

 The purpose of this study was to investigate the cytotoxic effects of tributylphosphate (TBP) and tris (2-butoxyethyl) phosphate (TBEP) and to explore the underlying molecular mechanism focusing on oxidative stress, apoptosis, and cell cycle arrest.
The purpose of this study was to investigate the cytotoxic effects of tributylphosphate (TBP) and tris (2-butoxyethyl) phosphate (TBEP) and to explore the underlying molecular mechanism focusing on oxidative stress, apoptosis, and cell cycle arrest.
Abstract
The purpose of this study was to investigate the cytotoxic effects of tributylphosphate (TBP) and tris (2-butoxyethyl) phosphate (TBEP) and to explore the underlying molecular mechanism focusing on oxidative stress, apoptosis, and cell cycle arrest. The results showed that TBP and TBEP could inhibit cell proliferation, induce cellular reactive oxidative stress, and suppress the mitochondrial membrane potential in HepG2 cells. TBP and TBEP could induce both mitochondrial and p53 mediated apoptosis through different mitogen-activated protein kinase (MAPK) signal pathways. TBP activated the c-Jun N-terminal kinase (JNK) and extracellular regulated protein kinase (ERK1/2) pathways, while TBEP activated the JNK pathway. Furthermore, TBP and TBEP caused a concentration-dependent decrease of cyclin D1 expression and an increase of cyclin-dependent kinase (CDK) inhibitor proteins such as p21 and p27, resulting in significant cell cycle arrest in the G0/G1 phase. Taken together, the toxicity of TBP and TBEP on the HepG2 cells was associated with apoptosis and cell cycle arrest induced by oxidative stress.
Introduction
Flame retardants (FRs) are widely used around the world to meet the strict flammability standards for building materials, textiles and electric appliances. Certain additive FRs such as polybrominated diphenyl ethers (PBDE, penta-BDE, octa-BDE, and deca-BDE) and hexabromocyclododecane (HBCD) have been banned or voluntarily phased out in many countries due to their toxicity in organisms, persistence in the environment, and bioaccumulation in food chains.1,2 Organophosphate esters are introduced as their potential replacements and commonly known as organophosphate flame retardants (OPFRs). Presently, OPFRs have accounted for approximately 15% of the total amount of flame retardants employed around the world.3,4
OPFRs can migrate to the appliance surface over time and be emitted into the environment through volatilization, leaching and/or abrasion.3–5 The ubiquitous existence of OPFRs in various environmental media such as soil, water, sediment, and air might result in OPFR exposure through ingestion, inhalation, and dermal contact.4,6–8 Marklund et al. reported that adults and children in the sampled environments would be exposed up to 5.8 mg kg–1 day–1 and 57 mg kg–1 day–1 of total OPFRs, respectively.9 Practically, OPFRs and their metabolites have been detected at high concentrations in various environmental samples, including household dust, indoor air, drinking water, and sediment,4,8,10,11 as well as biotic samples, including fishes, mussels, birds, human breast milk, and human urine samples.12–14 It is reported that OPFRs can cause adverse effects to the environment and human health.15–17 Although the toxicity of OPFRs is comparatively low in mammals (as compared with PBDE or HBCD), recent studies have shown that OPFRs have the potential to cause oxidative stress, endocrine disruption, neurological disorder, and even carcinogenic effects in different organisms.18–22
Tributylphosphate (TBP) and tris (2-butoxyethyl) phosphate (TBEP) are usually used as plasticizers in unsaturated polyester resins, cellulose acetate, polyvinylchloride, acrylonitrile-butadienestyrene, synthetic rubber, floor wax, and rubber stoppers.23 TBP is one of the most abundant OPFRs in air and water environments,9,23 with the amount reaching 0.5–120 ng m–3 in the indoor air of domestic and occupational environments.23 The concentration of TBP at 19 Waste Water Processing Stations around the Pearl River Delta (China) ranged from 7.1 μg per kg to 804.9 μg per kg (dw).24 TBEP has been detected in freshwater fish and invertebrates in Lake Ontario, as well as European perch in Swedish lakes and herring gull eggs in the Great Lakes.13,25
TBEP had endocrine disrupting potential in human adrenocarcinoma (H295R) cells, increasing the concentration of both 17 b-estradiol and testosterone and the transcription of major steroidogenic genes.26In vitro reporter gene assays also showed that both TBP and TBEP had pregnane X receptor (PXR) agonistic activity, and TBP could antagonize the activity of estrogen receptors and androgen receptors.27 Sun et al. demonstrated the developmental neurotoxicity of TBEP in the early life stages of Japanese medaka.28 Now, TBEP has been classified as a suspected carcinogenic compound (IPCS, 2000).29 However, the toxicity and health risk data available for TBP and TBEP are still limited, and the mechanisms behind their toxicity are less well understood.
It was reported that OPFRs could be extensively metabolized by liver enzymes30 and increasingly accumulated in the liver depending on the exposure dose.31 The HepG2 cell is a suitable in vitro model system for chemical and environmental risk assessments.32,33 The expressions of antioxidant and xenobiotic metabolizing enzymes usually influenced by various chemical substances are similar in HepG2 cells and primary human hepatocytes.34 In this study, the HepG2 cell was used to investigate the in vitro hepatic toxicity of TBP and TBEP. The oxidative stress response is a cellular self-defending system upon stimulation of various types of exogenous chemical and contaminants. Mitochondria are sensitive targets for oxidative damage. Reactive oxygen species (ROS) can directly activate the mitochondrial permeability transition, leading to the loss of mitochondrial membrane potential (MMP) and mitochondrial membrane integrity, release of proapoptotic factors, suppression of cell proliferation and induction of cell apoptosis.35 Thus, cell proliferation, MMP, cell apoptosis and cell cycles were assayed in this study to evaluate the cytotoxicity of OPFRs in the HepG2 cells. Then, the expression of related proteins of different signaling pathways was measured to explore the potential mechanism. These results will provide further supplemental information for a better evaluation of the health risk assessment of OPFR exposure.
