural menopause loci (Panels B,C: FRMD5, GPRC5B) using a modified random-effects trans-ethnic meta-analysis of more than 31,000 women, and showing independence from previously published cardiometabolic SNP associations (shown in gray if missing).
Secondary signal analysis
As shown in S7 Fig, three AAM loci (SEC16B, BDNF, CUX2) had suggestive evidence of secondary signals, which were in low LD (r2<0.2) with the primary AAM signal observed in our unconditional trans-ethnic analyses.
At SEC16B, variation at the frequency of the SNP representing the possible AAM secondary signal (rs114548967-G the decreasing allele; p-valueconditional = 2.18x10-4) was driven by African and Hispanic/Latina ancestries and varied between from 0.3% to 18.6% across the 12 samples contributing to the trans-ethnic meta-analysis. In the case of BDNF, this Bonferroni-significant secondary signal (rs113940328-C; p-valueconditional = 3.90x10-5) was monomorphic in 1000 Genomes EUR, but varied in MAF from 0.4% to 7.4% across the 15 samples contributing to the trans-ethnic meta-analysis. This secondary SNP was independent from the previously established BMI primary and secondary signals (r2<0.01 with other SNPs in AFR) [51,52]. At CUX2 the array-wide significant secondary signal (rs10849931-C; p-valueconditional = 1.05x10-7) was in weak LD with a previously described SNP for coronary artery disease (rs886126, r2 = 0.5 in 1000 Genomes EUR) [53] and weakly associated with the other previously described trait associations in the region (r2≤0.2). However, unlike the primary signal at this region (rs76455660-T, which is monomorphic in 1000 Genomes EUR) the lead conditional SNP was present in all race/ethnic groups, with remarkable variation in allele frequency across the 21 ancestry and study-specific groups analyzed jointly (9.3% to 62.3%).
Discussion
Our trans-ethnic meta-analysis of reproductive traits has expanded our understanding of the transferability of reproductive trait loci discovered in women of European descent to other race/ethnic groups. First, we observed more directionally consistent trans-ethnic associations than we expected by chance across all 32 AAM and 14 ANM loci on the MetaboChip (p-valuesbinomial of 1.2x10-6 and 0.01, respectively). Second, we generalized six AAM loci (NUCKS1, LIN28B TMEM38; SEC16B, BDNF, FTO), and two ANM loci (BRSK1, MCM8) to African, Hispanic/Latina, Asian or American Indian/Alaskan Native women, observing at each locus directional consistency between our trans-ethnic risk alleles and previous reports among European descent women [13,15,17–19]. This suggests that much of the currently known genetic architecture for AAM and ANM appears to be transferable across ancestrally diverse racial/ethnic groups.
Additionally, we conducted an array-wide analysis of AAM and ANM, and identified array-wide significant SNPs at three novel loci (AAM: CUX2; ANM: FRMD5, GPRC5B), which were most frequent in populations with African and Asian ancestry (CUX2, GPRC5B; and FRMD5, respectively). Even though previous studies have associated variation in CUX2 with Type 1 diabetes, coronary artery disease, atrial fibrillation and most recently with AAM [22,53–55], our novel signals appear to be distinct and infrequent in populations of European descent. According to HaploReg v4.1, the novel SNPs at CUX2 and FRMD5 are both predicted to be enhancers in the brain, which is consistent with the modulatory effect that neurotransmitter and gonadal hormones may have on each other [56]. A SNP in FRMD5 has been previously associated with triglycerides [57], but it is 6kb downstream and in weak LD with our SNP in 1000 Genomes CHB+JPT (r2 = 0.01). Common genetic variation near GPRC5B has been previously associated with both BMI and AAM [20,51,52], but has not previously been associated with ANM. In HaploReg, the lead SNP intronic to GPRC5B was a predicted histone mark, promoter and/or enhancer in brain and ovary tissue, as well as having DNAase activity in ovary and several other tissues and binding affinity to neuron-restrictive silencer factor.
