

Abstract
This tutorial presents the workflow of adapting an adult physiologically based pharmacokinetic (PBPK) model to the pregnant populations using the Open Systems Pharmacology (OSP) software suite (http://www.open-systems-pharmacology.org). This workflow is illustrated using a previously published PBPK model for metronidazole that is extrapolated to pregnancy by parameterizing and extending the model structure in terms of pregnancy‐induced physiological changes. Importantly, this workflow can be applied to other scenarios where PBPK models need to be re‐parameterized or structurally modified.
Physiologically based pharmacokinetic modeling
Over the past years, there was a surge in the interest in physiologically based pharmacokinetic (PBPK) models and the applications in academia, health, and regulatory authorities, as well as industry focusing on healthcare or environmental risk assessment. PBPK models are mathematical models that mechanistically describe the pharmacokinetics (PKs) of xenobiotics based on their physicochemical properties and the physiology of the species, such as rodents or humans. They are typically composed of multiple compartments, each representing a separate organ or tissue, interconnected via transport rate equations representing the circulatory system of the body. Relying on a priori knowledge on partly independent physiological processes integrated within a mechanistic framework, PBPK models allow the prediction and description of absorption, distribution, metabolism, and excretion (ADME) properties of a drug.1, 2
The PBPK models can be applied for different purposes in drug research and development. For example, they are used at the preclinical/clinical interface of drug development to support dose finding for first‐in‐human studies by predicting the PK in humans prior to in vivo studies.3, 4 PBPK models are frequently applied to evaluate metabolite disposition and predict the risk and magnitude of drug‐drug interactions,5 an application scenario that is also accounted for in the recent regulatory guidelines on drug‐drug interaction by the European Medicines Agency (EMA)6 and the US Food and Drug Administration (FDA).7 PBPK models can also be used to scale the PK understanding in adults to a special population, for which information on the drug's PK is sparse or lacking. Examples include preterm neonates,8 children,9 pregnant women,10 critically ill patients with sepsis,11 and renally or hepatically impaired patients.12 In these scenarios, PBPK models may ultimately contribute to optimizing dosing strategies or to individualize drug therapy. Generally, PBPK models can be used to integrate and analyze diverse data from preclinical and clinical development and challenge PK understanding. The increasing importance is also reflected by recent regulatory guideline drafts dedicated to PBPK.13, 14
Integral to PBPK models is the vast amount of anatomic and physiological data, often referred to as system‐specific parameters, which underlie the model structure. Different software has become available that already include these system‐specific data along with generic model structures for several populations. Notable among these are GastroPlus (SimulationsPlus, Lancaster, CA), PKQuest (University of Minnesota), Simcyp (Simcyp, Sheffield, UK), and the Open Systems Pharmacology (OSP) software suite.15 The OSP suite includes PK‐Sim, a comprehensive software tool for PBPK modeling that contains databases summarizing relevant general physiological knowledge for humans and common animal species along with predefined generic model structures that can be combined with knowledge about the drug and study setting of interest to parameterize a substance PBPK model. The OSP suite further includes MoBi, a software tool for multiscale physiological modeling and simulation that allows users to access, manipulate, and extend PK‐Sim models as well as build (systems pharmacology) models from scratch. Irrespective of the software in use, PBPK models generally separate the system‐specific parameters from substance‐specific parameters (physicochemical properties of the drug; e.g., among others, molecular weight, lipophilicity, solubility, or pKa values) from other parameter groups related to, for example, formulation or administration. This separation of parameters supports an easy reparameterization of certain groups to extrapolate to yet unknown scenarios.
Aiming for PK predictions of a drug in a new population, the system‐specific parameters of the standard population (in general adults) are typically substituted with those of the population or scenario of interest. Intrinsic factors (e.g., age, gender, weight, pregnancy, and disease) or extrinsic factors (e.g., concomitant intake of drugs or herbal products, diet, and smoking) can thereby be incorporated in the model and their effect on drug disposition investigated in silico. Several databases have been published in this context that support the incorporation of population‐specific physiological values in PBPK models, in particular for preterm neonates,8 children,16 pregnant women,17 or elderly individuals.18
In contrast, drug‐specific parameters can also be exchanged and different sets of physicochemical properties can be overlaid onto the model, which enables PK predictions for unknown drugs in a well‐defined population. Both kinds of extrapolations are sometimes termed parametric, because the model structure remains unchanged.19 Nonparametric extrapolations, in contrast, necessitate a modification of the model structure and, hence, of the underlying ordinary differential equations. Often, such nonparametric extrapolations also require the definition of new system‐specific parameters that are not part of the default model structure. Well‐known examples for nonparametric extrapolations include the development of different pregnancy PBPK models10, 20, 21 because such models often account for compartments that are not part of the standard model structure, such as the placenta and fetus.
A general introduction to PBPK/pharmacodynamic (PD) modeling using the OSP software has been presented by Kuepfer et al.,22 in which an introduction to the software is presented along with relevant points to consider as well as parameters and workflows for establishing a respective model. Maharaj & Edginton23 presented an application to pediatric drug development and procedural steps to developing a pediatric PBPK model exemplified in PK‐Sim as part of OSP. Pediatrics is currently conveniently considered in most PBPK platforms and mainly requires platform‐guided parametric extrapolations. The objective of this tutorial is to introduce the workflow and technical procedure of a nonparametric extrapolation of an adult PBPK model to populations of pregnant women using the OSP software suite. More generally, points to consider for more complex model extensions as well as nonstandard parameter changes and additions will be introduced. To illustrate this workflow, a previously published PBPK model for i.v. administered metronidazole is constructed24 and PK predictions are performed in a population of term pregnant women.
Using the pregnancy feature implemented in the OSP software suite
There are different options to use the pregnancy feature implemented in the OSP software suite that differ in their degree of flexibility and complexity. The choice of which of the following options is used depends on the question at hand. There are three options to use the implemented pregnancy feature:
Option 1: For currently 11 compounds, substance PBPK models for pregnancy have been described in the literature10, 24 and are freely available on the OSP website as consolidated PK‐Sim files25 that can be directly used for certain research questions related to these drugs. With the new release of the OSP software suite (version 7.2.0), these files can be opened and the simulations in populations of pregnant women can be reproduced in PK‐Sim. This option is convenient for users who wish to familiarize with the new pregnancy feature and are interested in the published substance PBPK model for pregnancy. However, this option provides limited flexibility. For example, physiological or clearance values and the dose can be manually changed and the model can be parameterized for another stage of pregnancy by exchanging the pregnant population with a new one defined for another fertilization age. In addition, the physicochemical properties of the drug, such as lipophilicity, molecular weight, and fraction unbound, can be modified in any of the 11 substance models. However, it is not possible to define new liberation, absorption, distribution, metabolism, and excretion processes in these model files, which may be required when modeling other substances.
Option 2: This option is suited for users wishing to create new substance PBPK models for pregnant women or for extending the published models to another stage of pregnancy. This option combines the flexibility of the OSP software suite with a straightforward modeling approach by making use of predefined building blocks for the spatial structure and passive transports in pregnancy. These building blocks can be used in MoBi to build a pregnancy simulation that can subsequently be exported to PK‐Sim for population simulations. The workflow of this option is extensively described in this article.
Option 3: The last option addresses users who wish to structurally extend or change the pregnancy (or exemplary any other) model in MoBi. This option makes full use of the flexibility of the OSP software suite and requires a proficient knowledge and experience with the software as the increased gain in flexibility comes with an increased complexity. This option relies fully on MoBi and is described at the end of this article.
