

Abstract
Despite hope that a cure was imminent when the causative gene was cloned nearly 30 years ago, cystic fibrosis (CF [MIM: 219700]) remains a life-shortening disease affecting more than 70 000 individuals worldwide. However, within the last 6 years the Food and Drug Administration's approval of Ivacaftor, the first drug that corrects the defective cystic fibrosis transmembrane conductance regulator protein [CFTR (MIM: 602421)] in patients with the G551D mutation, marks a watershed in the development of novel therapeutics for this devastating disease. Here we review recent progress in diverse research areas, which all focus on curing CF at the genetic, biochemical or physiological level. In the near future it seems probable that development of mutation-specific therapies will be the focus, since it is unlikely that any one approach will be efficient in correcting the more than 2000 disease-associated variants. We discuss the new drugs and combinations of drugs that either enhance delivery of misfolded CFTR protein to the cell membrane, where it functions as an ion channel, or that activate channel opening. Next we consider approaches to correct the causative genetic lesion at the DNA or RNA level, through repressing stop mutations and nonsense-mediated decay, modulating splice mutations, fixing errors by gene editing or using novel routes to gene replacement. Finally, we explore how modifier genes, loci elsewhere in the genome that modify CF disease severity, may be used to restore a normal phenotype. Progress in all of these areas has been dramatic, generating enthusiasm that CF may soon become a broadly treatable disease.
Introduction
Cystic fibrosis [CF (MIM: 219700)] is an autosomal recessive genetic disease caused by mutations in the gene encoding the CF transmembrane conductance regulator [CFTR (MIM: 602421)] protein. The CFTR protein is found in the cell membrane of epithelial cells and acts as an ion channel. Since the initial discovery of CFTR and the most common mutation F508del (p.Phe508del; legacy names for mutations are used here with the variant names in parentheses) in 1989, over 2000 variants have been identified. A systematic effort to distinguish disease-causing from neutral variants (CFTR2.org) has identified 327 mutations that cause CF (1) and have varying effects on CFTR protein function. Several mutations are now classified based on the functional abnormality seen within the protein (Table 1).
Table 1.
Classification of CFTR mutations based on the impact of the CFTR protein
| Class | How CFTR protein is affected | Examples |
|---|---|---|
| I | No functional CFTR protein is made | G542X, W1282X, 621 + 1G>T |
| II | CFTR protein is misfolded and does not reach the cell membrane | F508del, G85E |
| III | CFTR protein reaches the cell membrane but the channel gate does not open | G551D |
| IV | CFTR protein reaches the cell membrane but the channel does not move chloride efficiently | R117H, R334W, R347P |
| V | The CFTR protein is made and works properly but the amount is insufficient | 3849 + 10kb C>T, 2789 +5 G>A |
| VI | The CFTR protein is made and is in the correct location but has reduced stability and accelerated turnover | rF508del, 4326del, 4279insTC |
rF508 denotes the rescued protein after drug treatment.
CF is a multi-organ system disease though the major causes of morbidity and mortality arise from progressive lung disease. CFTR mutations lead to aberrant chloride transport in epithelial tissues resulting in altered hydration and pH of airway surface fluid, and viscous mucus. Within the lungs, this leads to an increased susceptibility to infections and inflammation, further damaging the airways (Fig. 1). This cycle of infection and inflammation results in bronchiectasis and end-stage lung disease, the leading cause of premature death in patients living with this disease. Until recently, therapeutic options for CF focused on the downstream effects once damage has occurred, in an effort to delay the progression of the disease.
Figure 1.

Pathophysiology of CF Disease. Process by which mutations in CFTR ultimately lead to reduced lung functional capacity. Therapeutic strategies currently target all phases of this process: Gene therapy and gene editing target the gene defect; RNA and protein therapies and modulating alternative channels aim to improve ion transport; early therapeutic paradigms targeted the downstream consequences of this process with airway clearance techniques, mucolytics, antibiotics and lung transplantation.
Over the past decade, significant therapeutic advances have been made in CF with new drugs targeting the defective protein to enhance its function. Depending on the functional abnormality, the therapeutic target may involve one or more steps. In situations where the CFTR protein is in the correct cellular location but the channel does not function optimally, so-called gating mutations, small molecule drugs have been developed to open the channel. A more complex situation exists when the CFTR protein is not properly processed and in these circumstances two drugs are necessary to enhance synthesis of functional protein and then further increase the channel opening once it is in the right location. The first drug ivacaftor (KalydecoTM) was used in a landmark trial of patients with G551D, a class III mutation. Ivacaftor therapy resulted in an average absolute increase of 10.5% in percent predicted forced expiratory volume in 1 s (FEV1pp), 55% reduction in pulmonary exacerbations, improved quality of life and a 48 mmol/L reduction in sweat chloride testing compared with placebo with relatively minimal side effects (2). (A diagnosis of CF correlates with sweat chloride levels of >60 mmol/L.) There is evidence for sustained benefits using ivacaftor in those individuals with gating mutations (3). Although there are country-specific differences in prevalence, less than 5% of the CF population worldwide carries this mutation. More recently, ivacaftor was approved for use in patients with class IV mutations that have residual CFTR function.
The most common mutation, F508del, became the focus of future clinical trials using combination therapy with both a corrector (which moves the protein to the cell surface) and a potentiator (which improves channel function, such as ivacaftor) due to the complex nature of the protein dysfunction with this mutation. The original corrector molecule, lumacaftor, in combination with ivacaftor (OrkambiTM), was shown to modestly improve lung function and reduce pulmonary exacerbations, however, significant side effects were seen with worsening shortness of breath and chest tightness, particularly in those with low baseline lung function as well as drug–drug interactions (4). A newer corrector molecule, tezacaftor, in combination with ivacaftor (SymdekoTM), appears to have a better side effect profile and showed improvements in lung function, reduction in pulmonary exacerbations in individuals who were homozygous for F508del or heterozygous with a second mutation which had residual function (5,6). Even more exciting are preliminary results from phase 1 and 2 studies using triple combination therapy for patients who are homozygous for F508del and those who carry one F508del mutation and a residual function mutation. These drugs utilize the combination therapy of ivacaftor-tezacaftor with a third molecule aimed at stabilizing the protein within the cell membrane. Phase 3 trials are ongoing with triple combination therapy, with results expected later this year (7).
Although there have been significant advances in CFTR protein modulator therapies, challenges still exist. First, not all patients with eligible mutations respond to treatment. Second, many mutations that lack treatments are rare and our understanding of their functional consequences is incomplete. Third, mutations resulting in premature termination of the protein due to stop codons, Class I mutations, are an even more complex problem to address than Class II or III mutations since no functional protein is made. Therefore, alternative approaches to therapy are needed beyond potentiators and correctors.
The objective of this review is primarily to focus on innovative therapies for CF, many of which are still at preclinical stages. These therapies build upon advances in genetics, genomics and recent insights into CF pathology to develop strategies to make CF a treatable disease for all patients, irrespective of their specific CFTR mutations.
Correcting the Gene
Despite the remarkable advances in developing new drugs to circumvent the impairment of the CFTR protein and chloride ion channel, there is still a need to correct mutations associated with loss or greatly reduced amounts of mature CFTR transcript or protein (e.g. Classes I and V). The most frequent of these errors include ‘stop/nonsense’ mutations that introduce a premature termination codon (PTC) into the CFTR mRNA, alterations in splice sites and gross lesions deleting parts of the locus. Here we consider targeted therapeutics for each of these mechanisms and also mutation agnostic approaches that will use gene editing to correct or replace the endogenous CFTR locus in somatic cells.
Targeted Therapeutics
Stop mutations and “read through” drugs
About 70 ‘stop’ mutations are recorded in CFTR2 (December 2017) (http://cftr2.org; date last accessed June 2018), the most common of these include G542X (p.Gly542X), R553X (p.Arg553X) and W1282X (p.Trp1282X) all of which have more than 1000 alleles documented in CFTR2. Therapeutic approaches targeting these mutations will likely be applicable more broadly if proven to be effective. Earlier observations showing that aminoglycoside antibiotics enhanced read through of nonsense mutations (8,9) led to hopes that these would provide valuable therapeutics, if renal and ototoxic side-effects could be overcome. This approach was evaluated for CF and also other common genetic disorders such as Duchenne Muscular Dystrophy [DMD (MIM: 310200)] and Spinal Muscular Atrophy [SMA (MIM: 253300)] among others. Ataluren, a small molecule read through therapy developed by PTC Therapeutics showed early promise, but failed to meet its endpoints of improved lung function and reduced pulmonary exacerbation in a later clinical trial (10). Several related aminoglycosides are included in pharmaceutical grade gentamycin and recent studies show that only the minor gentamycin B1 component has robust PTC read through activity, while the major gentamycins do not and further may repress B1 (11). Perhaps new aminoglycoside formulations will prove more effective. Of note, in addition to enhancing read through, therapeutics for stop mutations may also target nonsense-mediated decay, which profoundly impacts the levels of nonsense transcripts available for correction (reviewed in 12).