Our results, however, are limited by the imbalance in sample sizes available for the AAM and ANM analyses, and the relatively low proportion of established AAM and ANM loci from studies of European descent women to date available on the MetaboChip (9% and 32%, respectively). Although our exclusion of extreme values restricted our analytic sample size, it allowed us to report on the common genetic causes of normal variation in AAM and ANM. Additionally, our use of the MetaboChip as a common dense-genotyping array to all PAGE studies, and with consistent genotype calling and quality control applied to each study, is a key strength of this study. Nonetheless our ascertainment of AAM and ANM did rely primarily on self-report. Lastly, the available sample size of racial/ethnic minority women who experienced these reproductive milestones is only a fraction of the samples of European descent women published on previously [22]. Due to relative paucity of genetic data on minority women, we did not have sufficient sample sizes to achieve statistical power to identify genetic associations of low frequency variants (MAF<5%) in most of our analyses, to seek independent replication of our novel significant findings (Table 4), or to systematically explore the role of gene-environment interactions on our findings. Yet, our observation of enrichment of directional consistency (S1 Table) suggests that given sufficient power or more comprehensive genotyping arrays, additional AAM and ANM loci may be significantly associated with reproductive traits in minority women. Larger samples of diverse women are needed to investigate all currently known AAM and ANM loci, establish statistical significance and describe the magnitude of the novel genetic effects on reproductive traits with more precision.
Our findings advance our current understanding of the scope of race/ethnic groups, to which previously reported reproductive traits may be generalized, albeit often at another SNP within the previous association signal. Specifically, we were able to generalize the widely-replicated LIN28B association (e.g. in African, Hispanic/Latina and East Asian studies) [24,26,28,31] to a more diverse group of Asian ancestries including Native Hawaiian women from the MEC (p-value = 1.0x10-10), as well as to American Indian/Alaskan Native women from WHI, albeit at nominal significance (p-value = 0.01). We also extended evidence of the NUCKS1 association with AAM beyond women of European descent to a trans-ethnic sample of women for the first time. Although all race/ethnic groups in our study had effects that were directionally consistent with previous reports at NUCKS1, only African American and Hispanic/Latina women were nominally associated with AAM (p-value≤0.02; Table 2).
Even though several studies have previously generalized reproductive trait associations at TMEM38B (AAM), BRSK1 and MCM8 (ANM) to African, Hispanic/Latina and East Asian ancestries [24–26,28,31], our study is the first to investigate heterogeneity within and across populations with distinct ancestries. As illustrated by our heterogeneous findings at MCM8 (S3 Fig), the role of within group heterogeneity should be investigated in future studies of populations with European admixture, like Hispanic/Latinos [25]. Similar to previous work, we noted that MCM8 also replicated in our sample of Hispanic/Latinas [31], even though it did not generalize to any other racial/ethnic group. This finding and the generalization of several other loci to Hispanic/Latinas may be due to their European admixture [58], or perhaps a less similar genetic architecture of reproductive traits between European and the other race/ethnic groups analyzed herein. Using the densely genotyped regions of the MetaboChip, we also demonstrated how diverse samples can help identify potential independent signals and putative variants/regions of interest for future functional follow up (S5 Table). For example, we also observed evidence that the MCM8 region may harbor two independent signals for ANM in Hispanic/Latinas [23]. Yet, the role of ancestral differences or environmental exposures/interactions in the observed findings warrants further research [25].
Lastly, we also observed that our Bonferroni-significant AAM associations were in moderate to strong LD (r2>0.2; Fig 1) with the previously reported putative variants of the primary association signals at BDNF and FTO (Fig 1B and 1C). Previously, secondary signals have not been described at SEC16B for BMI [52], and the secondary signal we observe with AAM at BDNF appears to be distinct from BMI secondary signals based on our trans-ethnic LD estimates (r2<0.2; Fig 1A). These findings suggest that the study of AAM may yield additional insights into the genetic architecture of growth and development than studying BMI alone. The co-localization of genetic signals further supports the shared genetic underpinnings of early life growth and development in both females and males using various methodologies [59–63]. A recent study highlighted the extent of overlapping genetic loci involved in these interrelated traits, observing that the genetics of age at first birth positively correlated with the genetics of birth weight, AAM and age at voice breaking, and negatively with the genetics of smoking, BMI and ANM [64]. Even though we did not data necessary to disentangle the genetic effects on AAM and BMI in early and late life in this current PAGE Study, an increasing body of work suggests that early life growth can influence both puberty and downstream cardiometabolic consequences [22]. Future trans-ethnic research should leverage longitudinal data or casual inference methods, when attempting to further decompose the complex relationship between the genetics of growth and development across the life course.