Materials and methods
Chemicals and reagents
Tributylphosphate (TBP, Cas: 126-73-8) and tris (2-butoxyethyl) phosphate (TBEP, Cas: 78-51-3) were purchased from the Lab of Dr Ehrenstorfer (Augsburg, Germany). Dulbecco's modified Eagle's medium (DMEM), D-Hank's buffer and fetal bovine serum (FBS) were obtained from Invitrogen (Paisley, UK). Cell counting kit-8 (CCK-8) was purchased from Dojindo (Kumamoto, Japan). Rhodamine 123 (Rh123) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). An annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit was purchased from BD Biosciences (Sparks, MD, USA). The mammalian protein extraction reagent (M-PER) and the bicinchoninic acid (BCA) protein assay kit were purchased from Thermo Fisher Scientific Inc (Waltham, MA, USA). The nitrocellulose membrane was purchased from Millipore (Darmstadt, Germany). All other reagents were from Sigma-Aldrich (Saint Louis, MO, USA) and were of analytical grade, if not stated otherwise.
All antibodies were purchased commercially as follows: anti-phosph-p38, anti-p38, anti-phosph-p44/42 extracellular regulated protein kinases 1/2 (ERK1/2), anti-p44/42 ERK1/2, anti-apoptotic protease activating factor-1 (Apaf 1), anti-Bcl-2 associated X protein (Bax), anti-caspase 3, anti-caspase 9, anti-B cell lymphoma 2 (Bcl-2), anti-p21, anti-p27, and anti-cyclin D1 (Cell Signaling, Beverly, MA, USA), anti-phosph-c-Jun N-terminal kinase 1/2/3 (JNK1/2/3) (T183 + T183 + T221), anti-JNK1/2/3 and anti-heme oxygenase-1 (HO-1) (Abcam, Cambridgeshire, UK), anti-p53 (Proteintech, Chicago, IL, USA), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Multisciences Biotechnology, Hangzhou, China), anti-rabbit IgG (H + L)/HRP and anti-mouse IgG (H + L)/HRP (Dingguo, China).
Cell culture and treatments
Human hepatoma cells (HepG2) were obtained from ATCC (American Type Culture Collection). HepG2 cells were cultured in DMEM (high glucose concentration) supplemented with 10% FBS, 0.33% sodium bicarbonate, and 100 units per ml penicillin, 0.1 mg ml–1 streptomycin. All cells were maintained at 37 °C in a 95 : 5 air/CO2 water-saturated atmosphere. For all experiments, cells were passaged every two days and used for experiments at an exponential growth phase. Before exposure to OPFRs, the cells were seeded into 35 mm culture dishes, six-well plates or 96-well plates and cultured for 24 hours. Then the fresh culture medium containing different concentrations of TBP or TBEP (50, 100 and 200 μM) was supplemented and co-incubated for different lengths of time. TBP and TBEP (200 mM) were dissolved in DMSO and the working solutions were freshly diluted in DMEM with 10% FBS. The final concentration of DMSO was 0.1% (v/v). The control cells were treated with the corresponding vehicle alone. All experiments were repeated three times, with at least three replicates for each sample.
Cell viability
CCK-8 assay, a sensitive colorimetric assay to determine the number of viable cells, was used to measure the cell proliferation. In brief, the HepG2 cells were seeded in 96-well plates with 6 replicates for 24 h to allow cell adhesion at a cell density of 3000 cells per well. Then the cells were exposed to various concentrations of TBP or TBEP (50, 100 and 200 μM) for 24 and 48 h. After OPFR treatment, the treatment medium was removed and 10 μL of the CCK-8 reagent in 90 μL of fresh DMEM per well was added, and then incubated at 37 °C for 30–60 min. Finally, the absorbance of each well was measured with a Multiscan Mk3 plate reader (Thermo Electron Corporation, Waltham, MA, USA) at 450 nm to ascertain the number of viable cells indirectly.
Measurement of the mitochondrial membrane potential (MMP)
The level of the MMP was determined by using the fluorescent dye Rh123, which can aggregate in the mitochondria and emit yellow green fluorescence. The uptake of Rh123 into the mitochondria is dependent on the mitochondrial membrane potential. The cells cultured in 35 mm culture dishes were treated with different concentrations of TBP or TBEP (50, 100 μM) for 24 h, then washed with warm D-Hank's solution, and 10 μM Rh123 buffer solution diluted with culture medium was added. After incubation at 37 °C for 30 min, the medium was removed, and the cells were washed three times with D-Hank's solution. The cells were observed under a fluorescence microscope (Olympus BX-51, Japan). The intensity of fluorescence was analyzed by using Image-pro plus 6.0 software and the fluorescence of the control group was normalized to 100%.
Cell apoptosis assay
An annexin V-FITC/PI detection kit was used to identify and quantify the apoptotic cells. After treatment with OPFRs for 24 h, the cells were rinsed twice with warm D-Hank's buffer, then harvested with 0.25% trypsin and washed twice with cold D-Hank's buffer. Apoptotic cells were stained according to the kit instructions. Approximately 106 cells from each sample were suspended in 400 μL binding buffer. The cells were incubated with 5 μL of annexin V-FITC at 37 °C for 15 min, and then with 10 μL of PI for 5 min. The stained cells were immediately analyzed by flow cytometry. Notably, the staining procedure was conducted in the dark.
