

A transition metal–assisted carbonization process can convert small organic molecules to functional carbon materials directly.
Abstract
Nanostructured carbon materials with large surface area and desired chemical functionalities have been attracting considerable attention because of their extraordinary physicochemical properties and great application potentials in catalysis, environment, and energy storage. However, the traditional approaches to fabricating these materials rely greatly on complex procedures and specific precursors. We present a simple, effective, and scalable strategy for the synthesis of functional carbon materials by transition metal–assisted carbonization of conventional small organic molecules. We demonstrate that transition metals can promote the thermal stability of molecular precursors and assist the formation of thermally stable polymeric intermediates during the carbonization process, which guarantees the successful preparation of carbons with high yield. The versatility of this synthetic strategy allows easy control of the surface chemical functionality, porosity, and morphology of carbons at the molecular level. Furthermore, the prepared carbons exhibit promising performance in heterogeneous catalysis and electrocatalysis.
INTRODUCTION
With high electric conductivity, good chemical stability, and unique microstructure, carbon materials (CMs) exhibit great application potentials in diverse fields, such as environmental treatment, polymer science, energy storage, catalysis, and advanced electronic devices (1–5). In particular, when CMs have high surface area, porous structure, and the desired heteroatom doping, their performance in certain applications becomes especially outstanding and competitive (6–9). For example, transition metal/nitrogen codoped nanostructured carbons with high surface area displayed a comparable electrocatalytic performance to that of noble metal catalysts (10–12). Traditionally, CMs were prepared by the carbonization of low–vapor pressure natural products, such as cellulose (3, 13), chitin (14), starch (15), alginic acid (16), and chitosan (17), and synthetic polymers, such as polyacrylonitrile (18), polyaniline (11), and phenolic resins (6, 19). The main drawback of natural precursors for CM synthesis is the difficulty to tailor the microstructures and chemical compositions of the obtained products because of the poor solubility and limited heteroatom contents and species in natural precursors. As for synthetic polymers, the specific polymerization initiators, complicated and slow polymerization processes cause much inconvenience in CM synthesis and hinder their large-scale production. Therefore, substantial efforts have been dedicated to the search for simple and feasible methods and suitable precursors for preparing CMs (1, 7, 20).
In this regard, Dai and Antonietti’s groups (21–23) independently discovered that ionic liquid, a semiorganic salt, was an excellent precursor for preparing CMs via a simple carbonization process, although this strategy only worked for a few expensive ionic liquid with cross-linkable functional groups (that is, cyano/nitrile groups) that could form stable intermediate polymeric structures during thermal decomposition of such precursor. Moreover, Watanabe’s group (24, 25) recently reported that protic ionic liquid and protic salts, which were prepared by protonating N-containing bases with sulfuric acid, could be versatile precursors to prepare a family of N-doped CMs. Although it is possible to obtain desired CMs with tailored structures and properties by carefully choosing specific base-acid pairs, the protonation strategy was limited to synthesizing N-doped CMs, as the protonation is exclusively effective for N-containing bases.
Here, we report a simple, effective, and versatile method to prepare a series of functional CMs from small organic molecules (SOMs) by a transition metal–assisted carbonization process. Compared to natural products, synthetic polymers, or ionic liquids, the advantage of SOMs as precursors for preparing CMs includes common availability, relatively low cost, and diverse element species with various contents. Previous efforts on the transformation of SOMs into CMs almost relied on harsh synthesis conditions, for example, pyrolysis in sealed reactors (26), chemical vapor deposition (27), or salt melt–based ionothermal carbonization (28), due to the high volatility of SOMs at evaluated temperatures. Our synthetic strategy involves the pyrolysis of a mixture of SOMs and transition metal salts (TMSs) in a conventional tubular furnace, which does not need complicated equipment and harsh conditions and is therefore easy to scale up. The transition metals catalyze the preferential formation of thermally stable intermediate polymeric structures and thus avoid the direct sublimation of SOMs during the heating process, which guarantees the successful preparation of CMs with high carbon yield. The versatility of our strategy makes it easy to effectively control the surface chemical functionality (that is, N, O, S, and metal doping), porosity, and morphology of CMs at the molecular level. Furthermore, the prepared functional CMs exhibit great application potentials for heterogeneous catalysis, for example, selective oxidization of ethylbenzene and hydrogenation of nitrobenzene, and electrocatalysis, for example, hydrogen evolution reaction (HER) and oxygen reduction reaction (ORR).
RESULTS
Preparation of CMs
Figure 1A illustrates the typical preparation process of CMs from SOMs. Briefly, we first obtained a homogeneously mixed powder by dissolving both SOMs and TMSs in a suitable solvent before drying the solution with a rotary evaporator. Then, we subjected the mixed powder to carbonization under N2 atmosphere. We finally leached the carbonized product by dilute sulfuric acid to remove metallic species to afford CMs. To demonstrate the versatility of the synthesis, we used a total of 15 SOMs and 9 TMSs as carbon precursors and catalysts, respectively, to prepare CMs (Fig. 1B). The prepared CMs are referred to as CM-x/y, where x and y are SOM and transition metal in TMS, respectively.
Fig. 1. Preparation of CMs.
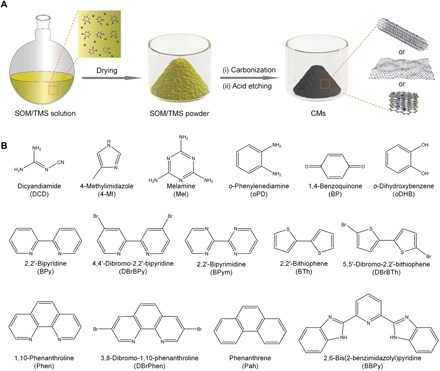
(A) Schematic illustration of the preparation process of CMs. (B) Structures of the investigated SOMs for the CM preparation.
We first investigated the thermal decomposition of SOMs with or without TMSs by thermogravimetric analysis (TGA). As expected, the TGA curves of TMS-free SOMs under N2 atmosphere showed that all of the SOMs were completely evaporated or decomposed below 700°C without any carbon residue left (Fig. 2A). In contrast, considerable solid residues were obtained for the carbonization of all SOMs with Co(NO3)2 (10%, wCo/wSOM), as manifested by TGA analyses (Fig. 2B and fig. S1A). Correspondingly, we successfully prepared a series of CMs by carbonization of SOMs with Co(NO3)2 in the oven at 900°C (table S1). We found that a variety of TMSs could effectively assist the thermal transformation of SOMs into CMs. Taking oPD as a typical example, nine kinds of TMSs, including Co(NO3)2, Fe(NO3)3, Cr(NO3)3, H2PtCl6, Cu(NO3)2, Mn(NO3)2, AgNO3, Ni(NO3)2, and Zn(NO3)2, were proved to be feasible catalysts for preparing CMs in this work (Fig. 2C, fig. S1B, and table S2). However, the non-TMSs, for example, SnCl2, NaNO3, and Ca(NO3)2, failed to catalyze the thermal transformation of oPD to CMs (fig. S1C and table S2). It was believed that the transition metal ions containing vacant d orbitals could accept lone pair electrons of N atoms in oPD, which facilitated the formation of a homogeneous and thermally stable oPD/TMS mixture. During the carbonization process, the oPD in the mixture could survive until the temperature of ca. 200°C at which cobalt catalyzed the thermal transformation of oPD into a stable polymeric intermediate that finally was converted to CMs at high temperature. In the case of the mixture containing non-TMSs, however, the non-TMSs without vacant d orbital could not change the thermal stability of oPD, which caused the direct sublimation of oPD before the occurrence of polymerization (fig. S1C).
Fig. 2. TGA analyses.
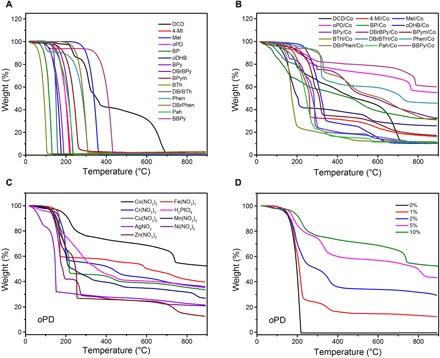
(A) TGA curves of different SOMs (TMS-free) in N2 atmosphere. (B) TGA curves of different SOMs with Co(NO3)2 in N2 atmosphere. (C) TGA curves of oPD with different TMSs in N2 atmosphere. (D) TGA curves of oPD with different Co(NO3)2 contents in N2 atmosphere.