Conclusions
Our study is the first trans-ethnic analysis of female reproductive traits to our knowledge. Future trans-ethnic meta-analyses should include large, diverse samples with dense genotyping 1) to fine-map the reproductive trait association signals described herein, 2) to examine the joint role of functional genetic variants and environmental risk factors, and 3) to describe genetic risk factors for extreme AAM or ANM and predict their effects on the reproductive windows of women of diverse race/ethnic groups. Our findings provide support for the relevance of multiple reproductive loci to racially/ethnically diverse groups of women, and the presence of a complex genetic architecture underpinning female growth and development across the life course.
Supporting information
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Acknowledgments
The authors gratefully acknowledge Dr. Ben Voight for sharing the MetaboChip SNP linkage disequilibrium and minor allele frequency statistics estimated in the Malmö Diet and Cancer Study. The PAGE Study thanks the staff and participants of all PAGE studies for their important contributions.
Data Availability
The datasets generated as part of the PAGE study can be accessed through the dbGaP repository (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000356).
Funding Statement
LFR was supported by the Cardiovascular Disease Epidemiology Training Grant from the National Heart, Lung, and Blood Institute (T32HL007055), from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (T32-HD007168), and the Carolina Population Center (P2C-HD050924). SFC was supported by the American Heart Association Go Red for Women Strategically Focused Research Network Grant 16SFRN27940007. KEN was supported by R01-DK089256; 2R01HD057194; U01HG007416; R01DK101855, and AHA grant 13GRNT16490017. The Population Architecture Using Genomics and Epidemiology (PAGE) program was funded by the National Human Genome Research Institute (NHGRI), supported by U01HG004803 (CALiCo), U01HG004798 (EAGLE), U01HG004802 (MEC), U01HG004790 (WHI), and U01HG004801 (Coordinating Center), and their respective NHGRI ARRA supplements. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. The complete list of PAGE members can be found at PAGE website (http://www.pagestudy.org). The data and materials included in this report result from a collaboration between the following studies: The "Epidemiologic Architecture for Genes Linked to Environment (EAGLE)" was funded through the NHGRI PAGE program (U01HG004798 and its NHGRI ARRA supplement). The dataset(s) used for the analyses described were obtained from Vanderbilt University Medical Center’s BioVU which was supported by institutional funding and by the Vanderbilt CTSA grant UL1 TR000445 from NCATS/NIH. The Vanderbilt University Center for Human Genetics Research, Computational Genomics Core provided computational and/or analytical support for this work. The Multiethnic Cohort study (MEC) characterization of epidemiological architecture was funded through NHGRI (HG004802, and HG007397) and the NHGRI PAGE program (U01HG004802 and its NHGRI ARRA supplement). The MEC study was funded through the National Cancer Institute (CA164973, R37CA54281, R01 CA 063464, P01CA33619, U01CA136792, and U01CA98758). Funding support for the “Epidemiology of putative genetic variants: The Women’s Health Initiative” study was provided through the NHGRI PAGE program (U01HG004790 and its NHGRI ARRA supplement). The WHI program was funded by the National Heart, Lung, and Blood Institute; NIH; and U.S. Department of Health and Human Services through contracts N01WH22110, 24152, 32100-2, 32105-6, 32108-9, 32111-13, 32115, 32118-32119, 32122, 42107-26, 42129-32, and 44221. Funding support for the Genetic Epidemiology of Causal Variants Across the Life Course (CALiCo) program was provided through the NHGRI PAGE program (U01HG004803 and its NHGRI ARRA supplement). The following studies contributed to this manuscript and were funded by the following agencies: The Atherosclerosis Risk in Communities Study (ARIC) was carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C), R01HL087641, R01HL59367 and R01HL086694; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. Infrastructure partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research. The Coronary Artery Risk Development in Young Adults Study (CARDIA) is supported by contracts HHSN268201300025C, HHSN268201300026C, HHSN268201300027C, HHSN268201300028C, HHSN268201300029C, and HHSN268200900041C from the National Heart, Lung, and Blood Institute (NHLBI), the Intramural Research Program of the National Institute on Aging (NIA), and an intra agency agreement between NIA and NHLBI (AG0005). The