Splice mutations and antisense oligonucleotides
Another common class of errors in CFTR involves splicing of one of the 27 exons in the gene, either by reducing the efficiency of a splice donor or acceptor or through the creation of a novel cryptic splice site within an intron. Inspection of CFTR2 reveals four particularly common splice site mutations with more than 1000 alleles carrying one of the 621+1G>T (c.489+1G>T), 1717–1G>A (c.1585–1G>A), 2789+5G>A (c.2657+5G>A) and 3849+10C>T (c.3718–2477C>T) mutations. Again therapeutic approaches that build on progress in other common genetic disorders are in development for CF splicing mutations. For both SMA and DMD antisense oligonucleotides (ASOs) carefully targeted to the splice acceptor site of specific exons can induce their skipping to circumvent inclusion of an exon carrying a deleterious mutation. Chemical modification of the oligonucleotides reduces their intracellular degradation. Though the impact of ASOs in clinical trials for DMD were modest and variable (reviewed in 13), they are now an accepted therapeutic for type I SMA, where ASOs enhance inclusion of exon 7 of the Survival of Motor Neuron 2 [SMN2 (MIM: 601627)] transcript to produce a full-length functional SMN2 protein (reviewed in 14). To date the ASO approach to restore CFTR function is less well advanced and has not yet moved beyond preclinical studies. In vitro proof-of-concept studies showed that ASOs could effectively restore normal splicing of the 3849+10C>T mutation in CFTR. This error creates a cryptic splice site resulting in the inclusion of an extra 84 bp exon that causes a premature stop codon in the transcript (15). More recently evidence for an effective ASO approach to correct the 2789+5G>A mutation in vitro was reported (16). Of note, though the Food and Drug Administration (FDA) in the United States approved the use of ivacaftor for the treatment of five common splice mutations in CFTR, based on its potency in activating the residual normally spliced mRNA, anecdotal evidence suggests that it may not always be clinically effective. More recently the combination of tezacaftor and ivacaftor was also approved for these splice mutations.
Gene Editing
Targeting specific mutations
The possibility of somatic cell editing of specific CFTR mutations fuelled many in vitro approaches to achieve this, even prior to the widespread use of CRISPR/Cas9 protocols. Earlier work with zinc finger nucleases (17,18) showed that correcting mutations in the CFTR locus in airway cells and induced pluripotent stem cells was efficient. Similarly CRISPR/Cas9 correction of the F508del by homology-directed repair (HDR) was achieved in intestinal organoids from a CF patient (19). Another group of disease-associated mutations in CFTR, which are particularly amenable to CRISPR/Cas9 editing, are those deep within introns that create cryptic splice sites. These sites are utilized instead of normal splice sites in the gene and thus incorporate additional pseudo-exons into the CFTR transcript, many of which contain stop codons. Examples are the 1811+1.6kbA>G (c.1679+1634A>G) error and the 3849+10kbC>T (c.3718–2477C>T) mutation mentioned above. These variants were effectively corrected by nonhomologous end joining (NHEJ) using CRISPR/Cas9 with guide RNAs flanking the mutation to delete it from the genomic sequence (20). The same approach was effective for correcting an out-of-frame 5′ extension to an existing exon 3272–26A>G (c.3140–26A>G) (20). Restoration of normal CFTR splicing was observed in targeted airway cell lines. Clearly this approach would not be applicable to the more common pathogenic splice variants in CFTR such as 621+1G>T, 1717–1G>A, 2789+5G>A, since these errors lie very close to intron/exon boundaries.
Fixing all CFTR mutations
In light of the more than 2000 reported CFTR variations, a gene editing approach that can correct all mutations is an attractive goal. Since gene therapy approaches to deliver a CFTR cDNA transgene driven by a heterologous promoter to the airway have, until now, proven at best only marginally effective (with gradual loss of transgene expression being a common problem), alternative protocols are being developed. A particularly attractive approach is to use CRISPR/Cas9, zinc finger nuclease or TALEN technologies to introduce a complete CFTR cDNA into the endogenous CFTR genomic locus, within an intron close to the 5′ end of the gene. The major knowledge gap in this method is whether the regulation of the integrated transgene will be subject to the normal regulatory mechanisms of the locus. We, and others, have shown that CFTR has a complex tissue-specific control mechanism whereby intronic and intergenic enhancers (identified by open chromatin mapping, Fig. 2) are recruited to the gene promoter by chromatin looping to drive gene expression (21–24). This cell type-specific pattern of chromatin looping is clearly illustrated by circular chromatin conformation capture protocols, such as the 4C-seq shown in Figure 3. The CFTR promoter alone does not drive tissue-specific expression of the gene, so it may be essential for efficient transgene expression to not impact the recruitment of activating transcription factors, bound to distal enhancers, to the gene promoter. If the looping mechanisms are not impaired by insertion of a transgene into one of the upstream introns of the gene, then it may be possible to express a normally regulated CFTR transcript and protein while bypassing all other mutations located further downstream in the locus.
Figure 2.

Open chromatin/DNase I hypersensitive sites (DHS) across the CFTR locus in CF-relevant cell types. DNase-seq data from Caco2 (colon carcinoma), Calu3 (lung adenocarcinoma) cell lines and HTE (primary human tracheal epithelial cells) (30,31). DHS are seen at the CFTR promoter and at several cell-type selective sites (23). Airway sites: teal dashed box (−44kb, −35 kb from the CFTR translational start site). Intestinal sites: purple boxes (introns 1, 10, 11). Ubiquitous sites; black boxes (−80.1kb, +15.6 kb, +48.9 kb 3′ to the CFTR last coding base). Each lane shows combined data from two biological replicas of the cell type. Pink arrow denotes a potential location of cDNA insertion in intron 1 of CFTR.
Figure 3.

Cell-type-specific chromatin structure at the CFTR locus. 4C-seq profiles with a CFTR promoter viewpoint. Genomic location of CFTR and adjacent genes (chr 7) (top) and known cis-regulatory elements for the CFTR locus (below). The sites are named as in Figure 2. Open chromatin mapped by DNase-seq and 4C-seq data are shown for Caco2, Calu3 and skin fibroblasts. Representative data from one replica are shown. DNase-seq peaks show our data visualized on the UCSC genome browser. 4C-seq data are presented in alignment with the DNase-seq data and have two parts. The upper panel indicates the main trend of the contact profile using a 5-kb window size. Relative interactions are normalized to the strongest point (which is set to 1) within each panel. The lower panel is a domainogram with color-coded intensity values (see main text) to show relative interactions with window sizes varying from 2 to 50 kb. Arrows denote key data features: turquoise, known intestinal cis-regulatory elements in CFTR; peaks of interaction with CFTR promoter of known (black) cis-elements; orange, airway-selective regulatory elements and their interaction with the CFTR promoter in Calu3.
Common challenges of gene editing approaches
Significant challenges of all gene editing approaches include delivery, toxicity and potential off-target effects. These will not be considered in detail here as they are not unique to CF therapies, but deserve some comment. The development and refinement of highly efficient nanoparticle formulations (25) and adeno-associated virus (AAV) vectors (reviewed in 26) provide some optimism that even the mucus-obstructed CF lung epithelium may be accessible to these reagents. The active debate on the dangers of potential off-target gene editing by CRISPR/Cas9 is as relevant to CF as to many other genetic diseases. Perhaps more unique to CF is what fraction of cells in the airway epithelium need to be corrected in order to restore normal lung function and whether it will be possible to achieve this by focusing on stem cells in the lung. Also, it may be detrimental to induce high levels of CFTR expression in all cells in the lung epithelium, when expression of the gene is normally subject to tight cell-specific control mechanisms, including both activation (27,28) and repression (29).
Alternative Targets
An alternative strategy to repairing CFTR is to modulate other targets that can compensate for CFTR dysfunction. This mutation-agnostic approach could complement approved or advanced combination therapies that modulate F508del-CFTR (Introduction) by addressing the variation in response, and also provide a therapeutic strategy that benefits all patients.
The impact of CFTR loss on epithelial function in the airways (and several other tissues) has been reviewed elsewhere (32,33). As mentioned, abnormal CFTR function results in defective apical chloride and bicarbonate transport leading to altered airway surface liquid (ASL) that is dehydrated, reducing mucociliary clearance and innate immune defense mechanisms (34). The ASL is a fluid layer that covers the apical surface of airway epithelia and it has been proposed that modulating other ion channels, transporters and pumps that contribute to the delicate balance of ASL volume, ionic and nutrient content could compensate for loss of CFTR (33,35) (Fig. 4).
Figure 4.