Table S1 summarizes the carbon yields of different SOMs catalyzed by Co(NO3)2 under TGA and oven experiments. Note that the TGA carbon yield and oven carbon yield are very close, confirming the reliability of both methods for investigating carbon yield. The carbon yields are highly dependent on the structures of SOMs. For example, the BPy only afforded an oven yield of 4.20% and a relative yield (oven/theoretical) of 5.46%, while the oven yield and relative yield of BBPy reached 67.20 and 91.70%, respectively, approaching the theoretical yield of BBPy. In general, the SOMs with the structural features discussed below would exhibit a high CM yield. First, SOMs that have thermally stable domains, such as fused aromatic groups, give a high carbon yield (for example, much higher CM yields of Phen and BBPy than that of BPy). This can be explained by the fact that the thermally stable aromatic domains in these SOMs can survive harsh carbonization conditions and generate CMs (25). Second, heteroatoms in SOMs (for example, nitrogen, oxygen, or sulfur) can donate electrons to the vacant d orbitals of cobalt ions, which significantly promotes the thermal stability of SOMs and avoids their sublimation at low temperature, therefore resulting in relatively higher CM yields. For example, Phen, as a typical bidentate ligand, afforded an oven relative carbon yield of 67.06%, which is much higher than that of N-free analog, Pah, with an oven relative carbon yield of 12.40%. Besides, for the SOMs with a similar molecular structure, the heteroatom content also affects the carbon yield. For example, the oPD with two amine groups gave a high oven relative carbon yield of 83.44%, while aniline with one amine group could not yield any CM residue. Similar results were also found between BPy and BPym. This point is not unexpected, given that more N atoms in SOMs would result in a stronger interaction between SOMs and transition metal ions. Third, the bromo functional groups on a SOM are also beneficial to improving the carbon yield, as confirmed by comparing the carbon yields between BPy and DBrBPy and between BTh and DBrBTh. It is well known that TMSs can catalyze the C–C coupling by dehalogenation of halogen-functionalized molecules (for example, DBrBPy and DBrBTh) at a relatively low temperature and lead to the formation of thermally stable polymeric structures (26, 29). For the cases of halogen-free SOMs with similar structures (for example, BPy and BTh), it is more difficult to trigger the C–C coupling reaction to form stable intermediates by the dehydrogenation of SOMs (30), which is catalyzed by the transition metal as well.
The types and contents of TMSs also significantly affected the carbon yields (Fig. 2, C and D, and fig. S1, B and D). When oPD was chosen as a typical precursor, the efficiency of different TMSs in catalyzing carbonization of oPD was in the order of Co(NO3)2 > Fe(NO3)3 > Cr(NO3)3 > H2PtCl6 > Cu(NO3)2 > Mn(NO3)2 > AgNO3 > Ni(NO3)2 > Zn(NO3)2 (table S2). Moreover, the relative carbon yield of oPD increased with the cobalt content from 17.11% for 1 weight % (wt %) Co to 98.44% for 10 wt % Co (Fig. 2D, fig. S1D, and table S3), indicating that nearly all of the carbon elements in oPD were converted into CMs at 10 wt % Co.
Characterization of CMs
We performed scanning electron microscopy (SEM) and transmission electron microscopy (TEM) to investigate the morphology of the prepared CMs. Fifteen investigated CMs exhibited three different prominent microstructures that were highly dependent on the molecular structures of SOMs (Fig. 3 and fig. S2). First, CM-DCD/Co and CM-Mel/Co displayed a typical bamboo-like multiwalled carbon nanotube (CNT) structure with a diameter of 20 to 200 nm and a length of several micrometers (Fig. 3, A and B, and fig. S2), accompanying with some encapsulated metallic nanoparticles that were not removed by acid leaching. The second structure was the micrometer-sized nanosheets for the CM-4-MI/Co, CM-oPD/Co, CM-Phen/Co, and CM-Pah/Co (Fig. 3, C and D, and fig. S2). In addition, we found Co nanoparticles to be embedded within the carbon sheet structures. The third microstructure was irregular particles, as shown by the microscopic images of CM-BP/Co, CM-oDHB/Co, CM-BPy/Co, CM-DBrBPy/Co, CM-BPym/Co, CM-BTh/Co, CM-DBrBTh/Co, CM-DBrPhen/Co, and CM-BBPy/Co (Fig. 3, E and F, and fig. S2). The different microstructures may result from different polymerization and pyrolysis processes for various SOMs during carbonization (note S1). Moreover, it was found that TMSs had little influence on the microstructures of the CMs, as indicated by the TEM observation of CM-oPd/y samples (fig. S3). High-resolution TEM (HRTEM) images revealed the highly graphitic structures of the prepared CMs. In particular, the graphitic structure was mainly in the growth direction of CNTs for CM-DCD/Co, while the CM-oPD/Co was predominantly made up of randomly orientated graphitic layers (Fig. 3, G and H). The highly graphitic structures in CMs would be crucial for practical applications in terms of electric conductivity and chemical stability (24, 25).
Fig. 3. Microscopic characterization of CMs.

(A to F) SEM and TEM images of CM-DCD/Co (A and B), CM-oPD/Co (C and D), and CM-BPy/Co (E and F). (G and H) HRTEM images of CM-DCD/Co (G) and CM-oPD/Co (H).
We investigated the textural properties of CMs by nitrogen adsorption/desorption measurements, and table S1 summarizes the results. The isotherms for the typical CM-x/Co samples exhibit type IV curves with a distinct hysteresis loop in the range of 0.4 to 1.0 P/P0, indicating the mesoporous nature of these CMs (Fig. 4A), which is confirmed by the pore size distribution (PSD) curve (Fig. 4B, left). These mesopores mainly come from the relocation and growth of Co-based nanoparticles during the carbonization process and subsequent removal of these nanoparticles by acid leaching (31). The apparent Brunauer-Emmett-Teller (BET) surface areas of the CMs range from 65 to 696 m2 g−1, strongly depending on the SOM precursors, although there is no clear correlation between BET surface area/pore volume and SOM structure. Accordingly, the pore volume of the CMs varies from 0.03 to 0.46 cm3 g−1 (table S1). In particular, the CM-DBrBPy/Co displayed a high BET surface area of 696 m2 g−1, which can compete with reported porous carbon nanomaterials, such as SiO2-templated mesoporous Co/N codoped carbons (568 m2 g−1) (32), graphene aerogel (584 m2 g−1) (33), porous N-doped carbon nanofibers (563 m2 g−1) (34), and mesocrystal-derived hierarchically porous carbons (842 m2 g−1) (35).
Fig. 4. Textural properties and chemical compositions of CMs.
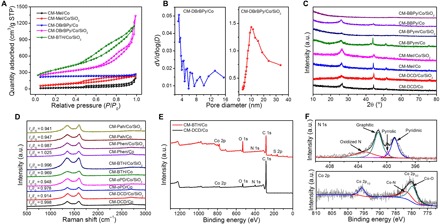
(A) Nitrogen adsorption/desorption isotherms of selected CMs. STP, standard temperature and pressure. (B) PSD of CM-DBrBPy/Co and CM-DBrBPy/Co/SiO2. (C) XRD patterns of CMs. a.u., arbitrary units. (D) Raman spectra of CMs. (E) XPS survey spectra of CM-DCD/Co and CM-BTH/Co. (F) High-resolution XPS spectra of the deconvoluted N 1s peak and Co 2p peak for CM-DCD/Co.
With the aim to further increase the porosity of CMs, we used SiO2 nanoparticles (fumed powder, 7 nm) as hard templates for the synthesis. The CMs prepared with SiO2 templates are referred to as CM-x/y/SiO2, where x and y are SOM and transition metal in TMS, respectively. Microscopic observations revealed the highly porous carbon structures for most CM-x/Co/SiO2 samples, except CM-DCD/Co/SiO2 and CM-Mel/Co/SiO2 that still exhibited CNT structures (fig. S4). As expected, the CM-x/Co/SiO2 samples showed a more pronounced hysteresis loop in the range of 0.4 to 1.0 P/P0 for their isotherms (Fig. 4A), confirming the well-defined mesoporous structures. Different from the broad mesopore distribution of CM-DBrBPy/Co prepared without SiO2 templates, the CM-DBrBPy/Co/SiO2 sample exhibited a single mesopore size (Fig. 4B, right), suggesting a good hard template effect of SiO2. Both BET surface area and pore volume for all CMs increased greatly after incorporation of SiO2 templates in the synthesis (table S4). For example, the BET surface area of CM-BTH/Co/SiO2 reaches 1202 m2 g−1, nearly six times that of CM-BTH/Co. The pore volume of CM-x/Co/SiO2 is as high as 2.16 cm3 g−1, also much larger than that of CM-x/Co. Besides SiO2 templates, other hard templates, such as SBA-15, were also applicable to preparation of high–surface area CMs due to the versatility of the synthetic strategy (fig. S5 and note S2).