Hispanic Community Health Study/Study of Latinos (SOL) was carried out as a collaborative study supported by contracts from the National Heart, Lung, and Blood Institute (NHLBI) to the University of North Carolina (N01-HC65233), University of Miami (N01-HC65234), Albert Einstein College of Medicine (N01-HC65235), Northwestern University (N01-HC65236), and San Diego State University (N01- HC65237). Additional support was provided by 1R01DK101855-01 and 13GRNT16490017. The following Institutes/Centers/Offices contributed to the HCHS/SOL through a transfer of funds to the NHLBI: National Center on Minority Health and Health Disparities, the National Institute of Deafness and Other Communications Disorders, the National Institute of Dental and Craniofacial Research, the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute of Neurological Disorders and Stroke, and the Office of Dietary Supplements. The Mount Sinai BioMe Biobank was supported by The Andrea and Charles Bronfman Philanthropies. The Hypertension Genetic Epidemiology Network (HyperGEN) study was supported by National Heart, Lung, and Blood Institute contracts HL086694 and HL055673. MESA and the MESA SHARe project are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts HHSN268201500003I, N01- HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC- 95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC- 95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420, UL1-TR-001881, and DK063491. MetaboChip genotyping data was supported in part by grants and contracts R01HL98077, N02-HL-64278, HL071205, UL1TR001881, DK063491, RD831697, and P50 ES015915. Additional support was provided by MESA Family, which is funded by grants and contracts R01HL071051, R01HL071205, R01HL071250, R01HL071251, R01HL071258, R01HL071259, by the National Center for Research Resources, Grant UL1RR033176, and the National Center for Advancing Translational Sciences, Grant UL1TR001881. Although the research described in this manuscript has been funded in part by the United States Environmental Protection Agency through RD831697 to the University of Washington, it has not been subjected to the Agency’s required peer and policy review and therefore does not necessarily reflect the views of the Agency and no official endorsement should be inferred. Assistance with phenotype harmonization, SNP selection and annotation, data cleaning, data management, integration and dissemination, and general study coordination was provided by the PAGE Coordinating Center (U01HG004801-01 and its NHGRI ARRA supplement). The National Institutes of Mental Health also contributed support for the Coordinating Center. Contributions made by JRM to this manuscript were performed while as a graduate student at Vanderbilt University. Write InSciTe, LLC, did not play a role in the study design and data analysis of this work. Write InSciTe provided support to JRM in the form of salary at the time of manuscript preparation and revision, but did not play any other role in the study design, data collection and analysis, decisions to publish or about the specific content of the manuscript. Analytic contributions made by JMJ to this manuscript were performed as a Postdoctoral Research Fellow at the Icahn School of Medicine at Mount Sinai. Illumina Inc. provided support in the form of salary for JMJ at the time of manuscript revisions, but Illumina did not play a role in the study design, data collection and analysis, decision to publish or the preparation of the manuscript. The specific roles of these authors are articulated in the ‘author contributions’ section. NHGRI collaborators (LH) assisted in the study design, analysis, and preparation of the manuscript. All other funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.McDowell MA, Brody DJ, Hughes JP (2007) Has age at menarche changed? Results from the National Health and Nutrition Examination Survey (NHANES) 1999–2004. J Adolesc Health 40: 227–231. 10.1016/j.jadohealth.2006.10.002 [DOI] [PubMed] [Google Scholar]
- 2.Buttke DE, Sircar K, Martin C (2012) Exposures to endocrine-disrupting chemicals and age of menarche in adolescent girls in NHANES (2003–2008). Environ Health Perspect 120: 1613–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGuinn LA, Ghazarian AA, Joseph Su L, Ellison GL (2015) Urinary bisphenol A and age at menarche among adolescent girls: evidence from NHANES 2003–2010. Environ Res 136: 381–386. 10.1016/j.envres.2014.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canoy D, Beral V, Balkwill A, Wright FL, Kroll ME, Reeves GK, et al. (2015) Age at menarche and risks of coronary heart and other vascular diseases in a large UK cohort. Circulation 131: 237–244. 10.1161/CIRCULATIONAHA.114.010070 [DOI] [PubMed] [Google Scholar]
