

SUMMARY
Chromatin regulators play a broad role in regulating gene expression, and when gone awry, can lead to cancer. Here we demonstrate that ablation of the histone demethylase LSD1 in cancer cells increases repetitive element expression, including ERVs, and decreases expression of RNA-induced silencing complex (RISC) components. Significantly, this leads to dsRNA stress and activation of type 1 interferon, which stimulates anti-tumor T cell immunity and restrains tumor growth. Furthermore, LSD1 depletion enhances tumor immunogenicity and T cell infiltration in poorly immunogenic tumors, and elicits significant responses of checkpoint blockade-refractory mouse melanoma to anti-PD-1 therapy. Consistently, TCGA data analysis shows an inverse correlation between LSD1 expression and CD8+ T cell infiltration in various human cancers. Our study identifies LSD1 as a potent inhibitor of anti-tumor immunity and responsiveness to immunotherapy, and suggests LSD1 inhibition combined with PD-(L)1 blockade as a novel cancer treatment strategy.
In brief
Ablating the histone demethylase LSD1 genetically or pharmacologically enhances tumor immunogenicity by stimulating endogenous retrovirus expression and downregulating RNA-induced silencing complex, supporting the promise of LSD1 inhibition in overcoming resistance to checkpoint blockade in cancer treatment.
INTRODUCTION
Chromatin modifications play a broad and general role in regulating gene expression, and when they go awry, can lead to a variety of diseases (Brookes and Shi, 2014) Consistent with this notion, recent cancer genome sequencing efforts have identified mutations in chromatin regulators, and in the case of hematopoietic cancers, chromatin regulators are one of the most frequently mutated groups of genes (Dawson and Kouzarides, 2012). In addition, the epigenome roadmap has revealed dysregulation of chromatin states in cancer (Baylin and Jones, 2011; Flavahan et al., 2017). The roles of chromatin regulators in cancer have been investigated both at the levels of cancer cell proliferation and impact on the human immune system. However, the relative effects of chromatin regulation on cancer cell-intrinsic functions versus T cell functions, as well as on the overall responses of tumors to the immune system, are underexplored.
Recent human clinical trials using PD-(L)1 directed immunotherapy have reported promising results, leading to FDA approval of PD-(L)1 pathway inhibitors for multiple tumor types (Sharpe and Pauken, 2018). However, a majority of cancer patients do not respond to anti-PD-(L)1 therapy, due to multiple immunosuppressive mechanisms in the tumor microenvironment including dysfunctional T cells and lack of T cell infiltration or recognition by T cells (Sharma et al., 2017; Zou et al., 2016). Recent studies found that inhibition of DNA methylation alone, or together with HDAC inhibitors, leads to tumor interferon (IFN) pathway activation and increased responses to cancer immunotherapy (Chiappinelli et al., 2015; Topper et al., 2017). Blocking de novo DNA methylation in T cells enhances anti-PD-L1-mediated T cells rejuvenation and tumor control (Ghoneim et al., 2017). However, how the full spectrum of chromatin regulators modulates tumor responses to cancer immunotherapy is still poorly understood.
In this study, we provide evidence that the histone H3K4 demethylase LSD1 (KDM1A) (Shi et al., 2004) plays a critical role in suppressing endogenous double stranded RNA (dsRNA) levels and IFN responses in tumor cells, and demonstrate that dsRNA stress resulted from LSD1 inhibition leads to potent anti-tumor T cell immunity. Furthermore, LSD1 depletion renders refractory B16 tumors significantly responsive to anti-PD-1 therapy. Our findings have important implications for harnessing chromatin and epigenetic regulators for cancer immunotherapy.
RESULTS
LSD1 represses ERV expression and IFN activation in human cancer cells
To identify chromatin regulators that control tumor responses to host immunity, we initiated a curated screen with compounds targeting chromatin factors, with up-regulation of endogenous retroviral element (ERV) transcripts and activation of type 1 IFNs as readouts, as both factors are implicated in regulating tumor responses to host immunity (Kassiotis and Stoye, 2016; Parker et al., 2016; Rooney et al., 2015). In this screen, we discovered that a LSD1 catalytic inhibitor, GSK-LSD1, induced the up-regulation of a few randomly selected ERVs, which were examined with PCR primers that detected overall transcript levels of the corresponding ERV subfamilies transcribed from multiple genomic loci (Figure S1A). Type 1 and III IFNs, as well as interferon-stimulated genes (ISGs) were also induced by GSK-LSD1 (Figure S1A). To ascertain that this was caused by a GSK-LSD1 on-target effect, we performed shRNA-mediated LSD1 knockdown (KD) (Figure S1B), which confirmed these results (Figures S1C and S1D). Furthermore, introduction of wild type (WT) FHLSD1 but not catalytically compromised LSD1 (LSD1-K661A) into LSD1 KD cells fully restored the repression of the four tested ERVs as well as IFN-β and IL-28 activation (Figures S1E–S1G). These results demonstrate that demethylase activity of LSD1 is necessary for ERV and IFN repression. We further showed that neither DNMT protein expression nor global DNA methylation was affected by LSD1 inhibition (Figures S1H and S1I), suggesting a DNA methylation-independent pathway. We recapitulated these observations in T47D (Figure S1J) and 293T cell lines (Figure S1K). Together, these results suggest that LSD1 may repress the expression of a group of ERVs and regulate IFN activation in human cancer cells.
We next carried out transcriptomic analysis to comprehensively explore how LSD1 regulates ERV expression and IFN activation. We observed a significant impact of LSD1 inhibition on gene expression in MCF-7 cells (Figure S1L). Gene ontology (GO) enrichment analysis of the differentially expressed genes revealed that the up-regulated genes were significantly enriched in GO terms related to type 1 IFN response and antiviral response (Figure 1A), which were further confirmed by GSEA analysis (Figure 1B). However, almost none of the up-regulated IFN/antiviral responsive genes (125 in total) appeared to be direct targets of LSD1, as ChIP-seq analysis failed to identify LSD1 occupancy at their promoters, while in contrast genes directly regulated by LSD1 showed LSD1 occupancy at their promoters and H3K4me2 increase when LSD1 was knocked down (Figure 1C). We therefore speculated that a main mode of IFN/antiviral responses induced by LSD1 inhibition was through activation of an upstream event, such as ERV transcript expression. We thus analyzed the expression of repetitive elements in the RNA-seq data, and found many of them were up-regulated by LSD1 inhibition (Figure S1M), including a number of ERVs that were transcribed in either sense or antisense transcripts (Figure 1D). Furthermore, many ERVs appeared to be direct targets of LSD1, as they were bound by LSD1 and showed elevated H3K4me2 levels upon LSD1 KD (Figures 1E and S1N). Importantly, a number of up-regulated ERVs were expressed in both sense and antisense directions with overlapping sequences potentially allowing for the formation of dsRNAs (Figure 1F). We further confirmed this observation by analyzing a few selected ERVs using strand-specific PCR (Figure S1O). Thus, these findings demonstrate that LSD1 is important for transcriptionally silencing ERVs, consistent with a previous report in mouse embryonic stem cells (mESCs) (Macfarlan et al., 2011).
Figure 1. LSD1 restrains intracellular dsRNA stress and IFN activation.

(A) A dot map showing top 10 terms in GO analysis of up-regulated genes (log2(FC-KD/Ctrl) > 1 and FDR < 0.05) in LSD1 KD versus control MCF-7 cells. FC-fold change. Background color and dot size represent FDR and log2 transformed odds ratio separately.
(B) GSEA analysis in LSD1 KD versus control MCF-7 cells.
(C) LSD1 and H3K4me2 ChIP-seq signals at promoter regions of 125 induced IFN/antiviral responsive genes (IFN-gene, log2(FC-KD/Ctrl) > 0 and FDR < 0.05) or selected 537 genes with LSD1 peaks as positive control (Pos-gene) in control and LSD1 KD cells.
(D) Heatmaps for differential expression (FDR < 0.05) of sense or antisense transcripts of ERVs between control and LSD1 KD cells.
(E) LSD1 and H3K4me2 ChIP-seq signals at genomic loci of 8593 individual ERVs from 279 ERV subfamilies in control and LSD1 KD cells.
(F) Fold changes of a number of representative bi-directional ERV transcripts in LSD1 KD versus control cells determined by directional RNA-seq.
(G) Immunoblots of TLR3, MDA5 and RIG-I in MCF-7 cells.
(H–J) The RT-qPCR analysis of transcripts of selected ERVs, IFNs and ISGs in MCF-7 cells transduced with shRNA against scramble, LSD1 or LSD1 plus TLR3 (H), MDA5 (I) or RIG-I (J). The RT-qPCR data were normalized to GAPDH and presented as fold changes of gene expression in the test sample compared to the control, representing two to three independent experiments. Error bars represent standard deviation (SD) between two replicate samples in one experiment. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant, as determined by unpaired t-test.