Accidentally, it was found that the SiO2 templates could improve the carbon yields of SOMs (table S4). Taking Pah for example, the oven carbon yield increased greatly from 11.70 to 67.13% after incorporating SiO2 templates in the synthesis. Note that the carbonization of SOMs/SiO2 mixture without TMSs did not yield any CMs, further confirming the key role of TMSs in the CM synthesis. It is believed that the SiO2 templates could prevent the metal species from aggregating into big nanoparticles during the thermal decomposition of SOMs; the better dispersed metal species were more effective at catalyzing the polymerization and carbonization of SOMs. Besides, the confined effect of SiO2, which was found by Wang and Dai (22) in the carbonization of ionic liquids, is considered another factor in the increase of carbon yield.
We then conducted x-ray diffraction (XRD) to analyze the composition of CMs (Fig. 4C). For the typical CMs, a peak centered at 26.2° that was assigned to the (002) planes of graphitic carbon and three peaks at 44.2°, 51.5°, and 75.8° that were assigned to the (111), (200), and (220) planes of metallic Co (JCPDS 15-0806) were observed in their XRD patterns (Fig. 4C). After the incorporation of SiO2 for the CM-BPym/Co and CM-BBpy/Co precursors, the prepared CMs were free of any metallic phase, which indicated that the SiO2 templates were beneficial for the formation of removable Co species during the thermal decomposition of these SOMs with Co(NO3)2. TGA analyses verified that the Co contents of most SiO2-templating CMs (except CM-DCD/Co/SiO2, CM-Mel/Co/SiO2, and CM-DBrBPy/Co/SiO2) were much lower than those of CMs prepared without SiO2 templates (tables S1 and S4). We also collected Raman spectra of the samples to further assess the graphitic structure of the CMs (Fig. 4D). We observed a typical D band (1350 cm−1) resulting from the disordered carbon atoms and a G band (1590 cm−1) from sp2-hybridized graphitic carbon atoms for all of the investigated samples. The ID/IG values of the investigated CMs are about 1, indicating the high graphitic degree of these CMs (36). We found that heteroatom-free Pah-derived CM (that is, CM-Pah/Co) had a lower ID/IG (0.947) than the CMs derived from heteroatom-containing SOM precursors, such as CM-DCD/Co (0.998), CM-oPD/Co (0.978), CM-BTH/Co (0.969), and CM-Phen/Co (1.025), revealing more defects in these CMs derived from heteroatom-containing SOMs. Moreover, except CM-BTH/Co, the ID/IG values of the investigated CM-x/Co are more than that of the corresponding CM-x/Co/SiO2, suggesting that the introduction of SiO2 templates could improve the graphitic degree of CMs. The results can also be explained by the fact that SiO2 templates could make Co species be better dispersed during the thermal decomposition of SOMs, resulting in more effective graphitization of SOMs at high temperature.
We further probed the chemical compositions and element bonding configurations of the CMs by x-ray photoelectron spectroscopy (XPS) measurements. Tables S1 and S4 summarize the types and contents of various elements in CMs based on the XPS analyses. Figure 4E shows the survey spectra of two typical CMs (that is, CM-DCD/Co and CM-BTh/Co). Most CMs consist of C, O, N, and Co elements. The N element in CMs that we prepared with N-free SOMs (that is, BP, oDHB, BTh, DBrBTh, and Pah) originated from nitrate ion in Co(NO3)2. The N contents of the CMs ranged from 0.31 to 6.44%, depending on the molecular precursors. Moreover, considerable S doping was achieved for the CMs prepared from BTh (13.35% S for CM-BTH/Co) and DBrBTh (11.73% S for CM-DBrBTH/Co). We analyzed the high-resolution N 1s and Co 2p spectra of CM-DCD/Co to reveal the N and Co chemical environments (Fig. 4F). The N 1s spectra could be deconvoluted into four peaks assignable to the pyridinic N (398.9 eV), pyrrolic N (399.9 eV), graphitic N (401.2 eV), and oxidized N (403.1 eV) (31, 32, 37), indicating that N was structurally integrated into the carbon matrix. The high-resolution Co 2p3/2 spectrum of CM-DCD/Co consisted of two peaks at the binding energies of 781.8 and 779.8 eV, which correspond to nitrogen- and oxygen-coordinated Co, respectively (32). In addition, scanning TEM–energy dispersive spectroscopic (STEM-EDS) elemental mapping revealed the homogeneous distribution of N and/or S over the whole carbon matrix in CMs (figs. S6 to S8). Note that the Co element was also uniformly distributed on carbon framework for the Co particle–free samples (for example, CM-oPD/Co/SiO2). Previous researches have revealed that the heteroatom doping in CMs could effectively modify their electronic characteristics, spin structures, and surface chemical properties, which frequently resulted in enhanced performances in electrocatalysis, supercapacitors, lithium-ion batteries, etc. (1, 9). In particular, the nitrogen-metal coordinated structures in CMs were believed to be highly active in various electrocatalysis and heterogeneous processes (10–12, 38–40).
Overall, as stated above, the versatility of the synthetic strategy allowed us to produce distinct CMs in terms of yield, surface chemical functionality (that is, N, O, S, and metal doping), porosity, and graphitic structure. Although it is impossible to figure out a detailed carbonization process of SOMs/TMSs, which may differ substantially depending on the molecular structures of SOMs and the type of TMSs, we would conclude that TMSs played a key role for the synthesis of CMs in two aspects. First, TMSs greatly improved the thermal stability of SOMs by donating electrons from heteroatoms in SOMs (for example, oPD, oDHB, BPy, BPym, BTh, etc.) to the vacant d orbitals in transition metal ions. This made the SOMs survive harsh carbonization conditions, at which the SOMs were thermally polymerized and cross-linked into polymeric intermediates that finally formed CMs at high temperature. Second, transition metals could catalyze the polymerization of the SOMs by dehalogenation C–C coupling (for example, DBrBPy and DBrBTh) (26, 29), dehydrogenation C–C coupling (for example, BPy, BTh, Phen, etc.) (30), or cross-linking of –NH2 and –CN functional groups (for example, oPD, DCD, and Mel) at a relatively low temperature (21, 23), which therefore eventually avoided the sublimation of the SOMs and resulted in CMs. XPS analyses of DBrPhen/Co(NO3)2 system reveal that the atom ratios of Br/C gradually decrease with the increase of carbonization temperatures, from 16.24% for the DBrPhen/Co(NO3)2 precursor to 2.50% for the carbonized product at 500°C (fig. S9), indicating the removal of bromine groups and the possible occurrence of dehalogenation C–C coupling to form the conjugated polymer, as demonstrated by numerous studies in solution or on metal surface (29). We detected high–molecular weight organic species by gel permeation chromatography in the soluble part of the carbonization product that was obtained by heating DBrPhen with Co(NO3)2 at 250°C for 0.5 hours under N2 (fig. S10). When the pyroysis temperature increased to 400°C, the carbonized products could not be soluble in various solvents anymore (fig. S11), revealing the formation of highly cross-linked polymers at an elevated temperature. Furthermore, we tried to get soluble components from the products after pyrolysis of DBrBTH/Co(NO3)2 at a relatively low temperature of 200°C and analyzed them by ultraviolet-visible (UV-vis) absorption spectroscopy (fig. S12). The DBrBTh molecule shows a maximum absorption at ca. 320 nm, while the maximum absorption of DBrBTh/Co-200 generates bathochromic shift to ca. 395 nm, indicating the formation of thiophene oligomers with a bigger conjugated system (41, 42).
Note that we failed to get CMs from low–boiling point SOMs that are liquids at room temperature (fig. S13). One possible reason was that these SOMs in the mixtures were not thermally stable enough to endure high temperature for the occurrence of polymerization before they escaped out in the form of volatile vapors.
Application of CMs in heterogeneous catalysis
Catalytic oxidization and hydrogenation reactions represent important industrial processes that can produce key feedstocks for the synthesis of many fine chemicals (43–45). Therefore, development of low-cost, efficient, and recyclable catalysts for these reactions is highly desired but challenging. Recent research has revealed that heteroatom-doped, nanostructured CMs were promising heterogeneous catalysts for various catalytic reactions (39, 40, 45, 46). Four typical CMs, including CM-Phen/Co, CM-Phen/Co/SiO2, CM-DBrPhen/Co, and CM-DBrPhen/Co/SiO2, were chosen as the catalysts for heterogeneous catalysis reactions (see figs. S14 to S16 and note S3 for the detailed characterizations of these catalysts).