- 5.Bodicoat DH, Schoemaker MJ, Jones ME, McFadden E, Griffin J, Ashworth A, et al. (2014) Timing of pubertal stages and breast cancer risk: the Breakthrough Generations Study. Breast Cancer Res 16: R18 10.1186/bcr3613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ali AT (2014) Reproductive factors and the risk of endometrial cancer. Int J Gynecol Cancer 24: 384–393. 10.1097/IGC.0000000000000075 [DOI] [PubMed] [Google Scholar]
- 7.Gold EB (2011) The timing of the age at which natural menopause occurs. Obstet Gynecol Clin North Am 38: 425–440. 10.1016/j.ogc.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gold EB, Bromberger J, Crawford S, Samuels S, Greendale GA, Harlow SD, et al. (2001) Factors associated with age at natural menopause in a multiethnic sample of midlife women. Am J Epidemiol 153: 865–874. [DOI] [PubMed] [Google Scholar]
- 9.Henderson KD, Bernstein L, Henderson B, Kolonel L, Pike MC (2008) Predictors of the timing of natural menopause in the Multiethnic Cohort Study. Am J Epidemiol 167: 1287–1294. 10.1093/aje/kwn046 [DOI] [PubMed] [Google Scholar]
- 10.Campbell Jenkins BW, Addison C, Wilson G, Liu J, Fortune M, Robinson K, et al. (2011) Association of the joint effect of menopause and hormone replacement therapy and cancer in African American women: the Jackson Heart Study. Int J Environ Res Public Health 8: 2491–2504. 10.3390/ijerph8062491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kallen AN, Pal L (2011) Cardiovascular disease and ovarian function. Curr Opin Obstet Gynecol 23: 258–267. 10.1097/GCO.0b013e3283488a21 [DOI] [PubMed] [Google Scholar]
- 12.Nichols HB, Trentham-Dietz A, Hampton JM, Titus-Ernstoff L, Egan KM, Willett WC, et al. (2006) From menarche to menopause: trends among US Women born from 1912 to 1969. Am J Epidemiol 164: 1003–1011. 10.1093/aje/kwj282 [DOI] [PubMed] [Google Scholar]
- 13.Elks CE, Perry JR, Sulem P, Chasman DI, Franceschini N, He C, et al. (2010) Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat Genet 42: 1077–1085. 10.1038/ng.714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ong KK, Elks CE, Li S, Zhao JH, Luan J, Andersen LB, et al. (2009) Genetic variation in LIN28B is associated with the timing of puberty. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He C, Kraft P, Chen C, Buring JE, Pare G, Hankinson SE, et al. (2009) Genome-wide association studies identify loci associated with age at menarche and age at natural menopause. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He C, Zhang C, Hunter DJ, Hankinson SE, Buck Louis GM, Hediger ML, et al. (2010) Age at menarche and risk of type 2 diabetes: results from 2 large prospective cohort studies. Am J Epidemiol 171: 334–344. 10.1093/aje/kwp372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perry JR, Stolk L, Franceschini N, Lunetta KL, Zhai G, McArdle PF, et al. (2009) Meta-analysis of genome-wide association data identifies two loci influencing age at menarche. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stolk L, Perry JR, Chasman DI, He C, Mangino M, Sulem P, et al. (2012) Meta-analyses identify 13 loci associated with age at menopause and highlight DNA repair and immune pathways. Nat Genet 44: 260–268. 10.1038/ng.1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stolk L, Zhai G, van Meurs JB, Verbiest MM, Visser JA, Estrada K, et al. (2009) Loci at chromosomes 13, 19 and 20 influence age at natural menopause. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perry JR, Day F, Elks CE, Sulem P, Thompson DJ, Ferreira T, et al. (2014) Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature 514: 92–97. 10.1038/nature13545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu YZ, Guo YF, Wang L, Tan LJ, Liu XG, Pei YF, et al. (2009) Genome-wide association analyses identify SPOCK as a key novel gene underlying age at menarche. PLoS Genet 5: e1000420 10.1371/journal.pgen.1000420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day FR, Thompson DJ, Helgason H, Chasman DI, Finucane H, Sulem P, et al. (2017) Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day FR, Ruth KS, Thompson DJ, Lunetta KL, Pervjakova N, Chasman DI, et al. (2015) Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat Genet 47: 1294–1303. 10.1038/ng.3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demerath EW, Liu CT, Franceschini N, Chen G, Palmer JR, Smith EN, et al. (2013) Genome-wide association study of age at menarche in African-American women. Hum Mol Genet 22: 3329–3346. 10.1093/hmg/ddt181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen CT, Liu CT, Chen GK, Andrews JS, Arnold AM, Dreyfus J, et al. (2014) Meta-analysis of loci associated with age at natural menopause in African-American women. Hum Mol Genet 23: 3327–3342. 10.1093/hmg/ddu041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanikawa C, Okada Y, Takahashi A, Oda K, Kamatani N, Kubo M, et al. (2013) Genome wide association study of age at menarche in the Japanese population. PLoS One 8: e63821 10.1371/journal.pone.0063821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pyun JA, Kim S, Cho NH, Koh I, Lee JY, Shin C, et al. (2014) Genome-wide association studies and epistasis analyses of candidate genes related to age at menarche and age at natural menopause in a Korean population. Menopause 21: 522–529. 10.1097/GME.0b013e3182a433f7 [DOI] [PubMed] [Google Scholar]
