

Abstract
Background—
Regulation of vascular endothelial growth factor receptor-2 (VEGFR2) signaling is a control point that determines the extent of vascular tree formation. Recent studies demonstrated an important role played by VEGFR2 endothelial trafficking in control of its activity and suggested the involvement of a phosphotyrosine phosphatase 1b (PTP1b) in this process. This study was designed to define the role of PTP1b in endothelial VEGFR2 signaling and its role in regulation of angiogenesis and arteriogenesis.
Methods and Results—
We generated mice carrying an endothelial-specific deletion of PTP1b and examined the effect of this knockout on VEGF signaling, angiogenesis, and arteriogenesis in vitro and in vivo. PTP1b knockout endothelial cells had increased VEGF-dependent activation of extracellular signal-regulated kinase signaling, sprouting, migration, and proliferation compared with controls. Endothelial PTP1b null mice had increased retinal and Matrigel implant angiogenesis and accelerated wound healing, pointing to enhanced angiogenesis. Increased arteriogenesis was demonstrated by observations of faster recovery of arterial blood flow and large numbers of newly formed arterioles in the hindlimb ischemia mouse model. PTP1b endothelial knockout also rescued impaired blood flow recovery after common femoral artery ligation in synectin null mice.
Conclusions
PTP1b is a key regulator of endothelial VEGFR2 signaling and plays an important role in regulation of the extent of vascular tree formation.
Keywords: angiogenesis, arteriogenesis, extracellular signal-regulated kinases, PTP1b, VEGF, VEGF receptor-2
Recent studies involving signal transduction by vascular endothelial growth factor (VEGF)-A in endothelial cells demonstrated the existence of an important regulatory mechanism involving intracellular trafficking of VEGF receptor-2 (VEGFR2).1 The presumed role of this event is movement of a VEGFR2-containing endosome from a phosphatase-rich environment to the part of the cytoplasm where VEGFR2 is not being actively dephosphorylated. In particular, this mechanism plays a critical role in VEGF-induced extracellular signal-regulated kinase (ERK)-1/2 activation that has been linked to the development of arterial circulation (arteriogenesis).2–5
Clinical Perspective on p 909
VEGF-induced ERK activation involves ligand-driven dimerization of the receptor leading to trans- phosphorylation of C-loop activation loop tyrosines 1054 and 1059. This, in turn, leads to phosphorylation of a number of cytoplasmic domain tyrosines including the Y1175 site, phosphorylation of which is required for ERK activation.6,7 Once phosphorylated, Y1175 serves as a binding site for phospholipase Cγ and subsequent activation of the Raf1/MEK/ERK cascade. One unusual aspect of this process is the requirement for VEGFR2 phosphorylation and trafficking. In the absence of VEGFR2 endocytosis, ERK activation is practically absent as seen after knockouts of EphrinB2, phospholipase C alpha, or protease activated receptor 3. A complete, or nearly complete, lack of ERK activation leads to a total failure of vascular development as seen in VEGFR2 knockouts, mice carrying a Y1175F knock-in mutation, or mice with knockouts of EphrinB2, phospholipase C alphas, or protease activated receptor 3.8–10
Once endocytosed, VEGRF2 undergoes an important trafficking step that is essential for full activation of the ERK signaling cascade. Delayed trafficking of the receptor from clathrin-coated pits to EEA1 early endosomes reduces the extent of ERK activation attributable to partial dephosphorylation of its Y1175 site. The trafficking event requires formation of a neuropilin-1 (Nrp1, another VEGF-A receptor)-synectin-myosin-VI complex. A knockout of any of these genes or a knock-in of Nrp1 missing its PDZ domain (Nrp1cyto) that is required for its binding to the PDZ scaffold protein synectin (Gipc1) results in reduced Y1175 phosphorylation and decreased ERK activation. In vivo this manifests itself as impaired embryonic and adult arteriogenesis, including reduced arterial vessel density, branching, and decreased lumen diameter.4,5
This trafficking-dependent phosphorylation of VEGFR2 suggests the existence of a phosphatase that dephosphorylates the Y1175 site. Studies using in vitro siRNA knockouts of various protein tyrosine phosphatases in endothelial cells with impaired VEGFR2 trafficking (synectin or myosin-VI null knockouts) demonstrate that only phosphotyrosine phosphatase 1b (PTP1b) knockdown rescues VEGF-induced ERK activation.5 In agreement with this is the observation that overexpression of PTP1b in normal endothelial cells reduces, whereas knockdown increases, VEGF-induced ERK activation11 while inhibition of PTP1b activity using a chemical inhibitor rescues blood flow recovery and arteriogenesis in the hindlimb ischemia model in Gipc1−/− mice.5 Finally, VEGFR2 can be immunoprecipitated with PTP1b,11 and the 2 proteins have been visualized together on Rab5 endosomes using structured illumination microscopy (SIM) super-resolution imaging.4
These data point to PTP1b as the likely phosphatase regulating VEGF-induced ERK activation. However, whether PTP1b actually normally regulates VEGFR2 signaling in vivo in an endothelial cell–specific manner and plays a role in its various biological activities remains unknown. In this study we set out to examine this question by selectively deleting the phosphatase in mouse endothelial cells. We find that endothelial-specific PTP1b (PtpN1ECKO) deletion is associated with increased ERK signaling and increased angiogenesis and arteriogenesis. These results demonstrate that PTP1b is an important endogenous endothelial cell regulator of VEGF-A signaling.
Materials and Methods
Reagents and Antibodies
Human fibronectin (BD Biosciences#356008), Matrigel Basement Membrane Matrix Growth Factor Reduced (BD#354230), IGF-1(Sigma), VEGF-A165 (#293-VE R&D systems); Antibodies used included the following: anti-CD31 (BD#55370), anti–VE-cadherin (Santa Cruz#SC6458), antiphospho p44/42 MAP kinase (phospho-ERK, Cell Signaling#9106), anti-p44/42 MAP kinase (total ERK, Cell Signaling#9102), anti-PTP1b (Santa Cruz#sc-1718), anti phospho IGF-1 receptor β (Tyr1131) (CS#3921), anti IGF-1 receptor β (CS#3027). We used appropriate secondary antibodies that were conjugated to horseradish peroxidase (Vector Laboratories) or fluorescently labeled (Life Technologies). IsolectinB4 was purchased from Molecular Probes (Invitrogen).
