

Human β-defensin 2 permeabilizes and kills fungal cells by specifically targeting the important plasma membrane lipid PIP2.
Abstract
Human defensins belong to a subfamily of the cationic antimicrobial peptides and act as a first line of defense against invading microbes. Their often broad-spectrum antimicrobial and antitumor activities make them attractive for therapeutic development; however, their precise molecular mechanism(s) of action remains to be defined. We show that human β-defensin 2 (HBD-2) permeabilizes Candida albicans cell membranes via a mechanism targeting the plasma membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2). We determined the structure of HBD-2 bound to PIP2, which revealed two distinct PIP2-binding sites, and showed, using functional assays, that mutations in these sites ablate PIP2-mediated fungal growth inhibition by HBD-2. Our study provides the first insight into lipid-mediated human defensin membrane permeabilization at an atomic level and reveals a unique mode of lipid engagement to permeabilize cell membranes.
INTRODUCTION
Cationic antimicrobial peptides (CAPs) are integral host innate defense molecules in the protection against invading pathogens and often display potent broad-spectrum antimicrobial activity (1, 2). Defensins are a large subfamily of CAPs and are ubiquitous in plants, fungi, invertebrates, and vertebrates. Human β-defensin 2 (HBD-2) is widely expressed by epithelial cells (3, 4) and is often up-regulated as a direct result of microbial activity or as part of the inflammatory response (5). HBD-2 exhibits broad-spectrum antimicrobial capabilities (3, 6), lyses anionic liposomes (7), and permeabilizes cell membranes of Escherichia coli (8). However, the precise mechanism of action for HBD-2–mediated permeabilization of membranes, including the identity and role of specific ligands, remains to be defined.
RESULTS
To define the mechanism of membrane permeabilization of HBD-2, we first confirmed that HBD-2 inhibits the growth of the human fungal pathogen Candida albicans at low micromolar concentrations (Fig. 1A). Flow cytometry analysis of propidium iodide (PI) uptake assays revealed significant PI uptake by 40% of the fungal cell population within 2.5 min, followed by rapid cell fragmentation over the course of 30 min at which point 65% of the cells were permeabilized (Fig. 1B).
Fig. 1. HBD-2 kills C. albicans via rapid induction of cell permeabilization and fragmentation.
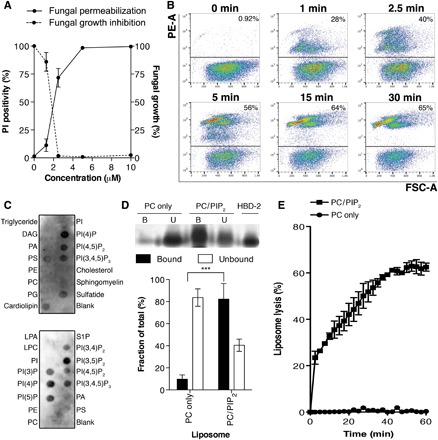
(A) Dose-dependent cell permeabilization and growth inhibition activities of HBD-2 on C. albicans, measured by PI uptake assay and fungal growth inhibition assay, respectively. Data are means ± SEM of three independent experiments, each in triplicate. (B) Time-course PI uptake assay. Data are representatives of three independent experiments. PE-A, phycoerythrin area; FSC-A, forward scatter area. (C) HBD-2 preferentially binds PIP2 and lyses liposomes. Immunodetection of lipid binding by HBD-2 using protein-lipid overlay assays is shown. Relative binding intensity was determined by densitometry analysis of chemiluminescence signals. DAG, diacylglycerol; PA, phosphatidic acid; PS, phosphatidylserine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; S1P, sphingosine 1-phosphate; PI, phosphatidylinositol; PI(3)P, phosphatidylinositol 3-phosphate; PI(4)P, phosphatidylinositol 4-phosphate; PI(5)P, phosphatidylinositol 5-phosphate; PI(3,4)P2, phosphatidylinositol 3,4-bisphosphate; PI(3,5)P2, phosphatidylinositol 3,5-bisphosphate; PI(4,5)P2, phosphatidylinositol 4,5-bisphosphate; PI(3,4,5)P3, phosphatidylinositol 3,4,5-trisphosphate. (D) Liposome pulldown assay. Unilamellar liposomes containing PC only or PC/PIP2 (95:5 molar ratio) were incubated with 1 μg of HBD-2, followed by centrifugation to separate supernatant (unbound fraction) and pellet (bound fraction) before SDS–polyacrylamide gel electrophoresis (PAGE) analysis and silver staining. Protein band intensity was quantified densitometrically using ImageJ software and normalized against HBD-2 loading control. (E) Liposome lysis by 50 μM HBD-2 using calcein-encapsulated liposomes. Liposome lysis was normalized against Triton X-100 treatment. Data in (C) and (D) are means ± SEM of at least three independent experiments. ***P < 0.001, unpaired t test.