We first studied selective oxidization of ethylbenzene to acetophenone as the probe reaction to demonstrate the potential of our CMs as heterogeneous catalysts (Fig. 5A). As shown in Fig. 5B, the CM-Phen/Co exhibited a conversion of 52.4% and a good selectivity of 97.7% toward acetophenone, while a much lower conversion of 8.1% and a poor selectivity of 27.4% were achieved in the blank experiments. The highly porous CM-Phen/Co/SiO2 displayed the highest conversion of 91.4% and a good selectivity of 97.9%. Aberration-corrected high-angle annular dark-field STEM (HAADF-STEM) and thiocyanate ion poisoning experiments identified the atomically dispersed cobalt as the main active sites (figs. S15, F and G, and S17 and note S3). The high BET surface area of CM-Phen/Co/SiO2 is beneficial to the exposure of more single-atom cobalt active sites for substrate adsorption and conversion, leading to good catalytic performance. We could easily separate the CM-Phen/Co/SiO2 from the reaction system and use it at least five times without obvious performance degradation in terms of both activity and selectivity (Fig. 5C). When we used O2 as the oxidant for this catalytic reaction, CM-Phen/Co/SiO2 also exhibited an outstanding selectivity of 99.1%, although the activity needs to be improved (fig. S18, A and B).
Fig. 5. Catalytic performance of CMs for selective oxidization of ethylbenzene and hydrogenation of nitrobenzene.
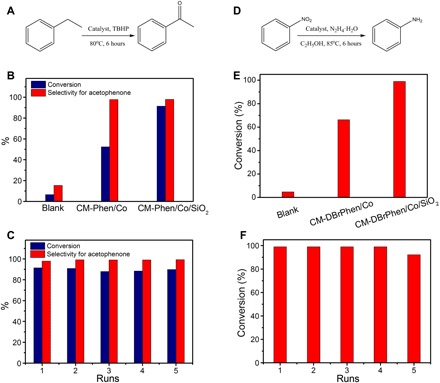
(A) Reaction equation of selective oxidization of ethylbenzene. TBHP, tert-butyl hydroperoxide. (B and C) Activity (B) and stability (C) tests of CM catalysts for ethylbenzene oxidization. (D) Reaction equation of hydrogenation of nitrobenzene. (E and F) Activity (E) and stability (F) tests of CM catalysts for nitrobenzene hydrogenation.
We then studied the hydrogenation of nitrobenzene to aniline to evaluate the catalytic performance of CMs for hydrogenation reactions (Fig. 5D). We observed a negligible conversion of 4.71% for the blank experiment, while the CM-DBrPhen/Co achieved 66.3% conversion after 6 hours of reaction. Similarly, the SiO2-templating CMs (that is, CM-DBrPhen/Co/SiO2) with higher BET surface area and abundant single-atom cobalt active sites exhibited an impressive conversion of 99.0% under the same experimental conditions, which can compete with reported carbon-based heterogeneous catalysts (44, 47). Moreover, only a slight activity decrease was observed after five runs of catalytic reactions, probably due to the unavoidable catalyst loss during the recycle process. Moreover, the CM-DBrPhen/Co also displayed a superior catalysis performance of ca. 100.0% conversion when using H2 as the reductant (fig. S18, C and D). These preliminary results undoubtedly demonstrate the great application potential of the CMs for catalytic oxidization and hydrogenation reactions, while the catalytic performance could be further improved in the future by optimizing the catalyst preparation.
Application of CMs in electrocatalysis
Electrochemical processes, such as HER and ORR, play an important role in future clean energy devices (48, 49). Although Pt and its alloys represent the most efficient electrocatalysts for HER and ORR, their scarcity and high cost hinder their large-scale commercial applications (49). Recently, metal-nitrogen–doped CMs (M-Nx/C, M = Fe, Co, Ni) have been proven to be promising non-noble metal catalysts for various electrochemical reactions (11, 12, 49). Considering the structural feature of CMs, that is, high surface area and porosity, graphitization degree, and considerable nitrogen/metal doping, we investigated the performance of the prepared CMs for catalyzing HER and ORR. For comparison, we also tested commercial 20 wt % Pt/C (Johnson Matthey) under the same conditions. As expected, the Pt/C catalyst exhibited excellent catalytic activity for HER in acidic medium with an onset potential of ca. 0 V (the potential at which the cathodic current density is 1 mA cm−2; Fig. 6A). Meanwhile, the CM-DBrPhen/Co/SiO2 catalyst also displayed a low onset overpotential of only 47 mV (Fig. 6A). To reach a current density of 10 mA cm−2, the CM-DBrPhen/Co/SiO2 needed an overpotential of 158 mV, which is comparable to that of reported highly active non-noble metal HER catalysts (table S5), such as Cu3P nanowire arrays (143 mV) (50), graphitic C3N4 nanoribbon/graphene hybrid (207 mV) (51), Co0.6Mo1.4N2 (200 mV) (52), and CoS2/reduced graphene oxide/CNT (142 mV) (53). Besides, thiocyanate ion poisoning experiments reveal that the single-atom cobalt active sites in CM-DBrPhen/Co/SiO2 are also the main active sites for HER (fig. S19). The Tafel slope value of the CM-DBrPhen/Co/SiO2 catalyst was 104.9 mV dec−1, falling within the range of 40 to 120 mV dec−1 (fig. S20A), suggesting that the HER taking place on the C-MDBrPhen/Co/SiO2 catalyst followed a Volmer-Heyrovsky mechanism and that the electrochemical desorption was the rate-limiting step (12, 54).
Fig. 6. Electrocatalytic performance of CMs for HER.
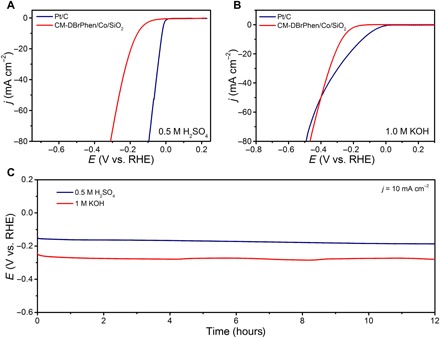
(A and B) Polarization curves of CM-DBrPhen/Co/SiO2 and commercial Pt/C catalyst in 0.5 M H2SO4 (A) and 1 M KOH (B). (C) Chronopotentiometry tests of CM-DBrPhen/Co/SiO2 at 10 mA cm−2 in 0.5 M H2SO4 and 1 M KOH, respectively.
Furthermore, we also studied the electrocatalytic HER activity of CM-DBrPhen/Co/SiO2 in basic media (1 M KOH). The polarization curve of CM-DBrPhen/Co/SiO2 showed a small overpotential of 271 mV for achieving a significant hydrogen evolution at the current density of 10 mA cm−2 (Fig. 6B). The CM-DBrPhen/Co/SiO2 electrocatalyst outperformed the Pt/C catalyst for overpotential higher than ca. 400 mV, making the CM-DBrPhen/Co/SiO2 catalyst an excellent nonprecious metal HER electrocatalyst in alkaline media (54). In addition, the CM-DBrPhen/Co/SiO2 catalyst exhibited a notably small Tafel slope of 78.8 mV dec−1 (fig. S20B), even less than that of Pt/C (83.0 mV dec−1), indicating a favorable kinetics for CM-DBrPhen/Co/SiO2. We finally used a chronopotentiometry method to evaluate the stability of CM-DBrPhen/Co/SiO2 for HER. Twelve hours of continuous tests at 10 mA cm−2 did not cause any noticeable increase of overpotential in both acidic and basic media (Fig. 6C), indicating the excellent durability of CM-DBrPhen/Co/SiO2 catalyst for HER. Besides catalyzing HER, the obtained CM was also highly efficient in catalyzing ORR in acidic medium (fig. S21 and note S4).
DISCUSSION
To summarize, we report a facile, effective, and scalable approach for the synthesis of CMs by a transition metal–assisted thermal transformation of a variety of SOMs. In particular, the methodology allows effective control of the surface chemical functionality (that is, heteroatom and metal doping), porosity, and morphology of CMs at the molecular level for various applications. The prepared heteroatom/metal-doped CMs with high surface area and graphitic structure exhibited promising performance for catalytic selective oxidization of ethylbenzene and hydrogenation of nitrobenzene, as well as eletrocatalytic HER and ORR. There is still a large room for the further improvement of the catalytic performance by the exploration of numerous molecular precursors and transition metals. Moreover, dual heteroatom–doped (for example, N/S) and bimetallic (for example, Co/Cu) CM catalysts that we can easily obtain with our approach would also potentially lead to an enhancement in the catalytic activity for various reactions (fig. S22). The transition metal–assisted thermal transformation of molecular precursor into CMs described in the present work sheds new light on the future development of functional carbon-based materials with promising applications in heterogeneous catalysis, electrocatalysis, and supercapacitors. Future work will be dedicated to better understanding the thermal decomposition mechanism of various SOMs and figure out a clear correlation between molecules, carbon structures, and performances.