Mouse Generation
To obtain mice homozygous for a conditional allele of PTP1b (Ptpn1), Ptpn1loxP/loxP mice13 were crossed with Cdh5-CreERT2 mice (gift of R. Adams, Max-Planck-Institute for Molecular Biomedicine).10 Cre activity was induced in 3-week-old mice by 5 consecutive daily intra peritoneal injections of tamoxifen (0.1 mL of a 20 mg/mL solution in corn oil). The phenotypes of mutant mice were analyzed at 6 to 10 weeks of age, and littermate animals were used as controls. All animal experiments were performed under a protocol approved by the Institutional Animal Care and Use Committee of Yale University.
Isolation of Murine Endothelial Cells
Primary arterial endothelial cells were isolated from adult mouse dorsal aorta as previously described.14 Arteries from 4 mice were harvested, pooled, minced with scissors, and then digested in 25 mL collagenase 0.2% (wt/vol) at 37°C for 45 minutes. The crude cell preparation was pelleted and resuspended in Dulbecco’s phosphate buffered saline. The cell suspension was incubated with platelet endothelial cell adhesion molecule-1–coated beads (IgG Dynal beads from Dynal Corp, Great Neck, NY) at room temperature for 10 min. with end-over-end rotation. Using a magnetic separator, the bead-bound cells were recovered, washed, and resuspended in complete culture medium (DMEM containing 20% fetal calf serum, supplemented with 100 ug/mL heparin, 100 ug/mL endothelial cell growth factor growth supplement [endothelial cell growth factor: Biomedical Technologies, Stoughton, MA], and nonessential amino acids, sodium pyruvate, L-glutamine, and antibiotics at standard concentrations), and then plated in a 0.1% gelatin-coated 10-cm tissue culture dish.
Migration and Proliferation of Endothelial Cells
The xCELLigence RTCA DP analyzer was used to measure migration, adhesion, and proliferation of wild-type and PTP1b knockout cells in response to 0.5% FBS, 20% FBS, VEGF-A (100ng/mL), and FGF2 (100ng/mL). For migration the CIM-Plate 16 was used, the bottom chamber contained the growth factor, and 40 000 cells were added to the top chamber and the plate monitored every 5 minutes for 24 hours. For proliferation and adhesion the E-Plate 16 was used, the plates were coated with 0.1% gelatin, and 5000 cells per well were added to media containing the growth factor. For proliferation the plate was monitored every 15 minutes for 80 hours, and for adhesion the plate was monitored every 5 minutes for 4 hours. For each condition at least 4 replicate wells were used.
siRNA Transfection
siRNAs (FlexiTube siRNA) were purchased from Qiagen. Human umbilical vein endothelial cells were transfected with 25 pmol siRNA per 6 wells with 2.5 mL RNAiMax (Invitrogen) according to the instructions of the manufacturer. Cells were used for experiments 48 hours after transfection.
In Vitro EC Cord Formation
Cells were starved overnight in 0.5% FBS, detached with trypsin, and seeded at a density of 60 000 cells per well in a 24-well plate containing 0.5 mL per well of reduced-growth factor matrigel (BD Biosciences). Cells were in media containing 0.5% FBS, VEGF-A (100 ng/mL), or FGF2 (100 ng/mL), 1 mL per well. After 8 hours cells were fixed with 4% paraformaldehyde and 5 random images were taken of each well (×10 objective) and the total length of cord formation was quantified for each field. The mean of 10 total lengths per well represent an experimental point. Cord length was assessed using the NeuronJ plug-in of ImageJ.
In Vitro Sprouting Assay
After siRNA transfection, human umbilical vein endothelial cells (250 000 cells per well in 6-well plates) were resuspended in fibrinogen solution (2.5 mg/mL fibrinogen [Sigma-Aldrich] in EBM-2 [Lonza] supplemented with 2% FBS and 50 mg/mL aprotinin [Sigma-Aldrich]), and plated on top of a precoated fibrin layer (fibrinogen solution clotted with 1 U thrombin [Sigma-Aldrich] for 20 minutes at 37°C). The second layer of fibrin was clotted for 1 hour at 37°C. Wi-38 cells (250 000 cells per well), in EBM-2 supplemented with 2% FBS and 25 ng/mL VEGF, were then plated on top of the fibrin layers. Cultures were incubated at 37°C, 5% CO2. After 4 to 6 days, cultures were labeled with 4 mg/mL Calcein AM for 1 hour, and imaged by fluorescence using a standard FITC filter.
Hindlimb Ischemia Model
As previously described,14 the femoral artery was ligated at 2 positions spaced 5 mm apart and the arterial segment between the ligatures was excised. Tissue perfusion was assessed preoperatively, immediately postoperatively, and 3, 7, and 14 days after surgical intervention. Flow images of the foot were acquired using a Moor Infrared Laser Doppler Imager (LDI; Moor Instruments Ltd) at 37.0°C to 38.0°C under ketamine/xylazine (80/5 mg/kg) anesthesia. Data were analyzed with Moor LDI image processing software V3.09 and reported as the ratio of flow in the right/left (R/L) hindlimb after background subtraction.
Micro-CT Angiography
2D mCT scans were acquired with a GE eXplore MS Micro-CT System, using a 400 cone-beam with angular increment of 0.5 degrees and 8 to 27 μm slice thickness at a voltage of 163.2 mAs, 80 kVp. mCT data were transferred to a Dell Dimension computer with 3D volume rendering software (version 3.1, Vital Images Inc, Plymouth, MN) and microview software (version 1.15, GE medical system). NIH ImageJ (National Institutes of Health, Bethesda, MD) and Image Pro Plus (Media Cybernatics) software were used to analyze vessel number, diameter, area, volume, and arterial density.
Wound Healing Assay and Analysis
Wound healing studies were provided by the microsurgery core at Yale Cardiovascular Research Center. Mice were anaesthetized by intraperitoneal injection of a ketamine (100 mg/kg)/xylazine (10 mg/kg) solution. Wounds were created with a sterile 6-mm biopsy punch in the back skin (Miltex Inc, PA) without injuring the underlying muscle. Wound regions were photographed using a Leica M125 microscope with an HC80 HD camera (Leica, Germany) on days 0, 1, 3, 5, and 7. Wound area was calculated using NIH ImageJ software. Wound sizes at different time points were expressed as percentage of the wound area on day 0.
In Vivo Matrigel Assay and Analysis
Growth factor-depleted Matrigel plugs (0.5 mL) containing either FGF2 (100ng/mL) or heparin (10U) with VEGF-A165 (100 ng/mL) were injected subcutaneously on both sides of C57BL/6 mice. Seven days later, Matrigel plugs were excised and stained with a rat antimouse monoclonal CD31 antibody. CD31-positive vessels were counted in randomly acquired images using ImageJ.