We have previously established that plant defensins targeting phosphatidylinositol 4,5-bisphosphate (PIP2) exhibit potent antimicrobial and antitumor activities (9–13). Furthermore, we have also shown that HBD-3 binds PIP2 to mediate tumor cell permeabilization (14). Therefore, we examined the ability of HBD-2 to bind a range of phospholipids as part of its membrane-permeabilizing activity. Protein-lipid overlay assays revealed that HBD-2 bound all the major cell membrane phosphatidylinositol lipids including PIP2 (Fig. 1C). Using liposome pulldown assays, we then established that HBD-2 binds to liposomes enriched in PIP2 but not to phosphatidylcholine (PC)–only liposomes (Fig. 1D). HBD-2 readily lysed PIP2-enriched unilamellar liposomes but had no effect on PC-only liposomes, as measured by calcein release assays (Fig. 1E), suggesting that the ability of HBD-2 to bind PIP2 is crucial for its ability to permeabilize membranes.
To understand the structural basis of PIP2 interaction with HBD-2 and its role in fungal cell permeabilization and fragmentation, we crystallized HBD-2 in complex with PIP2. The HBD-2–PIP2 complex was solved by molecular replacement using monomeric HBD-2 as a search model (7) and was refined to a resolution of 1.85 Å (Table 1). Two HBD-2 monomers and two PIP2 molecules were identified in the asymmetric unit (Fig. 2A), with Fo-Fc difference maps allowing for unambiguous placement of both PIP2 molecules (Fig. 3A). The two PIP2 molecules are bound to HBD-2 in two distinct binding sites (designated site 1 and site 2). Site 1 is located vertically between two symmetry-related dimers (chain [A1,B1] and chain [A2,B2]) (Fig. 2B), and the phosphates of the bound PIP2 molecule form a network of ionic interactions and hydrogen bonds with K10, K25, R22, as well as the backbone of R23, from three different HBD-2 chains. Specifically, the 1′-phosphate engages K10 from chain [A1] and R22 from chain [B1], and the 5′-phosphate group engages K25 and the backbone of R23 from chain [A2] (Figs. 2C and 3B). In contrast, site 2 is positioned perpendicular to site 1 and creates a layer of PIP2 molecules between individual rows of HBD-2 dimers (Fig. 2B). Here, the 1′-phosphate group of PIP2 engages K36 and the backbone of G34 of chain [B1], and the 5′-phosphate engages the backbone of G34 from chain [A3] (Figs. 2D and 3B). Both PIP2 molecules are also involved in a substantial number of hydrophobic contacts with numerous side chains via their acyl chains (Fig. 3B). Compared to the crystal structure of apo–HBD-2 (7), the PIP2-bound HBD-2 dimer is rotated around its dimer axis. Instead of two hydrogen bonds between the backbones of C15, the interface comprises two hydrogen bonds between H16 and the backbone nitrogen of C15 (Fig. 4, A and B). Analysis of the electrostatic surface interface of the PIP2-bound dimer revealed two distinct interaction surfaces, one that is highly charged and flat, with the other side being hydrophobic and featuring a groove (Fig. 3C). The headgroup of PIP2 bound to site 1 is sandwiched between two dimers on the positively charged side of HBD-2, whereas acyl chains of PIP2 in site 2 are lodged in the hydrophobic “grip” on the opposite side, creating a continuous PIP2 layer throughout the crystal (Fig. 3D). The change in dimer formation from apo–HBD-2 to PIP2-bound HBD-2 results in the surface exposure of several hydrophobic residues, including P5, V6, L9, and L32, that were previously buried in the apo–HBD-2 dimer (Fig. 4, C and D), enabling these residues to interface with the acyl chain of the bound PIP2. This change results in an exposure of an additional 194 Å2 of hydrophobic surface area that appears to be driven by the need to partially sequester hydrophobic elements of PIP2, and we speculate that this would be assisted by the presence of a membrane bilayer during the initial HBD-2–PIP2 encounter. While we used shorter-chain PIP2 molecules with fully saturated C8 acyl chains for the crystallization of the HBD-2–PIP2 complex, the PIP2-binding configuration we observed is expected to be representative of complexes with longer and partly unsaturated acyl chains, since the contact region between HBD-2 and PIP2 only involves the first four acyl chain bonds.
Table 1. Data collection and refinement statistics.