Hypothetical graphic of candidate therapeutic targets for CF airway disease. Figure adapted from (32,35,36). CFTR dysfunction in CF leads to decreases in chloride and bicarbonate secretion, which in turn lower ASL height due to impaired anion and fluid secretion (32). The impaired fluid secretion leads to severe mucus plugging and susceptibility to infection (37). Proteins shown in red are CF modifiers identified by Genome-Wide Association Studies (GWAS) (38–41). SLC6A14 stimulation has been shown to impact Pseudomonas aeruginosa infection (42). Stimulation of SLC26A9 and or ANO-1 (anoctamin-1; also known as TMEM16A) may restore chloride secretion into the lumen and thus fluidity. Inhibition of ENaC, either directly or indirectly by targeting proteolytic enzymes may also restore ASL hydration and height by preventing reabsorption of sodium ions. Lastly, ATP12A, a subunit of the nongastric H+/K+-ATPase, impairs the functioning of innate immune defenses, likely due to an increase in H+ secretion, and thus, both ATP12A and SLC9A3 (aka. NHE3) could be inhibited in the apical membrane (and or stimulated in the basolateral membrane for SLC9A3) to prevent secretion of hydrogen ions into the ASL. Inhibition of SLC9A3 at the apical membrane is also beneficial due to reduced sodium reabsorption, which improves ASL fluidity. SLC, solute carrier; CA, carbonic anhydrase; ENaC, epithelial sodium channel; ASL, airway surface liquid; Arg+, arginine; NHE3, Na+/H+ exchanger 3; CFTR, cystic fibrosis transmembrane conductance regulator.
Targeting other known channels
Other channels that inhibit excess sodium absorption or improve chloride transport such as the epithelial sodium channel (ENaC) and the calcium-dependent chloride channel (TMEM16A a.k.a anoctamin-1 or ANO-1, respectively) have been proposed as candidate therapeutic targets (33,35,36). Indeed, a review of the literature to catalogue ongoing clinical (or pre-clinical) trials of modulators of alternative targets identified several companies focusing on the same target, ENaC (Table 2).
Table 2.
Currently known active pharmacological agents targeting alternatives to CFTR and undergoing testing in patients with CF
| Agent | Class | Company | References | Status |
|---|---|---|---|---|
| QUB-TL1 | Inhibition of ENaC-activating CAPs | – | (43) | Pre-clinical |
| NAP858 | Inhibition of ENaC-activating CAPs | – | (44) | Pre-clinical |
| SPX-101 | SPLUNC-1 peptide mimetic (stimulates ENaC) | Spyryx Biosciences | (45) | Phase 2 |
| GSK2225745 | ENaCα siRNA | GlaxoSmithKline Plc | (46) | Pre-clinical |
| IONIS-ENAC-2.5Rx | Antisense oligonucleotides (ASOs) against ENaCα | Ionis Pharmaceuticals | (47) | Pre-clinical |
| NVP-QBE170 | ENaC inhibitor | Novartis | (48) | Pre-clinical |
| AZD5634 | ENaC inhibitor | AstraZeneca | (49–51) | Phase 1b |
| ETD-001 | Long-acting ENaC inhibitor | Enterprise Therapeutics | (52) | Pre-clinical |
| QBW276 | ENaC inhibitor | Novartis | NA | Phase 2 |
| ET000516-A-2 | TMEM16A activator | Enterprise Therapeutics | (53) | Pre-clinical |
Table was compiled in May 2018 from the review of ClinicalTrials.gov and the US Cystic Fibrosis Foundation website (https://www.cff.org/Trials/Pipeline/) of ongoing clinical trials as well as industry-released public announcements and a PubMed search. CAP, channel activating protease. NA, not available.
ENaC is composed of three subunits (α, β and γ) (54) encoded by SCNN1A, SCNN1B, SCNN1G, and is regulated by SPLUNC-1 (short palate, lung and nasal epithelial clone-1), which protects ENaC from proteolytic cleavage, preventing its activation (55–57). In CF, ENaC contributes to dehydration of the ASL by mediating sodium absorption. More than 20 years ago, the ENaC blocker amiloride was tested in clinical trials by inhalation (58,59). These trials were not successful possibly due to the short half-life and potency of amiloride (60,61). More potent inhibitors such as benzamil and phenamil also proved unsuccessful (62). Later pre-clinical studies showed that early treatment (from birth) in β−ENaC-Tg mice (a CF mouse model) could prevent progression of CF lung disease (63). Direct inhibition of ENaC, however, led to hyperkalemia and sudden cardiac arrest because of effects on renal function (64). Current therapeutic paradigms aim to inhibit excessive ENaC function mainly through inhibition of channel activating proteases (CAPs) (Table 2). Broad inhibition of trypsin-like serine proteases (aprotinin and Camostat) is effective in attenuating ENaC and improving mucociliary clearance (65,66). However, more specific ENaC inhibitors are currently being evaluated (48–51). Candidate compounds that aim to inhibit ENaC-activating CAPs include QUB-TL1 (43) and NAP858 (44,67). Both compounds improve ASL height, mucociliary clearance and purport to delay CF lung disease. Another approach includes the use of a peptide mimetic of SPLUNC-1 (SPX-101), which was shown to greatly improve the survival of β-ENaC-transgenic mice (45). Other approaches targeting ENaC include nanoparticle formulations containing siRNA for ENaCα (46,68), and aerosolized ASOs (47).
TMEM16A is the channel responsible for the alternate Ca2+-activated Cl− channel (CaCC)-mediated secretion of chloride ions in the airways (69–71). Reports also show this channel is permeable to bicarbonate (72), and its stimulation may improve ASL fluidification and pH regulation (35,73,74). In the past, indirect TMEM16A stimulation using denufosol in CF patients failed to improve respiratory function in phase 3 clinical trials (75,76) likely due to the shorter half-life of the drug in vivo (74,77). Thus recent proposals aim to directly target the channel (78). While much progress has been made elucidating TMEM16A as an alternate chloride channel therapy in CF, more recent reports reveal an intricate connection and cross-talk of TMEM16A and CFTR. Specifically, ablation of TMEM16A in the airways (or gut) of mice obliterates CFTR-mediated chloride currents and induces a CF-like lung phenotype (79,80).
Alternative approaches such as the use of anionophores have been proposed as potential therapeutics as they confer anion channel activity (81). Cholapods (81) and decalins (e.g. bis-ureiododecalin) (82,83) show good promise in cell lines. In vivo testing, however, has not been carried out.
Targets identified through genome-wide studies
A potential new set of CF therapeutic targets arises through genome-wide association studies (GWAS) in CF population cohorts. These studies identify genetic loci that associate with the severity of disease in the presence of dysfunctional CFTR (i.e. modifier genes), irrespective of biological hypotheses. Notably, the clinical success rate of drugs in development is appreciably higher when there is human genetic evidence (from GWAS) that ties a target gene to a disease (84).
Several genome-wide studies (using exome sequencing and SNP arrays) have been carried out in CF. Modifier genes were identified that contribute to different phenotypes in the CF-affected organs and span the course of disease (Table 3). These include GWAS for intestinal obstruction at birth (meconium ileus) (39,40), infection with Pseudomonas aeruginosa (85,86), lung disease severity (87–89) and CF-related diabetes (38). These genome-wide studies identified common contributors [e.g. Solute Carrier Family 26 member 9 (SLC26A9) contributes to meconium ileus (40), CF-related diabetes (38) and CF lung disease (41)] and unique factors [e.g. EHF (ETS homologous factor)/APIP (Apoptotic peptidase activating factor 1 (APAF1) interacting protein) appears specific to lung disease severity] (Table 3). Consolidating evidence from studies of multiple CF phenotypes informs the potential broad or narrow impact should these be targeted as therapies, and provides insights into the differential relationships of modifiers with CFTR over developmental time and tissue (41).
Table 3.
Loci identified through genome-wide studies to contribute to CF co-morbidities and their functional characterization in CF
| Posited gene at associated locusa | GWAS/Exome-associated phenotypes | GWAS/Exome reference | Functional characterisation in CF |
|---|---|---|---|
| MUC4/20 | Lung function | (89) |
|
| SLC9A3 (a.k.a. NHE3) | Lung function, Meconium ileus | (40,89) |
|
| HLA-DRA | Lung function | (89) | |
| EHF | Lung function | (89) |
|
| AGTR2 | Lung function | (89) | |
| SLC6A14 (a.k.a. ATB0,+) | Lung function, Meconium ileus | (40,89) | |
| SLC26A9 | Meconium ileus, CF-related diabetes | (38,40) | |
| DCTN4 | PsA infection | (85) | |
| CAV2 | PsA infection | (86,126) |
|
| TMC6 | PsA infection | (86,126) |
|
| ATP12A | Meconium ileus | (39) |
|
PsA, Pseudomonas aeruginosa; DKO, double knockout; GWAS, genome-wide association study; GTEx, genotype-tissue expression project.
Based on evidence from published functional studies or associated protein-coding variation from exome analysis.
Interestingly, 6 of the 10 loci identified through GWAS contained genes suggested to contribute to ASL volume, pH or bacterial defence, with several having already been investigated functionally as candidates contributing to CF including SLC26A9, Solute Carrier Family 6 Member 14 (SLC6A14), Solute Carrier Family 9 Member 3 (SLC9A3) and ATPase H+/K+ Transporting Non-Gastric Alpha2 Subunit (ATP12A) (Table 3). Of note, GWAS did not provide association evidence for the ENaC subunits (SCNN1A, SCNN1B and SCNN1G) or TMEM16A.