Immunohistochemistry of Whole-Mount Retinas
P5 pups were euthanized and the eyes removed and prefixed in 4% PFA for 20 minutes at room temperature. The retinas were dissected out and blocked overnight at 4°C in blocking buffer (0.1 mol/L Tris-HCl, 150 mmol/L NaCl, 1% Blocking Reagent [PerkinElmer)] 0.5% Triton X-100). After washing with Pblec (1 mmol/L MgCl2, 1 mmol/L CaCl2, 0.1 mmol/L MnCl2, 1% Triton X-100 in PBS), the retinas were incubated with IsolectinB4 in Pblec overnight followed by incubation with the corresponding secondary antibody for 2 hours at room temperature. Then the retinas were mounted in fluorescent mounting medium (DAKO, Carpinteria, CA). Images were acquired using a Perkin Elmer UltraVIEW VoX spinning disc confocal microscope. Quantification was performed on wild-type and PTP1bECKO neonates; 20 to 25 images per group were acquired, and Biological CMM Analyzer software16 was used to quantify vascular area and number of vessel branch points per image.
Statistical Analysis
All data are shown as mean±standard error of the mean (SEM) or standard deviation (SD) as bars in the histograms. Differences were considered statistically significant if P≤0.05 by Student t test in case of 2-group comparison or by Mann–Whitney U, Kruskal-Wallis, and 2-way ANOVA followed by Bonferonni analysis as appropriate.
Results
In Vitro Effects of Endothelial Cell PTP1b Deletion
To test the effect of PTP1b deletion on the response of endothelial cells to various growth factors, primary mouse endothelial cells (EC) from PtpN1fl/fl mice were treated with an adenovirus carrying the Cre gene (Ad-Cre) or control virus (Ad-CMV), growth arrested, and then stimulated with IGF-1, VEGF-A165, or FGF2. As expected, PTP1b deletion resulted in enhanced activation of IGF1 receptor (IGF1Rβ) and ERK signaling (Figure 1A and 1B). Consistent with the hypothesis that PTP1b downregulates VEGF signaling, ERK activation by VEGF was also increased in knockout compared with wild-type EC (Figure 1C and 1D). On the other hand, stimulation with FGF2 detected no differences in ERK activation, suggesting that PTP1b is not involved in FGF signaling (Figure 1E and 1F). In agreement with increased ERK activation by VEGF, VEGFR2 phosphorylation was also increased in PTP1b−/− compared with control endothelial cells (Figure IA and IC in the online-only Data Supplement), despite a decrease in total VEGFR2 levels (Figure IB in the online-only Data Supplement). Furthermore, there were no differences in Akt or p38MAPK activation in response to VEGF in PTP1b−/− versus control endothelial cells (Figure I in the online-only Data Supplement).
Figure 1.
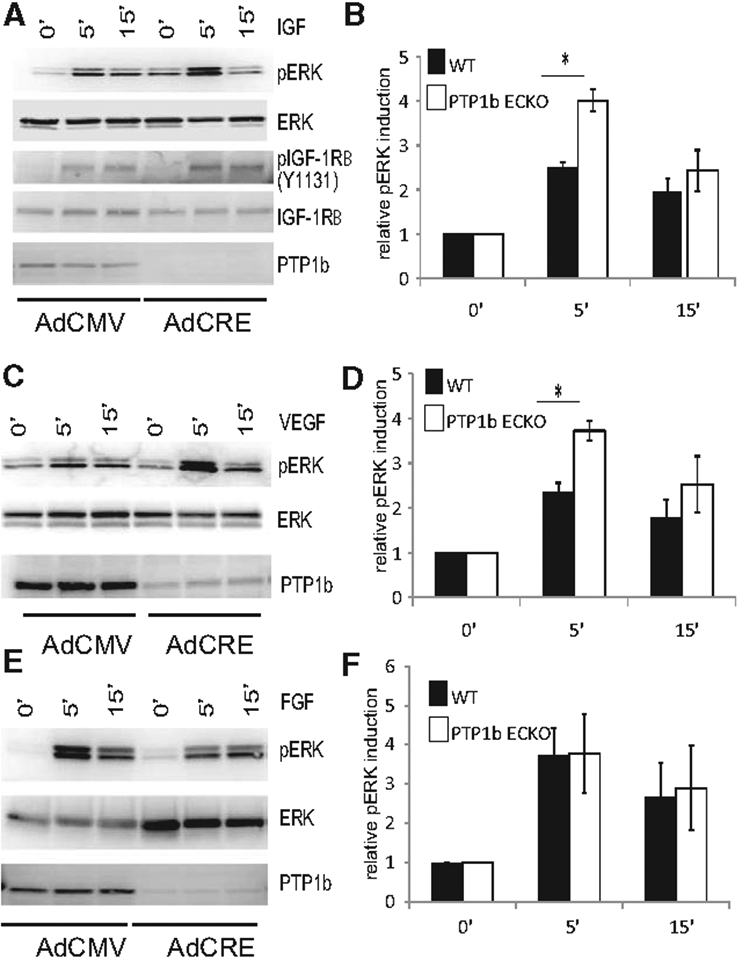
Increased IGF-1 and VEGF-A signaling in PTP1bECKO cells relative to WT. A, C, and E, IGF1, FGF2, and VEGF-A signaling in wild-type and phosphotyrosine phosphatase 1b (PTPIb)–null endothelial cells. Western blotting of total cell lysates isolated from floxed PTP1b cells treated with control CMV adenovirus or CRE adenovirus for 3 days and then starved overnight. Confluent, serum-starved cells were stimulated for the times indicated with 50 ng/mL of IGF-1, FGF2 or VEGF-A. A, Phosphorylation of the tyrosine residue 1131 of the IGF1 receptor and p44/p42 MAP kinase is increased in PTP1bECKO cells relative to WT after IGF-1 treatment. C, Phosphorylation of p44/p42 MAP kinase is increased in PTP1bECKO cells relative to wild-type after VEGF-A treatment. E, Phosphorylation of p44/p42 MAP kinase is not changed in PTP1bECKO cells relative to wild-type following FGF2 treatment. B, D, and F, Quantification of relative pERK induction by IGF1, FGF2, and VEGF-A in wild-type and PTP1bECKO cells (n=3; SD, *P<0.05).
In agreement with these data, PTP1b−/− endothelial cells demonstrated faster proliferation (Figure 2A–2C) and migration (Figure 2D–2F) in response to VEGF but not FGF2 or fetal bovine serum stimulation. Furthermore, cord formation in the in vitro Matrigel assay was also accelerated by VEGF-A, but not FGF2 or fetal bovine serum (Figure 2G–2I).
Figure 2.