PDB, Protein Data Bank.
| HBD-2–PIP2 | |
| PDB ID | 6CS9 |
| Data collection | |
| Space group | P21 |
| No. of molecules in asymmetric unit |
2 + 2 |
| Cell dimensions | |
| a, b, c (Å) | 32.87, 25.54, 40.17 |
| α, β, γ (°) | 90.00, 98.64, 90.00 |
| Wavelength (Å) | 0.9537 |
| Resolution (Å)* | 39.71–1.85 (1.89–1.85) |
| Rsym or Rmerge* | 0.057 (0.639) |
| I/σI* | 10.5 (1.1) |
| CC(1/2) | 0.997 (0.509) |
| Completeness (%)* | 98.4 (87.0) |
| Redundancy* | 3.5 (2.5) |
| Wilson B-factor (Å2) | |
| Refinement | |
| Resolution (Å) | 39.71–1.85 |
| No. of reflections | 5693 |
| Rwork/Rfree | 0.1947/0.2267 |
| No. of nonhydrogen atoms | |
| Protein | 605 |
| PIP2 | 94 |
| Water | 28 |
| B-factors | |
| Protein | 33.86 |
| PIP2 | 48.87 |
| Water | 42.89 |
| Root-mean-square deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angle (°) | 0.809 |
| Ramachandran plot (%) | |
| Favored | 94.87 |
| Allowed | 5.13 |
| Disallowed | 0.00 |
*Values in parentheses are for highest-resolution shell.
Fig. 2. Crystal structure of HBD-2 in complex with PIP2.
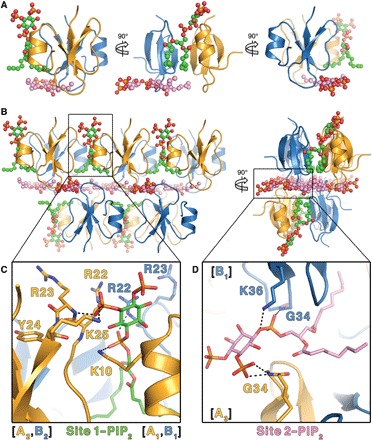
(A) The asymmetric unit comprises two HBD-2 molecules (cartoon representation; chain A in gold and chain B in blue) and two PIP2 molecules (ball and stick representations; oxygen in red, phosphorus in orange, and carbon in green for site 1 and pink for site 2). (B) Crystal packing of HBD-2 represented as in (A) as seen from the front (left) and from the side (right). (C) Close-up of site 1 represented as above, with PIP2 and all interacting residues shown as sticks. (D) Close-up of site 2 represented as above, with PIP2 and all interacting residues shown as sticks.
Fig. 3. Molecular details of PIP2 binding and electrostatic surface of HBD-2–PIP2 complex.
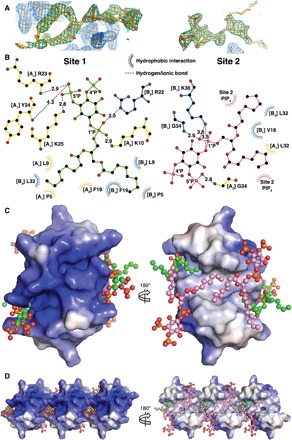
(A) Omit maps of the PIP2 binding sites. The maps were generated by rerunning the final structure refinement with both lipid molecules removed from the model. The Fo-Fc maps are contoured in PyMOL at 2 sigma (green), while the 2Fo-Fc maps are contoured at 1 sigma (blue). The 2Fo-Fc maps around the PIP2 molecules are omitted for clarity. (B) Schematic representation of PIP2 site 1 (left) and site 2 (right). Residues near the binding site are depicted as sticks and dashed lines for ionic and hydrogen bonds and as eyelashes for hydrophobic interactions. (C) Qualitative electrostatic surface representation (blue is positive charge, red is negative charge, and white is uncharged or hydrophobic) of the HBD-2–PIP2 dimer form viewed from the top (left) and from the bottom (right). PIP2 molecules are shown as balls and sticks, with oxygen in red, phosphorus in orange, and carbon in green for site 1 and in pink for site 2. (D) Qualitative electrostatic surface representation of three dimers in the crystal, depicted as in (B), highlighting the site 1 binding pocket (left) and the continuous hydrophobic network of site 2 PIP2 molecules (right).
Fig. 4. Dimers of HBD-2 in the PIP2 bound and unbound state.
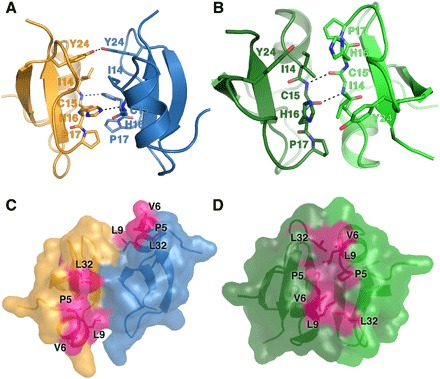
The dimer interfaces of HBD-2 are shown as cartoons, with residues 14 to 17 and 24 depicted as sticks and hydrogen bonds as dashed lines. (A) The PIP2 -bound HBD-2 dimer with chain A in gold and chain B in blue (PIP2 molecules are omitted for clarity). Two hydrogen bonds between H16 and the backbone of C15 are observed, as well as between the hydroxyl groups of Y24. (B) Apo–HBD-2 dimer with its two chains colored light green and dark green. Two hydrogen bonds between the backbones of C15 are shown. (C) Surface representation of (A). Exposed hydrophobic residues are shaded in pink. (D) Surface representation of (B). Exposed hydrophobic residues are shaded in pink.