MATERIALS AND METHODS
Materials
SiO2 fumed powder and SBA-15 were purchased from Sigma-Aldrich and ShangHai JiangGe Chem Co. Ltd., respectively. Other chemicals were commercially available from Sinopharm Chemical Reagent Co. Ltd. and used as received without further purification.
Synthesis of CMs
In a typical synthesis, 0.5 g of SOMs and 0.25 g of Co(NO3)2·6H2O (WCo/WSOM is 1/10) were first dissolved in suitable solvents, including deionized water (for dissolving DCD, 4-MI, and Mel), ethanol (for dissolving oPD, BP, oDHB, Phen, and DBrPhen), N,N′-dimethylformamide (DMF) (for dissolving BPy, DBrBPy, BPym, Pah, and BBPy), and tetrahydrofuran (for dissolving BTh and DBrBTh). The solvent was then removed by rotary evaporation. The obtained dried powder was subsequently carbonized under flowing N2 for 2 hours at 900°C. Finally, the carbonized product was leached in 0.5 M H2SO4 at 90°C for 4 hours to remove unstable metallic species and afford CMs. For the typical templating synthesis, 0.5 g of SiO2 fumed powder (7 nm; Sigma-Aldrich S5130) or 0.5 g of SBA-15 (ShangHai JiangGe Chem Co. Ltd.) was added into the solution dissolved with SOMs and Co(NO3)2·6H2O. After drying and carbonization of the mixture under the same conditions, the product then underwent alkaline (2.0 M NaOH) and acidic (0.5 M H2SO4) leaching successively to remove SiO2 templates and unstable metallic species, respectively.
Instrumentation
SEM images were obtained on a Zeiss Supra 40 scanning electron microscope at an acceleration voltage of 5 kV. TEM was carried out using a Hitachi H7650 or Hitachi H7700 transmission electron microscope with a charge-coupled device imaging system and an accelerating voltage of 100 kV. XRD data were recorded on a Philips X’Pert PRO SUPER X-ray diffractometer equipped with graphite monochromatic Cu Kα radiation (λ = 1.54056 Å). XPS were acquired on an x-ray photoelectron spectrometer (ESCALAB MKII) with an excitation source of Mg Kα radiation (1253.6 eV). TGA was measured by a TGA Q5000IR analyzer under N2 or O2 flow with a temperature ramp of 10°C min−1. HRTEM was performed on JEOL-2100F with an acceleration voltage of 200 kV. STEM-EDS element mapping was carried out on a JEOL-2100F equipped with Oxford Inca.Aberration-corrected HAADF-STEM images were acquired using a JEM-ARM 200F atomic resolution analytical microscope operating at an accelerating voltage of 200 kV. Raman scattering spectra were recorded with a Renishaw System 2000 spectrometer using the 514.5-nm line of an Ar+ laser for excitation. N2 sorption analysis was conducted using an ASAP 2020 accelerated surface area and porosimetry instrument (Micromeritics), equipped with automated surface area, at 77 K using BET calculations for the surface area. The PSD plot was recorded from the adsorption branch of the isotherm based on the Barrett-Joyner-Halenda model. Gel permeation chromatography (GPC) measurements were conducted by using a multiangle laser light scattering (MALLS) detector. DMF containing LiBr (10 mM) was used as the eluent at a flow rate of 1.0 ml min−1. The Wyatt MALLS detector (120 mW solid-state laser; λ = 658 nm; DawnHeleos S/N342-H) measures the excess Rayleigh ratios (related to the scattered light intensity) at different angles for each slice of the chromatogram. Raw data were processed with the Astra V software (Wyatt Technology). The UV-vis absorption spectra were recorded on a PerkinElmer Lambda 950 spectrophotometer.
Catalytic oxidation of ethylbenzene
In a typical catalytic experiment, 61 μl of ethylbenzene (0.5 mmol), 1100 μl of tert-butyl hydroperoxide (70 wt % water), and 5.0 mg of CM catalyst were added in a glass reaction tube sealed with a Teflon lid. Then, the mixture was heated under stirring at the 80°C for 6 hours. Afterward, 77 μl of anisole was added into the reaction system and used as an internal standard. Ten milliliters of ethyl acetate was used to extract the organic compounds in the reaction system. Finally, the reaction products were analyzed using a Shimadzu gas chromatograph with a flame ionization detector and high-purity nitrogen as the carrier gas. For the stability tests, the CM catalyst was recovered by centrifugation under 9000 rpm for 9 min, thoroughly washed with ethanol and water, and dried under vacuum at 60°C overnight.
The oxidation of ethylbenzene using O2 as the oxidant was carried out in a steel Parr autoclave. Typically, 1 ml of substrate and 10 mg of CM catalyst were added into the reaction glass vial, and the reaction vial was placed into a Parr autoclave reactor. Then, the autoclave was sealed and purged with O2 to replace the air three times to reach 1 MPa. Subsequently, the reactor was heated to 120°C in 20 min. The reaction was carried out for 10 hours with magnetic stirring (stirring rate, 1500 rpm). After the reaction, the reactor was placed in ice water to quench the reaction, and the products were analyzed by glassy carbon (GC) with the same method as above.
Catalytic hydrogenation of nitrobenzene
In a typical experiment, 102 μl of nitrobenzene (1 mmol), 200 μl of hydrazine hydrate (4 mmol), 5 mg of CM catalyst, and 5 ml of ethanol were introduced into 25-ml round flasks. The reaction was refluxed at 85°C for 6 hours with magnetic stirring at 1000 rpm. After cooling at room temperature, 46 μl of n-dodecane was then added in the reaction system as an internal standard. Ten milliliters of ethyl acetate was used to extract the organic compounds in the reaction system. For the stability tests, the catalyst was separated by centrifugation, washed with ethanol and water several times, and then dried under vacuum at 60°C overnight.
The hydrogenation of nitrobenzene using H2 as the reductant was carried out in a steel Parr autoclave. Typically, 102 μl of substrate (1 mmol), 2 ml of tetrahydrofuran, 100 μl of H2O, and 10 mg of CM catalyst were added into a reaction glass vial, and the reaction vial was placed into a Parr autoclave reactor. Then, the autoclave was sealed and purged with H2 to replace the air three times to reach 4.9 MPa. Subsequently, the reactor was heated to 110°C. The reaction was carried out for 10 hours with magnetic stirring (stirring rate, 1500 rpm). After reaction, the reactor was placed in ice water to quench the reaction, and the products were analyzed by GC with the same method as above.
Electrocatalytic HER measurements
All electrochemical measurements were performed in a conventional three-electrode cell using a WaveDriver 20 bipotentiostat (Pine Instrument Company) controlled at room temperature. Ag/AgCl (3.5 M KCl) and platinum foil were used as reference and counter electrodes, respectively. For HER stability test, a graphite rod was used as the counter electrode to avoid the possible contribution of dissolved Pt species. All potentials in this study refer to reversible hydrogen electrode (RHE). The potential difference between Ag/AgCl and RHE was determined on the basis of the calibration measurement in H2-saturated electrolyte. A 5.0-mm-diameter GC disk (disk geometric area, 0.196 cm2) was used as the substrate of working electrode, with a thorough clean and a mirror-finished polish before use. To prepare the working electrode for electrochemical measurements, the catalyst inks were prepared by blending 10 mg of each catalyst with 100 μl of Nafion solution (0.5 wt %) and 920 μl of ethanol. Then, the inks were ultrasonicated to generate a homogeneous dispersion. Twelve microliters of the dispersion was pipetted onto the GC disk and dried at room temperature, resulting in the catalyst loading of 0.6 mg cm−2. For comparison, the working electrode of commercial 20 wt % Pt/C was obtained as follows: 5 mg of Pt/C and 25 μl of Nafion solution (0.5 wt %) were added in 5 ml of isopropanol by sonication for 0.5 hours to obtain a well-dispersed ink. Twelve microliters of ink was then pipetted on the GC surface, leading to a loading of 0.06 mg cm−2. The electrocatalytic HER measurements were performed in N2-saturated 0.5 M H2SO4 or 1 M KOH solution. Linear sweep voltammetry was measured at 1600 rpm [to remove the in situ–formed H2 bubbles on the rotating disk electrode (RDE)] with a sweep rate of 2 mV s−1. To test the durability of catalysts, a 12-hour chronopotentiometry test was conducted at 10 mA cm−2.