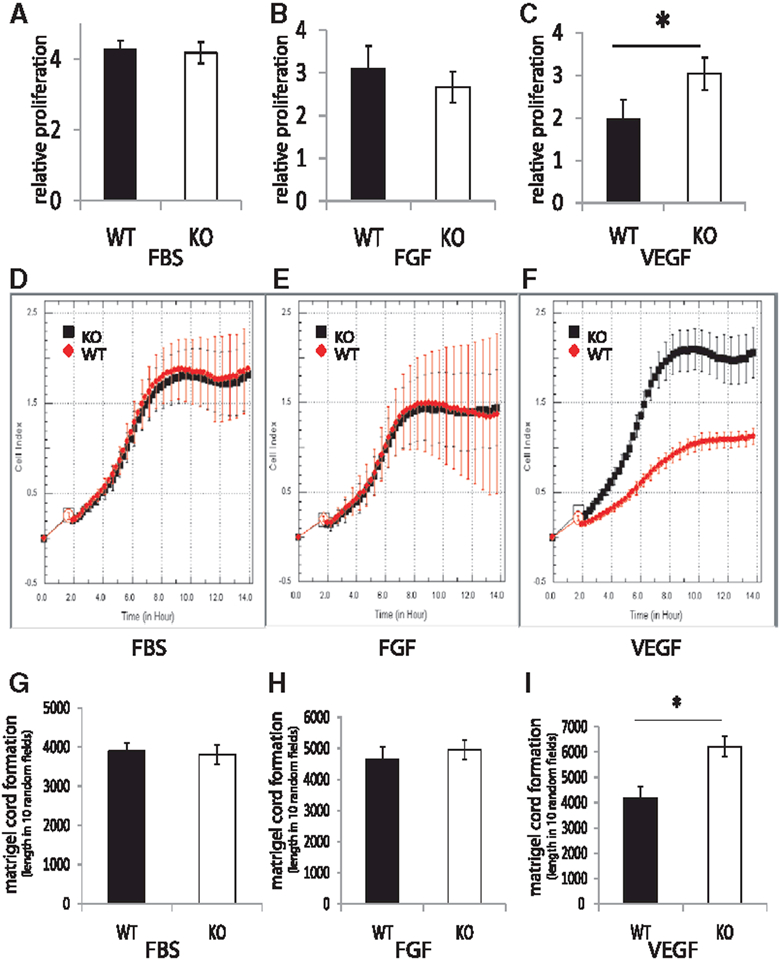
Proliferation, migration, and in vitro matrigel analysis of phosphotyrosine phosphatase 1b (PTP1b)ECKO primary endothelial cells. A–C, Proliferation of wild-type (WT) and PTP1bECKO cells after treatment with FBS, FGF2, or VEGF-A. Proliferation rates of WT and PTP1bECKO cells after serum starvation and stimulation with FBS, FGF2, or VEGF-A for 72 hours measured using Roche xCELLigence system (n=5000 cells, repeated three times). Note the increase in proliferation of PTP1bECKO cells relative to WT when treated with VEGF-A (SD, *P<0.05, Student t test). D–F, Migration of Wt and PTP1bECKO cells after treatment with FBS, FGF2, or VEGF-A. Migration rates of WT and PTP1bECKO cells after serum starvation and stimulation with FBS, FGF2, or VEGF-A for 14 hours measured using Roche xCELLigence system (n=5000 cells, repeated 3 times). Note the increase in migration of PTP1bECKO cells in response to treatment with VEGF-A (SD). G–I, In vitro matrigel analysis of WT and PTP1b ECKO cells after treatment with FBS, FGF2, or VEGF-A. Matrigel cord formation after starvation of WT or PTP1bECKO and plating for 8 hours on matrigel containing either 0.5% FBS, FGF2 (100ng/mL), or VEGF-A (100ng/mL). Note the increase in cord formation of PTP1bECKO cells in response to treatment with VEGF-A (SD, * P<0.05, Student t test).
In Vivo Effects of Endothelial Cell PTP1b Deletion: Angiogenesis
Endothelial-specific deletion of PTP1b was achieved by crossing PtpN1fl/fl mice with Cdh5-CreERT2 line as described in the Methods section. The effectiveness of deletion in the resulting PtpN1ECKO mice was assessed by isolating endothelial cells from the heart, lungs, and aorta of Ptpn1ECKO and control littermate mice and testing them for PTP1b expression. As previously reported for this Cre line, all endothelial cells tested demonstrated nearly complete target gene deletion15 (Figure II in the online-only Data Supplement).
To test the role of PTP1b in angiogenesis, we first used an in vitro sprouting assay. Human umbilical vein endothelial cells that were exposed to either control or PTP1b siRNA were treated with VEGF-A or saline and the extent of sprouting was determined as outlined in the Methods. PTP1b knockdown was associated with a highly significant increase in sprouting both in the absence or presence of VEGF-A (Figure 3A and 3B). To check the validity of this result in vivo, we analyzed vascular sprouting in the retinas of P5 PtpN1ECKO and control littermates pups (Figure 3C–3E). As in the case of the in vitro assay, endothelial PTP1b knockout was associated with significantly increased sprouting as demonstrated by increased number of branch points and vessel density. At the same time, there was no change in the extent of vascular coverage of the retinas (Figure 3F). Finally, Ptpn1ECKO mice had larger numbers of tip cells along the vascular front as assessed by the number of filopodia per vessel length (Figure 3G and 3H).
Figure 3.
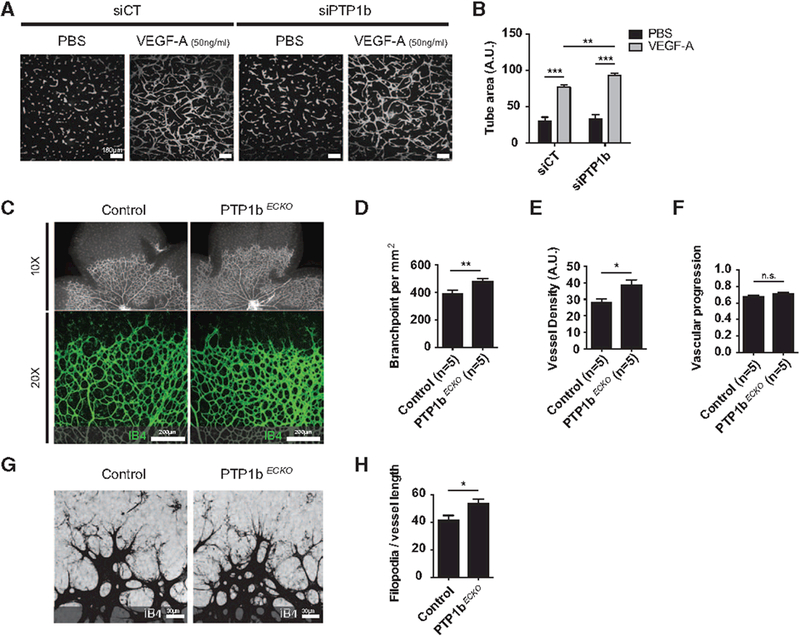
In vitro and in vivo increased angiogenesis in response to phosphotyrosine phosphatase 1b (PTP1b) inhibition. A, Representative images of in vitro sprouting cells treated with a PTP1b or control siRNA in response to VEGF. B, Quantification of sprouting: note the increase in sprouting in cells treated with the PTP1b siRNA (n=6, mean±SEM). C, Representative images of P5 retinas from wild-type (WT) and PTP1bECKO mice. D–F, Quantification of sprouting of P5 retinas from WT and PTP1bECKO mice. Note the increase in sprouting in retinas of P5 PTP1bECKO mice (n=5, mean±SEM). G, Vascular sprouts in PTP1bECKO present increased filopodia projections. H, Quantification of filopodia bursts per sprouting front length. (n=18, mean±SEM). Statistical significance was assessed using a Mann–Whitney U test. *P<0.05, **P<0.01, ***P<0.001.