Our structure of HDB-2 bound to PIP2 suggests that a dimer of HBD-2 is required for successful PIP2 binding. However, considering that the previously determined crystal structure of apo–HBD-2 revealed a different dimeric configuration, we examined the oligomeric state of HBD-2 in solution using small-angle x-ray scattering (SAXS) combined with in-line size exclusion chromatography (SEC) (fig. S1A and table S1). These data revealed that at a concentration of 5 mg/ml, HBD-2 elutes as a broad peak featuring a constant variation of Rg, indicative of the presence of both monomers and dimers of HBD-2. Scattering data extracted from the peak were of high quality, as shown by a Guinier plot (fig. S1, B and D), and indicate a monomer/dimer ratio of 70:30 at pH 7.5 (fig. S1C), based on the molecular weight calculated from the forward scattering intensity [I(0)] (15). HBD-2 has been found to exist as a mixture of aggregated molecules (mostly dimers) using dynamic light scattering at 30 mg/ml (7), suggesting that multimerization may be concentration-dependent.
To examine the role of both PIP2-binding sites for the ability of HBD-2 to kill fungal cells, we generated a panel of six mutants that specifically targeted key HBD-2–PIP2 interactions in both PIP2-binding sites and evaluated the ability of each mutant in C. albicans growth inhibition assays. While wild-type HBD-2 inhibited C. albicans growth at a half maximal inhibitory concentration (IC50) of 2.2 ± 0.2 μM, the site 1 HBD-2 mutants K10A, R22A, R23A, and K25A displayed higher IC50 values (3.0 ± 0.3, 5.5 ± 0.2, 3.0 ± 0.1, and 4.0 ± 0.4 μM, respectively), whereas the site 2 HBD-2 mutant K36A had no effect on fungal growth up to 10 μM (Fig. 5, A and B). We also generated an alanine mutation of a residue in the β1-β2 loop that does not contact PIP2 via its side chain Y24A as a control mutant, which displayed activity comparable to the wild type (2.1 ± 0.2 μM) (Fig. 5, A and B). Furthermore, PI uptake assays revealed a decrease in fungal cell permeabilization for HBD-2 mutants K10, R22A, and K25A and a complete lack of permeabilization for K36A, whereas Y24A and R23A demonstrated no significant difference compared to wild type (Fig. 5C). In addition, reduced and alkylated HBD-2 [HBD-2(R&A)] was not able to permeabilize C. albicans (fig. S2, A and C), suggesting that the three-dimensional structure of HBD-2 is critical for antifungal activity.
Fig. 5. Effects of HBD-2 mutation on fungal cell killing, PIP2 binding, and liposomal lysis.
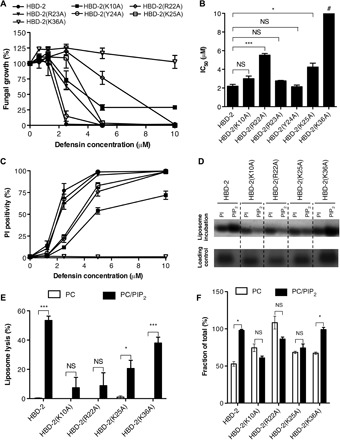
Data are means ± SEM of three independent experiments. NS, not significant. *P < 0.05, **P < 0.01, and ***P < 0.001, unpaired t test. (A) Fungal growth inhibition by HBD-2 and mutants. C. albicans was incubated with titrations of HBD-2 or mutant for 24 hours, followed by OD600 (optical density at 600 nm) measurement to determine fungal growth. (B) IC50 values for the fungal growth inhibitory effect of HBD-2 and mutants. #The value for HBD-2(K36A) could not be determined within the tested range of 0 to 10 μM and designated as greater than 10 μM. (C) Fungal permeabilization by HBD-2 and mutants. C. albicans was treated with HBD-2 or mutants at indicated concentrations for 1 hour, followed by the addition of PI before flow cytometry analysis. (D) Liposome pulldown assay. Unilamellar liposomes containing PC only or PC/PIP2 (95:5 molar ratio) were incubated with 1 μg of HBD-2 or mutants, followed centrifugation to collect pellets (bound fraction) before SDS-PAGE analysis and silver staining. (E) Protein band intensity of (D) was quantified densitometrically using ImageJ software and normalized against corresponding protein loading control. (F) Liposome lysis by 50 μM HBD-2 or mutants using calcein-encapsulated liposomes. Lyposome lysis was normalized against Triton X-100 treatment.