SLC26A9 is an anion chloride channel that contributes to constitutive apical chloride conductance and enhances cAMP-regulated CFTR currents (90–92). It has been suggested to partially compensate for loss of CFTR channel function in the intestine in in vivo studies of Cftr−/− mice (93). In genome-wide studies of CF patients with severe CF-causing CFTR genotypes including F508del homozygosity, the same noncoding variation in the SLC26A9 gene is associated with meconium ileus (40), CF-related diabetes (38) and early exocrine pancreatic disease (94). The role of SLC26A9 in these early-onset phenotypes is presumed to be mediated through exocrine pancreatic damage in utero (39,94–96). SLC26A9 was not a modifier of CF lung disease when the majority of patients included were homozygous for F508del [(89); Fig. 5]. However, in untreated and Kalydeco-treated patients with CFTR gating mutations, and in CF F508del/F508del primary bronchial cultures treated with the CFTR corrector component of ORKAMBITM (lumacaftor or VX-809), there was a dose–response relationship with the improvement in CFTR function dependent on SLC26A9 SNP genotype (41,91). Bertrand et al. (91) show that SLC26A9 does not confer chloride currents in epithelial cells expressing F508del. Taken together, the working hypothesis is that in the CFTR-F508del lungs, the SLC26A9 benefit to CFTR function is only achieved alongside correction of CFTR-F508del (91,97). The interplay between SLC26A9 and other CFTR mutations requires investigation.
Figure 5.
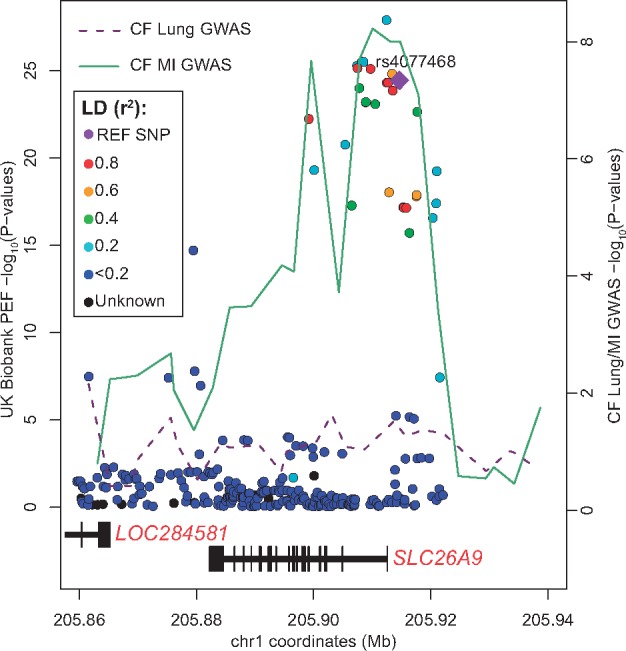
Genetic association of Peak Expiratory Flow (PEF) and Meconium Ileus (MI) co-localize at the SLC26A9 locus. The lines connect the lowest P-value at 3.2 kb windows across the genomic region from the GWAS of the International CF Gene Modifier Consortium for MI (green line) (40) and lung function (magenta line) (89). Dots in the figure represent the genetic association with PEF in 307 638 unrelated individuals from the UK Biobank resource [analyzed and available in the Global Biobank Engine, Stanford, CA (http://gbe.stanford.edu) accessed January 2018]. The marked SNP (purple diamond shaped) was the top SNP in the MI GWAS (40). These individuals are sampled across the United Kingdom and are aged between 40 and 69 at recruitment (98). PEF is measured in L/min and measures the maximal exhalation rate during an expiratory effort. The MI genetic association at the SLC26A9 locus mirrors the association pattern observed for PEF in the UK population, whereas there is not evidence of genetic association for lung function in patients with CF. The CF MI and lung GWAS P-values may be accessed from http://lab.research.sickkids.ca/strug/publications-software/; date last accessed June 2018.
Further supporting the complex relationship between CFTR and SLC26A9 in the lungs are the GWAS results for the airway obstruction lung function measure of peak expiratory flow (PEF) in individuals from the UK Biobank (98) (Fig. 5). The UK Biobank is a study following more than 500 000 volunteers (http://www.ukbiobank.ac.uk/; date last accessed June 2018). These individuals were sampled from across the United Kingdom and were between the ages of 40 and 69 at recruitment. Many of the phenotypes collected have been analyzed genome-wide and results made publicly available by the Global Biobank Engine (131) (http://gbe.stanford.edu; date last accessed June 2018). PEF was measured in 307 638 of the more than 500 000 participants. In these 307 638 individuals presumed to have functional CFTR, variants 5′ of SLC26A9 are associated with PEF (Fig. 5, colored dots; min p=1.27e-28 for rs1342062), and the association pattern mirrors the GWAS results for meconium ileus in CF (Fig. 5, solid green line). This is in stark contrast to the lack of association demonstrated for CF lung disease in individuals predominantly homozygous for F508del (Fig. 5, magenta dotted line) and supports the functional studies showing differential SLC26A9 interaction with wild-type CFTR and with F508del-CFTR in human bronchial epithelia (41,91). Furthermore, the SLC26A9 relationship with pulmonary function in the general population highlights the potential of SLC26A9 as a therapeutic target to enhance lung function in other pulmonary obstructive diseases; indeed in asthma SLC26A9 has demonstrated prevention of airway obstruction and mucus plugging (99). There is no association evidence for the ENaC subunits (SCNN1A, SCNN1B and SCNN1G) or for TMEM16A with PEF in the UK Biobank (http://gbe.stanford.edu).
The mechanism of action and direction that SLC6A14 should be modulated is less clear. The SLC6A14 locus was associated with both meconium ileus and CF lung disease (Table 3) in GWAS. Expression quantitative trait locus (eQTL) analysis in lung tissue provided by the Genotype Tissue Expression Consortium [GTEx; version v6p (100)] show the risk allele for both phenotypes to be associated with increased SLC6A14 messenger RNA (39). This appears inconsistent with results in primary bronchial epithelial cell cultures from patients with CF and ex vivo lungs and trachea explants of Slc6a14-/y mice showing greater attachment of Pseudomonas aeruginosa in the presence of SLC6A14 inhibitor α-methyl-l-tryptophan (α-MT), or greater bacterial burden in the absence of the transporter (42). SLC6A14 expression and amino acid uptake activity are upregulated via inflammatory cytokines (101–103), suggesting a role in reducing the viability of pathogens (42,102).
SLC9A3 and ATP12A inhibition may also improve ASL height and pH in the airways. It has been proposed that SLC9A3 inhibition at the apical membrane and/or stimulation at the basolateral membrane would help with ASL pH, and inhibition of sodium absorption would help with ASL height (35). Indeed, Cftr-null mice lacking one or more copies of Slc9a3 have improved fluidity of intestinal contents (104). Given the role of ATP12A in secreting protons to the ASL (105), drugs that aim to inhibit the nongastric H+/K+ ATPase are suggested (35).
Although the list of ongoing clinical (or pre-clinical) trials (Table 2) is certainly not exhaustive, efforts targeting the CF modifier loci (Table 3) were not identified, possibly due to early stages of investigation.
Concluding Remarks
There is significant allelic heterogeneity in the causal CF gene, CFTR. The different mutations pose a variety of challenges for restoring protein function, which cannot be addressed by a single therapeutic strategy. We reviewed the therapeutic paradigm for the F508del mutation, which now combines several molecules to address the folding, stability and gating limitations of the protein. Phase 3 trials show significant promise for these combination therapies that may enhance the average effect size of the approved drug ORKAMBITM, but many mutations remain for which there are no treatment options. Alternative strategies are required to ensure effective treatments are available for all patients. These alternatives include correcting, editing or replacing CFTR, or modulating alternative channels, transporters or pumps to compensate for dysfunctional CFTR. Though many elegant approaches to editing or circumventing CFTR mutations at the DNA or RNA level are currently being developed, some may be dependent on passing necessary ethical and regulatory hurdles before they become a reality. To our knowledge, therapies targeting alternatives to CFTR have largely focused on ENaC and TMEM16A, while several other relevant targets have been identified through genome-wide studies in CF. Genome-wide data from CF and population-based studies can be leveraged to guide future efforts to prioritize alternative therapeutic targets.
Acknowledgements
We thank the Rivas laboratory for making the Global Biobank Engine resource available, Bill Skach, Katherine Tuggle, Brian R. Davis and Daniela Rotin for helpful discussions and our funders.
Conflict of Interest statement: None declared.
Funding
(L.J.S.): Canadian Institutes of Health Research (CIHR MOP 258916), Cystic Fibrosis Canada (CFC; #2626), Genome Canada through the Ontario Genomics Institute (2018-OGI), and Cystic Fibrosis Foundation (STRUG17PO) (A.H.): Cystic Fibrosis Foundation (Harris14G0, Harris 16G0 and 17XX0); National Institutes of Health: R01HL094585, R01HD068901 and R01HL117843.