Workflow for establishing a pregnancy PBPK model (option 2)
This chapter refers to option 2 from the list above and describes how a pregnancy PBPK model can be built for a new compound of interest. PK‐Sim and MoBi will both be used along with predefined building blocks for the spatial structure and passive transports in pregnancy that can be freely downloaded from the OSP website26 will also be used in the workflow.
Overview
Previously, a tutorial published in this journal has exemplarily described the general workflow and recommended best practices for building a PBPK model.22 The herein described model for nonpregnant women is constructed following the recommended practices. First, available physicochemical data of metronidazole and in vivo PK data for both nonpregnant and pregnant subjects are gathered from the literature. On the basis of these data, a reference PBPK model is first developed for a virtual population of nonpregnant healthy women that matches the anthropometric measures of the in vivo PK study group obtained from the literature. Once this model describes the in vivo PK data in nonpregnant populations reasonably well, it is scaled to pregnancy.
The first step for developing a pregnancy PBPK model is to inform the model structure with data on changes in system‐specific parameters. Such data have been compiled from the literature and thoroughly analyzed17 before being incorporated in a PBPK framework.10 Furthermore, the standard model structure needs to be exchanged with the pregnancy‐specific model structure that includes, in addition to the compartments of the standard model structure, nine pregnancy‐specific compartments, namely the amniotic fluid, breasts, endometrium, fetus, myometrium, the maternal and fetal tissue proportion of the placenta, as well as the arterial and venous blood pool of the umbilical cord. Figure 1 shows the model structure of an adult individual together with that of a pregnant woman as implemented in PK‐Sim. The exchange of the model structure is carried out in MoBi making use of the extensive flexibility provided there (which is fully used in option 3). The herein presented workflow involves the following consecutive steps:
Figure 1.
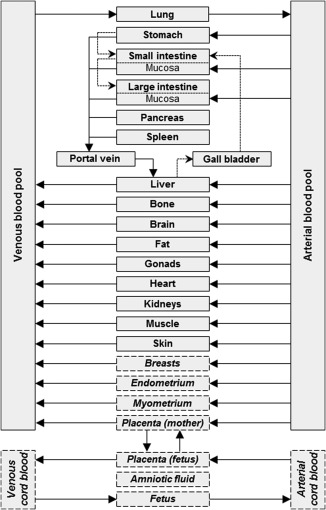
Structure of the physiologically‐based pharmacokinetic (PBPK) model for nonpregnant adults and pregnant women implemented in the Open Systems Pharmacology software suite. Compartments that are exclusively part of the pregnancy PBPK model structure are shown in italics and with dashed borders. In the pregnancy model structure, the amniotic fluid was included as a separate compartment without intercompartmental exchange to facilitate future considerations, such as fetal excretion in the amniotic fluid, and to obtain a representative body weight when summing all organ weights.
the collection and meta‐analysis of relevant, quantitative information on pregnancy‐induced changes in system‐specific parameters;
the integration of this information into the database of system‐specific parameters in PK‐Sim;
the construction of a prototype simulation in PK‐Sim;
the export of the prototype simulation from PK‐Sim to MoBi, where it can be further modified to obtain a valid whole‐body pregnancy PBPK model. This procedure comprises several steps, in particular (i) the import of the pregnancy model structure; (ii) the import of the passive transports building block and linkage of drug molecule to passive transport clearance processes; (iii) the update of the molecule start values building block; and (iv) the creation of a new parameter start values building block. After these modifications, a simulation can be created in MoBi:
the simulation is then send back to PK‐Sim, where simulations can be performed in populations of pregnant women.
Although simpler workflows may be envisioned when designing software dedicated to the question at hand, the current workflow illustrates the flexible modeling environment in PK‐Sim/MoBi. In the following sections, all of these steps are separately addressed and explained in detail using a previously published PBPK model for metronidazole as an illustrative example.24
Compilation of available data and information
The first step for building a PBPK model in a special population (e.g., pregnant women) is to establish a reference model in nonpregnant healthy women that is then scaled to pregnancy. For such a reference model, physicochemical data of the probe substance and in vivo PK data for the healthy population and the pregnant population are collected from the literature.
In this tutorial, metronidazole, a narrow‐spectrum antibiotic and antiprotozoal agent, was chosen as a substance example because of its various elimination pathways (renal excretion and metabolism via multiple enzymes) that have to be implemented in the model. Metronidazole is a relatively hydrophilic compound with low protein binding.27 Elimination occurs predominantly via biotransformation (∼85% of the administered dose), whereas a small fraction is excreted unchanged in urine (10–15% of the administered dose).28, 29 At least three different metabolites have been identified in humans. About 53% of the dose are metabolized to 2‐hydroxymetronidazole via cytochrome P450 (CYP)2A6 and CYP3A4, whereas 15–20% are oxidized to 1‐metronidazole acetic acid and ∼7% to metronidazole‐glucuronide.28, 30 The involved enzyme in metronidazole oxidation to 1‐metronidazole acetic acid has not yet been clearly identified in humans, but CYP2E1 was suggested to play a major role.28 In healthy adult women, metronidazole displays linear PK over the dose range from 0.2–2 g.31 The physicochemical properties of metronidazole are summarized in Table 1.32, 33, 34 In vivo PK data following i.v. administration in healthy nonpregnant and in term pregnant women have been reported in the literature.29, 35
Table 1.
Summary of physicochemical properties of metronidazole
| Parameter | Value | Unit | Reference |
|---|---|---|---|
| Molecular weight | 171 | g/mol | 32 |
| Lipophilicity | −0.02 | log units | 32 |
| pK a (basic) | 2.49 | 33 | |
| Fraction unbound | 0.89 | 34 | |
| Major binding protein | Albumin |
pK a, acid dissociation constant.
In the pregnancy PBPK model, system‐specific parameters have to be redefined to reflect anatomic and physiological changes during pregnancy. Given the multitude of system‐specific parameters, starting this procedure from scratch can be very time‐consuming because it entails screening the literature for data on pregnancy‐induced changes, compiling relevant data from different sources, subjecting the gathered data to thorough quality appraisal, and construing consistent trends from the cleaned dataset (e.g., through nonlinear regression analysis). Previously, a meta‐analysis has been published that reports the results of these steps and enables its implementation in a database for pregnancy PBPK modeling purposes.17 The proposed regression equations describe pregnancy‐relevant changes as a function of the fertilization age and are implemented in the PK‐Sim database, as described further below.
Establishment of a reference PBPK model for nonpregnant individuals
Recently, an i.v. metronidazole PBPK model for nonpregnant and pregnant women has been described in the literature,24 which is also used in this tutorial. Briefly, this model uses the physicochemical properties given in Table 1 and the approach by Rodgers et al.36 and Rodgers & Rowland37 for estimating the organ‐to‐plasma partition coefficients. Minor contribution to total elimination through unchanged renal excretion was modeled as a kidney plasma clearance with a dose fraction excreted unchanged in urine ( ) of around 0.15.28, 29 Tissue‐specific abundance of CYP2A6 and CYP3A4 was calculated from an internal database for real‐time polymerase chain reaction protein expression implemented in the software.38 Owing to the lack of quantitative information on the specific contribution of CYP2A6 and CYP3A4 to overall 2‐hydroxymetronidazole formation, roughly equal dose fractions metabolized ( ) via these enzymes were assumed in the presented model.28, 30 Metronidazole metabolism via CYP2E1 and uridine 5′‐diphospho‐glucuronosyltransferase (UGT) was lumped in a total hepatic plasma clearance process. On the basis of literature data,28, 30 the combined dose fraction metabolized via CYP2E1 and UGT was estimated to be ∼0.26, with CYP2E1 contributing around 71% to this lumped total hepatic plasma clearance process.