We then performed liposome pulldown and calcein release assays to examine the effect of the mutations in both PIP2-binding sites. The K25A, K10A, and R22A mutants that targeted site 1 showed no significant PIP2 binding, whereas the site 2 K36A mutant displayed PIP2-binding levels comparable to that of wild-type HBD-2 (Fig. 5, D and E). Furthermore, neither the K10A nor the R22A mutant harbored significant liposomal lysis activity, while K25A displayed significantly reduced lysis activity (P < 0.05). In contrast, K36A lysed PIP2-containing liposomes at similar levels compared to wild-type HBD-2 (P < 0.001) (Fig. 5F). HBD-2(R&A) showed only low levels of lytic activity against PIP2-containing liposomes (fig. S2B), again suggesting that the three-dimensional structure of HBD-2 is important for this activity. These data suggest that the site 1 PIP2-binding site is critical for HBD-2 activity, with each residue identified as part of the site 1 PIP2-binding site being important for PIP2 binding, PIP2-mediated liposome lysis, and fungal growth inhibition. The role of site 2 is more elusive; K36 appears to have no involvement in PIP2-mediated liposome lysis while being crucial for fungal cell killing at the same time. In contrast to artificial liposomes, yeast cell membranes only present PIP2 on the inside of their plasma membrane. We speculate that, since HBD-2(K36A) still binds PIP2 and lyses PIP2-enriched liposomes, the functional importance of K36 is associated with a different stage of the mechanism of action of HBD-2, such as the initial contact with the fungal cell wall, outer cell membranes, or entry into the cytoplasm. While our structure reveals a novel dimeric configuration for HBD-2 that is critical for binding PIP2, we note that the PIP2 site 1 comprises residues originating from more than a single HBD-2 dimer. Since all identified key residues in site 1 were shown to be biologically significant in our mutagenesis assay, this raises the possibility that multiple dimers cooperate during PIP2 binding and, ultimately, membrane permeabilization. Such a scenario has previously been observed for the plant defensins NaD1 and NsD7 (9, 12).
DISCUSSION
Our data identify PIP2 binding as a critical feature of HBD-2 antifungal activity and establish a molecular mechanism of action for human defensins where HBD-2 dimers specifically engage PIP2 to permeabilize target cell membranes. While targeting of PIP2 for antimicrobial defense is conserved among plants and mammals, the detailed mechanism of action is strikingly different, with HBD-2 using different binding sites to engage PIP2 compared to plant defensins (9, 12, 13). This suggests that sequestration of PIP2 or related phospholipids is the primary mechanism underlying HBD-2–mediated membrane permeabilization. These findings indicate that phospholipids such as PIP2 play a significant role as target molecules in CAP-mediated innate defense and that this role is evolutionarily conserved across species. Furthermore, with the increasing interest in exploiting CAPs as therapeutic agents (9, 12–14, 16), our findings provide a platform for the design of defensin-based targeted strategies against human fungal pathogens.
MATERIALS AND METHODS
Recombinant expression and purification of HBD-2 and mutants in Pichia pastoris
The DNA fragment encoding the mature HBD-2 protein was polymerase chain reaction–amplified from human blood–derived genomic DNA using primers 5′-CTCGAGAAAAGAGGTATAGGCGATCCTGTTACC-3′ (forward; introduced Xho I restriction site underlined) and 5′-GCGGCCGCTTATGGCTTTTTGCAGCATTTT-3′ (reverse; introduced Not I restriction site underlined). It was subsequently ligated into the pPIC9 vector (Thermo Fisher Scientific) at the Xho I and Not I sites for expression in the methylotrophic yeast P. pastoris, followed by protein purification using SP Sepharose cation exchange chromatography, as described previously (12). HBD-2 point mutations were generated using the QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies), as per manufacturer’s instructions. The purified proteins were concentrated using Amicon Ultra 3000 MWCO centrifugal filters (Merck Millipore) and desalted into water using the same centrifugal filters. The protein concentration was determined using the BCA assay (Thermo Fisher Scientific). Protein purity and identity were confirmed by electrospray ionization quadrupole time-of-flight mass spectrometry analysis performed at the Comprehensive Proteomic Platform, La Trobe Institute for Molecular Science, La Trobe University.
Crystallization and structure determination
The HBD-2–PIP2 crystals were grown over 2 days in sitting drops at 20°C by mixing 200 nl of well solution containing 0.697 M sodium malonate–malonic acid, 0.1 M glycine-glycine (pH 8.38), with 50 nl of PIP2 solution (6.27 mM in water), and 150 nl of protein solution (8.3 mg/ml). Crystals were flash-cooled at 100 K in mother liquor supplemented with 25% ethylene glycol, and data were collected at the Australian Synchrotron (MX2 beamline) and processed using XDS (17). The structure was solved by molecular replacement using Phaser (18), with the structure of HBD-2 in its monomeric form as a search model (PDB ID: 1FD4, chain F) (7). The final model was built with Coot (19) and refined with Phenix (20) to a resolution of 1.85 Å. All data collection and refinement statistics are summarized in Table 1. Refinement yielded Rwork and Rfree values of 0.1944 and 0.2298, respectively. All programs were accessed via the SBGrid suite (21). Figures were prepared using PyMOL.