References
- 1. Sosnay P.R., Siklosi K.R., Van Goor F., Kaniecki K., Yu H., Sharma N., Ramalho A.S., Amaral M.D., Dorfman R., Zielenski J.. et al. (2013) Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet., 45, 1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ramsey B.W., Davies J., McElvaney N.G., Tullis E., Bell S.C., Dřevínek P., Griese M., McKone E.F., Wainwright C.E., Konstan M.W.. et al. (2011) A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med., 365, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sawicki G.S., McKone E.F., Pasta D.J., Millar S.J., Wagener J.S., Johnson C.A., Konstan M.W. (2015) Sustained benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am. J. Respir. Crit. Care Med., 192, 836–842. [DOI] [PubMed] [Google Scholar]
- 4. Wainwright C.E., Elborn J.S., Ramsey B.W., Marigowda G., Huang X., Cipolli M., Colombo C., Davies J.C., De Boeck K., Flume P.A.. et al. (2015) Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med., 373, 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Taylor-Cousar J.L., Munck A., McKone E.F., van der Ent C.K., Moeller A., Simard C., Wang L.T., Ingenito E.P., McKee C., Lu Y.. et al. (2017) Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N. Engl. J. Med., 377, 2013–2023. [DOI] [PubMed] [Google Scholar]
- 6. Rowe S.M., Daines C., Ringshausen F.C., Kerem E., Wilson J., Tullis E., Nair N., Simard C., Han L., Ingenito E.P.. et al. (2017) Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N. Engl. J. Med., 377, 2024–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martiniano S.L., Toprak D., Ong T., Zemanick E.T., Daines C.L., Muhlebach M.S., Esther C.R. Jr, Dellon E.P. (2018) Highlights from the 2017 North American Cystic Fibrosis Conference. Pediatr. Pulmonol, 53, 979–986. [DOI] [PubMed] [Google Scholar]
- 8. Howard M., Frizzell R.A., Bedwell D.M. (1996) Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med., 2, 467–469. [DOI] [PubMed] [Google Scholar]
- 9. Wilschanski M., Yahav Y., Yaacov Y., Blau H., Bentur L., Rivlin J., Aviram M., Bdolah-Abram T., Bebok Z., Shushi L.. et al. (2003) Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N. Engl. J. Med., 349, 1433–1441. [DOI] [PubMed] [Google Scholar]
- 10. Aslam A., Jahnke N., Remmington T., Southern K.W. (2017) Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis. Paediatr. Respir. Rev., 24, 32–34. [DOI] [PubMed] [Google Scholar]
- 11. Baradaran-Heravi A., Niesser J., Balgi A.D., Choi K., Zimmerman C., South A.P., Anderson H.J., Strynadka N.C., Bally M.B., Roberge M. (2017) Gentamicin B1 is a minor gentamicin component with major nonsense mutation suppression activity. Proc. Natl. Acad. Sci. U S A, 114, 3479–3484. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Linde L., Kerem B. (2011) Nonsense-mediated mRNA decay and cystic fibrosis. Methods Mol. Biol., 741, 137–154. [DOI] [PubMed] [Google Scholar]
- 13. Aartsma-Rus A., Straub V., Hemmings R., Haas M., Schlosser-Weber G., Stoyanova-Beninska V., Mercuri E., Muntoni F., Sepodes B., Vroom E.. et al. (2017) Development of exon skipping therapies for duchenne muscular dystrophy: a critical review and a perspective on the outstanding issues. Nucleic Acids Ther., 27, 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bowerman M., Becker C.G., Yanez-Munoz R.J., Ning K., Wood M.J.A., Gillingwater T.H., Talbot K., Consortium U.S.R. (2017) Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis. Model. Mech., 10, 943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Friedman K.J., Kole J., Cohn J.A., Knowles M.R., Silverman L.M., Kole R. (1999) Correction of aberrant splicing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by antisense oligonucleotides. J. Biol. Chem., 274, 36193–36199. [DOI] [PubMed] [Google Scholar]
- 16. Igreja S., Clarke L.A., Botelho H.M., Marques L., Amaral M.D. (2016) Correction of a cystic fibrosis splicing mutation by antisense oligonucleotides. Hum. Mutat., 37, 209–215. [DOI] [PubMed] [Google Scholar]
- 17. Lee C.M., Flynn R., Hollywood J.A., Scallan M.F., Harrison P.T. (2012) Correction of the DeltaF508 mutation in the cystic fibrosis transmembrane conductance regulator gene by zinc-finger nuclease homology-directed repair. Biores. Open Access, 1, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crane A.M., Kramer P., Bui J.H., Chung W.J., Li X.S., Gonzalez-Garay M.L., Hawkins F., Liao W., Mora D., Choi S.. et al. (2015) Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Rep., 4, 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schwank G., Koo B.K., Sasselli V., Dekkers J.F., Heo I., Demircan T., Sasaki N., Boymans S., Cuppen E., van der Ent C.K.. et al. (2013) Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell, 13, 653–658. [DOI] [PubMed] [Google Scholar]
- 20. Sanz D.J., Hollywood J.A., Scallan M.F., Harrison P.T. (2017) Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS One, 12, e0184009.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ott C.J., Blackledge N.P., Kerschner J.L., Leir S.H., Crawford G.E., Cotton C.U., Harris A. (2009) Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus. Proc. Natl. Acad. Sci. U S A, 106, 19934–19939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gheldof N., Smith E.M., Tabuchi T.M., Koch C.M., Dunham I., Stamatoyannopoulos J.A., Dekker J. (2010) Cell-type-specific long-range looping interactions identify distant regulatory elements of the CFTR gene. Nucleic Acids Res., 38, 4325–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang R., Kerschner J.L., Gosalia N., Neems D., Gorsic L.K., Safi A., Crawford G.E., Kosak S.T., Leir S.H., Harris A. (2016) Differential contribution of cis-regulatory elements to higher order chromatin structure and expression of the CFTR locus. Nucleic Acids Res., 44, 3082–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smith E.M., Lajoie B.R., Jain G., Dekker J. (2016) Invariant TAD boundaries constrain cell-type-specific looping interactions between promoters and distal elements around the CFTR locus. Am. J. Hum. Genet., 98, 185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schneider C.S., Xu Q., Boylan N.J., Chisholm J., Tang B.C., Schuster B.S., Henning A., Ensign L.M., Lee E., Adstamongkonkul P.. et al. (2017) Nanoparticles that do not adhere to mucus provide uniform and long-lasting drug delivery to airways following inhalation. Sci. Adv., 3, e1601556.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loring H.S., ElMallah M.K., Flotte T.R. (2016) Development of rAAV2-CFTR: history of the first rAAV vector product to be used in humans. Hum. Gene Ther. Methods, 27, 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Z., Leir S.H., Harris A. (2013) Immune mediators regulate CFTR expression through a bifunctional airway-selective enhancer. Mol. Cell. Biol., 33, 2843–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Z., Leir S.H., Harris A. (2014) Oxidative stress regulates CFTR gene expression in human airway epithelial cells through a distal antioxidant response element. Am. J. Respir. Cell. Mol. Biol., 33, 2843–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mutolo M.J., Leir S.H., Fossum S.L., Browne J.A., Harris A. (2018) A transcription factor network represses CFTR gene expression in airway epithelial cells. Biochem. J., 475, 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bischof J.M., Ott C.J., Leir S.H., Gosalia N., Song L., London D., Furey T.S., Cotton C.U., Crawford G.E., Harris A. (2012) A genome-wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax, 67, 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gillen A.E., Yang R., Cotton C.U., Perez A., Randell S.H., Leir S.H., Harris A. (2018) Molecular characterization of gene regulatory networks in primary human tracheal and bronchial epithelial cells. J. Cyst. Fibros., 17, 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saint-Criq V., Gray M.A. (2017) Role of CFTR in epithelial physiology. Cell. Mol. Life Sci., 74, 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haq I.J., Gray M.A., Garnett J.P., Ward C., Brodlie M. (2016) Airway surface liquid homeostasis in cystic fibrosis: pathophysiology and therapeutic targets. Thorax, 71, 284–287. [DOI] [PubMed] [Google Scholar]
- 34. Knowles M.R., Boucher R.C. (2002) Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Invest., 109, 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martin S.L., Saint-Criq V., Hwang T.C., Csanady L. (2018) Ion channels as targets to treat cystic fibrosis lung disease. J. Cyst. Fibros, 17, S22–S27. [DOI] [PubMed] [Google Scholar]
- 36. Mall M.A., Galietta L.J. (2015) Targeting ion channels in cystic fibrosis. J. Cyst. Fibros., 14, 561–570. [DOI] [PubMed] [Google Scholar]
- 37. Inglis S.K., Corboz M.R., Ballard S.T. (1998) Effect of anion secretion inhibitors on mucin content of airway submucosal gland ducts. Am. J. Physiol., 274, L762–L766. [DOI] [PubMed] [Google Scholar]
- 38. Blackman S.M., Commander C.W., Watson C., Arcara K.M., Strug L.J., Stonebraker J.R., Wright F.A., Rommens J.M., Sun L., Pace R.G.. et al. (2013) Genetic modifiers of cystic fibrosis-related diabetes. Diabetes, 62, 3627–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Strug L.J. (2016) Meta-GWAS identifies modifiers of meconium ileus susceptibility in cystic fibrosis. In The 30th Annual North American Cystic Fibrosis Conference, Orlando, FL. [Google Scholar]
- 40. Sun L., Rommens J.M., Corvol H., Li W., Li X., Chiang T.A., Lin F., Dorfman R., Busson P.F., Parekh R.V.. et al. (2012) Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nat. Genet., 44, 562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Strug L.J., Gonska T., He G., Keenan K., Ip W., Boelle P.Y., Lin F., Panjwani N., Gong J., Li W.. et al. (2016) Cystic fibrosis gene modifier SLC26A9 modulates airway response to CFTR-directed therapeutics. Hum. Mol. Genet., 25, 4590–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Di Paola M., Park A.J., Ahmadi S., Roach E.J., Wu Y.S., Struder-Kypke M., Lam J.S., Bear C.E., Khursigara C.M. (2017) SLC6A14 is a genetic modifier of cystic fibrosis that regulates Pseudomonas aeruginosa attachment to human bronchial epithelial cells. MBio, 8, e02073–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reihill J.A., Walker B., Hamilton R.A., Ferguson T.E., Elborn J.S., Stutts M.J., Harvey B.J., Saint-Criq V., Hendrick S.M., Martin S.L. (2016) Inhibition of protease-epithelial sodium channel signaling improves mucociliary function in cystic fibrosis airways. Am. J. Respir. Crit. Care Med., 194, 701–710. [DOI] [PubMed] [Google Scholar]
- 44. Douglas L., Ferguson T., Reihill J., Martin L. (2017) An Investigation into novel inhibitors of channel activating proteases with implicators for cystic fibrosis lung disease. In 14th European Cystic Fibrosis Society Basic Science Conference, Albufeira, Portugal.