We next determined whether ERV transcript up-regulation caused by LSD1 inhibition was a causal factor for the induction of IFN/antiviral responsive genes. To this end, we ectopically expressed an engineered 4 kb ERV fragment without protein coding capacity, derived from HERV-K and HERV-E, and found that its RNA overexpression readily caused the induction of IFNs and ISGs in MCF-7 cells (Figure S1P), which demonstrates the sufficiency of ERV up-regulation in triggering IFN activation.
TLR3 and MDA5 sense dsRNA accumulation, caused by LSD1 abrogation, and trigger IFN activation
We next investigated the mechanism by which ERV up-regulation causes IFN activation. We hypothesized that the expression of ERVs as well as other retrotransposons in both sense and antisense directions may contribute to the generation of dsRNAs, which may then trigger IFN activation. To test this, we used RNase A, which cleaves single stranded (ss) RNA and preserves dsRNA under high salt condition (Roulois et al., 2015), and also a dsRNA-specific antibody (J2) (White et al., 2014) to measure the presence of dsRNAs. We found that dsRNA enrichment for a number of ERVs as well as a few other retrotransposons was much higher in LSD1 KD samples compared to control samples (Figure S2A). Using the dsRNA-specific J2 antibody, we also captured more transcripts of selected retrotransposons in LSD1 KD samples (Figure S2B). These results provide evidence for the elevation of intracellular dsRNA levels as a result of LSD1 inhibition.
Intracellular dsRNAs are recognized by pattern recognition receptors, TLR3, MDA5 (encoded by IFIH1) and RIG-I (encoded by DDX58), which are involved in subsequent activation of the IFN pathways (Takeuchi and Akira, 2010). In LSD1 KD cells, all three dsRNA sensors were among the up-regulated genes (Figure S2C and 1G). To identify which sensor is essential for recognizing dsRNAs to elicit cellular responses, we inhibited the expression of individual sensors by shRNA-mediated knockdown prior to knocking down LSD1 (Figure S2D), and then assessed the impact on IFN activation. Importantly, abrogation of TLR3 and MDA5, but not RIG-I, significantly diminished the induction of IFN-β, IL-28 as well as ISGs without altering ERV expression level (Figures 1H–1J and S2E). In addition, abrogation of MAVS (Figure S2F), which is a downstream adaptor of the MDA5 pathway (Kawai et al., 2005), also blocked IFN activation in response to LSD1 inhibition (Figure S2G). As a further control, we knocked down cGAS and STING, two key sensors for the cytoplasmic DNA (Chen et al., 2016), to the extent that cytoplasmic DNA recognition pathway was significantly impaired, and showed that cytoplasmic DNA is unlikely the trigger of IFN responses in LSD1 KD cells (Figures S2H–S2J). Therefore, dsRNA recognition by TLR3 and MDA5 is essential for IFN activation upon LSD1 inhibition, consistent with the observation that the up-regulated IFN/antiviral responsive genes are indirect targets of LSD1 (Figure 1C). Previous studies show that TLR3 and MDA5 recognize dsRNAs that are at least 40bp in length or longer, respectively, whereas RIG-I prefers ssRNA or short dsRNA with 5’ triphosphate ends (Takeuchi and Akira, 2010). Thus, our finding of the involvement of TLR3 and MDA5, but not RIG-I, suggests that long dsRNAs, rather than ssRNAs or short dsRNAs, are responsible for the generation of an IFN response when LSD1 is ablated.
Decreased expression of RISC components due to loss of LSD1 contributes to dsRNA stress and promotes IFN activation
Double stranded RNAs derived from ERV transcripts can go on to trigger IFN responses or be processed by the RISC complex, which includes DICER and AGO proteins to mediate RNA interference (Berrens et al., 2017; Okamura and Lai, 2008). The steady state of the dsRNA pool is determined not only by ERV transcription but also the action of the RISC complex. Interestingly, we found that LSD1 KD led to a reduced protein expression of key components (DICER, AGO2 and TRBP2) of RISC (Figure 2A). The regulation of RISC was dependent on LSD1 catalytic activity, as reintroduction of WT FH-LSD1 but not the catalytic compromised LSD1-K661A back into LSD1 KD cells restored the protein expression of DICER, AGO2 and TRBP2 (Figure 2A). In contrast, we did not observe an obvious impact of LSD1 on the expression of Drosha, which acts at the initiation step of miRNA processing (Lee et al., 2003) (Figure S3A). Consistent with the above expression analysis, LSD1 KD also resulted in an elevated expression of a GFP reporter (Figures 2B–2D), whose expression was under the control of let-7 miRISC activity (Qi et al., 2008). This finding suggests that RISC may also be involved in dsRNA stress and IFN responses. Indeed, when we inhibited AGO2 expression, we also observed increased dsRNA abundance derived from a few retrotransposons tested (Figure 2E), and concomitant induction of IFN-β and IL-28 as well as ISGs assayed at both RNA and protein levels (Figures 2F and 2G). Similarly, inhibition of either DICER or TRBP2 also activated the IFN pathway (Figures S3B–S3E), consistent with a previous report showing that DICER restricts endogenous dsRNA and IFN activation (White et al., 2014). Therefore, disruption of the RISC complex, which perturbs intracellular dsRNA homeostasis, also induced IFN activation. To confirm that suppression of the RISC complex is involved in LSD1 inhibition-stimulated IFN activation, we compensated for the reduction of RISC by overexpressing AGO2 in LSD1 KD cells. We found that AGO2 overexpression significantly diminished dsRNA accumulation caused by LSD1 inhibition, leading to a reduction in some IFN/ISG activation (Figures 2H–2J). Collectively, these findings suggest that, in addition to regulating ERV transcription, LSD1 also regulates the expression of RISC components and consequently RISC function. Both actions of LSD1 contribute to its suppression of dsRNA accumulation.
Figure 2. LSD1 inhibition suppresses the expression of RISC components, contributing to dsRNA stress.

(A) Immunoblots of RISC components in MCF-7 cells transduced with shRNA against scramble or LSD1, and rescued with either WT LSD1 or catalytically compromised LSD1-K661A.
(B–D) U2OS stable cell line expressing dual reporters GFPL/GFP-let-7 was transduced with shRNA against scramble, LSD1 or AGO2. The expression of GFPL and GFP was measured by immunoblot (B) and RT-qPCR (C). The ratios of GFPL over GFP protein in different samples from five repeats for sh-LSD1 and two repeats for sh-AGO2 were calculated and the ratio in sh-Ctrl sample was considered as 100% miRISC activity (D).
(E) The dsRNA enrichment of a few retrotransposons assessed by RNase A digestion and RT-qPCR analysis.
(F and G) The expression of IFNs and ISGs in control and AGO2 KD MCF-7 cells analyzed by RT-qPCR (F) and immunoblot (G).
(H–J) The measurement of AGO2 and LSD1 protein expression by immunoblot (H), dsRNA enrichment of a few retrotransposons (I), and RNA levels of IFNs and ISGs by RT-qPCR (J) in MCF-7 cells with indicated manipulations.
Error bars represent SD between duplicates (C, E and F) or triplicates (J) in one of two experiments, or standard error of the mean (SEM) from five experiments (I). *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant, as determined by unpaired t-test.
Also see Figure S3.
LSD1 regulates AGO2 methylation status and stability
In order to understand the mechanism by which LSD1 regulates the expression of RISC components, we first examined whether LSD1 inhibition-induced dsRNA stress, which was previously reported to decrease DICER protein expression (Wiesen and Tomasi, 2009), is involved. To this end, we mitigated dsRNA stress by blocking its recognition by dsRNA sensors and observed that TLR3 KD fully rescued the protein level of DICER but not AGO2 or TRBP2 in LSD1 KD cells (Figure S3F). This result confirms that the regulation of DICER expression by LSD1 is through dsRNA stress and therefore is indirect, while it also suggests that the regulation of AGO2 and TRBP2 expression is likely independent of dsRNA. Given that AGO2 is the central component responsible for target RNA cleavage, we decided to focus our investigations on AGO2. We first analyzed AGO2 RNA level but detected no obvious alterations upon LSD1 KD (Figure S3G), suggesting the regulation occurs at posttranscriptional level. Indeed, in a cyclohexamide (CHX) chase assay, we detected a substantial decrease in AGO2 protein half-life when LSD1 was inhibited (Figures 3A and 3B), implicating a regulatory role of LSD1 in AGO2 protein stability. Interestingly, we found that LSD1 physically interacted with the RISC complex (Figures 3C and S3H). This physical interaction likely occurred in the nucleus, because LSD1 is exclusively localized in the nucleus and a portion of RISC components was also detected in the nuclear fraction (Figure S3I), in line with a recent report (Gagnon et al., 2014). As further evidence, we performed reciprocal co-immunoprecipitation with either whole cell lysate (WCL) or nuclear extract (NE) of MCF-7 cells stably expressing FH-LSD1, and detected a much stronger association between LSD1 and AGO2 in NE compared with that in WCL (Figure 3D).