In-line SEC-SAXS
In-line SEC-SAXS on HBD-2 was performed as described previously (22). Briefly, HBD-2 samples at 10 mg/ml (75 μl) were subjected to SEC using a Superdex 75 5/150 GL equilibrated with 50 mM sodium phosphate (pH 7.5) and 50 mM NaCl at a flow rate of 0.2 ml/min. SAXS analyses were conducted in-line during elution via a coupled coflow sample sheath flow environment run at a fractional sample flow rate of 0.5 (23). Data were acquired on the SAXS/wide-angle x-ray scattering (WAXS) beamline at the Australian Synchrotron (24), which has been optimized for low instrument background, using a camera length of 1.430 m, providing q ranges of 0.011 to 0.65 Å−1 at 12 keV at a flux 6 × 1012 photons/s, and continuous exposures with a 1-s integration time using a Pilatus 1M detector. Images were inspected, averaged, and subtracted using PRIMUS from the ATSAS suite of SAXS data analysis tools (25). Data analysis was carried out, as described by the 2017 publication guidelines, using the Rg and the I(0) normalized over concentration scattering data selection over a given peak (26), with only frames representative of the higher protein concentration and consistent scattering profile used. Initial analysis based on reduced data in the form of data files using Guinier plots to calculate the radius of gyration was conducted using AUTORG from the ATSAS suite of SAXS data analysis tools (25). Uncertainties from Guinier fits are 2 SEs of the slope of fitted linear regressions of ln(I) versus q2. HBD-2 scattering curve was retrieved from the main peak as used for oligomerization analysis using MULCh (27) for a protein sequence–based estimate of partial specific volume and contrast. All data collection and analysis statistics are summarized in table S1.
PI uptake assays
C. albicans (LTUMC001 isolate, provided by the Department of Microbiology, La Trobe University, Melbourne, Australia) was inoculated overnight in yeast peptone dextrose (YPD) medium [30°C with shaking (160 rpm)]. Cells were pelleted, washed with 1× phosphate-buffered saline (PBS) buffer and counted using a hemocytometer before resuspending in half-strength potato dextrose broth (BD Biosciences). The fungal cells (1 × 106 cells/ml) were treated with different concentrations of HBD-2 or mutants for 30 min at 30°C with shaking (160 rpm), followed by PI staining (3 μg/ml; 5 min). The samples were diluted fivefold with ice-cold PBS before the flow cytometry analysis. For time-course experiments, C. albicans was resuspended in PI-containing potato dextrose broth and incubated for 5 min at 30°C and 160 rpm before HBD-2 treatment. At an indicated time point, the samples were immediately diluted with ice-cold PBS and analyzed by flow cytometry.
Fungal growth inhibition assays
C. albicans suspension in potato dextrose broth was prepared from overnight YPD culture, as described above. The fungal cells (400 cells per well) were incubated with a titration of sterile-filtered proteins in a sterile 96-well plate. After 24 hours, cell growth was determined by measuring absorbance at 600 nm in a SpectraMAX M5e plate reader (Molecular Devices) in nine-well scan mode.
Protein-lipid overlay assays
Protein-lipid overlay assays were performed using Membrane Strip or PIP Strip (Echelon Biosciences) using defensin (1 μg/ml), as described previously (14). Defensin bound to lipids was immunodetected using rabbit anti–HBD-2 antibodies (0.5 μg/ml), followed by horseradish peroxidase–conjugated donkey anti-rabbit immunoglobulin antibodies (0.5 μg/ml; Abcam). Quantitation of chemiluminescence signal intensity was determined by densitometric analysis using ImageJ (National Institutes of Health; http://imagej.nih.gov/ij/).
Liposome pulldown assays
Liposome pulldown assays were carried out as described previously (14). Liposomes were generated (28) with natural PIP2 (porcine brain, in chloroform/methanol/water solution at 20:9:1 molar ratio) and PC (chicken egg, in chloroform) from Avanti Polar Lipids. PC-only or PC/PIP2 (95:5 molar ratio) solutions were dried under a stream of nitrogen gas, followed by drying overnight in a vacuum. The lipid films were rehydrated to 10 mg/ml in 20 mM Hepes (pH 7.4) at 60°C for 2 hours, followed by three cycles of freezing in liquid nitrogen and thawing at 60°C. After washing with 20 mM Hepes (pH 7.4) (16,500g, 4°C, 10 min), the liposomes were incubated with 1 μg of defensin for 30 min. The pellet (bound fraction) and the supernatant (unbound fraction) were separated by centrifugation at 16,500g and 4°C for 10 min, then analyzed by SDS-PAGE, and silver-stained (Thermo Fisher Scientific) as per the manufacturer’s instructions. The intensity of protein bands was determined by densitometric analysis using ImageJ and normalized against the appropriate protein loading control.