We next studied the effect of this knockout in the skin wound healing response that is heavily, but not exclusively, angiogenesis-dependent. In agreement with in vitro results showing increased proliferation and migration of PtpN1−/− endothelial cells, wounds closed significantly faster in PtpN1ECKO compared with littermate control mice (Figure 4A and 4B)
Figure 4.
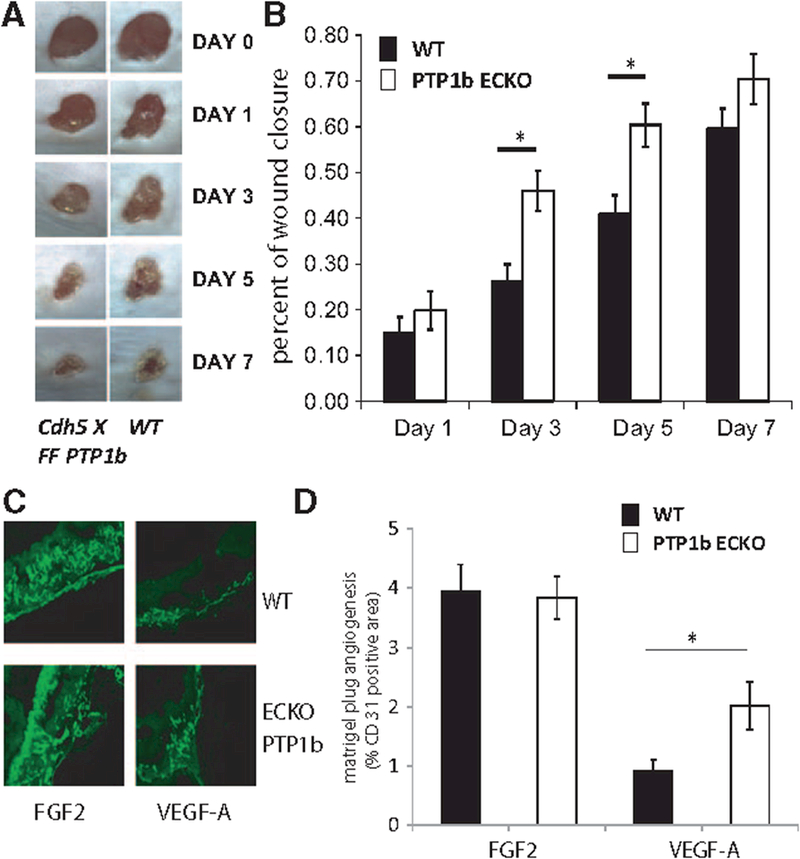
Phosphotyrosine phosphatase 1b (PTP1b)ECKO mice show increases relative to wild type (WT) in wound healing and matrigel angiogenesis assays. A, Representative images of wound healing in WT and PTP1bECKO mice after wounding by punch biopsy. B, Quantification of area of wound closure, note increased rate of wound closure of PTP1bECKO mice relative to WT (n=15, WT and n=16 PTP1bECKO mice, mean±SEM; *P<0.05, Student t test; Repeated measures analysis using Kruskal-Wallis 1-way nonparametric ANOVA, K.W. statistics 30.08, P<0.001). C, Representative images of CD31 stained Matrigel sections from Wt (and PTP1bECKO mice with FGF2 and VEGF treatment. D, Quantification of CD31 staining of Matrigel sections, note difference in VEGF response between WT and PTP1bECKO mice but not to FGF2 (n=10 each of WT and PTP1bECKO mice, mean±SEM,;*P<0.05, Student t test).
To more specifically explore the effect of this deletion on VEGF-driven angiogenesis, we used an in vivo Matrigel model. PtpN1ECKO and littermate control mice were implanted with Matrigel pellets containing VEGF-A or FGF2. Quantification of capillary ingrowth 7 days later demonstrated increased angiogenesis in implants containing VEGF-A but not FGF2 in PtpN1ECKO mice (Figure 4C and 4D).
In Vivo Effects of Endothelial Cell PTP1b Deletion: Arteriogenesis
We next evaluated whether PTP1b deletion has an effect on arteriogenesis in adult tissues. To this end, hindlimb ischemia was induced in PtpN1ECKO and littermate control mice and the kinetics, extent of blood flow recovery, and anatomic extent of arteriogenesis were evaluated. PTP1b knockout was associated with accelerated blood flow recovery as early as day 3 but the final extent was the same as in controls (Figure 5A and 5B). Micro-CT analysis of the hindlimb vasculature at 7 days demonstrated a significantly larger number of small arteries in PtpN1ECKO compared with control mice both above and below the knee (Figure 5C–5E). There were no differences in arterial vessel density at 14 days when the extent of blood flow recovery was the same in both groups (data not shown). These results suggest that PTP1b affects the rate but not the extent of new artery formation.
Figure 5.