Cell cycle assay
The cell cycle analysis in HepG2 was carried out using flow cytometry. In brief, the cells were plated in six-well plates at 105 cells per well for 24 h to allow cell adhesion, and then treated with OPFRs for 24 or 48 h. The cells were harvested with 0.25% trypsin and fixed with 70% ethanol at –20 °C for 24 h. The cell pellets were washed with D-Hank's buffer and resuspended in 1 mL of D-Hank's buffer containing 0.1 mg mL–1 RNase A at 37 °C for 30 min. Before analysis, PI was added to the cell samples to a final concentration of 50 μg mL–1. Finally, the cell samples were filtered with a 300 mesh nylon filter before the test.
Western blotting
Western blotting was performed according to our previous study.36 After treatment with OPFRs as described above, the HepG2 cells were washed with warm D-Hank's buffer, and lysed using M-PER to collect the total protein. Protein concentrations were determined by using the BCA protein assay using a Multiscan Mk3 plate reader (Thermo Electron Corporation, Waltham, MA, USA) at 590 nm. After denaturation, equal amounts of protein samples (60–80 μg) mixed with 6× Laemmli buffer were subjected to 8–12% sodiumdodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) at 70 V for 30 min and then at 100 V until the bromophenol blue dye reached the bottom, and transferred from the gel phase to a nitrocellulose membrane (Bio-Rad Hercules, CA, USA). The nitrocellulose membrane was then blocked with 5% non-fat milk in 3% Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h at room temperature. The membrane was incubated with primary antibodies overnight at 4 °C, washed with TBST, incubated with secondary antibodies for 1 h, and finally washed with TBST. The blots were visualized using chemiluminescence, and the optical densities of individual bands were quantitated using the Chemi-Imager digital imaging system (Alpha Innotech, San Leandro, CA, USA). GAPDH, a house-keeping protein was used as the internal reference.
Statistical analyses
All experiments were performed in triplicate and the data were expressed as means ± standard error of the mean (SEM). All data were subjected to one-way ANOVA analysis and Tukey's post hoc test for comparison. A p value <0.05 was considered to indicate the statistical significance.
Results
TBP and TBEP could inhibit the proliferation of HepG2 cells
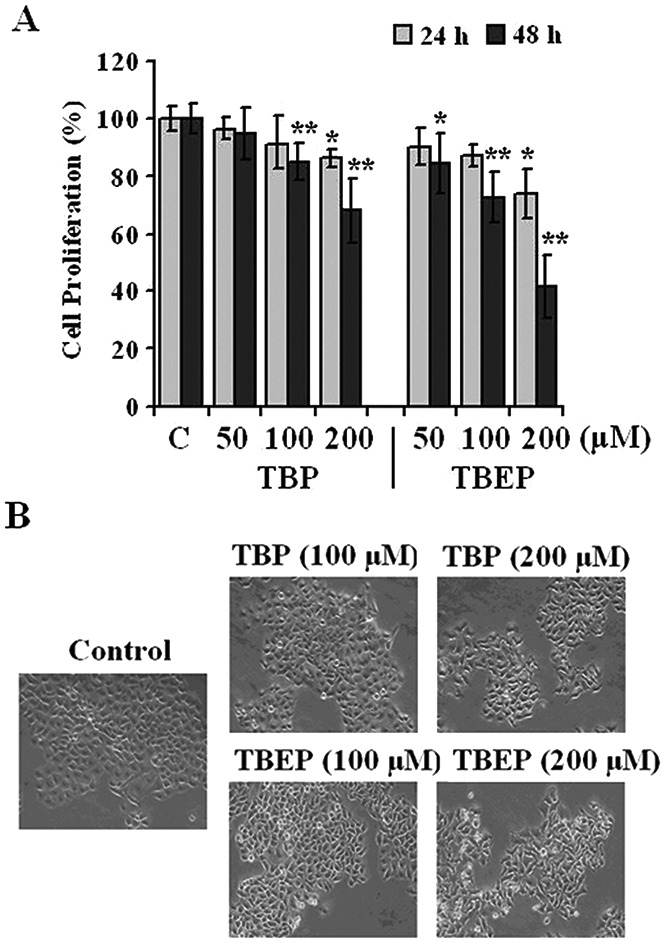
In the present study, it is shown that TBP and TBEP could inhibit the cell proliferation (Fig. 1A) in a concentration-dependent manner. The LC50 of TBP and TBEP is 299 and 151 μM for 48 h treatment in HepG2 cells, respectively. The morphological changes were observed using an inverted phase contrast microscope (Fig. 1B), showing obvious deformed cell shapes and disrupted cell junctions at a fairly high concentration range.
Fig. 1. Effects of TBP and TBEP on (A) cell proliferation and (B) morphological changes in HepG2 cells. (A) HepG2 cells were treated with different concentrations of TBP and TBEP (50, 100, and 200 μM) for 24 or 48 h. The cell proliferation was determined by using the CCK-8 assay. (C) Control groups. HepG2 cells without OPFR treatment. *p < 0.05, **p < 0.01 compared with the control (C) groups. (B) HepG2 cells were treated with different concentrations of TBP and TBEP (100 and 200 μM) for 48 h. The morphological changes were observed using an inverted phase contrast microscope (200×).

TBP and TBEP induced oxidative stress and reduced MMP
The outcomes of the immunoblot indicated that the exposure of TBP and TBEP resulted in a significant accumulation of HO-1 with a constant GAPDH expression (Fig. 2A). Furthermore, as shown in Fig. 2B, TBP and TBEP could significantly decrease the MMP (p < 0.05) level. After treatment with 200 μM of TBP and TBEP, the MMP reduced to 31.8% and 41.2%, respectively.