- 28.Shi J, Zhang B, Choi JY, Gao YT, Li H, Lu W, et al. (2016) Age at menarche and age at natural menopause in East Asian women: a genome-wide association study. Age (Dordr) 38: 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spencer KL, Malinowski J, Carty CL, Franceschini N, Fernandez-Rhodes L, Young A, et al. (2013) Genetic variation and reproductive timing: African American women from the Population Architecture using Genomics and Epidemiology (PAGE) Study. PLoS One 8: e55258 10.1371/journal.pone.0055258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carty CL, Spencer KL, Setiawan VW, Fernandez-Rhodes L, Malinowski J, Buyske S, et al. (2013) Replication of genetic loci for ages at menarche and menopause in the multi-ethnic Population Architecture using Genomics and Epidemiology (PAGE) study. Human Reproduction 28: 1695–1706. 10.1093/humrep/det071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen CT, Fernandez-Rhodes L, Brzyski RG, Carlson CS, Chen Z, Heiss G, et al. (2011) Replication of loci influencing ages at menarche and menopause in Hispanic women: the Women’s Health Initiative SHARe Study. Hum Mol Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris AP (2011) Transethnic meta-analysis of genomewide association studies. Genet Epidemiol 35: 809–822. 10.1002/gepi.20630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zakharov S, Wang X, Liu J, Teo YY (2015) Improving power for robust trans-ethnic meta-analysis of rare and low-frequency variants with a partitioning approach. Eur J Hum Genet 23: 238–244. 10.1038/ejhg.2014.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matise TC, Ambite JL, Buyske S, Carlson CS, Cole SA, Crawford DC, et al. (2011) The Next PAGE in understanding complex traits: design for the analysis of Population Architecture Using Genetics and Epidemiology (PAGE) Study. Am J Epidemiol 174: 849–859. 10.1093/aje/kwr160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voight BF, Kang HM, Ding J, Palmer CD, Sidore C, Chines PS, et al. (2012) The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet 8: e1002793 10.1371/journal.pgen.1002793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pulley J, Clayton E, Bernard GR, Roden DM, Masys DR (2010) Principles of human subjects protections applied in an opt-out, de-identified biobank. Clin Transl Sci 3: 42–48. 10.1111/j.1752-8062.2010.00175.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR, et al. (2008) Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther 84: 362–369. 10.1038/clpt.2008.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malinowski J, Farber-Eger E, Crawford DC (2014) Development of a data-mining algorithm to identify ages at reproductive milestones in electronic medical records. Pac Symp Biocomput: 376–387. [PMC free article] [PubMed] [Google Scholar]
- 39.Liu EY, Buyske S, Aragaki AK, Peters U, Boerwinkle E, Carlson C, et al. (2012) Genotype imputation of Metabochip SNPs using a study-specific reference panel of ~4,000 haplotypes in African Americans from the Women’s Health Initiative. Genet Epidemiol 36: 107–117. 10.1002/gepi.21603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weale ME (2010) Quality control for genome-wide association studies. Methods Mol Biol 628: 341–372. 10.1007/978-1-60327-367-1_19 [DOI] [PubMed] [Google Scholar]
- 41.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin DY, Tao R, Kalsbeek WD, Zeng DL, Gonzalez F, Fernandez-Rhodes L, et al. (2014) Genetic Association Analysis under Complex Survey Sampling: The Hispanic Community Health Study/Study of Latinos. Am J Hum Genet 95: 675–688. 10.1016/j.ajhg.2014.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen MH, Yang Q (2010) GWAF: an R package for genome-wide association analyses with family data. Bioinformatics 26: 580–581. 10.1093/bioinformatics/btp710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patterson N, Price AL, Reich D (2006) Population structure and eigenanalysis. PLoS Genet 2: e190 10.1371/journal.pgen.0020190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909. 