Electrocatalytic ORR measurements
For ORR tests of CM catalyst, 0.5 M H2SO4 was saturated with N2/O2 by bubbling N2/O2 before the start of each experiment for 30 min. The cyclic voltammetry tests were performed in N2- and O2-saturated 0.5 M H2SO4 electrolyte solutions with a scan rate of 50 mV s−1. RDE tests were measured in O2-saturated 0.5 M H2SO4 at 1600 rpm with a sweep rate of 10 mV s−1. Pt/C catalyst was also measured under the same conditions, except that 0.1 M HClO4 solution was used to avoid performance loss caused by bisulfate adsorption. The ORR durability was evaluated by accelerated durability tests at 50 mV s−1 under oxygen atmosphere for 2000 cycles.
Supplementary Material
Acknowledgments
We thank J.-M. Hu for the assistance in the GPC test and Y.-Z. Tan for the helpful discussions in characterizing polymeric intermediates. Funding: S.-H.Y. acknowledges the funding support from the National Natural Science Foundation of China (grants 21431006 and 21761132008), the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (grant 21521001), Key Research Program of Frontier Sciences, Chinese Academy of Sciences (CAS) (grant QYZDJ-SSW-SLH036), the National Basic Research Program of China (grant 2014CB931800), and the Users with Excellence and Scientific Research Grant of Hefei Science Center of CAS (2015HSC-UE007). H.-W.L. is thankful for the support by “the Recruitment Program of Global Experts” and the National Natural Science Foundation of China (grant 21671184). Z.-Y.W. acknowledges the funding support from the National Postdoctoral Program for Innovative Talents (grant BX201600142), the China Postdoctoral Science Foundation (grant 2017M610383), the Fundamental Research Funds for the Central Universities (grant WK2060190077), and the National Natural Science Foundation of China (grant 21703229). Author contributions: S.-H.Y., H.-W.L., and Z.-Y.W. conceived and designed the experiments. Z.-Y.W., S.-L.X., Q.-Q.Y., and Z.-Q.C. conducted the synthesis of the CMs. S.-L.X., Y.-W.D., and C.L. performed the characterization of CMs. S.-L.X. performed the catalytic experiments. S.-H.Y., H.-W.L., Z.-Y.W., and S.-L.X. co-wrote the manuscript. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/7/eaat0788/DC1
Fig. S1. TGA analyses.
Fig. S2. SEM and TEM images of CM-x/Co samples.
Fig. S3. TEM images of oPD-derived CMs prepared with different TMSs.
Fig. S4. SEM and TEM images of SiO2-templating CMs.
Fig. S5. Characterizations of CM-oPD/Co/SBA-15, and CM-Phen/Co/SBA-15.
Fig. S6. STEM-EDS elemental mapping images of CM-DCD/Co and CM-DCD/Co/SiO2.
Fig. S7. STEM-EDS elemental mapping images of CM-oPD/Co and CM-oPD/Co/SiO2.
Fig. S8. STEM-EDS elemental mapping images of CM-BTH/Co and CM-BTH/Co/SiO2.
Fig. S9. XPS survey spectra of the DBrPhen/Co(NO3)2 precursor and its carbonization products obtained at 200° to 500°C.
Fig. S10. GPC result of the sample that prepared by heating DBrPhen with Co(NO3)2 at 250°C for 0.5 hours under N2.
Fig. S11. Images of dispersing carbonization product obtained by heating DBrPhen with Co(NO3)2 at 400°C for 2 hours under N2.
Fig. S12. UV-vis absorption spectra for DBrBTh and the carbonization product obtained by heating BTh/Co(NO3)2 at 200°C for 0.5 hours under N2.
Fig. S13. The structures of four SOMs that could not be converted into CMs.
Fig. S14. XRD patterns of CM-Phen/Co, CM-Phen/Co/SiO2, CM-DBrPhen/Co, and CM-DBrPhen/Co/SiO2.
Fig. S15. Detailed characterizations of CM-Phen/Co and CM-Phen/Co/SiO2 catalysts.
Fig. S16. Detailed characterizations of CM-DBrPhen/Co and CM-DBrPhen/Co/SiO2 catalysts.
Fig. S17. The poisoning experiments for ethylbenzene oxidization and nitrobenzene hydrogenation.
Fig. S18. Catalytic performance for selective oxidization of ethylbenzene using O2 as the oxidant and hydrogenation of nitrobenzene using H2 as the reductant.
Fig. S19. The poisoning experiments for HER.
Fig. S20. Tafel curves of CM-DBrPhen/Co/SiO2 and commercial Pt/C catalyst.
Fig. S21. Electrocatalytic performance of CMs for ORR in acidic medium.
Fig. S22. HER polarization curves of CM-oPD/Co/SiO2 and CM-oPD/CoCu/SiO2 in 0.5 M H2SO4.
Table S1. Summary of carbon yield, texture properties, elemental composition of CMs prepared with Co(NO3)2 as the catalyst.
Table S2. Carbon yields of oPD with different TMSs as catalysts.
Table S3. Carbon yields of oPD with different amounts of Co(NO3)2 as catalysts.
Table S4. Summary of carbon yield, texture properties, and elemental composition of CMs prepared with Co(NO3)2 as the catalyst and SiO2 nanoparticles as hard templates.
Table S5. HER performance comparison.
Note S1. The proposed mechanism for different microstructures of CM-x/Co samples.
Note S2. Synthesis of high–surface area CMs from SOMs with SBA-15 as templates.
Note S3. Detailed characterization analysis of CM-Phen/Co, CM-Phen/Co/SiO2, CM-DBrPhen/Co, and CM-DBrPhen/Co/SiO2.
Note S4. Electrocatalytic performance of CMs for ORR.
REFERENCES AND NOTES
- 1.Borchardt L., Zhu Q.-L., Casco M. E., Berger R., Zhuang X., Kaskel S., Feng X., Xu Q., Toward a molecular design of porous carbon materials. Mater. Today 20, 592–610 (2017). [Google Scholar]
- 2.Jariwala D., Sangwan V. K., Lauhon L. J., Marks T. J., Hersam M. C., Carbon nanomaterials for electronics, optoelectronics, photovoltaics, and sensing. Chem. Soc. Rev. 42, 2824–2860 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Wu Z.-Y., Liang H.-W., Chen L.-F., Hu B.-C., Yu S.-H., Bacterial cellulose: A robust platform for design of three dimensional carbon-based functional nanomaterials. Acc. Chem. Res. 49, 96–105 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Titirici M.-M., Antonietti M., Chemistry and materials options of sustainable carbon materials made by hydrothermal carbonization. Chem. Soc. Rev. 39, 103–116 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Yang S., Bachman R. E., Feng X., Müllen K., Use of organic precursors and graphenes in the controlled synthesis of carbon-containing nanomaterials for energy storage and conversion. Acc. Chem. Res. 46, 116–128 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Biener J., Stadermann M., Suss M., Worsley M. A., Biener M. M., Rose K. A., Baumann T. F., Advanced carbon aerogels for energy applications. Energy Environ. Sci. 4, 656–667 (2011). [Google Scholar]
- 7.White R. J., Budarin V., Luque R., Clark J. H., Macquarrie D. J., Tuneable porous carbonaceous materials from renewable resources. Chem. Soc. Rev. 38, 3401–3418 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Zhai Y., Dou Y., Zhao D., Fulvio P. F., Mayes R. T., Dai S., Carbon materials for chemical capacitive energy storage. Adv. Mater. 23, 4828–4850 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Paraknowitsch J. P., Thomas A., Doping carbons beyond nitrogen: An overview of advanced heteroatom doped carbons with boron, sulphur and phosphorus for energy applications. Energy Environ. Sci. 6, 2839–2855 (2013). [Google Scholar]
- 10.Lefèvre M., Proietti E., Jaouen F., Dodelet J.-P., Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells. Science 324, 71–74 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Wu G., More K. L., Johnston C. M., Zelenay P., High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt. Science 332, 443–447 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Liang H.-W., Brüller S., Dong R., Zhang J., Feng X., Müllen K., Molecular metal–Nx centres in porous carbon for electrocatalytic hydrogen evolution. Nat. Commun. 6, 7992 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshino K., Matsuoka R., Nogami K., Yamanaka S., Watanabe K., Takahashi M., Honma M., Graphite film prepared by pyrolysis of bacterial cellulose. J. Appl. Phys. 68, 1720–1725 (1990). [Google Scholar]
- 14.Nogi M., Kurosaki F., Yano H., Takano M., Preparation of nanofibrillar carbon from chitin nanofibers. Carbohydr. Polym. 81, 919–924 (2010). [Google Scholar]