- 45. Scott D.W., Walker M.P., Sesma J., Wu B., Stuhlmiller T.J., Sabater J.R., Abraham W.M., Crowder T.M., Christensen D.J., Tarran R. (2017) SPX-101 is a novel epithelial sodium channel-targeted therapeutic for cystic fibrosis that restores mucus transport. Am. J. Respir. Crit. Care Med., 196, 734–744. [DOI] [PubMed] [Google Scholar]
- 46. Clark K.L., Hughes S.A., Bulsara P., Coates J., Moores K., Parry J., Carr M., Mayer R.J., Wilson P., Gruenloh C.. et al. (2013) Pharmacological characterization of a novel ENaCalpha siRNA (GSK2225745) with potential for the treatment of cystic fibrosis. Mol. Ther. Nucleic Acids, 2, e65.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Crosby J.R., Zhao C., Jiang C., Bai D., Katz M., Greenlee S., Kawabe H., McCaleb M., Rotin D., Guo S.. et al. (2017) Inhaled ENaC antisense oligonucleotide ameliorates cystic fibrosis-like lung disease in mice. J. Cyst. Fibros, 16, 671–680. [DOI] [PubMed] [Google Scholar]
- 48. Coote K.J., Paisley D., Czarnecki S., Tweed M., Watson H., Young A., Sugar R., Vyas M., Smith N.J., Baettig U.. et al. (2015) NVP-QBE170: an inhaled blocker of the epithelial sodium channel with a reduced potential to induce hyperkalaemia. Br. J. Pharmacol., 172, 2814–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gardiner P., Malmgren A., Ersdal E., Goldwater R., Patel N. (2017) Enac inhibitor Azd5634 first in human trial reveals promising clinical profile for the treatment of cystic fibrosis. Am. J. Resp. Crit. Care, 195, A7306. [Google Scholar]
- 50. Libby E.F., Fortinberry H., Birket S., Astrand A., Patel N., Malmgren A., Tearney G.J., Rowe S. (2017) Enac inhibitor Azd5634 augments airway surface liquid and mucociliary transport in primary cystic fibrosis airway cells. Am J Resp Crit Care, 195, 6466. [Google Scholar]
- 51. Libby E.F., Fortinberry H.K., Adewale T., Fu I., Astrand A., Patel N., Malmgren A., Tearney G., Rowe S.M. (2017) Enac inhibitor Azd5634 increases mucociliary transport alone and in combination with lumacaftor/ivacaftor in primary Cf Hbe cells. Pediatr. Pulm., 52, S320–S320. [Google Scholar]
- 52. Enterprise Therapeutics. (2018) Identifying pathways regulating goblet cell metaplasia: phenotypic screening with bronchospheres. In European Cystic Fibrosis Society Basic Science Meeting, Loutraki, Greece.
- 53. Enterprise Therapeutics. (2016) Pharmacological characterisation of TMEM16A regulators. In The 13th European Cystic Fibrosis Basic Science Conference, Pisa, Italy.
- 54. Canessa C.M., Schild L., Buell G., Thorens B., Gautschi I., Horisberger J.D., Rossier B.C. (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature, 367, 463–467. [DOI] [PubMed] [Google Scholar]
- 55. Gaillard E.A., Kota P., Gentzsch M., Dokholyan N.V., Stutts M.J., Tarran R. (2010) Regulation of the epithelial Na+ channel and airway surface liquid volume by serine proteases. Pflugers Arch., 460, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Myerburg M.M., Harvey P.R., Heidrich E.M., Pilewski J.M., Butterworth M.B. (2010) Acute regulation of the epithelial sodium channel in airway epithelia by proteases and trafficking. Am. J. Respir. Cell. Mol. Biol., 43, 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Garcia-Caballero A., Rasmussen J.E., Gaillard E., Watson M.J., Olsen J.C., Donaldson S.H., Stutts M.J., Tarran R. (2009) SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc. Natl. Acad. Sci. U S A, 106, 11412–11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Graham A., Hasani A., Alton E.W., Martin G.P., Marriott C., Hodson M.E., Clarke S.W., Geddes D.M. (1993) No added benefit from nebulized amiloride in patients with cystic fibrosis. Eur. Respir. J., 6, 1243–1248. [PubMed] [Google Scholar]
- 59. Knowles M.R., Church N.L., Waltner W.E., Yankaskas J.R., Gilligan P., King M., Edwards L.J., Helms R.W., Boucher R.C. (1990) A pilot study of aerosolized amiloride for the treatment of lung disease in cystic fibrosis. N. Engl. J. Med., 322, 1189–1194. [DOI] [PubMed] [Google Scholar]
- 60. Hofmann T., Stutts M.J., Ziersch A., Ruckes C., Weber W.M., Knowles M.R., Lindemann H., Boucher R.C. (1998) Effects of topically delivered benzamil and amiloride on nasal potential difference in cystic fibrosis. Am. J. Respir. Crit. Care Med., 157, 1844–1849. [DOI] [PubMed] [Google Scholar]
- 61. Pons G., Marchand M.C., d’Athis P., Sauvage E., Foucard C., Chaumet-Riffaud P., Sautegeau A., Navarro J., Lenoir G. (2000) French multicenter randomized double-blind placebo-controlled trial on nebulized amiloride in cystic fibrosis patients. The Amiloride-AFLM Collaborative Study Group. Pediatr. Pulmonol., 30, 25–31. [DOI] [PubMed] [Google Scholar]
- 62. Hirsh A.J., Sabater J.R., Zamurs A., Smith R.T., Paradiso A.M., Hopkins S., Abraham W.M., Boucher R.C. (2004) Evaluation of second generation amiloride analogs as therapy for cystic fibrosis lung disease. J. Pharmacol. Exp. Ther., 311, 929–938. [DOI] [PubMed] [Google Scholar]
- 63. Zhou Z., Treis D., Schubert S.C., Harm M., Schatterny J., Hirtz S., Duerr J., Boucher R.C., Mall M.A. (2008) Preventive but not late amiloride therapy reduces morbidity and mortality of lung disease in betaENaC-overexpressing mice. Am. J. Respir. Crit. Care Med., 178, 1245–1256. [DOI] [PubMed] [Google Scholar]
- 64. O’Riordan T.G., Donn K.H., Hodsman P., Ansede J.H., Newcomb T., Lewis S.A., Flitter W.D., White V.S., Johnson M.R., Montgomery A.B.. et al. (2014) Acute hyperkalemia associated with inhalation of a potent ENaC antagonist: phase 1 trial of GS-9411. J. Aerosol. Med. Pulm. Drug Deliv., 27, 200–208. [DOI] [PubMed] [Google Scholar]
- 65. Bridges R.J., Newton B.B., Pilewski J.M., Devor D.C., Poll C.T., Hall R.L. (2001) Na+ transport in normal and CF human bronchial epithelial cells is inhibited by BAY 39-9437. Am. J. Physiol. Lung Cell. Mol. Physiol., 281, L16–L23. [DOI] [PubMed] [Google Scholar]
- 66. Coote K., Atherton-Watson H.C., Sugar R., Young A., MacKenzie-Beevor A., Gosling M., Bhalay G., Bloomfield G., Dunstan A., Bridges R.J.. et al. (2009) Camostat attenuates airway epithelial sodium channel function in vivo through the inhibition of a channel-activating protease. J. Pharmacol. Exp. Ther., 329, 764–774. [DOI] [PubMed] [Google Scholar]
- 67. Martin L., Reihill J., Douglas L., Ferguson T., Walker B. (2017) Detection and inhibition of ENaC-activating proteases associated with airway dehydration in COPD. Eur. Respir. J., 50, PA4933. [Google Scholar]
- 68. Manunta M.D.I., Tagalakis A.D., Attwood M., Aldossary A.M., Barnes J.L., Munye M.M., Weng A., McAnulty R.J., Hart S.L. (2017) Delivery of ENaC siRNA to epithelial cells mediated by a targeted nanocomplex: a therapeutic strategy for cystic fibrosis. Sci. Rep., 7, 700.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Caputo A., Caci E., Ferrera L., Pedemonte N., Barsanti C., Sondo E., Pfeffer U., Ravazzolo R., Zegarra-Moran O., Galietta L.J. (2008) TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science, 322, 590–594. [DOI] [PubMed] [Google Scholar]