Figure 3. LSD1 regulates AGO2 methylation status and stability.

(A and B) Representative immunoblot of AGO2 (A) and quantification of AGO2 signal from five experiments (B, mean±SEM) in MCF-7 cells treated with 50 µg/ml cycloheximide (CHX) in the presence or absence of 2 µM GSK-LSD1 for the indicated times.
(C and D) The physical interaction between LSD1 and AGO2 was examined by co-IP assay using whole cell lysate (WCL) of MCF-7 cells stably expressing FH-AGO2 (C), or reciprocally using WCL and nuclear extract (NE) of MCF-7 cells stably expressing FH-LSD1 (D). FT-flow through.
(E and F) Ectopically expressed WT FH-AGO2 and mutants in MCF-7 cells treated by LSD1 KD (E) or GSK-LSD1 (F) were immunoprecipitated, and then immunoblotted with mono-methyl AGO2 specific antibody and an AGO2 antibody.
(G) The immunoblot of K726me1 on endogenous AGO2 in control or LSD1 KD MCF-7 cells.
(H) The protein stability of transiently expressed WT FH-AGO2 and FH-AGO2-K726R in 293T cells was measured using CHX chase assay in the presence or absence of 2 µM GSK-LSD1. The averaged AGO2 quantification from two experiments is shown.
**p < 0.01, as determined by unpaired t-test.
Also see Figure S3.
These above findings led us to investigate whether LSD1 regulates AGO2 stability by controlling AGO2 methylation. To identify methylation sites on AGO2 that are regulated by LSD1, we purified the overexpressed FH-AGO2 from MCF-7 cells with or without LSD1 inhibition, and conducted mass spectrometry analysis. We found that a lysine residue at position 726 (K726) was consistently mono-methylated through mass spectrometry analysis of multiple biological samples when LSD1 was inhibited either by shRNA-mediated KD or by GSK-LSD1 (Figures S3J and S3K). To validate this finding, we raised an antibody that preferentially recognized AGO2 peptides mono-methylated at K726 (K726me1) compared with un-methylated or dimethylated peptides (Figure S3L). Furthermore, this antibody detected increased K726me1 on ectopically expressed AGO2 when LSD1 was inhibited, which can be abrogated by substituting K726 with arginine (K726R) or alanine (K726A) (Figures 3E and 3F). Importantly, this antibody detected increased methylation of endogenous AGO2 upon LSD1 inhibition (Figure 3G), suggesting that LSD1 regulates AGO2 demethylation in cells. To ascertain that K726 demethylation is responsible for sustaining AGO2 stability, we blocked its methylation by substituting lysine 726 for arginine, and as expected we observed an increased stability for FHAGO2- K726R compared to WT FH-AGO2 under physiological condition as well as in response to LSD1 inhibition (Figure 3H). We noticed that the ectopic AGO2 (FHAGO2) and the endogenous AGO2 showed different stabilities, which has also been noted before (Qi et al., 2008; Sahin et al., 2014). Taken together, LSD1 modulates AGO2 stability by regulating AGO2 demethylation at K726. The decreased AGO2 protein level (possibly Dicer and TRBP2 as well) in response to LSD1 inhibition, together with increased ERV transcription, leads to dsRNA stress and IFN activation in human cancer cells.
LSD1 abrogation-induced dsRNA stress suppresses tumor cell growth in vitro
We next investigated biological consequences of LSD1 inhibition-induced dsRNA stress, and in particular, whether dsRNA stress-triggered cellular responses can be harnessed for anti-tumor immunity. To address these questions, we first asked whether our observations made in human cells could be recapitulated in mouse cells. Lewis lung carcinoma (LLC), D4m.3A and B16 melanoma are mouse tumor cell lines with poor immunogenicity (Jenkins et al., 2014; Lechner et al., 2013). LSD1 inhibition by CRISPR/Cas9-mediated gene deletion resulted in up-regulation of retrotransposons and activation of IFN pathways in those cell lines (Figures 4A, S4A–S4C and S5A–S5G), which recapitulates our findings in human cancer cells. In addition, we also analyzed and observed dsRNA accumulation in B16 and D4m.3A cells in response to LSD1 loss (Figures 4B, 4C and S4D).
Figure 4. LSD1 abrogation-induced dsRNA stress suppresses tumor cell growth in vitro.

(A) The transcripts of selected retrotransposons, IFNs and ISGs in control and LSD1 KO (clone g5-4) B16 cells analyzed by RT-qPCR.
(B) The dsRNA enrichment of a few retrotransposons in control and LSD1 KO B16 cells assessed by RNase A digestion and RT-qPCR analysis.
(C) Total RNA extract treated with mock, RNase T1, RNase III or RNase A (350 mM NaCl) was dotted on Hybond N+ membranes, visualized by methylene blue staining and immunoblotted with J2 antibody.
(D and E) Colony formation (D) and quantification of colony areas (E) of scramble, LSD1 KO or LSD1/MDA5 DKO B16 cells.
(F) The expression of selected retrotransposons, IFNs and ISGs in scramble, LSD1 KO and LSD1/MDA5 DKO B16 cells analyzed by RT-qPCR.
(G) The expression of IFNs and ISGs in scramble, LSD1 KO and LSD1/IFNAR1 DKO B16 cells analyzed by RT-qPCR.
(H and I) Colony formation (H) and quantification of colony areas (I) of scramble, LSD1 KO or LSD1/IFNAR1 DKO B16 cells.
Error bars represent SEM from three (A and B) or two (F) experiments, or representing SD between duplicates (G), quadruplicates (E) or triplicates (I) in one of two experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant, as determined by unpaired t-test.
Also see Figures S4 and S5.
LSD1 inhibition by genetic perturbation resulted in compromised growth of B16 cells in vitro (Figures S4E–S4H). To determine whether the growth phenotype is due to LSD1 abrogation-induced dsRNA stress, we deleted dsRNA sensors MDA5 or TLR3 in LSD1 KO B16 cells (Figures S5E and S5H). The deletion of MDA5 significantly, albeit partially, rescued the growth defect of LSD1 KO B16 cells (Figures 4D and 4E), suggesting that the growth defect was in part due to dsRNA stress induced by LSD1 deletion. At the molecular level, the induction of IFNs and ISGs, but not dsRNA abundance, was significantly diminished in LSD1/MDA5 double knockout (DKO) cells (Figures 4F and S4I). As a control, deletion of MDA5 alone had minimal effects on B16 cell growth and IFN activation (Figures S4J–S4L). We didn’t observe an apparent rescue by TLR3 genetic deletion (Figure S4M), which could be explained by the observation that TLR3 protein was minimally or not expressed in B16 cells (data not shown).
We expected a similar rescue effect on cell growth by blocking the IFN pathway in LSD1 KO B16 cells. Indeed, deletion of IFNAR1, a crucial subunit for type 1 IFN receptor, also diminished IFN activation and partially restored cell growth (Figures 4G–4I and S5I), in line with the suppressive effect of type 1 IFNs on cell growth (Parker et al., 2016). In addition, IFN-β deletion also displayed a similar, albeit milder, rescue effect (Figures S4N–S4P and S5J). In summary, LSD1-abrogation in mouse cancer cells causes dsRNA stress and subsequent IFN activation, leading to cell growth inhibition in vitro.
LSD1 abrogation-induced dsRNA stress triggers anti-tumor T cell immunity in vivo
The role of LSD1 in basic cancer biology has been previously reported, including sustaining cancer stem cell self-renewal and suppressing differentiation, promoting cell proliferation, enhancing EMT as well as modulating metastasis (Hosseini and Minucci, 2017). These studies either used in vitro cell culture systems or transplanted human cancer cells into immunodeficient mice, but did not explore the role of LSD1 in regulating the tumor responses to host immunity. Based on our in vitro finding that LSD1 regulates cellular processes that are potentially related to tumor immune responses, we next determined whether LSD1 inhibition might trigger anti-tumor immunity in vivo. To interrogate this possibility, we used mouse syngeneic tumor models by subcutaneously inoculating B16 cells into C57BL/6 WT mice. The deletion of LSD1 in B16 cells significantly inhibited tumor growth in vivo, assessed by both tumor size and animal survival (Figures 5A and 5B), in agreement with our in vitro observations (Figures S4E–S4H). To distinguish the role of LSD1 in regulating tumor autonomous growth versus adaptive anti-tumor immunity, we compared tumor growth in immunocompetent (WT) and immunodeficient T-cell receptor α (TCRα) KO mice. Although LSD1 deletion inhibited B16 tumor growth in WT mice, there was no growth difference between LSD1 KO and control B16 tumors in TCRα KO mice (Figures 5C and 5D, compare blue and green lines). This result indicates that LSD1 inhibition in tumor cells elicits potent anti-tumor T cell immunity in vivo, rather than affecting tumor cell autonomous growth to restrain tumor burden.