All clearance‐specific parameters in the model were simultaneously fitted to the in vivo PK data in nonpregnant women.29 To avoid parameter identifiability issues, these parameters were also fitted to the dose fractions eliminated via the different pathways given above, assuming that these fractions were reached 48 hours after administration (approximately corresponding to six times the reported half‐life of metronidazole29). For the sake of simplicity, the intracellular amount of the CYP2A6‐ and CYP3A4‐metabolite in the hepatocytes were used as surrogate markers for total body CYP2A6 and CYP3A4 metabolism, respectively. The error introduced by this simplification is marginal because CYP2A6 and CYP3A4 are predominantly expressed in the liver with only minor expression in other tissues. Furthermore, small deviances from the above‐mentioned dose fractions metabolized may be outweighed by the relatively high uncertainty in the fractions metabolized itself. The fitted parameters and dose fractions eliminated via different pathways, as used in the nonpregnant PBPK model, are listed in Table 2.28, 29, 30, 38 The simulated concentration‐time profile in healthy nonpregnant women is shown in Figure 2. As can be seen in this figure, the simulated plasma concentration‐time profile is in good agreement with the observed in vivo data. Therefore, this model is subsequently used as the basis for extrapolation to pregnancy.
Table 2.
Summary of clearance‐specific data used in the PBPK model for metronidazole in non‐pregnant women
| Parameter | Value | Unit | Reference | |
|---|---|---|---|---|
| Dose fractions excreted unchanged in urine ( ) and metabolized ( ) via different enzymes | ||||
|
|
0.25 | 28, 30 | ||
|
|
0.24 | 28, 30 | ||
|
|
0.27 | 28, 30 | ||
|
|
0.10 | 28, 30 | ||
|
|
0.13 | 28, 29 | ||
| K m, CYP 2A6 | 0.383 | mM | 30 | |
| k cat, CYP 2A6 | 1.45a | min−1 | fitted | |
| K m, CYP 3A4 | 33.1 | µM | 30 | |
| k cat, CYP 3A4 | 72.5a | min−1 | fitted | |
| Protein expression profile | RT‐PCR | 38 | ||
| Specific intrinsic hepatic clearance | 0.0125a, b | min−1 | fitted | |
| Specific renal plasma clearance | 0.0222a | min−1 | fitted |
CYP, cytochrome P450; , dose fraction excreted unchanged in urine; , dose fraction metabolized via a specific enzyme; k cat, unimolecular rate constant; K m, Michaelis‐Menten constant; PBPK: physiologically based pharmacokinetic; RT‐PCR, real‐time polymerase chain reaction; UGT, uridine 5′‐diphospho‐glucuronosyltransferase.
Values simultaneously fitted to in vivo PK data of non‐pregnant women29 and to the contribution of each pathway to overall elimination.28, 30
Describes elimination via CYP2E1 and UGT.
Figure 2.
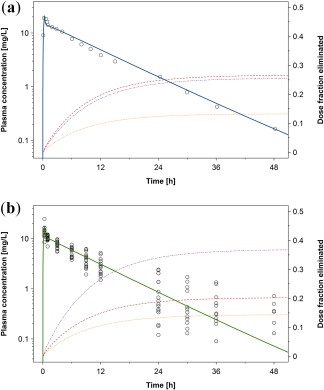
Metronidazole plasma concentration‐time profile after i.v. administration of 500 mg in nonpregnant women (a) and pregnant women (b). The solid line indicates the simulated or predicted median plasma concentration in populations of nonpregnant women or pregnant women; the dotted orange line indicates the simulated median dose fraction excreted unchanged in urine; the dash‐dotted purple line the simulated median dose fraction metabolized via CYP2A6; the dashed red line the simulated median dose fraction metabolized via CYP3A4; and symbols the observed data (mean values) taken from the literature.29, 35
BUILDING THE PBPK MODEL FOR PREGNANT WOMEN
Inclusion of physiological data in a database for pregnant women
The first step of building a PBPK model for pregnant women is the collection and inclusion of physiological data for parameters required for the PBPK model. The collection and analysis of these data has been described previously.17 Briefly, the literature was screened for relevant information in pregnant women and the collected data were quality appraised and analyzed via nonlinear regression analysis. For each parameter, pregnancy‐dependent changes in the mean and SD were described by one‐dimensional regression equations using the fertilization age as an independent variable. In contrast to the gestational age, which is estimated from the first day of the last menstrual period before conceiving, fertilization age considers the true embryonic or fetal age and refers to the estimated time passed since fertilization of the ovum. By convention, fertilization age is obtained by subtracting 14 days from the gestational age.
In PK‐Sim, the physiological parameter data of a virtual individual are taken from an incorporated database containing physiological values for all implemented populations. For every population, these values are defined for discrete age points, which serve as independent variables. For example, for the built‐in white population between 30 and 60 years of age, physiological values are defined in steps of 10 years (i.e., 30, 40, 50, and 60 years), with specific physiological parameter values being assigned to each of these age points. For ages falling between discrete age points (e.g., 35 years), the physiological values are automatically generated by the software through linear interpolation between the database entries for the lower and upper age point (i.e., in case of 35 years, the physiological values are obtained by linearly interpolating those at 30 and 40 years using the mean value in this specific case). In case of pregnant women, pregnancy‐induced physiological changes can be described as a function of the fertilization age. Instead of introducing fertilization age as another independent variable in the database, the age of a pregnant individual is used in PK‐Sim as dummy variable for fertilization age. Hence, in the current implementation, the age of 30 years of a pregnant individual corresponds to the very onset of pregnancy and the pregnancy‐induced physiological changes are assumed to be the same for a typical 20 year old and a typical 40 year old pregnant woman.
To include the physiological changes during pregnancy in PK‐Sim, data vectors were generated containing, for each parameter, the mean and SD values predicted by previously published regression equations.17 These values were generated between fertilization age 0 and 38 weeks, where full term pregnancy is reached, and were discretized in intervals of 1 day. Subsequently, to ensure a smooth transition between the nonpregnant and pregnant state, these values were scaled to join the standard level implemented in PK‐Sim using the scaling factors that have been described previously.10 Thereafter, these values were integrated into PK‐Sim as a new pregnancy population termed “Pregnant (Dallmann et al. 2017).”
Setting up a prototype individual simulation in PK‐Sim
In this section, a prototype simulation for a nonpregnant individual is set up in PK‐Sim. This prototype simulation is a kind of dummy simulation serving as a placeholder for the pregnancy building blocks. Alternatively, the respective nonpregnant building block may be cloned. The pregnancy building blocks will be later incorporated in the simulation in MoBi. Currently, a pregnant individual in PK‐Sim contains all pregnancy‐specific anatomic and physiological parameter values but the underlying model structure is the standard structure excluding pregnancy‐specific compartments. To overcome this hurdle, a prototype simulation has to be sent from PK‐Sim to MoBi. There, the model structure can be extended to pregnancy and can subsequently be reimported in PK‐Sim for running a population simulation in pregnant women.