- 70. Schroeder B.C., Cheng T., Jan Y.N., Jan L.Y. (2008) Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell, 134, 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yang Y.D., Cho H., Koo J.Y., Tak M.H., Cho Y., Shim W.S., Park S.P., Lee J., Lee B., Kim B.M.. et al. (2008) TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature, 455, 1210–1215. [DOI] [PubMed] [Google Scholar]
- 72. Jung J., Nam J.H., Park H.W., Oh U., Yoon J.H., Lee M.G. (2013) Dynamic modulation of ANO1/TMEM16A HCO3(-) permeability by Ca2+/calmodulin. Proc. Natl. Acad. Sci. U S A, 110, 360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sondo E., Caci E., Galietta L.J. (2014) The TMEM16A chloride channel as an alternative therapeutic target in cystic fibrosis. Int. J. Biochem. Cell Biol., 52, 73–76. [DOI] [PubMed] [Google Scholar]
- 74. Li H., Salomon J.J., Sheppard D.N., Mall M.A., Galietta L.J. (2017) Bypassing CFTR dysfunction in cystic fibrosis with alternative pathways for anion transport. Curr. Opin. Pharmacol., 34, 91–97. [DOI] [PubMed] [Google Scholar]
- 75. Accurso F.J., Moss R.B., Wilmott R.W., Anbar R.D., Schaberg A.E., Durham T.A., Ramsey B.W., Group T.-I.S. (2011) Denufosol tetrasodium in patients with cystic fibrosis and normal to mildly impaired lung function. Am. J. Respir. Crit. Care Med., 183, 627–634. [DOI] [PubMed] [Google Scholar]
- 76. Ratjen F., Durham T., Navratil T., Schaberg A., Accurso F.J., Wainwright C., Barnes M., Moss R.B.; TIGER-2 Study Investigator Group (2012) Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J. Cyst. Fibros, 11, 539–549. [DOI] [PubMed] [Google Scholar]
- 77. Mason S.J., Paradiso A.M., Boucher R.C. (1991) Regulation of transepithelial ion transport and intracellular calcium by extracellular ATP in human normal and cystic fibrosis airway epithelium. Br. J. Pharmacol., 103, 1649–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Namkung W., Yao Z., Finkbeiner W.E., Verkman A.S. (2011) Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J., 25, 4048–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Benedetto R., Ousingsawat J., Wanitchakool P., Zhang Y., Holtzman M.J., Amaral M., Rock J.R., Schreiber R., Kunzelmann K. (2017) Epithelial chloride transport by CFTR requires TMEM16A. Sci. Rep., 7, 12397.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lerias J., Pinto M., Benedetto R., Schreiber R., Amaral M., Aureli M., Kunzelmann K. (2018) Compartmentalized crosstalk of CFTR and TMEM16A (ANO1) through EPAC1 and ADCY1. Cell Signal., 44, 10–19. [DOI] [PubMed] [Google Scholar]
- 81. Valkenier H., Davis A.P. (2013) Making a match for Valinomycin: steroidal scaffolds in the design of electroneutral, electrogenic anion carriers. Acc. Chem. Res., 46, 2898–2909. [DOI] [PubMed] [Google Scholar]
- 82. Hussain S., Brotherhood P.R., Judd L.W., Davis A.P. (2011) Diaxial diureido decalins as compact, efficient, and tunable anion transporters. J. Am. Chem. Soc., 133, 1614–1617. [DOI] [PubMed] [Google Scholar]
- 83. Li H., Valkenier H., Judd L.W., Brotherhood P.R., Hussain S., Cooper J.A., Jurcek O., Sparkes H.A., Sheppard D.N., Davis A.P. (2016) Efficient, non-toxic anion transport by synthetic carriers in cells and epithelia. Nat. Chem., 8, 24–32. [DOI] [PubMed] [Google Scholar]
- 84. Nelson M.R., Tipney H., Painter J.L., Shen J., Nicoletti P., Shen Y., Floratos A., Sham P.C., Li M.J., Wang J.. et al. (2015) The support of human genetic evidence for approved drug indications. Nat. Genet., 47, 856–860. [DOI] [PubMed] [Google Scholar]
- 85. Emond M.J., Louie T., Emerson J., Zhao W., Mathias R.A., Knowles M.R., Wright F.A., Rieder M.J., Tabor H.K., Nickerson D.A.. et al. (2012) Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat. Genet., 44, 886–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Emond M.J., Louie T., Emerson J., Chong J.X., Mathias R.A., Knowles M.R., Rieder M.J., Tabor H.K., Nickerson D.A., Barnes K.C.. et al. (2015) Exome sequencing of phenotypic extremes identifies CAV2 and TMC6 as interacting modifiers of chronic Pseudomonas aeruginosa infection in cystic fibrosis. PLoS Genet., 11, e1005273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wright F.A., Strug L.J., Doshi V.K., Commander C.W., Blackman S.M., Sun L., Berthiaume Y., Cutler D., Cojocaru A., Collaco J.M.. et al. (2011) Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat. Genet., 43, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Soave D., Corvol H., Panjwani N., Gong J., Li W., Boelle P.Y., Durie P.R., Paterson A.D., Rommens J.M., Strug L.J.. et al. (2015) A joint location-scale test improves power to detect associated SNPs, gene sets, and pathways. Am. J. Hum. Genet., 97, 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Corvol H., Blackman S.M., Boelle P.Y., Gallins P.J., Pace R.G., Stonebraker J.R., Accurso F.J., Clement A., Collaco J.M., Dang H.. et al. (2015) Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat. Commun., 6, 8382.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bertrand C.A., Zhang R., Pilewski J.M., Frizzell R.A. (2009) SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J. Gen. Physiol., 133, 421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bertrand C.A., Mitra S., Mishra S.K., Wang X., Zhao Y., Pilewski J.M., Madden D.R., Frizzell R.A. (2017) The CFTR trafficking mutation F508del inhibits the constitutive activity of SLC26A9. Am. J. Physiol. Lung Cell. Mol. Physiol., 312, L912–L925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Salomon J.J., Spahn S., Wang X., Fullekrug J., Bertrand C.A., Mall M.A. (2016) Generation and functional characterization of epithelial cells with stable expression of SLC26A9 Cl- channels. Am. J. Physiol. Lung Cell. Mol. Physiol., 310, L593–L602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liu X., Li T., Riederer B., Lenzen H., Ludolph L., Yeruva S., Tuo B., Soleimani M., Seidler U. (2015) Loss of Slc26a9 anion transporter alters intestinal electrolyte and HCO3(-) transport and reduces survival in CFTR-deficient mice. Pflugers Arch., 467, 1261–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Miller M.R., Soave D., Li W., Gong J., Pace R.G., Boelle P.Y., Cutting G.R., Drumm M.L., Knowles M.R., Sun L.. et al. (2015) Variants in solute carrier SLC26A9 modify prenatal exocrine pancreatic damage in cystic fibrosis. J. Pediatr., 166, 1152–1157 e1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Soave D., Miller M.R., Keenan K., Li W., Gong J., Ip W., Accurso F., Sun L., Rommens J.M., Sontag M.. et al. (2014) Evidence for a causal relationship between early exocrine pancreatic disease and cystic fibrosis-related diabetes: a Mendelian randomization study. Diabetes, 63, 2114–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hart N.J., Aramandla R., Poffenberger G., Fayolle C., Thames A.H., Bautista A., Spigelman A.F., Babon J.A.B., DeNicola M.E., Dadi P.K.. et al. (2018) Cystic fibrosis-related diabetes is caused by islet loss and inflammation. JCI Insight, 3, e98240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sato Y., Robert R., Thomas D.Y., Hanrahan J.W. (2017) SLC26A9 is prematurely degraded along with misfolded F508del-CFTR. In the 14th European Cystic Fibrosis Society, Albufeira, Portugal.
- 98. Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J.. et al. (2017) Genome-wide genetic data on 500, 000 UK Biobank participants. bioRxiv. doi:10.1101/166298.