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- 46.Buyske S, Wu Y, Carty CL, Cheng I, Assimes TL, Dumitrescu L, et al. (2012) Evaluation of the metabochip genotyping array in African Americans and implications for fine mapping of GWAS-identified loci: the PAGE study. PLoS One 7: e35651 10.1371/journal.pone.0035651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26: 2190–2191. 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han B, Eskin E (2011) Random-Effects Model Aimed at Discovering Associations in Meta-Analysis of Genome-wide Association Studies. Am J Hum Genet 88: 586–598. 10.1016/j.ajhg.2011.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Lee SH, Goddard ME, Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88: 76–82. 10.1016/j.ajhg.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang J, Ferreira T, Morris AP, Medland SE, Madden PA, Heath AC, et al. (2012) Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 44: 369–375, S361-363. 10.1038/ng.2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernandez-Rhodes L, Gong J, Haessler J, Franceschini N, Graff M, Nishimura KK, et al. (2017) Trans-ethnic fine-mapping of genetic loci for body mass index in the diverse ancestral populations of the Population Architecture using Genomics and Epidemiology (PAGE) Study reveals evidence for multiple signals at established loci. Hum Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. (2015) Genetic studies of body mass index yield new insights for obesity biology. Nature 518: 197–206. 10.1038/nature14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee JY, Lee BS, Shin DJ, Woo Park K, Shin YA, Joong Kim K, et al. (2013) A genome-wide association study of a coronary artery disease risk variant. J Hum Genet 58: 120–126. 10.1038/jhg.2012.124 [DOI] [PubMed] [Google Scholar]
- 54.Huang J, Ellinghaus D, Franke A, Howie B, Li Y (2012) 1000 Genomes-based imputation identifies novel and refined associations for the Wellcome Trust Case Control Consortium phase 1 Data. Eur J Hum Genet 20: 801–805. 10.1038/ejhg.2012.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu L, Ebana Y, Nitta JI, Takahashi Y, Miyazaki S, Tanaka T, et al. (2017) Genetic Variants Associated With Susceptibility to Atrial Fibrillation in a Japanese Population. Can J Cardiol 33: 443–449. 10.1016/j.cjca.2016.10.029 [DOI] [PubMed] [Google Scholar]
- 56.Steiner M, Dunn E, Born L (2003) Hormones and mood: from menarche to menopause and beyond. J Affect Disord 74: 67–83. [DOI] [PubMed] [Google Scholar]
- 57.Global Lipids Genetics C, Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, et al. (2013) Discovery and refinement of loci associated with lipid levels. Nat Genet 45: 1274–1283. 10.1038/ng.2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Conomos MP, Laurie CA, Stilp AM, Gogarten SM, McHugh CP, Nelson SC, et al. (2016) Genetic Diversity and Association Studies in US Hispanic/Latino Populations: Applications in the Hispanic Community Health Study/Study of Latinos. Am J Hum Genet 98: 165–184. 10.1016/j.ajhg.2015.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang W, Zhao LJ, Liu YZ, Recker RR, Deng HW (2006) Genetic and environmental correlations between obesity phenotypes and age at menarche. Int J Obes (Lond) 30: 1595–1600. [DOI] [PubMed] [Google Scholar]
- 60.Fernandez-Rhodes L, Demerath EW, Cousminer DL, Tao R, Dreyfus JG, Esko T, et al. (2013) Association of adiposity genetic variants with menarche timing in 92,105 women of European descent. Am J Epidemiol 178: 451–460. 10.1093/aje/kws473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cousminer DL, Stergiakouli E, Berry DJ, Ang W, Groen-Blokhuis MM, Korner A, et al. (2014) Genome-wide association study of sexual maturation in males and females highlights a role for body mass and menarche loci in male puberty. Hum Mol Genet 23: 4452–4464. 10.1093/hmg/ddu150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Day FR, Bulik-Sullivan B, Hinds DA, Finucane HK, Murabito JM, Tung JY, et al. (2015) Shared genetic aetiology of puberty timing between sexes and with health-related outcomes. Nat Commun 6: 8842 10.1038/ncomms9842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. (2015) An atlas of genetic correlations across human diseases and traits. Nat Genet 47: 1236–1241. 10.1038/ng.3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barban N, Jansen R, de Vlaming R, Vaez A, Mandemakers JJ, Tropf FC, et al. (2016) Genome-wide analysis identifies 12 loci influencing human reproductive behavior. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Data Availability Statement
The datasets generated as part of the PAGE study can be accessed through the dbGaP repository (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000356).