The first step for setting up a prototype simulation in PK‐Sim is the creation of a new nonpregnant woman individual building block that includes all proteins intended to be used in the simulation (enzymes, transporters, binding‐proteins, etc.). Body weight and height can be chosen arbitrarily at this point because they will be overwritten later on when the pregnancy population is combined with the simulation exported from MoBi to PK‐Sim.
Thereafter, the compound of interest is created in the molecule building block. Here, all substance‐specific properties are defined, such as fraction unbound and the in silico model for estimating tissue‐to‐plasma partition coefficients. Furthermore, ADME‐relevant processes are defined, including metabolism via specific enzymes or renal excretion. In case of metronidazole, these processes encompass Michaelis‐Menten reactions via CYP2A6 and CYP3A4, a hepatic plasma clearance and a renal plasma clearance process, as described previously.24 All clearance values in the ADME tab of this building block should already be scaled to pregnancy. In case of metronidazole, the activity of CYP2A6 and CYP3A4 is assumed to be increased by 82% and 60% in late pregnancy, respectively.24 Biologically, the causative mechanism of this increase in activity is most likely the induction of enzyme expressions because the transcription of relevant genes is regulated by rising concentrations of hormones, mainly estradiol and progesterone, during pregnancy.39 Following this argumentation, system‐specific parameters of the pregnant individual that translate in increased CYP2A6 and CYP3A4 activity should be increased, in particular the reference concentration of the enzyme or the tissue‐specific expression of the enzyme relative to the reference concentration (termed “Relative Expression” in the software). However, such changes would later be overwritten by the default values when the simulation will be combined with the pregnant population in PK‐Sim because pregnancy‐specific enzyme expression profiles are not incorporated in the gene expression database implemented in PK‐Sim. Therefore, it may be more convenient to scale the kcat values to pregnancy. Even though it is a compound‐specific parameter lacking a direct biological rationale for scaling, enzyme kcat and expression enter as a product in the rate equation and changes are, therefore, mathematically exchangeable. An alternative to this approach is to adjust the enzyme expression in the individual in the last step after the simulation has been exported to PK‐Sim and combined with the pregnant population. In case the kcat values are adjusted in this step, values of 2.63 min−1 and 116 min−1 are obtained for CYP2A6 and CYP3A4, respectively. Furthermore, the value for the specific intrinsic hepatic clearance is 0.0125 min−1 in nonpregnant women in this example. Because the activity of CYP2E1 is assumed to be increased by 80% in late pregnancy, this value also has to be scaled to pregnancy, as described previously,24 again keeping in mind that scaling this value may lack a direct biological rationale as already discussed for CYP2A6 and CYP3A4. The last remaining clearance process for metronidazole is renal plasma clearance. Here, a value of 0.0222 min−1 is used for the specific renal clearance. This value is normalized to the kidney volume and does not need to be adjusted in pregnancy. Of note, because kidney volume increases in pregnancy, the absolute renal clearance value will also be increased. Of note, although this value may be specified here, it will be overwritten later in the workflow in MoBi, as further discussed in detail below. Therefore, at this step, the renal clearance rate may also be kept at its default value. In addition to the ADME processes, the fraction unbound, 0.89 in nonpregnant subjects,34 may also be scaled already at this step to pregnancy. Using a previously presented scaling approach,10 a value of 0.916 is obtained for late pregnant women. Of note, this value can also be specified later‐on in MoBi or in the last step when the simulation is re‐imported from MoBi to PK‐Sim; however, it is recommended doing that already here.
Subsequently, a formulation and administration protocol has to be defined in the respective building blocks. In the example of metronidazole, no formulation needs to be defined because it is administered i.v. Dose, dosing intervals, and duration of infusion can be defined in the administration protocol building block. Optionally, events, such as meal intake to account for food effects after oral administration, may be specified in the events building block. Here, no events are specified for metronidazole.
Finally, a new simulation can be created using the previously created building blocks. In this example, the simulation is arbitrarily called S1. Of note, the model for proteins and large molecules is currently not implemented for pregnancy and only small molecules can be simulated in pregnant women. After having followed these steps, the simulation is exported from PK‐Sim to MoBi, where fundamental modifications of the model structure and parameterization for pregnancy are carried out.
Customizing the simulation for pregnancy in MoBi
The modifications of the imported prototype simulation in MoBi comprise (i) the import of the pregnancy model structure; (ii) the import of the passive transports building block and linkage of drug molecule to passive transport clearance processes; (iii) the update of the molecule start values building block; and (iv) the creation of a new parameter start values building block. These steps will be consecutively addressed in the following.
In MoBi, the model structure is visible in the diagram view of the spatial structure building block. Compartments of the model structure are termed containers. There are two types of containers: physical containers can contain molecules in the simulation, whereas logical containers serve to facilitate the structural categorization of the model. Thus, logical containers can contain other containers or parameters but no molecules in the simulation. In this tutorial, the term “compartment” refers always to physical containers. The underlying model structure of the prototype simulation is the standard model structure that does not include pregnancy‐specific compartments, such as breasts, uterus, placenta, and fetus. These compartments have to be added to the model structure in MoBi.
The spatial structure building block for the pregnancy model structure is available on the OSP website from where it can be downloaded.26 Loading this spatial structure building block in MoBi is illustrated in Figure 3. Alternatively, the nonpregnant spatial structure can also be modified in MoBi by manually adding the pregnancy‐specific compartments to the model structure. The section “Adding a new compartment manually to the model structure” describes in detail how this can be done for any compartment of interest. However, as this approach is time‐consuming, it is recommended to use the spatial structure building block for pregnancy available on the OSP website, if this building block is suitable to address the question of interest.
Figure 3.
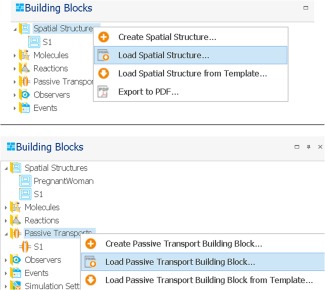
Importing the spatial structure (upper panel) and passive transports building block (lower panel) for pregnant women.
Apart from the spatial structure building block, the passive transports building block needs to be adjusted to pregnancy as well. This can again be done either manually or more conveniently by downloading the pregnancy passive transports building from the OSP website and loading it in MoBi, as shown in Figure 3. The pregnancy passive transports building block is freely available at the OSP website.26 The passive transports building block includes the description of all passive drug movements between two compartments (e.g., via blood flow, passive diffusion, or passive transport clearance processes). Except for passive transport clearance processes, the passive transports in the building block are per default activated for all molecules. In MoBi, the following clearance processes are technically defined as passive transports:
Glomerular filtration
Renal clearances − renal plasma clearance
Renal clearances − tubular secretion, first order
Renal clearances‐tubular secretion, Michaelis‐Menten type kinetics
Hence, after loading the pregnancy passive transports building block, some of the above‐listed clearance processes may have to be linked to the compound included in the prototype simulation. Linking one of the clearance processes above to the compound present in the molecule building block is done by adding the compound molecule in the “Include List” view, as shown in Figure 4. In the metronidazole example, the molecule “metronidazole” has to be linked to the renal plasma clearances process. Although it was recommended above to define the pregnancy‐specific clearance values already in PK‐Sim, the import of the pregnancy passive transports building block provided on the OSP website overwrites the values of the above‐listed passive transport clearance processes. In this pregnancy passive transports building block, all of the above‐listed clearances are set to zero. Hence, in addition to linking a molecule to a specific passive transport clearance process, the rate of that clearance process has to be set to the value observed or estimated in pregnancy. These rates can be defined in the tab “Parameters” in the upper right. In case of metronidazole, the value of the specific renal clearance has to be set at 0.0222 min−1.1 The advantage of importing the passive transports building block for pregnancy (and thereby overwriting the passive transport clearance values), is that all other pregnancy‐specific passive transports are already defined in this building block, in particular the passive transport describing drug diffusion over the placenta and those describing drug movements via the blood flow in the fetus. An alternative to importing the passive transports building block for pregnancy would be to use the original building block imported in MoBi and manually adding the new passive transports for pregnancy mentioned before.