- 99. Anagnostopoulou P., Riederer B., Duerr J., Michel S., Binia A., Agrawal R., Liu X., Kalitzki K., Xiao F., Chen M.. et al. (2012) SLC26A9-mediated chloride secretion prevents mucus obstruction in airway inflammation. J. Clin. Invest., 122, 3629–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. GTEx Consortium. (2013) The Genotype-Tissue Expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zegarra-Moran O., Folli C., Manzari B., Ravazzolo R., Varesio L., Galietta L.J. (2004) Double mechanism for apical tryptophan depletion in polarized human bronchial epithelium. J. Immunol., 173, 542–549. [DOI] [PubMed] [Google Scholar]
- 102. Gorrieri G., Scudieri P., Caci E., Schiavon M., Tomati V., Sirci F., Napolitano F., Carrella D., Gianotti A., Musante I.. et al. (2016) Goblet cell hyperplasia requires high bicarbonate transport to support mucin release. Sci. Rep., 6, 36016.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Galietta L.J., Folli C., Marchetti C., Romano L., Carpani D., Conese M., Zegarra-Moran O. (2000) Modification of transepithelial ion transport in human cultured bronchial epithelial cells by interferon-gamma. Am. J. Physiol. Lung Cell. Mol. Physiol., 278, L1186–L1194. [DOI] [PubMed] [Google Scholar]
- 104. Bradford E.M., Sartor M.A., Gawenis L.R., Clarke L.L., Shull G.E. (2009) Reduced NHE3-mediated Na+ absorption increases survival and decreases the incidence of intestinal obstructions in cystic fibrosis mice. Am. J. Physiol. Gastrointest. Liver Physiol., 296, G886–G898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Shah V.S., Meyerholz D.K., Tang X.X., Reznikov L., Abou Alaiwa M., Ernst S.E., Karp P.H., Wohlford-Lenane C.L., Heilmann K.P., Leidinger M.R.. et al. (2016) Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science, 351, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ma J., Rubin B.K., Voynow J.A. (2017) Mucins, mucus, and goblet cells. Chest, doi:10.1016/j.chest.2017.11.008. [DOI] [PubMed]
- 107. Schultheis P.J., Clarke L.L., Meneton P., Miller M.L., Soleimani M., Gawenis L.R., Riddle T.M., Duffy J.J., Doetschman T., Wang T.. et al. (1998) Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat. Genet., 19, 282–285. [DOI] [PubMed] [Google Scholar]
- 108. Donowitz M., Li X. (2007) Regulatory binding partners and complexes of NHE3. Physiol. Rev., 87, 825–872. [DOI] [PubMed] [Google Scholar]
- 109. O’Neal W.K., Gallins Paul., Pace R.G., Dang H., Wolf W.E., Jones L.C., Guo X.L., Zhou Y.-H., Madar V., Huang J.. et al. (2015) Gene expression in transformed lymphocytes reveals variation in endomembrane and HLA pathways modifying cystic fibrosis pulmonary phenotypes. Am. J. Hum. Genet., 96, 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Polineni D., Dang H., Gallins P.J., Jones L.C., Pace R.G., Stonebraker J.R., Commander L.A., Krenicky J.E., Zhou Y.H., Corvol H.. et al. (2018) Airway mucosal host defense is key to genomic regulation of cystic fibrosis lung disease severity. Am. J. Respir. Crit. Care Med., 197, 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Stolzenburg L.R., Yang R., Kerschner J.L., Fossum S., Xu M., Hoffmann A., Lamar K.M., Ghosh S., Wachtel S., Leir S.H.. et al. (2017) Regulatory dynamics of 11p13 suggest a role for EHF in modifying CF lung disease severity. Nucleic Acids Res., 45, 8773–8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Stanke F., van Barneveld A., Hedtfeld S., Wolfl S., Becker T., Tummler B. (2014) The CF-modifying gene EHF promotes p.Phe508del-CFTR residual function by altering protein glycosylation and trafficking in epithelial cells. Eur. J. Hum. Genet., 22, 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Fossum S.L., Mutolo M.J., Yang R., Dang H., O’Neal W.K., Knowles M.R., Leir S.H., Harris A. (2014) Ets homologous factor regulates pathways controlling response to injury in airway epithelial cells. Nucleic Acids Res., 42, 13588–13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Fossum S.L., Mutolo M.J., Tugores A., Ghosh S., Randell S.H., Jones L.C., Leir S.H., Harris A. (2017) Ets homologous factor (EHF) has critical roles in epithelial dysfunction in airway disease. J. Biol. Chem., 292, 10938–10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Joshi N., Darrah R., Mitchell A., Eastman J., Gopinath N., Jacono F., Drumm M. (2013) Alleles of the AGTR2 gene affect gene expression and airway mechanics in CF. Pediatr. Pulmonol., 48, 265.22528960 [Google Scholar]
- 116. Darrah R.J., Jacono F.J., Joshi N., Mitchell A.L., Sattar A., Campanaro C.K., Litman P., Frey J., Nethery D.E., Barbato E.S.. et al. (2018) AGTR2 absence or antagonism prevents cystic fibrosis pulmonary manifestations. J. Cyst. Fibros., doi: 10.1016/j.jcf.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Galietta L.J., Musante L., Romio L., Caruso U., Fantasia A., Gazzolo A., Romano L., Sacco O., Rossi G.A., Varesio L.. et al. (1998) An electrogenic amino acid transporter in the apical membrane of cultured human bronchial epithelial cells. Am. J. Physiol., 275, L917–L923. [DOI] [PubMed] [Google Scholar]
- 118. Loriol C., Dulong S., Avella M., Gabillat N., Boulukos K., Borgese F., Ehrenfeld J. (2008) Characterization of SLC26A9, facilitation of Cl(-) transport by bicarbonate. Cell. Physiol. Biochem., 22, 15–30. [DOI] [PubMed] [Google Scholar]
- 119. Ohana E., Yang D., Shcheynikov N., Muallem S. (2009) Diverse transport modes by the solute carrier 26 family of anion transporters. J. Physiol., 587, 2179–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chang M.H., Plata C., Sindic A., Ranatunga W.K., Chen A.P., Zandi-Nejad K., Chan K.W., Thompson J., Mount D.B., Romero M.F. (2009) Slc26a9 is inhibited by the R-region of the cystic fibrosis transmembrane conductance regulator via the STAS domain. J. Biol. Chem., 284, 28306–28318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Avella M., Loriol C., Boulukos K., Borgese F., Ehrenfeld J. (2011) SLC26A9 stimulates CFTR expression and function in human bronchial cell lines. J. Cell. Physiol., 226, 212–223. [DOI] [PubMed] [Google Scholar]
- 122. Ousingsawat J., Schreiber R., Kunzelmann K. (2012) Differential contribution of SLC26A9 to Cl(-) conductance in polarized and non-polarized epithelial cells. J. Cell. Physiol., 227, 2323–2329. [DOI] [PubMed] [Google Scholar]
- 123. Karki S., Tokito M.K., Holzbaur E.L. (2000) A dynactin subunit with a highly conserved cysteine-rich motif interacts directly with Arp1. J. Biol. Chem., 275, 4834–4839. [DOI] [PubMed] [Google Scholar]
- 124. Schroder J.M., Schneider L., Christensen S.T., Pedersen L.B. (2007) EB1 is required for primary cilia assembly in fibroblasts. Curr. Biol., 17, 1134–1139. [DOI] [PubMed] [Google Scholar]
- 125. King S.M. (2012) Integrated control of axonemal dynein AAA(+) motors. J. Struct. Biol., 179, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Emond M.J., Louie T., Emerson J., Chong J.X., Mathias R.A., Knowles M.R., Rieder M.J., Tabor H.K., Nickerson D.A., Barnes K.C.. et al. (2015) Correction: exome sequencing of phenotypic extremes identifies CAV2 and TMC6 as interacting modifiers of chronic pseudomonas aeruginosa infection in cystic fibrosis. PLoS Genet., 11, e1005424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zaas D.W., Swan Z.D., Brown B.J., Li G., Randell S.H., Degan S., Sunday M.E., Wright J.R., Abraham S.N. (2009) Counteracting signaling activities in lipid rafts associated with the invasion of lung epithelial cells by Pseudomonas aeruginosa. J. Biol. Chem., 284, 9955–9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lazarczyk M., Dalard C., Hayder M., Dupre L., Pignolet B., Majewski S., Vuillier F., Favre M., Liblau R.S. (2012) EVER proteins, key elements of the natural anti-human papillomavirus barrier, are regulated upon T-cell activation. PLoS One, 7, e39995.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lennox A.T., Coburn S.L., Leech J.A., Heidrich E.M., Kleyman T.R., Wenzel S.E., Pilewski J.M., Corcoran T.E., Myerburg M.M. (2018) ATP12A promotes mucus dysfunction during Type 2 airway inflammation. Sci. Rep., 8, 2109.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Novak I., Wang J., Henriksen K.L., Haanes K.A., Krabbe S., Nitschke R., Hede S.E. (2011) Pancreatic bicarbonate secretion involves two proton pumps. J. Biol. Chem., 286, 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. McInnes G., Tanigawa Y., DeBoever C., Lavertu A., Olivieri J.E., Aguirre M., Rivas M. (2018) Global Biobank Engine: enabling genotype-phenotype browsing for biobank summary statistics. bioRxiv, doi: https://doi.org/10.1101/304188. [DOI] [PMC free article] [PubMed]