Fig. 2. Effect of TBP and TBEP on oxidative stress and MMP in HepG2 cells. HepG2 cells were treated with different concentrations of TBP and TBEP (50, 100, and 200 μM) for 24. (A) Protein expression of HO-1 was measured by using the western blot assay, GAPDH protein expression was used as the internal reference. (B) The level of mitochondrial membrane potential (MMP) was determined by using the fluorescent dye Rh123. (C) Control groups. HepG2 cells without OPFR treatment. *p < 0.05, **p < 0.01 compared with the control (C) groups.

TBP and TBEP induced cell apoptosis
To further clarify the effect and mechanism of apoptosis induced by OPFRs, the HepG2 cells were treated with different concentrations of OPFRs, and then the apoptosis was evaluated by flow cytometry. The results showed that the apoptosis rate increased to 1.7, 4.4, and 13.3 folds after 50, 100, and 200 μM of TBP treatment (p < 0.01). With an increase in the concentration, a gradual increase in the apoptosis rate was also observed in the HepG2 cells treated with TBEP (Fig. 3, p < 0.05).
Fig. 3. Effect of TBP and TBEP on apoptosis in HepG2 cells. HepG2 cells were treated with different concentrations of TBP and TBEP (50, 100, and 200 μM) for 24 h. Apoptotic cells were stained using the annexin V-FITC/PI detection kit, and then identified and quantified by flow cytometry. (C) Control groups. HepG2 cells without OPFR treatment. R2: dead cells, R3: late apoptotic cells, R4: living cells, R5: early apoptotic cells. The rate of apoptosis is the sum of the early and late apoptotic cells (R3 + R5). *p < 0.05, **p < 0.01 compared with the control (C) groups.

TBP and TBEP could activate the mitochondrial and p53 mediated apoptosis
Our results suggested that TBP and TBEP could increase the expression of proapoptotic effectors Bax and inhibit the expression of anti-apoptotic protein Bcl-2. Furthermore, the expressions of caspase-3, caspase-9, p53, and Apaf-1 were also enhanced and the expression of GAPHD had no change in the treatment groups of TBP and TBEP (Fig. 4, p < 0.05).
Fig. 4. Effect of TBP and TBEP on the protein expression of apoptosis in HepG2 cells. (A) HepG2 cells were treated with different concentrations of TBP and TBEP (50, 100, and 200 μM) for 24 h (for Apaf-1, only 100 μM was applied). Protein expressions of caspase-3, caspase-9, Bax, Bcl-2, p53, and Apaf-1 were measured by using the western blot assay, GAPDH protein expression was used as the internal reference. (B) Quantization of the expression of caspase-3, caspase-9, Bax, Bcl-2 and p53 proteins. (C) Control groups. HepG2 cells without OPFR treatment. *p < 0.05, **p < 0.01 compared with the control (C) groups.

TBP and TBEP could activate the mitogen-activated protein kinase (MAPK) pathway
The western blotting results also showed that TBP and TBEP could activate different MAPK signal pathways. TBP activated JNK and ERK1/2, and inhibited p38 MAPK in a concentration-dependent manner. TBEP could significantly increase the phosphorylation of JNK1/2/3, and decrease the phosphorylation of ERK1/2 and p38 MAPK (Fig. 5A and B, p < 0.05). Meanwhile, neither TBP nor TBEP had effects on the expression levels of GAPDH. These findings suggested that TBP and TBEP targeted different proteins and MAPK signal pathways to regulate the cell proliferation and apoptosis.
Fig. 5. Effect of TBP and TBEP on the MAPK protein expression. (A) HepG2 cells were treated with different concentrations of TBP and TBEP (50, 100, and 200 μM) for 24 h. The expressions of MAPK pathway proteins were measured by using the western blot assay, and the GAPDH protein expression was used as the internal reference. (B) Quantization of the expression of proteins. (C) Control groups. HepG2 cells without OPFR treatment. *p < 0.05, **p < 0.01 compared with the control (C) groups.

TBP and TBEP could induce the expressions of cell cycle related proteins to regulate cell cycle arrest
TBP and TBEP could also induce significant cell cycle arrest in the G0/G1 phase. After the treatment of 200 μM of TBP and TBEP for 48 h, the ratio of cells in the G0/G1 phase increased from 67.8% (control groups) to 76.8% and 76.5% (p < 0.05), respectively (p < 0.05) (Table 1).
Table 1. The distribution of the cell cycle phase after OPFR treatment.
| Groups | Time dose | 24 h |
48 h |
||||
| G0/G1 | S | G2/M | G0/G1 | S | G2/M | ||
| Control | 61.5 ± 0.1 | 24.9 ± 0.2 | 13.6 ± 0.3 | 67.8 ± 1.2 | 19.4 ± 0.3 | 12.8 ± 0.4 | |
| TBP | 50 μM | 66.3 ± 0.1* | 20.2 ± 0.9 | 13.5 ± 0.9 | 71.0 ± 0.5* | 18.0 ± 0.1 | 11.0 ± 0.3 |
| 100 μM | 67.3 ± 0.9** | 21.9 ± 1.1 | 10.8 ± 2.0 | 73.5 ± 1.0** | 16.0 ± 1.0 | 10.5 ± 0.1 | |
| 200 μM | 71.7 ± 0.1** | 17.3 ± 0.5 | 11.0 ± 0.4 | 76.8 ± 0.1** | 14.2 ± 0.7 | 9.0 ± 0.6 | |
| TBEP | 50 μM | 65.2 ± 0.1* | 21.9 ± 0.7 | 12.9 ± 0.6 | 74.4 ± 0.8** | 15.5 ± 0.7 | 10.1 ± 0.1 |
| 100 μM | 67.0 ± 1.1** | 21.3 ± 1.0 | 11.7 ± 0.1 | 75.55 ± 0.4** | 14.7 ± 0.1 | 9.8 ± 0.5 | |
| 200 μM | 68.1 ± 0.8** | 19.7 ± 0.1 | 12.2 ± 0.8 | 76.5 ± 0.5** | 14.9 ± 1.3 | 8.6 ± 0.8 | |
HepG2 cells were treated with different concentrations of TBP and TBEP (50, 100, and 200 μM) for 24 or 48 h. The proportion of the cell cycle phase was analyzed using flow cytometry. *p < 0.05, **p < 0.01 compared with the control groups.