Figure 4.
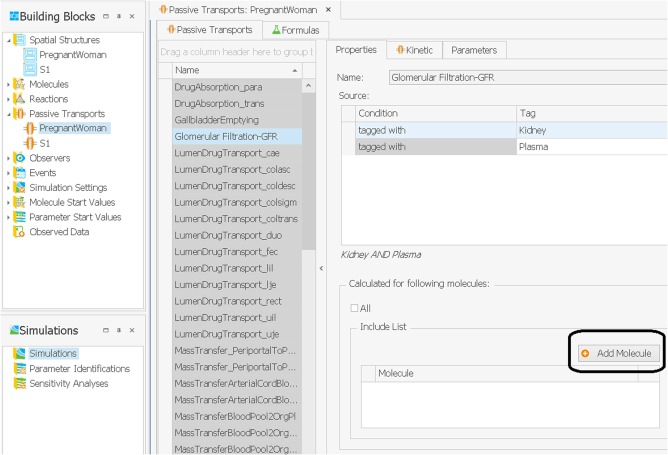
Linking a molecule to a specific clearance process, such as glomerular filtration.
In the next step, the molecule start values need to be adjusted. The molecule start values building block contains all values of molecule‐specific parameters in the model, such as physicochemical properties of metronidazole and also enzyme reference concentration values, which are used in the calculation of the tissue‐specific enzyme expression. A new molecule start values building block called “PregnancyTMP” has to be created based on the spatial structure “PregnantWoman” and the exported molecules building block “S1.” The created molecule start values building block needs to be saved as a PKML file. Subsequently, the saved molecule start values building block “PregnancyTMP” needs to be merged with the molecule start values building block “S1.” This is done by selecting “Merge” from the ribbon menu of the S1 building block (or by right‐clicking on the S1 building block and selecting “Merge”) and by selecting the saved PKML file. If the window “Resolve Merge Conflicts” appears, the option “Apply my choice to … remaining number of conflicts” should be checked and then “Keep existing” should be chosen, as illustrated in Figure 5. This step is necessary to propagate extensions in the spatial structure to the molecule start values building block.
Figure 5.
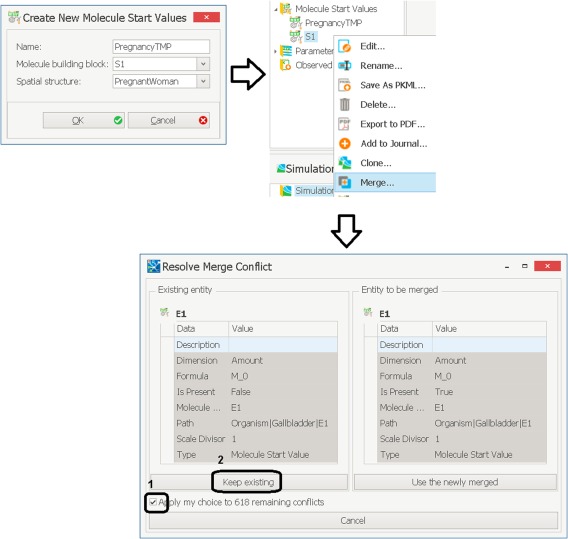
Creating a new molecule start values building block for a pregnant woman and merging conflicts.
Thereafter, one final step is required for obtaining a correct molecule start values building block for a pregnant woman. After opening the molecule start values building block S1, the following action needs to be performed for every protein molecule present in the simulation (e.g., in case of metronidazole, present protein molecules are CYP2A6 and CYP3A4) and additionally for a protein called “Undefined Liver” (the “Undefined Liver is a dummy enzyme technically created in the background when elimination is described via a total hepatic plasma clearance process, as is the case for metronidazole clearance via CYP2E1 and UGT). The first column showing the molecule name has to be filtered by the name of the protein (e.g., “CYP3A4,” “CYP2A6,” or “Undefined Liver”), as illustrated in Figure 6. Then, the second column with the header “Path Element 1” has to be filtered by all organ names that are only available in the pregnancy models (i.e., the nine additional pregnancy‐specific compartments) and additionally by one tissue organ of choice (e.g., Bone, as shown in Figure 6). Finally, the third column showing the “Path Element 2” has to be filtered for all subcompartments (i.e., “Blood Cells,” “Plasma,” “Interstitial,” and “Intracellular.”) Now, for every combination of a protein in the first column “Molecule name,” a pregnancy‐specific compartment in the column “Path Element 1,” and a subcompartment in the column “Path Element 2,” the box in the column “Is Present” has to be unticked so that the protein is not present and, hence, equal to that shown for “Bone.” This step is illustrated in Figure 6 and is necessary to deactivate the expression of relevant proteins in the subcompartments of all pregnancy‐specific compartments, unless the user would like to specifically evaluate a clearance contribution from one of these compartments.
Figure 6.
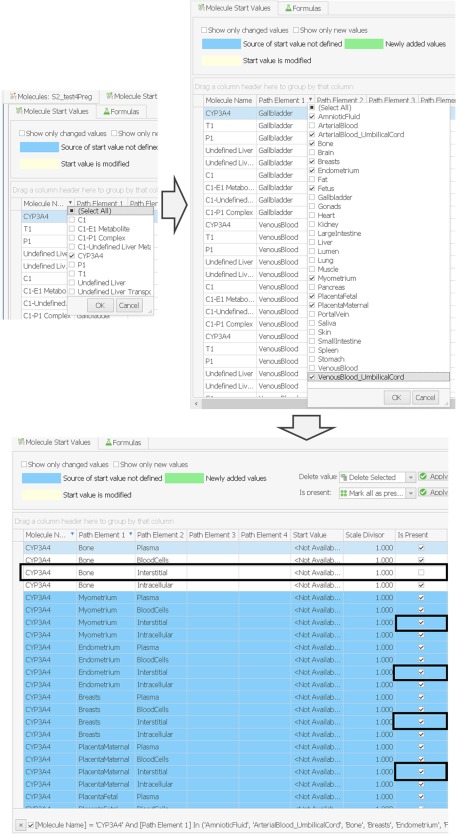
Adjusting the molecule start values building block for pregnancy. The molecule name has to be filtered for proteins in the model and the first path element for pregnancy‐specific compartments and additionally any standard compartment (e.g., Bone). Thereafter, the presence of the filtered proteins has to be deactivated in all subcompartments of all pregnancy‐specific compartments (in the figure shown for the interstitial).
In contrast to the molecule start values building block, the parameter start values building block can be left unchanged. Of note, the system‐specific parameters specified in the building block “S1” were exported from PK‐Sim and, hence, include the values for the prototype individual and a simulation in MoBi using this building block will not be properly parameterized for the pregnant woman of interest. However, when such a simulation is exported to PK‐Sim and combined with the pregnancy population building block, the correct system‐specific parameter values of the pregnant population will be transferred to the population simulation. This procedure is described in more detail below.