Calcein-encapsulated liposome assay
The lipid films were rehydrated to a final concentration of 10 mg/ml in 100 mM calcein (Sigma-Aldrich), 20 mM Hepes (pH 7.4), and 1 mM EDTA at 60°C for 2 hours. Following three freeze-thaw cycles (alternating between liquid nitrogen and 60°C water bath), unilamellar liposomes were extruded 15 to 20 times through a mini-extruder with a 100-μm membrane (Avanti Polar Lipids). Free calcein dye was removed by three centrifugal washes with 20 mM Hepes (pH 7.4) and 1 mM EDTA at 16,500g and 4°C for 10 min. The calcein-encapsulated liposomes were then incubated with HBD-2, HBD-2 mutants, or HBD-2(R&A) for 30 min, followed by fluorescence measurement with an excitation and emission at 495 and 515 nm, respectively. After correction for background reading in Hepes-only control, protein-induced liposome lysis was normalized against total lysis by 0.1% Triton X-100.
Supplementary Material
Acknowledgments
We thank the MX2 beamline staff at the Australian Synchrotron for the help with x-ray data collection, the Commonwealth Scientific and Industrial Research Organization C3 Collaborative Crystallization Centre for the assistance with crystallization, and the staff at the Comprehensive Proteomics Platform at the La Trobe Institute for Molecular Science for the assistance with mass spectrometry analyses. Funding: This work was supported by the Australian Research Council (Fellowship FT130101349 to M.K.). Author contributions: M.J.: experimental design, acquisition of data, analysis and interpretation of data, and drafting and revising of the article. T.K.P.: experimental design, acquisition of data, analysis and interpretation of data, and drafting and revising of the article. F.T.L.: acquisition of data, analysis and interpretation of data, and drafting and revising of the article. S.C.: acquisition of data, analysis and interpretation of data, and drafting and revising of the article. M.K.: conception and design, acquisition of data, analysis and interpretation of data, and drafting and revising of the article. M.D.H.: Conception and design, acquisition of data, analysis and interpretation of data, and drafting and revising of the article. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The coordinates have been deposited in the PDB (accession code 6CS9), and the raw diffraction data were deposited at the SBGridDB (accession code doi:10.15785/SBGRID/568) (29). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/7/eaat0979/DC1
Fig. S1. SEC-SAXS analysis of HBD-2.
Fig. S2. The three-dimensional structure of HBD-2 is important for its antifungal activity and liposome permeabilization.
Table S1. SAXS data collection and scattering-derived parameters.
REFERENCES AND NOTES
- 1.Ostaff M. J., Stange E. F., Wehkamp J., Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol. Med. 5, 1465–1483 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diamond G., Beckloff N., Weinberg A., Kisich K. O., The roles of antimicrobial peptides in innate host defense. Curr. Pharm. Des. 15, 2377–2392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bals R., Wang X., Wu Z., Freeman T., Bafna V., Zasloff M., Wilson J. M., Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J. Clin. Invest. 102, 874–880 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganz T., Lehrer R. I., Defensins. Curr. Opin. Immunol. 6, 584–589 (1994). [DOI] [PubMed] [Google Scholar]
- 5.Wehkamp J., Harder J., Wehkamp K., Wehkamp-von Meissner B., Schlee M., Enders C., Sonnenborn U., Nuding S., Bengmark S., Fellermann K., Schröder J. M., Stange E. F., NF-κB- and AP-1-mediated induction of human beta-defensin-2 in intestinal epithelial cells by Escherichia coli Nissle 1917: A novel effect of a probiotic bacterium. Infect. Immun. 72, 5750–5758 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vylkova S., Nayyar N., Li W., Edgerton M., Human β-defensins kill Candida albicans in an energy-dependent and salt-sensitive manner without causing membrane disruption. Antimicrob. Agents Chemother. 51, 154–161 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoover D. M., Rajashankar K. R., Blumenthal R., Puri A., Oppenheim J. J., Chertov O., Lubkowski J., The structure of human β-defensin-2 shows evidence of higher order oligomerization. J. Biol. Chem. 275, 32911–32918 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Mathew B., Nagaraj R., Variations in the interaction of human defensins with Escherichia coli: Possible implications in bacterial killing. PLOS ONE 12, e0175858 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poon I. K. H., Baxter A. A., Lay F. T., Mills G. D., Adda C. G., Payne J. A. E., Phan T. K., Ryan G. F., White J. A., Veneer P. K., van der Weerden N. L., Anderson M. A., Kvansakul M., Hulett M. D., Phosphoinositide-mediated oligomerization of a defensin induces cell lysis. eLife 3, e01808 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lay F. T., Veneer P. K., Hulett M. D., Kvansakul M., Recombinant expression and purification of the tomato defensin TPP3 and its preliminary x-ray crystallographic