In order to explore the underlying mechanism of OPFR-induced cell arrest, the effect of OPFR-treatment on the levels of specific cell cycle-regulatory proteins was evaluated. TBP and TBEP could cause a concentration-dependent decrease on the expression of cyclin D1 and an increase in the expression of CDK inhibitor proteins p21 and p27 (Fig. 6, p < 0.05).
Fig. 6. Effect of TBP and TBEP on the expressions of the cell cycle protein. (A) HepG2 cells were treated with different concentrations of TBP and TBEP (50, 100, and 200 μM) for 24 h. The expressions of the cell cycle protein were measured by using the western blot assay, and the GAPDH protein expression was used as the internal reference. (B) Quantization of the expression of proteins. (C) Control groups. HepG2 cells without OPFR treatment. *p < 0.05, **p < 0.01 compared with the control (C) groups.

Discussion
The toxic dose of OPFRs in organisms is at a higher level compared with the environmental concentration or previous FRs (for instance PBDEs). Kulkarni et al. observed that the DNA-damaging dose (500 μM) of TBP in the peripheral blood lymphocyte cells was threefold over the normal exposure level.37 The lowest observed effect concentration (LOEC, 50 μM) of TBEP to cause general toxicity in the developing zebrafish was significantly higher than the reported environmental concentration.38 Giraudo et al. also found that the mortality of 50% of individuals (LC50) due to TBEP in a 48 h acute toxicity testing in D. magna reached 147 mg L–1 (369 μM).39 Our previous study also showed that TBP and TBEP exposure (24 h) below 100 μM seems to have no remarkable proliferation-inhibiting effect on HepG2, human lung cancer (A549) and human colon cancer (Caco-2) cells.40
Reactive oxygen species (ROS) are well-established inducers of oxidative damage, which is closely related to cell viability, proliferation and cell death.41 ROS formation might be a pivotal mechanism behind the toxicity of multiple environmental chemical toxins, such as polychlorinated biphenyls (PCBs), PBDEs, organic phosphorus pesticides, etc.42,43 Research studies have shown that OPFRs could significantly elevate the oxidative stress in PC12 cells,19 and reduce the mitochondrial activity (concentrations ≥50 μM) in the MA-10 cell.44 The present study also found that TBP and TBEP induced the generation of the intracellular ROS in HepG2 cells in a dose-dependent manner. The level of ROS in the TBP or TBEP treatment groups (200 μM) increased over 20% as compared to the controls.39 The antioxidant genes are usually up-regulated in response to the oxidative stress to maintain a redox homeostasis. Heme oxygenase-1 (HO-1) is one of the major stress-responsive enzymes induced by oxidative stress.45 The increase of the HO-1 expression further confirmed the induction of ROS by OPFR exposure in this study (Fig. 2A). And the reduction of MMP suggested the mitochondrial damage caused by oxidative stress after TBP and TBEP treatment.
Apoptosis is involved in diverse biological processes such as development, cell differentiation, and proliferation, playing a central role in the elimination of unwanted, damaged, or infected cells in multicellular organisms. ROS generation is the triggering signal that leads to mitochondrial permeabilization and release of proapoptotic factors, such as caspase and Aparf-1. The loss of MMP is also a specific and early event in the mitochondrial pathway of apoptosis induced by various stimuli. Our results showed that OPFRs could reduce the MMP levels, and induce apoptosis in the HepG2 cells. This finding agreed with the report of Ta et al. that states that OPFRs could inhibit the cell growth and induce the apoptosis of PC12 cells in a dose-dependent manner.17
Two apoptotic pathways, the mitochondrial-dependent intrinsic pathway and the death receptor-mediated extrinsic pathway, have been elucidated to regulate the apoptosis process. Mitochondria-mediated apoptosis is a complicated procedure in which the cell undergoes an intrinsic program to determine the survival or death of cells in an appropriate cellular context. The mitochondria-mediated apoptosis is controlled by the key protein B cell lymphoma 2 (Bcl-2), which can directly bind with the activated proapoptotic effectors Bcl-2 associated X protein (BAX) and Bcl-2 homologous antagonist/killer (Bak) to regulate the mitochondrial membrane permeabilization and release of cytochrome c. And cytochrome c could interact with Apaf-1 to trigger caspase activation and consequent apoptosis. The interplay between the Bcl-2 and Bax complex, cooperating with the proapoptotic and antiapoptotic family members, tightly controlled the cell growth and apoptosis.46,47 P53 gene is a tumor suppressor gene which can regulate cell proliferation, cell growth, DNA repair, and apoptosis. The activation of the p53 pathway can lead to G1/S checkpoint control, cell cycle arrest, and apoptosis,48,49 which may partly explain the cell cycle and apoptosis changes induced by OPFRs in this study. The mechanism of apoptosis induced by OPFRs is so far not well-documented, but a number of studies have been conducted on apoptosis induced by other environmental chemicals. Abella et al. reported that non-dioxin-like (NDL)-PCBs 101, 153 and 180 could increase the caspase-3 activation and decrease the Bcl-2/Bax ratio to regulate the apoptosis.50 In this study, the decrease of the Bcl-2/Bax ratio and the elevation of caspase protein expression in the TBP and TBEP treatment groups suggested that TBP and TBEP could activate the mitochondrial apoptosis.