Now, a simulation for a pregnant woman can be set up using the following building blocks:
Spatial structure: PregnantWoman
Molecules: S1
Reactions: S1
Passive transports: PregnantWoman
Observers: S1
Events: S1
Simulation settings: S1
Molecule start values: S1
Parameter start values: S1
As already noted above, the system‐specific parameter values used in this simulation are mainly taken from the spatial structure available at the OSP website that has been loaded in MoBi, as described above. This spatial structure has been built for a typical pregnant woman in gestational week 20. To obtain a correct parameterization for the gestational week and body weight of the pregnant woman of interest, the simulation has to be exported from MoBi to PK‐Sim. This can be done by saving the simulation to a PKML file that can then be loaded in PK‐Sim.
Running a pregnancy population simulation in PK‐Sim
In order to run a pregnancy population simulation in PK‐Sim, a pregnant population has to be created. A pregnancy population is easily created by selecting this population from the drop‐down menu in the context menu (shown as “Pregnant (Dallmann et al. 2017)”). As already mentioned above, the fertilization age range of this population is defined via the age range, with an age of 30 years corresponding to the onset of pregnancy (i.e., the moment of conception). Thus, the maximum fertilization age of 38 weeks corresponds to an age of 30.75 years.
Once a pregnancy population has been created, the PKML file that has previously been saved in MoBi can be imported in PK‐Sim by combining it with the created pregnancy population building block, as described in the OSP suite manual (chapter 21.2 Importing Individual and Population Simulation). Thereafter, concentration‐time profiles in any compartment can be predicted in populations of pregnant women. The resulting concentration‐time profile in the peripheral venous blood plasma obtained in this example for metronidazole is shown in Figure 2.
As can be seen from Figure 2, the predicted metronidazole plasma concentrations are in good agreement with the observed in vivo data at term pregnancy. However, it should be noted that in spite of the good agreement with the in vivo data, uncertainty exists with respect to the underlying metabolic pathways. For example, metabolism via CYP2A6 and CYP3A4 has been implemented as a reaction following simple Michaelis‐Menten kinetics, yet a two‐site Michaelis‐Menten model might be more adequate.30 In addition to that, CYP2A6 is a polymorphic enzyme40 complicating extrapolations from one group of individuals to another. Such issues could be addressed by further in vitro and/or well‐designed in vivo studies and pharmacogenomic information can also be reflected in the model.41, 42, 43
Notably, the presented workflow for constructing a pregnancy PBPK model could now be applied to further investigations in this special population, for example, to predict pregnancy‐dependent PK changes of additional drugs or to delineate changes in enzyme activity where quantitative information are lacking in the literature. Further application scenarios might include detailing the fetal submodel (e.g., to embed a full‐blow fetal PBPK model) into the maternal model structure that would allow investigating fetal drug exposure in greater detail.
Adding a new compartment manually to the model structure (option 3)
This chapter refers to option 3 of the list above and describes how a pregnancy PBPK model can be built from scratch. The predefined building blocks for the spatial structure and passive transports in pregnancy will not be used here; instead, the standard nonpregnant model structure implemented in PK‐Sim will be manually extended by an exemplary compartment that will be connected via the blood flow to the arterial and venous blood pool of the mother. For obtaining a full pregnancy model structure, the workflow has to be repeated for every organ added to the model structure and has to be adjusted for some organs (e.g., fetal organs). However, the example is generic and familiarizes the user with the spatial structure concept in MoBi and the process of structural modifications. Modifications of the model structure may become necessary if the PK of a drug has to be characterized in a new tissue that is not explicitly represented in the model structure. Examples encompass a detailed description of the PK in different fetal tissues, but also, for example, in a tumor compartment. This chapter uses the pregnant woman as an example for adding a new compartment; however, the workflow description is generic and can be adapted for any scenario of interest.
In general, there are two ways to add a compartment to the model structure. Either a new compartment is created from scratch or a pre‐existing standard compartment, such as gonads, is saved and reimported in the model structure. Saving a compartment is done by right‐clicking on the compartment in the diagram view of the spatial structure building block and selecting “save to PKML,” Although the former procedure – creating a new compartment from scratch – may be appropriate for simple compartments in which only few parameters need to be specified, the latter procedure is more convenient for full organ compartments because all required parameters at the subcompartment level, such as tissue composition, volume, and blood flow, are kept during the reimport (one important exception is the partition coefficient between interstitial/water, which will be addressed below). In other words, these parameters need not be recreated in the new compartment – only the values of these parameters in the model have to be redefined to reflect the specific physiology of this new compartment. These values can be defined either directly in the menu context of the model structure or via the parameter start values building block, which is described in detail below.
When a compartment, such as the gonads, is saved and reimported in the spatial structure, all parameters of the old organ (i.e., gonads) are still present in the new compartment except one parameter, namely the partition coefficient (interstitial/water). This parameter needs to be manually added and defined in the neighborhood of the interstitial subcompartment and the intracellular subcompartment of the compartment. By changing to the parameter section of the molecule properties in the tree tab view, the parameter and its formula can be defined as described elsewhere.44 A Supplementary Information File of this article named “Extending Spatial Structures in MoBi” provides a detailed step‐by‐step guidance of the above‐described procedure.
Up to this point, the new compartment added to the model structure is not connected to any other compartments and, hence, dug movements to this compartment cannot be simulated. To enable passive intercompartmental drug transport via the blood flow, two input parameters have to be assimilated into the model: (i) a neighborhood with correct tags has to be created between the respective compartments in the spatial structure building block and (ii) the rate of the drug transport has to be defined by an ordinary differential equation (ODE) in the passive transports building block.
Neighborhoods between two specific compartments can be created in the diagram view of the spatial structure building block by drawing a line between the edges of the compartments to be connected. Of note, when the new compartment is wired to other compartments, it is important to correctly set the tag of the new compartment. For example, in the standard model structure exported from PK‐Sim to MoBi, a generic ODE system is per default included describing drug transport via the blood flow between the compartment of the arterial blood pool and all compartments tagged as “TissueOrgan.” Any new compartment added to the standard model structure can only receive drug molecules from the arterial blood pool via the blood flow if a neighborhood is created and if the compartment is appropriately tagged at the same time. If this tag is missing, no drug movements from the arterial blood pool can be simulated, even if the ODE for the drug transport is properly defined.
Furthermore, the ODEs for the intercompartmental drug transport have to be defined in the passive transport building block. In the example of a pregnant woman, ODEs have to be manually included for intercompartmental drug transport over the placental barrier and for all drug transport pathways in the fetal organism because these pathways differ from the standard pathways in many aspects. For example, in case of the standard model structure, the source and target of drug movements with the arterial blood flow are the maternal arterial blood pool and the tissue organs, respectively. Yet, in the fetus, the arterial blood pool of the umbilical cord has to be defined as the source for drug movements to fetal tissue organs via the arterial blood flow. The ODEs for these drug transport pathways have been described in detail in a previous publication.10
Similar to intercompartmental drug transport pathways between the arterial or venous blood pool and compartments tagged as tissue organs, generic ODEs are, per default, included in the standard model structure that describe the intracompartmental drug exchange between the different subcompartments within an organ via passive diffusion. These ODEs are also kept in the model structure when the simulation is exported from PK‐Sim to MoBi. Consequently, specific ODEs for passive diffusion pathways within an organ do need not to be defined if the new compartment belongs to be the same organism as the standard compartment. For example, when the breast tissue or endometrium is added to the model structure, passive diffusion pathways are already covered by the generic ODEs because the added compartments belong to the maternal organism. The respective tags, however, have to be correctly set in the added compartments. In contrast, if fetal compartments, such as fetal liver or kidneys, are added to the pregnancy model structure, the ODEs for passive diffusion pathways have to be redefined as the generic ODEs mentioned above all refer to the maternal organism. This can be done in a similar way to the procedure described above. All steps for implementing the outlined intercompartmental and intracompartmental drug transport pathways are described in detail in the Supplementary Information File of this article named “Extending Spatial Structures in MoBi.”