- 15.Budarin V., Clark J. H., Hardy J. J. E., Luque R., Milkowski K., Tavener S. J., Wilson A. J., Starbons: New starch-derived mesoporous carbonaceous materials with tunable properties. Angew. Chem. Int. Ed. 45, 3782–3786 (2006). [DOI] [PubMed] [Google Scholar]
- 16.White R. J., Antonio C., Budarin V. L., Bergström E., Thomas-Oates J., Clark J. H., Polysaccharide-derived carbons for polar analyte separations. Adv. Funct. Mater. 20, 1834–1841 (2010). [Google Scholar]
- 17.Primo A., Atienzar P., Sánchez E., Delgado J. M., Garcia H., From biomass wastes to large-area, high-quality, N-doped graphene: Catalyst-free carbonization of chitosan coatings on arbitrary substrates. Chem. Commun. 48, 9254–9256 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Chang Y., Hong F., He C., Zhang Q., Liu J., Nitrogen and sulfur dual-doped non-noble catalyst using fluidic acrylonitrile telomer as precursor for efficient oxygen reduction. Adv. Mater. 25, 4794–4799 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Yu Z.-L., Li G.-C., Fechler N., Yang N., Ma Z.-Y., Wang X., Antonietti M., Yu S.-H., Polymerization under hypersaline conditions: A robust route to phenolic polymer-derived carbon aerogels. Angew. Chem. Int. Ed. 55, 14623–14627 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Antonietti M., Fechler N., Fellinger T.-P., Carbon aerogels and monoliths: Control of porosity and nanoarchitecture via Sol–Gel routes. Chem. Mater. 26, 196–210 (2013). [Google Scholar]
- 21.Lee J. S., Wang X., Luo H., Baker G. A., Dai S., Facile ionothermal synthesis of microporous and mesoporous carbons from task specific ionic liquids. J. Am. Chem. Soc. 131, 4596–4597 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Wang X., Dai S., Ionic liquids as versatile precursors for functionalized porous carbon and carbon-oxide composite materials by confined carbonization. Angew. Chem. Int. Ed. 49, 6664–6668 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Paraknowitsch J. P., Zhang J., Su D., Thomas A., Antonietti M., Ionic liquids as precursors for nitrogen-doped graphitic carbon. Adv. Mater. 22, 87–92 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Zhang S., Miran M. S., Ikoma A., Dokko K., Watanabe M., Protic ionic liquids and salts as versatile carbon precursors. J. Am. Chem. Soc. 136, 1690–1693 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Zhang S., Dokko K., Watanabe M., Direct synthesis of nitrogen-doped carbon materials from protic ionic liquids and protic salts: Structural and physicochemical correlations between precursor and carbon. Chem. Mater. 26, 2915–2926 (2014). [Google Scholar]
- 26.Ito Y., Christodoulou C., Nardi M. V., Koch N., Kläui M., Sachdev H., Müllen K., Tuning the magnetic properties of carbon by nitrogen doping of its graphene domains. J. Am. Chem. Soc. 137, 7678–7685 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Chen Z., Ren W., Gao L., Liu B., Pei S., Cheng H.-M., Three-dimensional flexible and conductive interconnected graphene networks grown by chemical vapour deposition. Nat. Mater. 10, 424–428 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Chang Y., Antonietti M., Fellinger T.-P., Synthesis of nanostructured carbon through ionothermal carbonization of common organic solvents and solutions. Angew. Chem. Int. Ed. 54, 5507–5512 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Chen L., Hernandez Y., Feng X., Müllen K., From nanographene and graphene nanoribbons to graphene sheets: Chemical synthesis. Angew. Chem. Int. Ed. 51, 7640–7654 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Fan Q., Gottfried J. M., Zhu J., Surface-catalyzed C–C covalent coupling strategies toward the synthesis of low-dimensional carbon-based nanostructures. Acc. Chem. Res. 48, 2484–2494 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Wu Z.-Y., Xu X.-X., Hu B.-C., Liang H.-W., Lin Y., Chen L.-F., Yu S.-H., Iron carbide nanoparticles encapsulated in mesoporous Fe-N-doped carbon nanofibers for efficient electrocatalysis. Angew. Chem. Int. Ed. 54, 8179–8183 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Liang H.-W., Wei W., Wu Z.-S., Feng X., Müllen K., Mesoporous metal–nitrogen-doped carbon electrocatalysts for highly efficient oxygen reduction reaction. J. Am. Chem. Soc. 135, 16002–16005 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Worsley M. A., Pauzauskie P. J., Olson T. Y., Biener J., Satcher J. H. Jr, Baumann T. F., Synthesis of graphene aerogel with high electrical conductivity. J. Am. Chem. Soc. 132, 14067–14069 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Chen L.-F., Zhang X.-D., Liang H.-W., Kong M., Guan Q.-F., Chen P., Wu Z.-Y., Yu S.-H., Synthesis of nitrogen-doped porous carbon nanofibers as an efficient electrode material for supercapacitors. ACS Nano 6, 7092–7102 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Yang S. J., Antonietti M., Fechler N., Self-assembly of metal phenolic mesocrystals and morphosynthetic transformation toward hierarchically porous carbons. J. Am. Chem. Soc. 137, 8269–8273 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Pachfule P., Shinde D., Majumder M., Xu Q., Fabrication of carbon nanorods and graphene nanoribbons from a metal–organic framework. Nat. Chem. 8, 718–724 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Lin L., Zhu Q., Xu A.-W., Noble-metal-free Fe–N/C catalyst for highly efficient oxygen reduction reaction under both alkaline and acidic conditions. J. Am. Chem. Soc. 136, 11027–11033 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Li Y., Zhou W., Wang H., Xie L., Liang Y., Wei F., Idrobo J.-C., Pennycook S. J., Dai H., An oxygen reduction electrocatalyst based on carbon nanotube–graphene complexes. Nat. Nanotechnol. 7, 394–400 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Jagadeesh R. V., Surkus A.-E., Junge H., Pohl M.-M., Radnik J., Rabeah J., Huan H., Schünemann V., Brückner A., Beller M., Nanoscale Fe2O3-based catalysts for selective hydrogenation of nitroarenes to anilines. Science 342, 1073–1076 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Westerhaus F. A., Jagadeesh R. V., Wienhöfer G., Pohl M.-M., Radnik J., Surkus A.-E., Rabeah J., Junge K., Junge H., Nielsen M., Brückner A., Beller M., Heterogenized cobalt oxide catalysts for nitroarene reduction by pyrolysis of molecularly defined complexes. Nat. Chem. 5, 537–543 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Valencia D., Whiting G. T., Bulo R. E., Weckhuysen B. M., Protonated thiophene-based oligomers as formed within zeolites: Understanding their electron delocalization and aromaticity. Phys. Chem. Chem. Phys. 18, 2080–2086 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Imae I., Sagawa H., Mashima T., Komaguchi K., Ooyama Y., Harima Y., Synthesis of soluble polythiophene partially containing 3,4-ethylenedioxythiophene and 3-hexylthiophene by polycondensation. Open J. Polym. Chem. 4, 83–93 (2014). [Google Scholar]
- 43.He L., Weniger F., Neumann H., Beller M., Synthesis, characterization, and application of metal nanoparticles supported on nitrogen-doped carbon: Catalysis beyond electrochemistry. Angew. Chem. Int. Ed. 55, 12582–12594 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Jagadeesh R. V., Wienhöfer G., Westerhaus F. A., Surkus A.-E., Pohl M.-M., Junge H., Junge K., Beller M., Efficient and highly selective iron-catalyzed reduction of nitroarenes. Chem. Commun. 47, 10972–10974 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Deng D., Chen X., Yu L., Wu X., Liu Q., Liu Y., Yang H., Tian H., Hu Y., Du P., Si R., Wang J., Cui X., Li H., Xiao J., Xu T., Deng J., Yang F., Duchesne P. N., Zhang P., Zhou J., Sun L., Li J., Pan X., Bao X., A single iron site confined in a graphene matrix for the catalytic oxidation of benzene at room temperature. Sci. Adv. 1, e1500462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L., Wang A., Wang W., Huang Y., Liu X., Miao S., Liu J., Zhang T., Co–N–C catalyst for C–C coupling reactions: On the catalytic performance and active sites. ACS Catal. 5, 6563–6572 (2015). [Google Scholar]