Figure 5. LSD1 inhibition stimulates anti-tumor T cell immunity, which depends on dsRNA recognition and IFN-β production.

(A and B) Tumor growth (A) and survival curves (B) of immunocompetent mice inoculated with 500k scramble or LSD1 KO B16 cells.
(C and D) Tumor growth (C) and survival curves (D) of immunocompetent or immunodeficient (TCRα KO) mice inoculated with 500k scramble or LSD1 KO B16 cells.
(E and F) Tumor growth (E) and survival curves (F) of immunocompetent mice inoculated with 500k scramble, LSD1 KO, MDA5 KO, or LSD1/MDA5 DKO B16 cells.
(G) Tumor growth of immunocompetent mice inoculated with 500k scramble, LSD1 KO, IFN-β KO, or LSD1/IFN-β DKO B16 cells.
(H and I) Representative images (H) and quantification (I) of lung metastasis in immunocompetent mice receiving 200k scramble or LSD1 KO B16 cells intravenously.
Data represent two independent experiments (A–F). Error bars represent SEM of individual mice per group in one experiment. *p<0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant, as determined by unpaired t-test (A and I), Log-rank test (B, D and F), or 2-way ANOVA (C, E and G).
To confirm that host anti-tumor T cell immunity was boosted by tumor-intrinsic dsRNA stress, we compared the growth of LSD1 KO and LSD1/MDA5 DKO B16 tumors in immunocompetent mice. As expected, deletion of MDA5 was sufficient to diminish LSD1 inhibition-elicited anti-tumor immunity, as evidenced by the similar growth of LSD1/MDA5 DKO and control tumors targeted with scramble gRNA or MDA5 single KO tumors (Figures 5E and 5F). To further examine whether MDA5-associated type 1 IFN response is essential for LSD1 inhibition-elicited anti-tumor immunity, we genetically abrogated IFN-β production in LSD1 KO B16 tumors (Figures S5E and S5J), which completely reversed LSD1 KO-mediated inhibition of tumor growth in vivo to a level comparable to that of the control or IFN-β single KO tumors in immunocompetent mice (Figure 5G). This result demonstrates that tumor cell-derived IFN-β is essential for anti-tumor immunity against LSD1-null tumors, although IFN-β could be produced by multiple cell types of the host. In addition to controlling growth of tumors implanted subcutaneously, we further showed that LSD1 KO in B16 tumor cells markedly reduced tumor lung metastasis (Figures 5H and 5I). Thus, LSD1 inhibition-induced dsRNA stress and a resultant IFN response sensitizes tumors to T cell immunity, likely by increasing tumor immunogenicity.
LSD1 inhibition enhances tumor immunogenicity and increases T cell infiltration
To further elucidate the mechanism connecting LSD1 inhibition to enhanced antitumor T cell immunity, we investigated the effects of tumor cell-intrinsic LSD1 on T cell activity in the tumor microenvironment. We found a significant increase in CD4+ and CD8+ T cell numbers in LSD1 KO compared to control B16 tumors (Figure 6A), indicating a stronger ability to induce T cell immunity. Importantly, the increase in T cell infiltration was diminished when MDA5 was concurrently ablated (Figure 6A). In contrast, we did not detect any significant alteration in T cell populations in draining lymph nodes (dLNs) of B16 tumor-bearing mice (Figure S6A), suggesting tumor cell LSD1 ablation exerts effects locally within the tumor microenvironment. To assess the functional activity of CD8+ TILs (tumor infiltrating lymphocytes), we analyzed the expression of a proliferation marker, Ki-67, and a cytotoxic factor, Granzyme-B (GzmB), but neither showed a noticeable alteration when LSD1 was deleted in B16 tumor cells (Figure 6B). These results suggest that a major effect of LSD1 ablation is to promote T cell infiltration into tumors. To investigate whether the increased T cell infiltration is associated with increased TCR repertoire diversity of CD8+ TILs in LSD1 KO B16 tumors, we analyzed the clonality and entropy of these T cells by TCR sequencing, but did not find any significant changes compared to their counterparts in WT tumors (Figure 6C). Thus, the increased T cell infiltration in LSD1 KO tumors is unlikely due to changes in tumor antigenicity.
Figure 6. LSD1 inhibition enhances tumor immunogenicity.

(A) Tumor infiltrating lymphocytes (TILs) from transplanted B16 tumors (n=5 for scramble, n=5 for LSD1 KO and n=6 for LSD1/MDA5 DKO) in immunocompetent mice were analyzed by flow cytometry at day 14 post implantation when tumor sizes were comparable among the three groups.
(B) The expression of Ki-67 and GzmB by CD8+ TILs as in (A) was analyzed by flow cytometry.
(C) The clonality and entropy of CD8+ TILs isolated from transplanted B16 tumors (n=5 for scramble and n=3 for LSD1 KO) were analyzed by TCR sequencing.
(D–G) GFP-labeled B16 tumor cells (n=3 per group of scramble, LSD1 KO and LSD1/MDA5 DKO) were isolated from tumor-bearing immunocompetent mice and subjected to RNA-seq analysis. Differential gene expression was shown in volcano plots (D). Dots in red represent increased genes (log2(FC) > 1 and FDR < 0.05) and dots in blue represent decreased genes (log2(FC) < −1 and FDR < 0.05) in LSD1 KO versus scramble cells (left plot) or LSD1/MDA5 DKO versus scramble cells (right plot). GO analysis of up-regulated genes (log2(FC-KO/Ctrl) > 1 and FDR < 0.05) in LSD1 KO versus scramble cells was performed and top 10 terms were shown in a dot map (E). The up-regulated genes associated with top 10 GO terms (170 in total) were sorted out and log2(FC) of their expression in LSD1 KO and LSD1/MDA5 DKO versus scramble cells was plotted (F). All genes categorized in GO term “MHC protein complex” were displayed in a heatmap (G).
(H–J) Flow cytometry analysis of MHC-1 (H) and PD-L1 (J) expression and RNA-seq analysis of PD-L1 expression (I) by GFP-labeled B16 tumor cells (n=3 per group of scramble, LSD1 KO and LSD1/MDA5 DKO) isolated from tumor-bearing mice.
Data represent two independent experiments (A, B, H and J). Error bars represent SEM of individual mice per group in one experiment. *p < 0.05, ***p < 0.001, ****p < 0.0001, ns, not significant, as determined by unpaired t-test.
Also see Figures S6.
We next investigated if LSD1 inhibition-induced dsRNA stress altered tumor cell characteristics associated with tumor response to T cell immunity. Ex vivo tumor cell transcriptomic analysis revealed that LSD1 deletion significantly altered the gene expression profile in B16 tumor cells in vivo (Figures 6D and S6B). When MDA5 was simultaneously deleted, these transcriptional changes were reduced (Figures 6D and S6B). Consistent with the in vitro results with human cancer cells, LSD1 ablation also led to loss of LSD1 occupancy at ERVs, increased H3K4me2 as well as up-regulation of ERV transcription in B16 tumor cells in vivo (Figures S6C–S6E).
Differential expression analysis showed that immune response-related biological processes, including innate immune response, response to IFN-β, defense response to virus and MHC protein complex, were ranked among the top 10 GO terms in ex vivo LSD1 KO tumor cells (Figure 6E), providing evidence for increased tumor immunogenicity. In addition, genes associated with inflammatory responses were also enriched in LSD1 KO tumor cells as analyzed by GSEA (Figure S6F). Importantly, the induced expression of genes associated with the top 10 GO terms was significantly diminished by simultaneous MDA5 deletion in LSD1 KO cells (Figure 6F), suggesting a critical role of dsRNA recognition pathway in mediating tumor immunogenicity. Of note, we specifically looked at cell proliferation pathways by GSEA, but didn’t observe an apparent alteration by LSD1 deletion (Figure S6G), which further supports the notion that autonomous growth of B16 tumor cells in syngeneic mice is likely independent of LSD1 status.
In order to validate the findings from RNA-seq, we focused on the biological process “MHC protein complex” and assessed whether LSD1 KO affects MHC class 1 protein expression, because defects in MHC class 1 enable immune escape and are commonly found in solid tumors. In the RNA-seq analysis, most MHC-1 coding genes were up-regulated in LSD1 KO B16 cells, among which the induction of H2-D1 and H2-K1, which encode classical class 1 antigens, was largely dependent on the MDA5 pathway (Figure 6G, boxed). Consistent with the gene expression data, flow cytometry analysis of GFP-labeled B16 cells isolated from in vivo transplanted tumors revealed that LSD1 deletion caused a marked induction of MHC-1 expression on the tumor cell surface, which was completely abrogated by concurrent deletion of MDA5 (Figure 6H). Altogether, these results show that LSD1 inhibition activates the dsRNA recognition pathway and thereby enhances tumor immunogenicity, associated with increased T cell infiltration.