The MAPK signaling pathway constitutes a family of serine/threonine kinases involved in controlling various fundamental cellular processes, such as proliferation, differentiation, migration, metabolism, and apoptosis. However, the role of MAPK in cellular responses varies depending on the cell type and stimulus. The JNK family members (JNK1, JNK2, and JNK3) can phosphorylate the NH2 terminus of the transcription factor c-Jun, which is associated with the regulation of cell proliferation and apoptosis.51 The mitogen-activated protein kinase 4 (MEK4)/JNK/activator protein 1 (AP-1) pathway could regulate the intracellular stress response after exposure to nanomolar levels of PAHs in environmental mixtures.52 ERK1 and ERK2 activated by mitogens are also important regulators of differentiation and survival signals.51 TCDD treatments could induce a transient upshift in the ERK activity, followed by a decline, but a concomitant dramatic activation of p38 in RAW 264.7 cells to induce apoptosis.53 Our results also showed that TBP and TBEP could activate different MAPK signal pathways to regulate the cell proliferation and apoptosis.
On the other hand, cell proliferation is closely related to the rates of cell division, which consists of two main consecutive processes, DNA replication and segregation of replicated chromosomes. The cell division cycle was divided into two stages, mitosis (M phase) and interphase (including G1, S and G2 phase). Before commitment to DNA replication, cells enter a non-growing, non-proliferating resting G0 state.54 The replication of DNA occurs in the S phase, which is preceded by a gap called G1 preparing for DNA synthesis and then followed by a G2 gap preparing for mitosis. During the cell cycle, the cyclin protein periodically activates CDK, and further induces downstream processes. D-type cyclin proteins (D1, D2, and D3) can bind to CDK4 and CDK6, and the formed CDK-cyclin D complexes are essential for the cell entry to G1 from the G0 phase.55 Otherwise, the CDK activity can be counteracted by CDK inhibitors (CKI) which bind to CDK alone or to the CDK-cyclin complex to regulate the CDK activity. P21 and p27 belong to the Cip/Kip family, which can inhibit the G1 CDK-cyclin complexes.54 Our results showed that OPFRs could induce cell cycle arrest. The increase of p21 and p27 protein levels induced by OPFR treatment was expected to prevent the cell proliferation by mediating the cell cycle arrest in the G0/G1 and S phases by inhibiting the activation of cyclin A2 and cyclin D1. These consistent results had suggested that PCB29-pQ increased the S-phase cell population by down-regulating cyclins A/D1/E, CDK 2/4/6 and up-regulating p21/p27 protein expressions.56 Mattsson et al. also reported that PAHs induced the cell cycle arrest through decreased cyclin D1 and increased p21 responses.57 In addition, the activation of p53 induced by OPFR exposure in this study is also responsible for the G1 checkpoint block.
Conclusion
TBP and TBEP could induce cytotoxicity in HepG2 cells as evidenced by ROS generation, cell proliferation inhibition, apoptosis induction, mitochondrial membrane potential alteration, and cell cycle arrest. The toxic effects of TBP and TBEP are regulated by mitochondria- and p53-mediated apoptosis, and alteration of cyclin D1/CDK through different MAPK signaling pathways. To our knowledge, this is the first study to investigate the molecular mechanisms of apoptosis and cell cycle arrest induced by OPFRs. Nevertheless, more in-depth studies are required to confirm our results and to further clarify the underlying molecular mechanisms.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgments
This study was supported by grants from the National Natural Science Funds for Distinguished Young Scholars (41225013), the State Key Program of National Natural Science Foundation of China (41130752), the National Natural Science Foundation of China (No. 81072335; 41561144007), and Innovative Research Team in University (No. IRT13078).
References
- Covaci A., Harrad S., Abdallah M. A. E., Ali N., Law R. J., Herzke D., de Wit D. A. Environ. Int. 2011;37(2):532–556. doi: 10.1016/j.envint.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Darnerud P. O., Eriksen G. S., Johannesson T., Larsen P. B., Viluksela M. Environ. Health Perspect. 2001;109(Suppl. 1):49–68. doi: 10.1289/ehp.01109s149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Flame Retardant Association (EFRA), Flame Retardant Fact Sheet. Available at: http://www.cefic-efra.com, 2006.