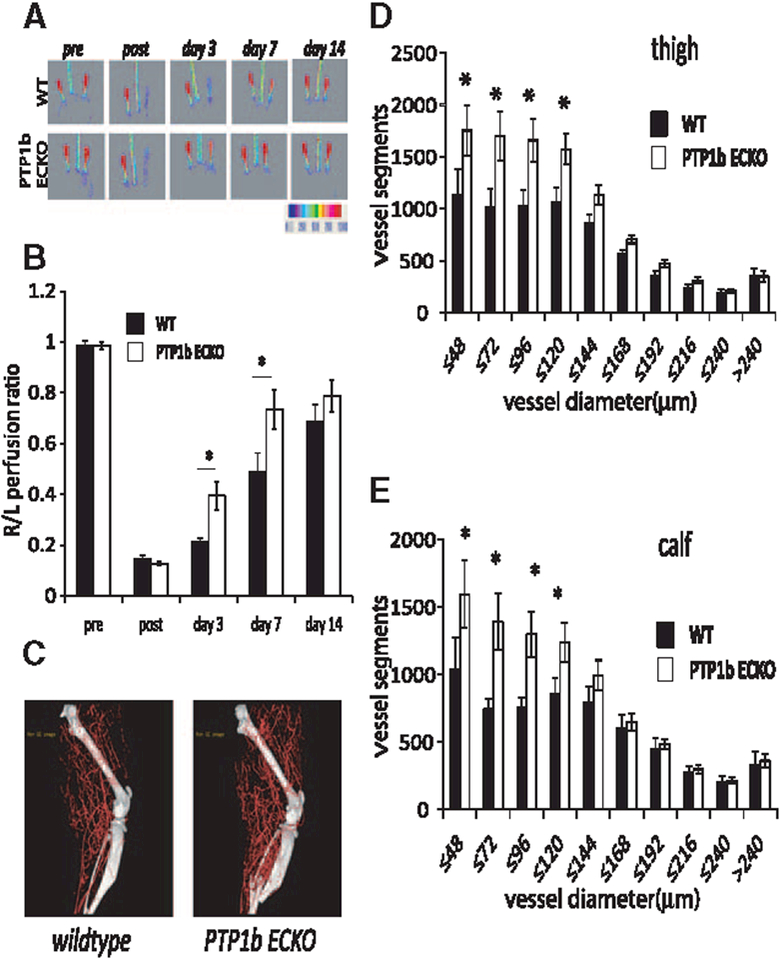
Phosphotyrosine phosphatase 1b (PTP1b) endothelial cell knockout (KO) mice show increased recovery in the hindlimb ischemia model. A, Representative laser Doppler images of the time course of perfusion in hindlimbs of PTP1bECKO and WT mice before and after hindlimb ischemia surgery. B, Quantitative analysis of laser Doppler images indicates significant alterations in hindlimb reperfusion starting at 3 days after femoral artery ligation in PTP1bECKO mice (white bars, n=7 mice) relative to WT mice (black bars, n= mice; mean±SEM, *P<0.05). C, Representative micro-CT images of wild-type (WT) and PTP1bECKO mice 7 days after HLI. D and E, Quantitative micro-CT analysis of arterial vasculature above and below the knee in PTP1bECKO mice (white bars, n=7500 cross-sections per mouse) and WT mice (black bars, n=5500 cross-sections per mouse) 7 days after common femoral artery ligation. Note a marked increase in total number of <120-μm-diameter vessels in PTP1bECKO mice relative to WT littermates in thigh and calf (mean±SEM, * P<0.05). Statistical significance was assessed using a Mann-Whitney U test and repeated measures analysis performed using 1-way nonparametric ANOVA (Kruskal Wallis test) P<0.001 for all trends.
Finally, we studied whether PTP1b deletion would restore blood flow in synectin knockout mice because impaired VEGF signaling in this strain is thought to be attributable to the reduced rate of VEGFR2 trafficking leading to increased PTP1b-driven receptor dephosphorylation. To this end, PtpN1ECKO mice were crossed onto the previously described Synectin null (Gipc1−/−) strain.14 The hindlimb ischemia model was then used to study arteriogenesis in double knockout mice (PtpN1−/−/Gipc1−/−) versus littermate controls (both single knockouts and wild-type animals).
In agreement with previous results, PTP1b deletion was associated with accelerated blood flow recovery and increased arteriogenesis, whereas both processes were reduced in synectin knockouts (Figure 6A and 6B). PTP1b knockout on the background of synectin deletion fully restored arteriogenesis, returning it to levels seen in control animals (Figure 6A and 6B).
Figure 6.
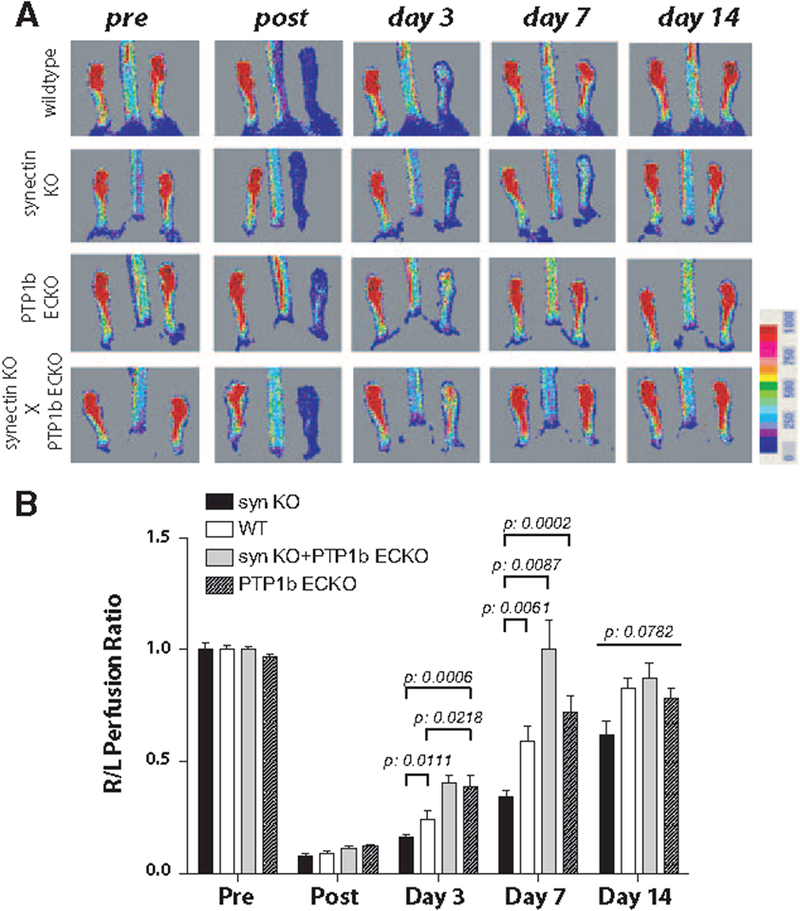
Phosphotyrosine phosphatase 1b (PTP1b) endothelial cell knockout in synectin knockout mice rescues blood flow recovery in the hindlimb ischemia model. A, Representative laser Doppler images of the time course of perfusion in hindlimbs of PTP1bECKO, synectin KO, PTP1bECKO/synectin KO and wild-type mice before and after hindlimb ischemia surgery. B) Quantitative analysis of laser Doppler images indicates a significant decrease in hindlimb reperfusion after femoral artery ligation in synectin KO mice (black bars, n=7) and an increase in reperfusion in PTP1bECKO mice (stippled bars, n=11) relative to wild-type mice (white bars, n=7). Note that the decrease in reperfusion in synectin KO mice is rescued by crossing with PTP1bECKO mice (grey bars, n=4; mean±SEM, *P<0.05). Statistical significance was assessed using Kruskal-Wallis and Mann–Whitney U tests and repeated measures analysis performed using 1-way nonparametric ANOVA (Kruskal Wallis test). P<0.001 for all trends.