To examine whether the enhanced tumor immunogenicity by LSD1 inhibition is a generalizable mechanism, we used another “cold” tumor model, D4m.3A melanoma (BrafV600E; Pten loss). LSD1 KO D4m.3A tumors grew slightly faster than control tumors in TCRα KO mice (Figure S6H). In contrast, in syngeneic immunocompetent mice, both tumors displayed slower growth than their counterparts in TCRα KO mice, and LSD1 KO D4m.3A tumors displayed much slower growth than WT control tumors (Figure S6H), suggesting enhanced T cell immunity against tumors lacking LSD1. Consistently, we also found increased T cell infiltration in LSD1 KO D4m.3A tumors (Figure S6I), and elevated MHC-1 expression on the surface of LSD1 KO D4m.3A tumor cells compared with control tumor cells (Figure 6J), indicating enhanced tumor immunogenicity. Thus, our results suggest that the enhanced tumor immunogenicity elicited by LSD1 inhibition is not limited to the B16 tumor model and may be of broad significance.
LSD1 inhibition overcomes tumor resistance to PD-1 blockade
Our observation of enhanced tumor cell immunogenicity associated with LSD1 KO tumors suggests that these otherwise refractory tumors may now be sensitized to checkpoint blockade therapy. Interestingly, our RNA-seq and flow cytometry analysis also revealed that PD-L1 expression was up-regulated in LSD1 KO B16 and D4m.3A tumors in vivo (Figures 6I, 6J and S6K). PD-L1 induction may suppress the functional activity of CD8+ TILs (Juneja et al., 2017), compromising the anti-tumor effect of increased TILs caused by LSD1 inhibition. These findings prompted us to determine whether PD-1 blockade would synergize with LSD1 ablation to promote anti-tumor immunity. We used B16 tumors for these studies because B16 tumors express PD-L1 but have poor immunogenicity, and are known to be non-responsive to PD-(L)1 blockade in the absence of vaccination (Chen et al., 2015; Curran et al., 2010; Juneja et al., 2017; Kleffel et al., 2015). As expected, PD-1 blockade alone had no overt effects on WT B16 tumor growth (Figures 7A and 7B). Strikingly, PD-1 blockade markedly reduced the growth of LSD1 KO B16 tumors when PD-1 blockade was initiated on day 14 after tumor implantation (Figures 7A and 7B). In addition, we administered anti-PD-1 to mice when LSD1 KO or control B16 tumors reached the same volume to ensure that effects of PD-1 blockade are not due to delayed kinetics of LSD1 KO tumor growth. In both scenarios, PD-1 blockade had a profound effect on decreasing growth of LSD1 KO tumors but not WT tumors (Figures 7C and 7D). These results demonstrate a potent synergy between LSD1 inhibition and PD-1 blockade in controlling tumor growth and suggest that targeting LSD1 bypasses the need for vaccination to achieve PD-1 blockade responsiveness in the B16 tumor model.
Figure 7. LSD1 inhibition overcomes tumor resistance to PD-1 blockade in a mouse model and LSD1 expression level is inversely correlated with T cell infiltration in human tumors.

(A and B) Tumor growth (A) and survival curves (B) of immunocompetent mice inoculated with 250k B16 cells, and treated with anti-PD-1 or isotype control. Arrows indicate time points of anti-PD-1 injection.
(C and D) Tumor growth (C) and survival curves (D) of immunocompetent mice inoculated with 500k B16 cells, and treated with anti-PD-1 or isotype control based on a set tumor size (~200 mm3) for initial treatment. Arrows indicate time points of initial anti-PD-1 injection into scramble tumor-bearing mice (in black) and LSD1 KO tumor-bearing mice (in red), followed by additional anti-PD-1 injections every other day.
(E) The analysis of LSD1 RNA expression in tumors and normal tissues from patients with indicated types of cancers in TCGA dataset.
(F and G) Correlation analysis for LSD1 expression level versus IFN signature (F) or CD8+ T cell infiltration (G) in tumors from indicated types of cancer patients in TCGA dataset.
Error bars represent SEM of individual mice per group in one experiment. ***p < 0.001, ****p < 0.0001, ns, not significant, as determined by 2-way ANOVA (A and C), or Log-rank test (B and D).
Also see Figure S7.
To determine the translational significance of these findings, we explored the public datasets on human cancer. We found that LSD1 was infrequently mutated, amplified or deleted in a majority of cancer types examined (Figure S7A). However, LSD1 was overexpressed in tumors compared with normal tissues in a variety of cancer types (Figure 7E). We then examined whether LSD1 expression level in tumors correlates with clinical outcome. We dichotomously divided patients of each cancer type by LSD1 expression median, and compared overall survival between two groups. Our analysis showed that the LSD1-high group had a significantly shorter overall survival time than the LSD1-low group for a number of cancer types (Figure S7B), suggesting the LSD1 overexpression is a poor prognostic factor. In line with the finding that LSD1 inhibition caused IFN/antiviral response in in vitro MCF-7 cells and in vivo B16 tumor cells (Figures 1A and 6E), LSD1 expression level was found to be inversely correlated with IFN/antiviral response in a variety of cancer types in the TCGA cancer patient dataset (Figure 7F). We also analyzed LSD1 expression level versus CD8+ T cell infiltration in multiple cancer types. Importantly, we found that LSD1 expression level was also inversely correlated with CD8+ T cell infiltration in most cancer types (Figure 7G), consistent with our finding of increased T cell infiltration by LSD1 inhibition in mouse models (Figures 6A and S6I).
We carried out further analysis on the TCGA skin cutaneous melanoma (SKCM) cohort, and found that the patient group with low LSD1 expression (LSD1-low) had better survival probability than that with intermediate or high LSD1 expression (LSD1-int/high) (Figure S7C). Consistently, the LSD1-low group was associated with increased expression of genes enriched in immune responses (Figure S7D). Specifically, both CD8α and GzmB were expressed at higher levels in the LSD1-low group than in the LSD1-int/high group, indicating increased CD8+ T cell infiltration (Figures S7E and S7F).
DISCUSSION
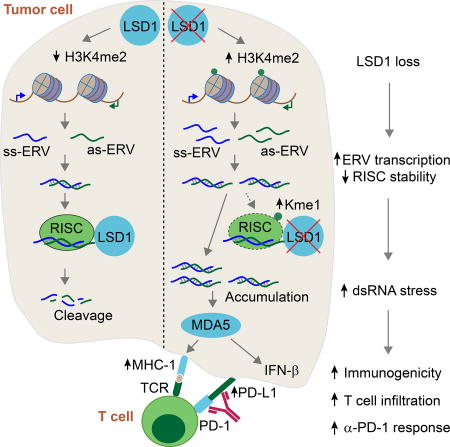
In this study, we have demonstrated that targeting LSD1 in tumor cells causes intracellular dsRNA stress and resultant cellular responses including IFN activation, which promotes anti-tumor T cell immunity and sensitizes refractory tumors to PD-1 blockade in vivo in a melanoma mouse model (Figure S7G). The remarkable ability of LSD1 inhibition to convert a tumor resistant to PD-1 blockade to a tumor responsive to PD-1 blockade may provide a means to increase the efficacy of anti-PD-1 cancer therapy and potentially turn “cold” tumors “hot” (Sharma et al., 2017).
Intracellular dsRNA homeostasis
The epigenetic regulation of repetitive element transcription in mammalian germ cells and early embryonic development is well documented (Feschotte and Gilbert, 2012; Leung and Lorincz, 2012), but much less is known in differentiated somatic cells. Our study shows that LSD1 represses the transcription of a subset of ERVs in human and mouse cancer cells. We further show that LSD1 inhibition also compromises the expression of RISC proteins and consequently RISC action. For instance, we showed that loss of LSD1 leads to a reduced expression of the dsRNA cleaving enzyme, DICER, which could explain how this regulation can contribute to the elevated level of dsRNAs in LSD1 KD/KO cells (White 2014). Loss of LSD1 also results in a reduction of AGO2 protein level, but how reduction of AGO2 contributes to a change in dsRNA level remains less clear. It's possible that AGO2 loaded with siRNA (generated from DICER) cleaves complementary RNA transcripts that form dsRNAs, thus reducing dsRNA formation (Berrens et al., 2017; Li et al., 2016). By coordinating ERV transcription and RISC component expression, LSD1 inhibition reinforces dsRNA stress and subsequent cellular responses.