- 47.Li Y., Zhou Y.-X., Ma X., Jiang H.-L., A metal–organic framework-templated synthesis of γ-Fe2O3 nanoparticles encapsulated in porous carbon for efficient and chemoselective hydrogenation of nitro compounds. Chem. Commun. 52, 4199–4202 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Jiao Y., Zheng Y., Jaroniec M., Qiao S. Z., Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 44, 2060–2086 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Liang Y., Li Y., Wang H., Dai H., Strongly coupled inorganic/nanocarbon hybrid materials for advanced electrocatalysis. J. Am. Chem. Soc. 135, 2013. –2036 ( 2013). [DOI] [PubMed] [Google Scholar]
- 50.Tian J., Liu Q., Cheng N., Asiri A. M., Sun X., Self-supported Cu3P nanowire arrays as an integrated high-performance three-dimensional cathode for generating hydrogen from water. Angew. Chem. Int. Ed. 53, 9577–9581 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Zhao Y., Zhao F., Wang X., Xu C., Zhang Z., Shi G., Qu L., Graphitic carbon nitride nanoribbons: Graphene-assisted formation and synergic function for highly efficient hydrogen evolution. Angew. Chem. Int. Ed. 53, 13934–13939 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Cao B., Veith G. M., Neuefeind J. C., Adzic R. R., Khalifah P. G., Mixed close-packed cobalt molybdenum nitrides as non-noble metal electrocatalysts for the hydrogen evolution reaction. J. Am. Chem. Soc. 135, 19186–19192 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Peng S., Li L., Han X., Sun W., Srinivasan M., Mhaisalkar S. G., Cheng F., Yan Q., Chen J., Ramakrishna S., Cobalt sulfide nanosheet/graphene/carbon nanotube nanocomposites as flexible electrodes for hydrogen evolution. Angew. Chem. Int. Ed. 53, 12594–12599 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Zou X., Zhang Y., Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 44, 5148–5180 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Zhang C., He Y., Mu P., Wang X., He Q., Chen Y., Zeng J., Wang F., Xu Y., Jiang J.-X., Toward high performance thiophene-containing conjugated microporous polymer anodes for lithium-ion batteries through structure design. Adv. Funct. Mater. 28, 1705432 (2018). [Google Scholar]
- 56.Qiao S., Du Z., Huang W., Yang R., Influence of aggregated morphology on carbon dioxide uptake of polythiophene conjugated organic networks. J. Solid State Chem. 212, 69–72 (2014). [Google Scholar]
- 57.Li J., Cao C., Zhu H., Synthesis and characterization of graphite-like carbon nitride nanobelts and nanotubes. Nanotechnology 18, 115605 (2007). [Google Scholar]
- 58.Zou X., Huang X., Goswami A., Silva R., Sathe B. R., Mikmeková E., Asefa T., Cobalt-embedded nitrogen-rich carbon nanotubes efficiently catalyze hydrogen evolution reaction at all pH values. Angew. Chem. Int. Ed. 53, 4372–4376 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Terrado E., Redrado M., Muñoz E., Maser W. K., Benito A. M., Martínez M. T., Carbon nanotube growth on cobalt-sprayed substrates by thermal CVD. Mater. Sci. Eng. C 26, 1185–1188 (2006). [Google Scholar]
- 60.Tessonnier J.-P., Su D. S., Recent progress on the growth mechanism of carbon nanotubes: A review. ChemSusChem 4, 824–847 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Yin P., Yao T., Wu Y., Zheng L., Lin Y., Liu W., Ju H., Zhu J., Hong X., Deng Z., Zhou G., Wei S., Li Y., Single cobalt atoms with precise N-coordination as superior oxygen reduction reaction catalysts. Angew. Chem. Int. Ed. 55, 10800–10805 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Liu W., Zhang L., Yan W., Liu X., Yang X., Miao S., Wang W., Wang A., Zhang T., Single-atom dispersed Co–N–C catalyst: Structure identification and performance for hydrogenative coupling of nitroarenes. Chem. Sci. 7, 5758–5764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Q., Zhou Z.-Y., Lai Y.-J., You Y., Liu J.-G., Wu X.-L., Terefe E., Chen C., Song L., Rauf M., Tian N., Sun S.-G., Phenylenediamine-based FeNx/C catalyst with high activity for oxygen reduction in acid medium and its active-site probing. J. Am. Chem. Soc. 136, 10882–10885 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Ding W., Li L., Xiong K., Wang Y., Li W., Nie Y., Chen S., Qi X., Wei Z., Shape fixing via salt recrystallization: A morphology-controlled approach to convert nanostructured polymer to carbon nanomaterial as a highly active catalyst for oxygen reduction reaction. J. Am. Chem. Soc. 137, 5414–5420 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Wang Y.-C., Lai Y.-J., Song L., Zhou Z.-Y., Liu J.-G., Wang Q., Yang X.-D., Chen C., Shi W., Zheng Y.-P., Rauf M., Sun S.-G., S-Doping of an Fe/N/C ORR catalyst for polymer electrolyte membrane fuel cells with high power density. Angew. Chem. Int. Ed. 54, 9907–9910 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/7/eaat0788/DC1
Fig. S1. TGA analyses.
Fig. S2. SEM and TEM images of CM-x/Co samples.
Fig. S3. TEM images of oPD-derived CMs prepared with different TMSs.
Fig. S4. SEM and TEM images of SiO2-templating CMs.
Fig. S5. Characterizations of CM-oPD/Co/SBA-15, and CM-Phen/Co/SBA-15.
Fig. S6. STEM-EDS elemental mapping images of CM-DCD/Co and CM-DCD/Co/SiO2.
Fig. S7. STEM-EDS elemental mapping images of CM-oPD/Co and CM-oPD/Co/SiO2.
Fig. S8. STEM-EDS elemental mapping images of CM-BTH/Co and CM-BTH/Co/SiO2.
Fig. S9. XPS survey spectra of the DBrPhen/Co(NO3)2 precursor and its carbonization products obtained at 200° to 500°C.
Fig. S10. GPC result of the sample that prepared by heating DBrPhen with Co(NO3)2 at 250°C for 0.5 hours under N2.
Fig. S11. Images of dispersing carbonization product obtained by heating DBrPhen with Co(NO3)2 at 400°C for 2 hours under N2.
Fig. S12. UV-vis absorption spectra for DBrBTh and the carbonization product obtained by heating BTh/Co(NO3)2 at 200°C for 0.5 hours under N2.
Fig. S13. The structures of four SOMs that could not be converted into CMs.
Fig. S14. XRD patterns of CM-Phen/Co, CM-Phen/Co/SiO2, CM-DBrPhen/Co, and CM-DBrPhen/Co/SiO2.
Fig. S15. Detailed characterizations of CM-Phen/Co and CM-Phen/Co/SiO2 catalysts.
Fig. S16. Detailed characterizations of CM-DBrPhen/Co and CM-DBrPhen/Co/SiO2 catalysts.
Fig. S17. The poisoning experiments for ethylbenzene oxidization and nitrobenzene hydrogenation.
Fig. S18. Catalytic performance for selective oxidization of ethylbenzene using O2 as the oxidant and hydrogenation of nitrobenzene using H2 as the reductant.
Fig. S19. The poisoning experiments for HER.
Fig. S20. Tafel curves of CM-DBrPhen/Co/SiO2 and commercial Pt/C catalyst.
Fig. S21. Electrocatalytic performance of CMs for ORR in acidic medium.
Fig. S22. HER polarization curves of CM-oPD/Co/SiO2 and CM-oPD/CoCu/SiO2 in 0.5 M H2SO4.
Table S1. Summary of carbon yield, texture properties, elemental composition of CMs prepared with Co(NO3)2 as the catalyst.
Table S2. Carbon yields of oPD with different TMSs as catalysts.
Table S3. Carbon yields of oPD with different amounts of Co(NO3)2 as catalysts.
Table S4. Summary of carbon yield, texture properties, and elemental composition of CMs prepared with Co(NO3)2 as the catalyst and SiO2 nanoparticles as hard templates.
Table S5. HER performance comparison.
Note S1. The proposed mechanism for different microstructures of CM-x/Co samples.
Note S2. Synthesis of high–surface area CMs from SOMs with SBA-15 as templates.
Note S3. Detailed characterization analysis of CM-Phen/Co, CM-Phen/Co/SiO2, CM-DBrPhen/Co, and CM-DBrPhen/Co/SiO2.
Note S4. Electrocatalytic performance of CMs for ORR.