- Reemtsma T., Quintana J. B., Rodil R., Lopez M. G., Rodrıguez I. Trends Anal. Chem. 2008;27:727–737. [Google Scholar]
- Marklund A., Andersson B., Haglund P. Chemosphere. 2003;53:1137–1146. doi: 10.1016/S0045-6535(03)00666-0. [DOI] [PubMed] [Google Scholar]
- Salamova A., Ma Y., Venier M., Hites R. Environ. Sci. Technol. 2014;1:8–14. [Google Scholar]
- Stapleton H. M., Sharma S., Getzinger G., Ferguson P. L., Gabriel M., Webster T. F., Blum A. Environ. Sci. Technol. 2012;46(24):13432–13439. doi: 10.1021/es303471d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Veen L., de Boer J. Chemosphere. 2012;88:1119–1153. doi: 10.1016/j.chemosphere.2012.03.067. [DOI] [PubMed] [Google Scholar]
- Marklund A., Andersson B., Haglund P. Environ. Sci. Technol. 2005;39(19):7423–7429. doi: 10.1021/es051013l. [DOI] [PubMed] [Google Scholar]
- Cao S. X., Zeng X. Y., Han S., Li H. R., Yu Z. Q., Sheng G. Y., Fu J. M. Environ. Toxicol. Chem. 2012;31:1478–1484. doi: 10.1002/etc.1872. [DOI] [PubMed] [Google Scholar]
- Li J., Yu N., Zhang B., Jin L., Li M., Hu M., Zhang X., Wei S., Yu H. Water Res. 2014;54:53–61. doi: 10.1016/j.watres.2014.01.031. [DOI] [PubMed] [Google Scholar]
- Kim J. W., Isobe T., Muto M., Tue N. M., Katsura K., Malarvannan G., Sudaryanto A., Chang K. H., Prudente M., Viet P. H., Takahashi S., Tanabe S. Chemosphere. 2014;116:91–97. doi: 10.1016/j.chemosphere.2014.02.033. [DOI] [PubMed] [Google Scholar]
- Sundkvist A. M., Olofsson U., Haglund P. J. Environ. Monit. 2010;12:943–951. doi: 10.1039/b921910b. [DOI] [PubMed] [Google Scholar]
- Van den Eede N., Neels H., Jorens P. G., Covaci A. J. Chromatogr., A. 2013;1303:48–53. doi: 10.1016/j.chroma.2013.06.042. [DOI] [PubMed] [Google Scholar]
- Fu J., Han J., Zhou B., Gong Z., Santos E. M., Huo X., Zheng W., Liu H., Yu H., Liu C. Environ. Sci. Technol. 2013;47(18):10574–10582. doi: 10.1021/es401265q. [DOI] [PubMed] [Google Scholar]
- Liu X., Ji K., Jo A., Moon H., Choi K. Aquat. Toxicol. 2013;134–135:104–111. doi: 10.1016/j.aquatox.2013.03.013. [DOI] [PubMed] [Google Scholar]
- Ta N., Li C., Fang Y., Liu H., Lin B., Jin H., Tian L., Zhang H., Zhang W. Z., Xi Z. Toxicol. Lett. 2014;227(3):164–171. doi: 10.1016/j.toxlet.2014.03.023. [DOI] [PubMed] [Google Scholar]
- Freudenthal R. I., Henrich R. Int. J. Toxicol. 2000;19:119–125. [Google Scholar]
- Dishaw L. V., Powers C. M., Ryde I. T., Roberts S. C., Seidler F. J., Slotkin T. A., Stapleton H. M. Toxicol. Appl. Pharmacol. 2011;256:281–289. doi: 10.1016/j.taap.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Liang K., Liu J., Yang L., Guo Y., Liu C., Zhou B. Aquat. Toxicol. 2013;126:207–213. doi: 10.1016/j.aquatox.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Wang Q., Lam J. C. W., Man Y. C., Lai N. L. S., Kwok K. Y., Guo Y. V., Lam P. K. S., Zhou B. Aquat. Toxicol. 2015;158:108–115. doi: 10.1016/j.aquatox.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Yuan L., Li J., Zha J., Wang Z. Environ. Pollut. 2016;208(Pt B):670–677. doi: 10.1016/j.envpol.2015.10.045. [DOI] [PubMed] [Google Scholar]
- Marklund A., Andersson B., Haglund P. J. Environ. Monit. 2005;7(8):814–819. doi: 10.1039/b505587c. [DOI] [PubMed] [Google Scholar]
- Zeng X., He L., Cao S., Ma S., Yu Z., Gui H., Sheng G., Fu J. Environ. Toxicol. Chem. 2014;33(8):1720–1725. doi: 10.1002/etc.2604. [DOI] [PubMed] [Google Scholar]
- Chen D., Letcher R. J., Chu S. J. Chromatogr., A. 2012;1220:169–174. doi: 10.1016/j.chroma.2011.11.046. [DOI] [PubMed] [Google Scholar]
- Liu X., Ji K., Choi K. Aquat. Toxicol. 2012;114–115:173–181. doi: 10.1016/j.aquatox.2012.02.019. [DOI] [PubMed] [Google Scholar]
- Kojima H., Takeuchi S., Van den Eede N., Covaci A. Toxicol. Lett. 2016;245:31–39. doi: 10.1016/j.toxlet.2016.01.004. [DOI] [PubMed] [Google Scholar]
- Sun L., Tan H., Peng T., Wang S., Xu W., Qian H., Jin Y., Fu Z. Environ. Toxicol. Chem. 2016;35(12):2931–2940. doi: 10.1002/etc.3477. [DOI] [PubMed] [Google Scholar]
- IPCS, Flame Retardants: Tris(2-Butoxyethyl) phosphate, tris (2-Ethylhexyl) phosphate and tetrakis(Hydroxymethyl) phosphonium salts, Environmental Health Criteria: volume 218, World Health Organization, Geneva, Switzerland, 2000. [Google Scholar]
- Van den Eede N., Erratico C., Exarchou V., Maho W., Neels H., Covaci A. Toxicol. Appl. Pharmacol. 2015;284(2):246–253. doi: 10.1016/j.taap.2015.01.021. [DOI] [PubMed] [Google Scholar]