Interestingly, two recent studies reported that DNMT inhibitors also lead to increased ERV transcription and IFN activation (Chiappinelli et al., 2015; Roulois et al., 2015). A previous study suggested that LSD1 regulates DNMT1 stability via demethylating DNMT1 in mESCs (Wang et al., 2009) but another study also using mESCs failed to find DNMT1 stability change (Macfarlan et al., 2011). In the context of cancer cells, we found that depletion of LSD1 had no overt effect on DNMT1 steady state level and global DNA methylation level, though our data cannot exclude locus-specific DNA methylation changes. Taken together, our findings suggest that the LSD1 effects we observed are probably not mediated by alterations in DNA methylation. It will be interesting to determine whether DNA and histone methylation regulate the same or different types of retroviral elements and whether inhibition of both will further elevate anti-tumor immunity.
Tumor cell-intrinsic LSD1 regulates host anti-tumor immunity
LSD1 regulates in vitro tumor cell growth through both dsRNA-IFN activation and possibly intrinsic proliferation program. The latter observation is consistent with previous reports that LSD1 promotes cell proliferation in cell culture and in mouse xenograft models (Mohammad et al., 2015; Zhang et al., 2013). However, since these studies transplanted human tumor cells into immunodeficient mice, the potential impact of lymphocytes was not taken into consideration. When syngeneic immunodeficient and immunocompetent mice were used in parallel, we found that LSD1 is not required for B16 tumor cell autonomous growth in vivo, revealing that LSD1 inhibition mainly elicits potent anti-tumor T cell immunity, which drastically reduces tumor growth. We speculate that host somatic cells such as stromal cells in the tumor microenvironment can foster cell proliferation that may circumvent the need of LSD1 for cell proliferation in vivo. On the other hand, it may also be possible that innate immune pressure, for example by secreting low amount of type 1 IFN acting on tumor cells, minimizes the growth difference between WT and LSD1-null tumors.
Given that our investigations are genetic in nature, we were able to conclude that the impact of LSD1 inhibition on anti-tumor immunity is due to alterations arising from the tumor cells, in which we further demonstrated dsRNA stress and resultant IFN-β as key mediators. Our study, together with those on the DNA methylation inhibitors, highlight an important role of chromatin modifications in regulating tumor cells and their behavior in vivo in the context of an intact immune system.
LSD1 inhibition and PD-(L)1 blockade
Our study shows that LSD1 inhibition elicits potent anti-tumor immunity associated with increased T cell infiltration and immunogenicity in low or non-immunogenic tumors. Accompanying the increased T cell infiltration, LSD1 KO B16 tumors also show elevated levels of PD-L1, a key inhibitor of T cell responses, which may compromise the efficacy of increased T cell infiltration. Indeed, the combination of LSD1 abrogation in B16 cells and PD-1 blockade showed a striking effect on controlling B16 tumor growth in the absence of vaccination. Therefore, we believe that LSD1 inhibition overcomes the resistance of B16 tumors to PD-1 blockade by increasing tumor immunogenicity and T cell infiltration (Chen et al., 2015; Juneja et al., 2017). By coordinating these two closely related processes, T cell infiltration and functional reinvigoration, LSD1 inhibition and PD-1 blockade achieve a potent synergy. Consistent with our findings in mouse models, the analysis of cancer patient data reveals an inverse correlation between LSD1 expression and CD8+ T cell infiltration and prognosis, pointing to the therapeutic potential of targeting LSD1 in combination with PD-(L)1 blockade for cancer treatment. The finding that high LSD1 expression is associated with poor prognosis in a variety of cancers further suggests LSD1 expression may be an informative biomarker.
In summary, our study provides the basis for developing the combinatorial use of LSD1 inhibitors and anti-PD-(L)1 for cancer therapy. Further work is needed to explore the function of LSD1 in immune cells. Given the general role of LSD1 in regulating dsRNA and IFN responses, targeting LSD1 in combination with anti-PD-(L)1 may prove to be a broadly applicable new strategy in cancer immunotherapy.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Yang Shi (yshi@hms.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
MCF-7, T47D, 293T, B16, LLC and D4m.3A cells were cultured in normal DMEM supplemented with 10% FBS and 1% penicillin/streptomycin in a 5% CO2 incubator at 37 °C.
Mice
6–10-wk-old female mice were used for all experiments. WT C57BL/6 mice were purchased from The Jackson Laboratory. Prior to all experiments, purchased mice were allowed one week to acclimate to housing conditions at the Harvard Medical School Animal Facility. For studies using immunodeficient mice, we compared mice from the same facility (in-house WT vs. in-house TCRα−/−) or (Jackson WT vs. Jackson TCRα−/−). The in-house strains of WT and TCRα−/− were originally purchased from The Jackson Laboratory. Colonies for each strain of mice were maintained in the same animal facility at Harvard Medical School. All experimental mice were housed in specific pathogen–free conditions and used in accordance with animal care guidelines from the Harvard Medical School Standing Committee on Animals and the National Institutes of Health. Animal protocols were approved by the Harvard Medical School Standing Committee on Animals.
METHOD DETAILS
Cell culture and chemical compound treatment
All cell lines were cultured in a 5% CO2 incubator at 37 °C, and passaged every 2–3 days. One day before compound treatment, cells were seeded in 6-well or 12-well plates, and then were treated with 2 µM GSK-LSD1, or DMSO as mock, in duplicates or triplicates for 5–6 days, during which cells were passaged once and replenished with fresh compound.
Gene knockdown by shRNA and ectopic expression
The shRNA oligos, with sequences for their respective target genes listed in Table S1, were annealed and cloned into a pLKO.1-Puromycin+ (Puro) or pLKO.1-Blasticidin+ (Bsd) lentiviral vector. Lentivirus carrying pLKO.1 plasmid was produced by co-transfecting HEK293T cells with four helper plasmids (pHDM-VSV-G, pHDMtat1b, pHDM-HgPM2, and pRC-CMVRaII) and by harvesting viral supernatant after 72 h by passing through a 0.45 µm filter. Collected lentivirus was used directly to infect cells with the addition of 8 µg/ml polybrene (Sigma-Aldrich, cat#H9268), or frozen at −80 °C for later use. Infected cells were selected and expanded with puromycin (Gold Biotechnology, cat#P-600-500) at 1 µg/ml or blasticidin (Sigma-Aldrich, cat#15205) at 5 µg/ml for 5 days before being used for subsequent assays. For double KD, MCF-7 cells were first transduced with lentiviral pLKO-sh-GFP-Puro as control or pLKO-sh-Sensor-Puro, and selected with puromycin for 3 days. Those cells were then transduced again with lentiviral pLKO-sh-Scramble-Bsd or pLKO-sh-LSD1-Bsd, and selected with both puromycin and blasticidin for 3–5 days to achieve double KD. In this context, pLKO-sh-GFP-Puro plus pLKO-sh-Scramble-Bsd was referred as sh-Ctrl, and pLKO-sh-GFP-Puro plus pLKO-sh-LSD1-Bsd was referred as sh-LSD1.
For LSD1 rescue assay, MCF-7 cells were first transduced with lentiviral pLKO-sh-Scramble-Bsd or pLKO-sh-LSD1-Bsd, and selected with blasticidin for 3 days. Those cells were then transduced again with lentiviral pHAGE-CMV-Flag-HAEV/ LSD1/LSD1-K661A (puromycin and sh-LSD1 resistant) (Mosammaparast et al., 2013), and selected with both blasticidin and puromycin for 5 days before subsequent analysis. In this context, pLKO-sh-Scramble-Bsd plus pHAGE-CMVFlag-HA-EV was referred as sh-Ctrl, and pLKO-sh-LSD1-Bsd plus pHAGE-CMVFlag-HA-EV was referred as sh-LSD1. For an alternative rescue method, MCF-7 cells were first transduced with lentiviral pHAGE-CMV-Flag-HA-EGFP/LSD1/LSD1-K661A, and selected with puromycin for 2 weeks to get stable cells, which were then transduced with lentiviral pLKO-sh-Scramble-Bsd or pLKO-sh-LSD1-Bsd followed by blasticidin selection for 5 days before subsequent analysis.
To establish stable cell lines ectopically expressing AGO2 or TRBP2, cDNAs were cloned into the pHAGE-CMV-Flag-HA vector, and then used to package virus and transduce MCF-7 cells, followed by puromycin selection for 2 weeks. To create GFP-labeled B16 cells, the MSCV-PIG retroviral vector (MSCV backbone with puromycin and IRES-GFP) was used with Phoenix cells to package virus and transduce B16 parent lines, followed by puromycin selection for one week. Afterwards, the expression of GFP was analyzed and confirmed by flow cytometry.