Discussion
Taken together, the data in this study point to a critical physiological role of PTP1b in the regulation of VEGFR2 signaling. Endothelial-specific deletion of this phosphatase resulted in a VEGF-driven increase in ERK activation that is central to arterial blood vessel growth and formation,6 and an increase in VEGF-driven cell proliferation and migration. In vivo this translated into increased angiogenesis and arteriogenesis.
VEGF signaling is one of the most tightly regulated processes in endothelial biology. Several tyrosine phosphatases have been reported to be associated with VEGFR2 including CD148/Dep1,16–18 VE-PTP,19–22 and PTP1b,4,11 the subject of the current study. As with all phosphatases, these molecules have a broad spectrum of activity and are capable of dephosphorylating a number of targets.23 However, their precise role in the vasculature has not been defined. Mice with an early embryonic inactivation of VE-PTP demonstrate normal vasculogenesis but defective yolk sac angiogenesis leading to midembryonic lethality.20 A knockout of CD148/DEP1 is also lethal (at E10.5) with the mice demonstrating enlarged aberrantly developed vasculature and impaired pericyte investment.18 In contrast, mice with global deletion of PTP1b appear grossly normal and no vascular defects have been reported to date.24,25
Nevertheless, recent studies have suggested that PTP1b should play an important role in VEGF signaling. In particular, we previously reported that PTP1b associates with early endosomes containing VEGFR2 and that VEGFR2-dependent activation of ERK is impaired in Synectin null (Gipc1−/−) or Nrp1cyto knock-in mice.4,5 This is thought to be attributable to prolonged exposure of VEGFR2-containing endosomes to PTP1b leading to a partial dephosphorylation of the VEGFR2 Y1175 site that is crucial for ERK activation. In vitro overexpression of PTP1b decreases VEGF-induced endothelial ERK activation,11 whereas impaired VEGF-induced ERK activation in synectin null endothelial cells can be restored by a knockdown of PTP1b expression. Finally, treatment of Gipc1−/− mice with a PTP1b inhibitor restored impaired arteriogenesis.5 These results suggest that PTP1b regulates the extent of VEGFR2 Y1175 site phosphorylation, thereby regulating the magnitude and duration of ERK activation. However, there is no direct genetic demonstration of the role of PTP1b in endothelial VEGF signaling, and it is not known how its deletion would affect embryonic vascular development or angiogenesis.
The current study was designed to address this gap. Endothelial cells derived from PtpN1ECKO mice demonstrated increased Y1175 VEGFR2 phosphorylation and enhanced ERK activation in response to VEGF-A165, consistent with the proposed role of PTP1b as a phosphatase controlling this site’s phosphorylation. At the same time, we observed no change in VEGF-driven Akt and p38-MAPK activation. The mechanism of VEGF-A-dependent activation of the former is not well established, but it is not dependent on Y1175 phosphorylation. The activation of the latter is dependent on Y1241 VEGFR2 phosphorylation,6 the site not controlled by PTP1b activity.4,5
Several assays, in vitro and in vivo, demonstrating increased endothelial sprouting, larger numbers of tip cells, faster wound healing, and more extensive neovascularization in Matrigel implants in PtpN1ECKO mice, are consistent with an enhanced angiogenic response. Accelerated blood flow recovery and increased arteriolar density in PtpN1ECKO mice in the hindlimb ischemia model and the rescue of impaired blood flow recovery in synectin null mice by endothelial PTP1b deletion demonstrate the role of PTP1b in regulation of arteriogenesis.
PTP1b activity is not specific to VEGFR2. Indeed, we observed increased IGF-induced ERK activation as well, a result consistent with the known role of this phosphatase in IGF receptor activation.24,25 At same time FGF signaling was not affected. One important difference between VEGF and IGF versus FGF signaling pathways is that whereas VEGFR2 undergoes clathrin-dependent endocytosis,26 FGFR1, the principle FGF receptor in endothelial cells, undergoes macropinocytic uptake.27 It is conceivable that differences in early trafficking of these receptors determine their susceptibility to PTP1b-induced dephosphorylation.
Another important consideration is that this study used an endothelial-specific deletion of PtpN1 because systemic inhibition of it activity may have affected the function of blood-derived macrophages, the cell type that is crucial to postnatal arteriogenesis. Finally, this study further supports the critical role played by endothelial cells in arteriogenesis,28 as selective manipulation of PTP1b expression in these cells had a profound effect on this process.
Overall, these studies illustrate that PTP1b-driven dephosphorylation of VEGFR2 is an important physiological regulator of vascular growth and emphasize the critical role endothelial VEGF signaling plays in angiogenesis and arteriogenesis.
Supplementary Material
CLINICAL PERSPECTIVE.
Vascular endothelial growth factor A (VEGF-A) is critical to formation of capillary (angiogenesis) and arterial (arteriogenesis) vasculature. VEGF-A accomplishes this by activating its key signaling receptor, VEGFR2. A failure to fully activate this receptor is likely the leading cause of poor arteriogenesis in patients with advanced atherosclerosis and diabetes mellitus. Recent studies have demonstrated that a full VEGFR2 activation requires its uptake into endothelial cells on ligand binding (endocytosis) and subsequent intracellular trafficking. In the absence of the latter step, the receptor is deactivated by an intracellular phosphotyrosine phosphatase 1b (PTP1b). This study shows that a disruption of PTP1b expression in endothelial cells leads to increased VEGFR2 activation that is strong enough to overcome poor arteriogenesis in a genetic model of reduced VEGFR2 activation. These findings demonstrate the critical role played by PTP1b in regulation of VEGF signaling and identify it as a potential new therapeutic target. The latter can be particularly important in patients with impaired VEGFR2 activation as frequently seen in advanced coronary and peripheral arterial disease.
Acknowledgments
The authors would like to thank Rita Webber, Nicole Copeland and Wayne Evangelisti for help with the mouse colony.
Footnotes
Disclosures
None.