- Farhat A., Crump D., Chiu S., Williams K. L., Letcher R. J., Gauthier L. T., Kennedy S. W. Toxicol. Sci. 2013;134:92–102. doi: 10.1093/toxsci/kft100. [DOI] [PubMed] [Google Scholar]
- Baderna D., Maggioni S., Boriani E., Gemma S., Molteni M., Lombardo A., Colombo A., Bordonali S., Rotella G., Lodi M., Benfenati E. Environ. Res. 2011;111(4):603–613. doi: 10.1016/j.envres.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Baderna D., Colombo A., Amodei G., Cantù S., Teoldi F., Cambria F., Rotella G., Natolino F., Lodi M., Benfenati E. Sci. Total Environ. 2013;463–464:790–801. doi: 10.1016/j.scitotenv.2013.06.088. [DOI] [PubMed] [Google Scholar]
- Wilkening S., Stahl F., Badera A. Drug Metab. Dispos. 2003;31(8):1035–1042. doi: 10.1124/dmd.31.8.1035. [DOI] [PubMed] [Google Scholar]
- Crompton M., Barksby E., Johnson N., Capano M. Biochimie. 2002;84(2–3):43–152. doi: 10.1016/s0300-9084(02)01368-8. [DOI] [PubMed] [Google Scholar]
- An J., Guo P., Shang Y., Zhong Y., Zhang X., Yu Y., Yu Z. Chemosphere. 2016;145:68–76. doi: 10.1016/j.chemosphere.2015.11.071. [DOI] [PubMed] [Google Scholar]
- Kulkarni S. V., Markad V. L., Melo J. S., D'Souza S. F., Kodam K. M. Appl. Microbiol. Biotechnol. 2014;98(2):919–929. doi: 10.1007/s00253-013-4938-2. [DOI] [PubMed] [Google Scholar]
- Han Z., Wang Q., Fu J., Chen H., Zhao Y., Zhou B., Gong Z., Wei S., Li J., Liu H., Zhang X., Liu C., Yu H. Aquat. Toxicol. 2014;150:175–181. doi: 10.1016/j.aquatox.2014.03.013. [DOI] [PubMed] [Google Scholar]
- Giraudo M., Douville M., Houde M. Chemosphere. 2015;132:159–165. doi: 10.1016/j.chemosphere.2015.03.028. [DOI] [PubMed] [Google Scholar]
- An J., Hu J. W., Shang Y., Zhong Y. F., Zhang X. Y., Yu Z. Q. J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng. 2016;51(11):980–988. doi: 10.1080/10934529.2016.1191819. [DOI] [PubMed] [Google Scholar]
- Foreman J., Demidchik V., Bothwell J. H., Mylona P., Miedema H., Torres M., Linstead P., Costa S., Brownlee C., Jones J. D., Davies J. M., Dolan L. Nature. 2003;422:442–446. doi: 10.1038/nature01485. [DOI] [PubMed] [Google Scholar]
- Valavanidis A., Vlahogianni T., Dassenakis M., Scoullos M. Ecotoxicol. Environ. Saf. 2006;64:178–189. doi: 10.1016/j.ecoenv.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Xu D., Li L., Liu L., Dong H., Deng Q., Yang X., Song E., Song Y. Environ. Toxicol. 2015;30(9):1063–1072. doi: 10.1002/tox.21979. [DOI] [PubMed] [Google Scholar]
- Schang G., Robaire B., Hales B. F. Toxicol. Sci. 2016;150(2):499–509. doi: 10.1093/toxsci/kfw012. [DOI] [PubMed] [Google Scholar]
- Bauer M., Bauer I. Antioxid. Redox Signaling. 2002;4:749–758. doi: 10.1089/152308602760598891. [DOI] [PubMed] [Google Scholar]
- Brown R. Br. Med. Bull. 1997;53:466–477. doi: 10.1093/oxfordjournals.bmb.a011624. [DOI] [PubMed] [Google Scholar]
- Wlodkowic D., Skommer J., Darzynkiewicz Z. Exp. Oncol. 2012;34(3):255–262. [PMC free article] [PubMed] [Google Scholar]
- Bi L., Yan X., Chen W., Gao J., Qian L., Qiu S. Integr. Cancer Ther. 2016;15(2):226–236. doi: 10.1177/1534735416637424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin M. F., Gueven N. Cell Death Differ. 2006;13:941–950. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- Abella V., Santoro A., Scotece M., Conde J., López-López V., Lazzaro V., Gómez-Reino J. J., Meli R., Gualillo O. Toxicol. Lett. 2015;234(1):13–19. doi: 10.1016/j.toxlet.2015.02.001. [DOI] [PubMed] [Google Scholar]
- Sun J., Nan G. J. Mol. Neurosci. 2016;59(1):90–98. doi: 10.1007/s12031-016-0717-8. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Dong S., Wang H., Tao S., Kiyama R. Environ. Pollut. 2016;213:809–824. doi: 10.1016/j.envpol.2016.03.050. [DOI] [PubMed] [Google Scholar]
- Park S. J., Yoon W. K., Kim H. J., Son H. Y., Cho S. W., Jeong K. S., Kim T. H., Kim S. H., Kim S. R., Ryu S. Y. Anticancer Res. 2005;25(4):2831–2836. [PubMed] [Google Scholar]
- Vermeulen K., Van Bockstaele D. R., Berneman Z. N. Cell Proliferation. 2003;36(3):131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr C. J. Cell. 1994;79:551–554. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- Song X., Li L., Shi Q., Lehmler H. J., Fu F., Su C., Xia X., Song E., Song Y. Chem. Res. Toxicol. 2015;28(11):2160–2169. doi: 10.1021/acs.chemrestox.5b00320. [DOI] [PubMed] [Google Scholar]
- Mattsson A., Lundstedt S., Stenius U. Environ. Mol. Mutagen. 2009;50(4):337–348. doi: 10.1002/em.20486. [DOI] [PubMed] [Google Scholar]