Gene deletion by CRISPR/Cas9
The gRNA oligos, with sequences for their respective target genes listed in Table S1, were annealed and cloned into the Lenti-CRISPR-v2-Puromycin+ vector. To delete target genes, B16, LLC and D4m.3A cells were transiently transfected with Lenti-CRISPR-v2 plasmid carrying respective gRNA, and selected with 1 µg/ml puromycin for 2 days. Cells were then transferred into fresh medium without puromycin and seeded at super-low density to allow colony formation from single cells. Colonies were then picked and expanded for KO validation by immunoblot or ELISA, and by sequencing of target genomic region. For double KO, LSD1 KO B16 cells (clone g5-4) were used for deleting the second target gene as described above.
Generation of ERV expression construct and transduction
Primers with sequences listed in table S1 were used to obtain a 2kb fragment of HERV-K and a 2kb fragment of HERV-E through PCR amplification with insertion of stop codons at 5’ ends and additional 30bp elongation primers at 3’ ends. HERV-K and HERV-E fragments with reverse complementary elongation primers at their 3’ ends were mixed, denatured, annealed and elongated, followed by PCR amplification to generate a 4kb HERV-(K+E) fusion fragment, which was further cloned into a pHAGE-CMV-Flag-HA lentiviral vector thus expressing sense transcript of HERV-K and antisense transcript of HERV-E. Viral packaging and transduction were performed as described above. MCF-7 cells transduced with HERV-(K+E) were cultured for 48 hours without drug selection before subsequent analysis.
RNA extraction and RT-qPCR
All reagents, buffers and containers used for RNA work were RNase-free grade or treated with 0.1% v/v DEPC (Sigma-Aldrich cat#D5758) if applicable, to eliminate RNase contaminants in experiments described in this section and other relevant sections. For total RNA extraction, cells in culture were directly lysed in TRIzol (Life Technologies, cat#15596018) after medium removal. RNA extraction was performed according to the manufacturer’s instructions. The extracted RNA was reversely transcribed into cDNA using the PrimeScript™ RT Reagent Kit (Clontech cat#RR037B) according to the manufacturer’s instructions, with the following modifications: 2 µg of RNA samples with the addition of primers were first denatured at 70 °C for 5 min and cooled down on ice before the addition of buffer and reverse transcriptase; incubation time (at 37 °C) was increased up to 30 min. The obtained cDNA samples were diluted and used for real-time quantitative PCR (RT-qPCR). SYBR green (Roche, cat#06649416001) and gene specific primers with sequences listed in Table S1 were used for PCR amplification and detection on a LightCycler 480 system (Roche). The RT-qPCR data were normalized to GAPDH and presented as fold changes of gene expression in the test sample compared to the control.
Strand-specific PCR for detection of sense and antisense ERV transcripts
The strand-specific PCR method was adapted from (Chiappinelli et al., 2015) and performed with the PrimeScript™ RT Reagent Kit (Clontech cat#RR037B) with modifications. In brief, gene- and strand-specific primers (GSP) were synthesized with an extra Tag sequence (listed in Table S1, which does not exist in the human genome) at the 5’-end to generate Tag-GSP (for example, HERV-E Tag-BR for sense strand, Tag-TF for antisense strand), and were used for reverse transcription, following these steps: 1 µg total RNA in 6 µl H2O was mixed with 1 µl Tag-GSP (10 µM stock), pre-heated at 65 °C for 5 min and cooled down on ice; then added 2 µl buffer (5X), 0.5 µl reverse-transcriptase and 120 ng actinomycin D (Sigma-Aldrich, cat#A9415) in 0.5 µl H2O to a total volume of 10 µl; incubated at 50 °C for 50 min for only first strand cDNA synthesis and deactivated at 85 °C for 5 min; finally 1 U of RNase H (New England Biolabs, cat#M0297S) was added, followed by incubation at 37 °C for 20 min and subsequent ethanol precipitati on for cDNA purification. The obtained cDNA was then used for PCR amplification with paired primers: Tag-primer in pair with TF-primer for amplifying sense strand and Tag-primer in pair with BR-primer for amplifying antisense strand. The amplicons were visualized on 1.5% agarose gels.
DsRNA analysis by RNase digestion and RT-qPCR
For dsRNA analysis by RNase A digestion, 5 µg total RNA extracted from MCF-7, B16 or D4m.3A cells was dissolved in 46 µl H2O and mixed well with 3.5 µl NaCl (5 M stock). Then 0.5 µl RNase A (10 mg/ml stock, Thermo Fisher Scientific, cat#EN0531) or H2O as mock was added to a total volume of 50 µl and mixed well, followed by incubation at room temperature for 10 min. Afterwards, 1 ml TRIzol was directly added to the mixture to terminate digestion, followed by RNA extraction. The RNA transcripts of selected retrotransposons were measured by RT-qPCR with GAPDH or Actb as an internal control. The ratios of (retrotransposon/GAPDH)RNase-A/(retrotransposon/GAPDH)mock were calculated as enrichment folds.
DsRNA analysis by J2 pulldown
Purified total RNA from control or LSD1 KD MCF-7 cells was used for the J2 pulldown assay. J2 antibody (Scicons, cat#10010200) and mouse IgG control (Santa Cruz Biotechnology, cat#sc-2025) were first conjugated (1 µg per pulldown) to Protein G dynabeads (Life Technologies, cat#10003D), respectively. For each pulldown, 30 µg RNA was mixed with 500 µl immunoprecipitation (IP) buffer (350 mM NaCl, 25 mM Tris pH7.4, 5 mM DTT and 0.5% NP-40), followed by the addition of 0.5 µl RNase A (10 mg/ml stock, Thermo Fisher Scientific, cat#EN0531) and thorough mixing. The addition of RNase A was to reduce the overwhelming ssRNA and enrich dsRNA for J2 capture. Then, the whole mixture was mixed with washed beads and rotated at 4 °C for 2h. Afterwards, the beads were washed with IP buffer and incubated in 50 µl Proteinase K digestion solution (1xTE, 100 mM NaCl, 1% SDS, and 1 µl Proteinase K (20 mg/ml stock, Thermo Fisher Scientific, cat#AM2546)) at 45 °C for 20 min. The eluate was directly added to 1 ml TRIzol for RNA purification and RT-qPCR analysis as described above.
DsRNA analysis by J2 immunoblot
Purified total RNA from B16 cells was subjected to digestion with mock, RNase T1 (Thermo Fisher Scientific, cat#AM2283) and RNase III (Thermo Fisher Scientific, cat#AM2290) in their respective buffers and according to the manufacturer’s instructions, or RNase A (Thermo Fisher Scientific, cat#EN0531) under high salt condition (350 mM NaCl). The digestion was deactivated by the addition of TRIzol and RNA was subsequently purified. Equal volumes (2.5 µl) of purified RNA was dotted on Hybond N+ membrane (GE Healthcare, cat#RPN119B), dried and autocrosslinked in a UV stratalinker 2400 (Stratagene) two times. The membrane was then blocked in 5% milk in PBS-T (0.1% Tween-20) for 30 min and probed with J2 antibody at 4 °C overnight. On the next day, the membrane was washed with PBS-T three times and probed with secondary goat-anti-mouse HRP antibody (Millipore cat#AP124P) in 5% milk at room temperature for 1 h. The membrane was washed again with PBS-T three times and ECL was applied for film development. Afterwards, the membrane was stained with methylene blue solution (0.3% w/v methylene blue + 30% v/v ethanol + 70% v/v H2O) to visualize the presence of RNA.
Protein extraction and immunoblot analysis
Cells in culture were washed with ice-cold PBS twice to completely remove residual medium. RIPA lysis buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and 50 mM Tris pH 8.0) supplemented with protease inhibitor (Roche, cat#04693132001) and phosphatase inhibitor (Roche, cat#04906837001) was directly added to cell layers and scraped on ice. Cell lysates were transferred to small tubes and lysed on ice for 10 min before being cleared by top-speed centrifugation (20, 000 g) at 4 °C. Protein concentrations in lysates were measured by Bio-Rad protein assay (Bio-Rad, Cat#5000006) and adjusted equally between samples, followed by the addition of SDS loading buffer (5X) and boiling at 95 °C for 5 min. Equal volume and equal quantity of protein samples were subjected to SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad, cat#162-0097). The membrane was blocked in 5% milk at room temperature for 1 h and incubated with appropriate antibodies at 4 °C overnight. On the next day, the membrane was washed with PBS-T three times and incubated with appropriate secondary HRP antibodies in 5% milk at room temperature for 1 h. The membrane was washed again with PBS-T three times and ECL was applied for film development.