In order to create a full‐blown pregnancy PBPK model, the steps described above have to be repeated for every pregnancy‐specific organ that is explicitly included in the model structure. As noted above, the building blocks for the spatial structure and passive transports of the recently published pregnancy PBPK model10 have been made available on the OSP website26 and can be directly loaded in MoBi. This avoids having to follow the above‐described steps. However, the above‐described steps aim to facilitate a detailed understanding about how any model structure can be mechanistically refined. This could, for example, be applied to further specify the fetal submodel of the published pregnancy PBPK model.10
SUMMARY
In conclusion, PBPK modeling can be a useful approach to scale the PK of a drug from well‐investigated populations to special population, for example, from healthy adults to pregnant women. Such extrapolations require the reparameterization of the underlying system‐specific data in the model to mimic the anatomy and physiology of the special population. In addition, the model structure might need to be adjusted to reflect specific physiological states, as is the case in pregnant women in which additional compartments representing the placenta and fetus should be included. This tutorial informs the reader how this can be modeled practically in the OSP software suite.
In a broader context, this workflow demonstrates how PK predictions can be theoretically performed in a new population of interest. Currently, first efforts are being made to facilitate the inclusion of new populations in the database of the OSP software suite.45 Until this feature becomes available, the description provided in this tutorial remains theoretical for populations not yet incorporated in the software or requires a deep‐dive and adaption of the software source code. Such populations encompass, for example, specific ethnicities not yet included in the software, the critically ill, or post‐partum women. In the latter case, such a model could also address potential drug transfer via beast‐feeding as exemplary investigated previously.46 These scenarios exemplify the usefulness of PBPK approaches for investigating PK changes in special populations.
Although several software platforms are available for PBPK modeling, few of them are freely available and/or open source. The technical steps of the herein presented workflow are specific to the OSP software suite and may not be fully applicable to other software platforms that presented different technical solutions for scaling from a normal to special populations, such as pregnant women. Whereas the technical steps described in this article focus on a pregnancy scenario, they can be generalized to other research questions, such as the inclusion of new compartments. Potential research questions requiring the inclusion of new compartments could focus on, for example, ophthalmic medicines, in which the drug concentrations in the eye need to be modeled or on oncologic substances, in which the drug concentration needs to be predicted in the tumor tissue. However, the presented conceptual workflow—establishment of a model for reference subjects using in vitro and clinical data, evaluation of this model against in vivo data, substitution of the system‐specific parameter values with values specific for other individuals, here, pregnancy‐specific values available as prior knowledge—is platform‐independent and common practice in PBPK research.9, 18, 22 Ideally, the model structures, parameterizations, and scaling workflows are comparable across different PBPK platforms and similar prediction results are obtained regardless of which platform is used as has recently been discussed in more detail elsewhere.47 The advantage of open source PBPK models is that the choice of the software to use may become secondary, allowing a concentrated focus on the method itself, including the assumptions and equations underlying the model structure. Therefore, this tutorial can be seen as an important contribution to familiarize readers with the OSP software suite. Being an open source, the OSP software suite can be extended by the interested scientific community and may be used for comparison and harmonization of PBPK models across different platforms.
Conflict of Interest
The authors declared no competing interests for this work.
Supporting information
Extending spatial structures in MoBi
Acknowledgment
The authors thank Jan‐Frederik Schlender, Paola Mian, and Xiaomei Liu for valuable feedback.
References
- 1. Gerlowski, L.E. & Jain, R.K. Physiologically based pharmacokinetic modeling: principles and applications. J. Pharm . Sci. 72, 1103–1127 (1983). [DOI] [PubMed] [Google Scholar]
- 2. Jones, H. & Rowland‐Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst. Pharmacol. 2, e63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jones, H.M. , Gardner, I.B. & Watson, K.J. Modelling and PBPK simulation in drug discovery. AAPS J. 11, 155–166 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thiel, C. et al A systematic evaluation of the use of physiologically based pharmacokinetic modeling for cross‐species extrapolation. J. Pharm . Sci. 104, 191–206 (2015). [DOI] [PubMed] [Google Scholar]
- 5. Wagner, C. et al Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin. Pharmacokinet. 54, 117–127 (2015). [DOI] [PubMed] [Google Scholar]
- 6. Medicines Agency European (EMA). Guideline on the investigation of drug interactions. <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf> (2012).
- 7. US Food and Drug Administration (FDA) . Clinical Drug Interaction Studies — Study Design, Data Analysis, and Clinical Implications Guidance for Industry. <http://formiventos.com/2017/10/26/clinical-drug-interaction-studies-study-design-data-analysis-and-clinical-implications/> (2017).
- 8. Claassen, K. et al Development of a physiologically‐based pharmacokinetic model for preterm neonates: evaluation with in vivo data. Curr. Pharm. Des. 21, 5688–5698 (2015). [DOI] [PubMed] [Google Scholar]
- 9. Maharaj, A.R. , Barrett, J.S. & Edginton, A.N. A workflow example of PBPK modeling to support pediatric research and development: case study with lorazepam. AAPS J. 15, 455–464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dallmann, A. et al Physiologically based pharmacokinetic modeling of renally cleared drugs in pregnant women. Clin. Pharmacokinet. 56, 1525–1541 (2017). [DOI] [PubMed] [Google Scholar]
- 11. Radke, C. et al Development of a physiologically based pharmacokinetic modelling approach to predict the pharmacokinetics of vancomycin in critically ill septic patients. Clin. Pharmacokinet. 56, 759–779 (2017). [DOI] [PubMed] [Google Scholar]
- 12. Edginton, A.N. & Willmann, S. Physiology‐based simulations of a pathological condition: prediction of pharmacokinetics in patients with liver cirrhosis. Clin. Pharmacokinet. 47, 743–752 (2008). [DOI] [PubMed] [Google Scholar]
- 13. European Medicines Agency (EMA). Guideline on the qualification and reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation (draft). <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500211315.pdf> (2016).
- 14.US Food and Drug Administration (FDA). Physiologically Based Pharmacokinetic Analyses‐Format and Content; Guidance for Industry; Availability. <https://www.federalregister.gov/documents/2016/12/02/2016-0000/physiologically-based-pharmacokinetic-analyses-format-and-content-draft-guidance-for-industry> (2016).
- 15.Open Systems Pharmacology. <http://www.open-systems-pharmacology.org>. Accessed 17 March 2018.