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA.114.009683/-/DC1.
References
- 1.Simons M An inside view: VEGF receptor trafficking and signaling. Physiology (Bethesda). 2012;27:213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deng Y, Larrivée B, Zhuang ZW, Atri D, Moraes F, Prahst C, Eichmann A, Simons M. Endothelial RAF1/ERK activation regulates arterial morphogenesis. Blood. 2013;121:3988–96, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hong CC, Peterson QP, Hong JY, Peterson RT. Artery/vein specification is governed by opposing phosphatidylinositol-3 kinase and MAP kinase/ERK signaling. Curr Biol. 2006;16:1366–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lanahan A, Zhang X, Fantin A, Zhuang Z, Rivera-Molina F, Speichinger K, Prahst C, Zhang J, Wang Y, Davis G, Toomre D, Ruhrberg C, Simons M. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev Cell. 2013;25:156–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, Carmeliet P, Simons M. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell. 2010;18:713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437:169–183. [DOI] [PubMed] [Google Scholar]
- 7.Shibuya M Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem. 2013;153:13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakayama M, Nakayama A, van Lessen M, Yamamoto H, Hoffmann S, Drexler HC, Itoh N, Hirose T, Breier G, Vestweber D, Cooper JA, Ohno S, Kaibuchi K, Adams RH. Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat Cell Biol. 2013;15:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, Acker-Palmer A. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465:487–491. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, Barberis A, Benjamin LE, Mäkinen T, Nobes CD, Adams RH. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura Y, Patrushev N, Inomata H, Mehta D, Urao N, Kim HW, Razvi M, Kini V, Mahadev K, Goldstein BJ, McKinney R, Fukai T, Ushio-Fukai M. Role of protein tyrosine phosphatase 1B in vascular endothelial growth factor signaling and cell-cell adhesions in endothelial cells. Circ Res. 2008;102:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bono F, De Smet F, Herbert C, De Bock K, Georgiadou M, Fons P, Tjwa M, Alcouffe C, Ny A, Bianciotto M, Jonckx B, Murakami M, Lanahan AA, Michielsen C, Sibrac D, Dol-Gleizes F, Mazzone M, Zacchigna S, Herault JP, Fischer C, Rigon P, Ruiz de Almodovar C, Claes F, Blanc I, Poesen K, Zhang J, Segura I, Gueguen G, Bordes MF, Lambrechts D, Broussy R, van de Wouwer M, Michaux C, Shimada T, Jean I, Blacher S, Noel A, Motte P, Rom E, Rakic JM, Katsuma S, Schaeffer P, Yayon A, Van Schepdael A, Schwalbe H, Gervasio FL, Carmeliet G, Rozensky J, Dewerchin M, Simons M, Christopoulos A, Herbert JM, Carmeliet P. Inhibition of tumor angiogenesis and growth by a small-molecule multi-FGF receptor blocker with allosteric properties. Cancer Cell. 2013;23:477–488. [DOI] [PubMed] [Google Scholar]
- 13.Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–924. [DOI] [PubMed] [Google Scholar]
- 14.Chittenden TW, Claes F, Lanahan AA, Autiero M, Palac RT, Tkachenko EV, Elfenbein A, Ruiz de Almodovar C, Dedkov E, Tomanek R, Li W, Westmore M, Singh JP, Horowitz A, Mulligan-Kehoe MJ, Moodie KL, Zhuang ZW, Carmeliet P, Simons M. Selective regulation of arterial branching morphogenesis by synectin. Dev Cell. 2006;10:783–795. [DOI] [PubMed] [Google Scholar]
- 15. Pitulescu ME, Schmidt I, Benedito R, Adams RH. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc. 2010;5:1518–1534. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez F, Vacaru A, Overvoorde J, den Hertog J. The receptor protein-tyrosine phosphatase, Dep1, acts in arterial/venous cell fate decisions in zebrafish development. Dev Biol. 2008;324:122–130. [DOI] [PubMed] [Google Scholar]
- 17.Spring K, Chabot C, Langlois S, Lapointe L, Trinh NT, Caron C, Hebda JK, Gavard J, Elchebly M, Royal I. Tyrosine phosphorylation of DEP-1/CD148 as a mechanism controlling Src kinase activation, endothelial cell permeability, invasion, and capillary formation. Blood. 2012;120:2745–2756. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi T, Takahashi K, St John PL, Fleming PA, Tomemori T, Watanabe T, Abrahamson DR, Drake CJ, Shirasawa T, Daniel TO. A mutant receptor tyrosine phosphatase, CD148, causes defects in vascular development. Mol Cell Biol. 2003;23:1817–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bäumer S, Keller L, Holtmann A, Funke R, August B, Gamp A, Wolburg H, Wolburg-Buchholz K, Deutsch U, Vestweber D. Vascular endothelial cell-specific phosphotyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood. 2006;107:4754–4762. [DOI] [PubMed] [Google Scholar]
- 20. Dominguez MG, Hughes VC, Pan L, Simmons M, Daly C, Anderson K, Noguera-Troise I, Murphy AJ, Valenzuela DM, Davis S, Thurston G, Yancopoulos GD, Gale NW. Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc Natl Acad Sci U S A. 2007;104:3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi M, Majumdar A, Li X, Adler J, Sun Z, Vertuani S, Hellberg C, Mellberg S, Koch S, Dimberg A, Koh GY, Dejana E, Belting HG, Affolter M, Thurston G, Holmgren L, Vestweber D, Claesson-Welsh L. VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nat Commun. 2013;4:1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winderlich M, Keller L, Cagna G, Broermann A, Kamenyeva O, Kiefer F, Deutsch U, Nottebaum AF, Vestweber D. VE-PTP controls blood vessel development by balancing Tie-2 activity. J Cell Biol. 2009;185:657–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matozaki T, Murata Y, Mori M, Kotani T, Okazawa H, Ohnishi H. Expression, localization, and biological function of the R3 subtype of receptor-type protein tyrosine phosphatases in mammals. Cell Signal. 2010;22:1811–1817. [DOI] [PubMed] [Google Scholar]
- 24. Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. [DOI] [PubMed] [Google Scholar]
- 26.Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174:593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elfenbein A, Lanahan A, Zhou TX, Yamasaki A, Tkachenko E, Matsuda M, Simons M. Syndecan 4 regulates FGFR1 signaling in endothelial cells by directing macropinocytosis. Sci Signal. 2012;5:ra36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moraes F, Paye J, Mac Gabhann F, Zhuang ZW, Zhang J, Lanahan AA, Simons M. Endothelial cell-dependent regulation of arteriogenesis. Circ Res. 2013;113:1076–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.