analysis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 314–316 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baxter A. A., Poon I. K. H., Hulett M. D., The lure of the lipids: How defensins exploit membrane phospholipids to induce cytolysis in target cells. Cell Death Dis. 8, e2712 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Järvå M., Lay F. T., Hulett M. D., Kvansakul M., Structure of the defensin NsD7 in complex with PIP2 reveals that defensin:lipid oligomer topologies are dependent on lipid type. FEBS Lett. 591, 2482–2490 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Baxter A. A., Richter V., Lay F. T., Poon I. K. H., Adda C. G., Veneer P. K., Phan T. K., Bleackley M. R., Anderson M. A., Kvansakul M., Hulett M. D., The tomato defensin TPP3 binds phosphatidylinositol (4,5)-bisphosphate via a conserved dimeric cationic grip conformation to mediate cell lysis. Mol. Cell. Biol. 35, 1964–1978 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phan T. K., Lay F. T., Poon I. K. H., Hinds M. G., Kvansakul M., Hulett M. D., Human β-defensin 3 contains an oncolytic motif that binds PI(4,5)P2 to mediate tumour cell permeabilisation. Oncotarget 7, 2054–2069 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mylonas E., Svergun D. I., Accuracy of molecular mass determination of proteins in solution by small-angle x-ray scattering. J. Appl. Crystallogr. 40, s245–s249 (2007). [Google Scholar]
- 16.Schneider T., Kruse T., Wimmer R., Wiedemann I., Sass V., Pag U., Jansen A., Nielsen A. K., Mygind P. H., Raventós D. S., Neve S., Ravn B., Bonvin A. M. J. J., De Maria L., Andersen A. S., Gammelgaard L. K., Sahl H.-G., Kristensen H.-H., Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science 328, 1168–1172 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Storoni L. C., McCoy A. J., Read R. J., Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60, 432–438 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., Sliz P., Cutting edge: Collaboration gets the most out of software. eLife 2, e01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banjara S., Mao J., Ryan T. M., Caria S., Kvansakul M., Grouper iridovirus GIV66 is a Bcl-2 protein that inhibits apoptosis by exclusively sequestering Bim. J. Biol. Chem. 293, 5464–5477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirby N., Cowieson N., Hawley A. M., Mudie S. T., McGillivray D. J., Kusel M., Samardzic-Boban V., Ryan T. M., Improved radiation dose efficiency in solution SAXS using a sheath flow sample environment. Acta Crystallogr. D Struct. Biol. 72, 1254–1266 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirby N. M., Cowieson N. P., Time-resolved studies of dynamic biomolecules using small angle x-ray scattering. Curr. Opin. Struct. Biol. 28, 41–46 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D. T., Konarev P. V., Svergun D. I., New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trewhella J., Duff A. P., Durand D., Gabel F., Guss J. M., Hendrickson W. A., Hura G. L., Jacques D. A., Kirby N. M., Kwan A. H., Pérez J., Pollack L., Ryan T. M., Sali A., Schneidman-Duhovny D., Schwede T., Svergun D. I., Sugiyama M., Tainer J. A., Vachette P., Westbrook J., Whitten A. E., 2017 publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: An update. Acta Crystallogr. D Struct. Biol. 73, 710–728 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whitten A. E., Cai S., Trewhella J., MULCh: Modules for the analysis of small-angle neutron contrast variation data from biomolecular assemblies. J. Appl. Crystallogr. 41, 222–226 (2008). [Google Scholar]
- 28.Zhang L., Rozek A., Hancock R. E. W., Interaction of cationic antimicrobial peptides with model membranes. J. Biol. Chem. 276, 35714–35722 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Meyer P. A., Socias S., Key J., Ransey E., Tjon E. C., Buschiazzo A., Lei M., Botka C., Withrow J., Neau D., Rajashankar K., Anderson K. S., Baxter R. H., Blacklow S. C., Boggon T. J., Bonvin A. M. J. J., Borek D., Brett T. J., Caflisch A., Chang C.-I., Chazin W. J., Corbett K. D., Cosgrove M. S., Crosson S., Dhe-Paganon S., Di Cera E., Drennan C. L., Eck M. J., Eichman B. F., Fan Q. R., Ferré-D’Amaré A. R., Fromme J. C., Garcia K. C., Gaudet R., Gong P., Harrison S. C., Heldwein E. E., Jia Z., Keenan R. J., Kruse A. C., Kvansakul M., McLellan J. S., Modis Y., Nam Y., Otwinowski Z., Pai E. F., Pereira P. J. B., Petosa C., Raman C. S., Rapoport T. A., Roll-Mecak A., Rosen M. K., Rudenko G., Schlessinger J., Schwartz T. U., Shamoo Y., Sondermann H., Tao Y. J., Tolia N. H., Tsodikov O. V., Westover K. D., Wu H., Foster I., Fraser J. S., Maia F. R. N. C., Gonen T., Kirchhausen T., Diederichs K., Crosas M., Sliz P., Data publication with the structural biology data grid supports live analysis. Nat. Commun. 7, 10882 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/7/eaat0979/DC1
Fig. S1. SEC-SAXS analysis of HBD-2.
Fig. S2. The three-dimensional structure of HBD-2 is important for its antifungal activity and liposome permeabilization.
Table S1. SAXS data collection and scattering-derived parameters.