- 16. Edginton, A.N. , Schmitt, W. & Willmann, S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin. Pharmacokinet. 45, 1013–1034 (2006). [DOI] [PubMed] [Google Scholar]
- 17. Dallmann, A. , Ince, I. , Meyer, M. , Willmann, S. , Eissing, T. & Hempel, G. Gestation‐specific changes in the anatomy and physiology of healthy pregnant women: an extended repository of model parameters for physiologically based pharmacokinetic modeling in pregnancy. Clin. Pharmacokinet. 56, 1303–1330 (2017). [DOI] [PubMed] [Google Scholar]
- 18. Schlender, J.F. et al Development of a whole‐body physiologically based pharmacokinetic approach to assess the pharmacokinetics of drugs in elderly individuals. Clin. Pharmacokinet. 55, 1573–1589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baeza‐Squiban, A. , Lacroix, G. & Bois, F.Y. Experimental models in nanotoxicology. (eds. Houdy, P., Lahmani, M. & Marano, F.) Nanoethics and Nanotoxicology, 1st edn 63–86 (Springer, New; York, NY, 2011). [Google Scholar]
- 20. De Sousa Mendes, M. et al Prediction of human fetal pharmacokinetics using ex vivo human placenta perfusion studies and physiologically based models. Br . J. Clin. Pharmacol. 81, 646–657 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schalkwijk, S. et al Prediction of fetal darunavir exposure by integrating human ex‐vivo placental transfer and physiologically based pharmacokinetic modeling. Clin. Pharmacokinet; e‐pub ahead of print 25 July 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuepfer, L. et al Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacometrics Syst. Pharmacol. 5, 516–531 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maharaj, A. & Edginton, A. Physiologically based pharmacokinetic modeling and simulation in pediatric drug development. CPT Pharmacometrics Syst. Pharmacol. 3, e150 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dallmann, A. , Ince, I. , Coboeken, K. , Eissing, T. & Hempel, G. A physiologically based pharmacokinetic model for pregnant women to predict the pharmacokinetics of drugs metabolized via several enzymatic pathways. Clin. Pharmacokinet; e‐pub ahead of print 18 September 2017. [DOI] [PubMed] [Google Scholar]
- 25.Open‐Systems‐Pharmacology: Pregnancy‐Models/Models. <https://github.com/Open-Systems-Pharmacology/Pregnancy-Models/tree/master/Models>. Accessed 17 March 2018.
- 26.Open‐Systems‐Pharmacology: Pregnancy‐Models/Building Blocks. < https://github.com/Open-Systems-Pharmacology/Pregnancy-Models/tree/master/BuildingBlocks>. Accessed 17 March 2018. [Google Scholar]
- 27. Ralph, E.D. Clinical pharmacokinetics of metronidazole. Clin. Pharmacokinet 8, 43–62 (1983). [DOI] [PubMed] [Google Scholar]
- 28. Loft, S. Metronidazole and antipyrine as probes for the study of foreign compound metabolism. Pharmacol. Toxicol. 66 Suppl 6, 1–31 (1990). [DOI] [PubMed] [Google Scholar]
- 29. Houghton, G.W. , Thorne, P.S. , Smith, J. , Templeton, R. & Collier, J. Comparison of the pharmacokinetics of metronidazole in healthy female volunteers following either a single oral or intravenous dose. Br. J. Clin. Pharmacol. 8, 337–341 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pearce, R.E. , Cohen‐Wolkowiez, M. , Sampson, M.R. & Kearns, G.L. The role of human cytochrome P450 enzymes in the formation of 2‐hydroxymetronidazole: CYP2A6 is the high affinity (low Km) catalyst. Drug Metab. Dispos. 41, 1686–1694 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wood, B.A. & Monro, A.M. Pharmacokinetics of tinidazole and metronidazole in women after single large oral doses. Br. J. Vener. Dis. 51, 51–53 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Machatha, S.G. & Yalkowsky, S.H. Comparison of the octanol/water partition coefficients calculated by ClogP, ACDlogP and KowWin to experimentally determined values. Int. J. Pharm. 294, 185–192 (2005). [DOI] [PubMed] [Google Scholar]
- 33. Shalaeva, M. , Kenseth, J. , Lombardo, F. & Bastin, A. Measurement of dissociation constants (pKa values) of organic compounds by multiplexed capillary electrophoresis using aqueous and cosolvent buffers. J. Pharm. Sci. 97, 2581–2606 (2008). [DOI] [PubMed] [Google Scholar]
- 34. Zhang, F. , Xue, J. , Shao, J. & Jia, L. Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov. Today 17, 475–485 (2012). [DOI] [PubMed] [Google Scholar]
- 35. Visser, A.A. & Hundt, H.K. The pharmacokinetics of a single intravenous dose of metronidazole in pregnant patients. J. Antimicrob. Chemother. 13, 279–283 (1984). [DOI] [PubMed] [Google Scholar]
- 36. Rodgers, T. , Leahy, D. & Rowland, M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate‐to‐strong bases. J. Pharm. Sci. 94, 1259–1276 (2005). [DOI] [PubMed] [Google Scholar]
- 37. Rodgers, T. & Rowland, M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 95, 1238–1257 (2006). [DOI] [PubMed] [Google Scholar]
- 38. Meyer, M. , Schneckener, S. , Ludewig, B. , Kuepfer, L. & Lippert, J. Using expression data for quantification of active processes in physiologically based pharmacokinetic modeling. Drug Metab. Dispos. 40, 892–901 (2012). [DOI] [PubMed] [Google Scholar]
- 39. Jeong, H. Altered drug metabolism during pregnancy: hormonal regulation of drug‐metabolizing enzymes. Expert Opin. Drug Metab. Toxicol. 6, 689–699 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McDonagh, E.M. et al PharmGKB summary: very important pharmacogene information for cytochrome P‐450, family 2, subfamily A, polypeptide 6. Pharmacogenet. Genomics 22, 695–708 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eissing, T. et al A computational systems biology software platform for multiscale modeling and simulation: integrating whole‐body physiology, disease biology, and molecular reaction networks. Front. Physiol. 2, 4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eissing, T. , Lippert, J. & Willmann, S. Pharmacogenomics of codeine, morphine, and morphine‐6‐glucuronide: model‐based analysis of the influence of CYP2D6 activity, UGT2B7 activity, renal impairment, and CYP3A4 inhibition. Mol. Diagn. Ther. 16, 43–53 (2012). [DOI] [PubMed] [Google Scholar]
- 43. Dickschen, K. , Willmann, S. , Thelen, K. , Lippert, J. , Hempel, G. & Eissing, T. Physiologically based pharmacokinetic modeling of tamoxifen and its metabolites in women of different CYP2D6 phenotypes provides new insight into the tamoxifen mass balance. Front. Pharmacol. 3, 92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Open Systems Pharmacology Suite Manual and Software, version 7.2. <https://github.com/Open-Systems-Pharmacology/Suite/releases/tag/v7.2.1> (2017).
- 45.Open‐Systems‐Pharmacology: ICRP‐Extended <https://github.com/Open-Systems-Pharmacology/ICRP-Extended>. Accessed 17 March 2018.
- 46. Willmann, S. , Edginton, A.N. , Coboeken, K. , Ahr, G. & Lippert, J. Risk to the breast‐fed neonate from codeine treatment to the mother: a quantitative mechanistic modeling study. Clin. Pharmacol. Ther. 86, 634–643 (2009). [DOI] [PubMed] [Google Scholar]
- 47. Dallmann, A. , Pfister, M. , van den Anker, J. , & Eissing, T. Physiologically Based Pharmacokinetic Modeling in Pregnancy: A Systematic Review of Published Models. Clinical Pharmacology & Therapeutics e‐pub ahead of print 10 April 2018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extending spatial structures in MoBi