Protein immunoprecipitation (IP)
Cells in culture were washed with ice-cold PBS twice to completely remove residual medium. IP lysis buffer (50 mM Tris PH 7.5, 150 mM NaCl, 0.1% NP-40, 0.1% TritonX-100, Glycerol 10%) supplemented with protease inhibitor (Roche, cat#04693132001) and phosphatase inhibitor (Roche, cat# 04906837001) was directly added to cell layers and scraped on ice. Cell lysates were transferred to small tubes and briefly sonicated to shear chromatin, followed by 10 min incubation on ice before being cleared by top-speed centrifugation (20, 000 g) at 4 °C. Protein concentrations in lysates were measured by Bio-Rad protein assay (Bio-Rad, #5000006) and adjusted equally between samples. For IP of Flag-HA-tagged proteins, cell lysates were incubated with anti-HA magnetic beads (Thermo Fisher Scientific, cat#88837) or anti-Flag M2 affinity gel (Sigma-Aldrich, cat#A2220) on a rotator at 4 °C overnight. For IP of endogenous AGO 2, protein L magnetic beads (Thermo Fisher Scientific cat#88849) conjugated with AGO2 antibody (Santa Cruz Biotechnology, cat#53521) were used. IP beads were then washed with IP lysis buffer three times, and boiled in SDS loading buffer (1X) at 95 °C for 5 min for SDSPAGE and immunoblot analysis.
ELISA
The ELISA assay was performed with a Human IFN Beta ELISA Kit (pbl assay science, cat#41415-1) and Mouse IFN Beta ELISA Kit (pbl assay science, cat#42410-1) according to the manufacturer’s instructions.
GFPL/GFP-let-7 dual reporter assay
The reporter assay for miRISC activity was performed as previously described (Qi et al., 2008). In brief, a U2OS cell line stably expressing dual reporters, GFPL and GFP-let-7, was transduced with lentiviral shRNA against scramble, LSD1 or AGO2. Cells were selected with puromycin at 1 µg/ml and G-418 (Research Products International, cat#G64500-20.0) at 200 µg/ml for 4 days before subsequent analysis. The expression of GFPL and GFP was measured by immunoblot and RT-qPCR as described in the above sections. Protein signals in immunoblot were quantified by ImageJ software according to the user manual. The ratios of GFPL over GFP protein signals in different samples were calculated and the ratio in control shRNA sample was considered as 100% miRISC activity.
Cell colony formation assay
B16 cells growing at 80% confluence were trypsinized and transferred into fresh medium in a single cell suspension. Cell numbers were counted and diluted appropriately for seeding on 6-well plates (500 cells per well) or 12-well plates (200 cells per well). Cells were allowed to grow for 6 days, with fresh medium addition at day 3 without removing old medium, before staining with crystal violet solution (0.5% w/v crystal violet powder, 80% v/v H2O and 20% v/v methanol). Colony areas were quantified by ImageJ software according to the user manual.
Mouse tumor models
Mice were anesthetized with Avertin (2.5%), shaved at the injection site, and then injected in the flank subcutaneously with 250,000 or 500,000 B16-F10 (scramble, LSD1 KO, MDA5 KO, IFN-β KO, LSD1/MDA5 DKO and LSD1/IFN-β DKO), or 250,000 or 500,000 D4m.3A (scramble and LSD1 KO) tumor cells. Tumors were measured every 2–3 days once palpable (long diameter and short diameter) with a caliper. Tumor volume was determined using the volume formula for an ellipsoid: 1/2 × D × d2 where D is the longer diameter and d is the shorter diameter. Mice were sacrificed when tumors reached 2000 mm3 or upon ulceration/bleeding.
For antibody treatments, mice were given 100 µg antibody via intraperitoneal injection at day 14, 16, 18, and 20 post tumor injection using the following antibodies: anti-PD-1 (clone 29F.1A12) or isotype (clone 2A3) (Liang et al., 2003). Rat IgG2a isotype control antibody was purchased from BioXCell (cat#BE0089). Prior to treatments mice were randomized such that treatment groups had similar average tumor volumes prior to treatment initiation.
B16 metastasis assay
200K B16.F10 (scramble or LSD1 KO) were transferred intravenously via tail vein injection. Lungs were removed 14 days post injection and fixed overnight in Fekete’s solution. Visible metastases were counted in a blinded fashion by two investigators.
Tumor infiltrating leukocyte flow cytometry
Tumors were excised on day 14 post injection and cut into 2 mm sized pieces in collagenase and DNase. Samples were dissociated with a Gentle MACS, incubated for 20 minutes at 37°C, dissociated with a Gentle MACS again, and passed through a 70 µm filter. To enrich for leukocytes, samples were spun through a Percoll gradient. Leukocytes were isolated from the interface of the 40 and 70% Percoll gradient, stained, and analyzed for fluorescent markers. For intracellular staining the eBioscience Foxp3 Fixation/Permeabilization Kit was used. All antibodies were purchased from Biolegend or BD Biosciences (CD45.2, CD11b, CD3, CD4, CD8b, Foxp3, Granzyme-B, Ki-67, CD44, PD-L1, MHC-1).
Tumor infiltrating lymphocyte TCR sequencing
T cells were enriched from tumors as above followed by sorting for CD8b+ T cells. Genomic DNA was extracted using a DNeasy Blood & Tissue kit (Qiagen, cat#69506) and submitted to Adaptive Biotechnologies for mouse TCRΒ CDR3 survey sequencing. Data were analyzed using Adaptive Biotechnologies’ online analysis platform.
Directional RNA-seq
Purified total RNA from MCF-7 cells cultured in vitro or GFP-labeled B16 cells isolated from tumor-bearing immunocompetent mice was quantified by Qubit (Invitrogen) and analyzed by an Agilent Bioanalyzer to assess RNA integrity. 1 µg RNA (RIN > 9) was used to generate rRNA-depleted RNA with a NEBNext® rRNA Depletion Kit (New England Biolabs, cat#E6310S) or poly(A)+ RNA with a Magnetic mRNA Isolation Kit (New England Biolabs, cat#S1550S) according to the manufacturer’s instructions. The rRNA-depleted or poly(A)+ RNA was analyzed in an Agilent Bioanalyzer to ensure the complete removal of rRNA, and then used to generate directional a RNA library with a NEBNext® Ultra™ II Directional RNA Library Prep Kit for Illumina® (New England Biolabs, cat#E7760L) and NEBNext® Multiplex Oligos for Illumina® (New England Biolabs, cat#E7335L) according to the manufacturer’s instructions. Library concentrations were quantified by Qubit (Invitrogen) and mixed equally for sequencing at HiSeq 2500 or Nextseq 500 (Illumina) to generate 50 bp reads (MCF-7) or 75 bp (B16) from paired-ends. The raw data are deposited at the Gene Expression Omnibus (GEO) under the subseries entry GSE112230.
ChIP-seq
For MCF-7 cells, cell nuclei were obtained by lysing whole cells in hypotonic buffer (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% v/v glycerol, 1 mM DTT, and 0.1% v/v Triton X-100) supplemented with protease inhibitor. For GFP-labeled B16 cells isolated from tumor-bearing immunocompetent mice, intact cells were used for the following steps. After washing with PBS, nuclei/cells were fixed in 1% formaldehyde for 10 min at room temperature, followed by quenching in 125 mM glycine. Nuclei/cells were then washed twice with ice-cold PBS, lysed in ChIP sonication buffer (50 mM HEPES pH7.9, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.2% SDS) supplemented with protease inhibitor, and were subjected to sonication to obtain DNA fragments of 300- 800 bp. Subsequent procedures were carried out by following the Epigenesys protocol (https://www.epigenesys.eu/en/). The following antibodies were used to ChIP: anti-LSD1 (Abcam, cat#ab17721), anti-H3K4me1 (Abcam, cat#ab8895), anti-H3K4me2 (EMD Millipore, cat#07-030), and mouse IgG (Santa Cruz Biotechnology, cat#sc-2025).
ChIP-seq libraries were prepared using a NEBNext® Ultra™ DNA Library Prep Kit for Illumina® (New England Biolabs, cat#E7370L) and a NEBNext® Multiplex Oligos for Illumina® (New England Biolabs, cat#E7335L) according to the manufacturer’s instructions. Library concentrations were quantified by Qubit (Invitrogen) and mixed equally for sequencing at HiSeq 2500 (Illumina) to generate 50 bp reads from single-end. The raw data are deposited at the Gene Expression Omnibus (GEO) under the subseries entry GSE112230.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses
Statistical analyses were performed using GraphPad Prism 6 software and statistical significance was determined by p < 0.05. Comparisons between two groups were made using an unpaired Student’s t test. For multiple comparisons of tumor growth, two-way ANOVA was used. For comparing mouse survival curves, a Log-rank (Mantel-Cox) test was used.
For ChIP-seq and RNA-seq data, all statistical analysis was performed with R (version 3.4.0) unless otherwise specified. Student’s t-test was used to determine whether a significant shift in mean occurred for all comparisons unless otherwise specified.
ChIP-seq analysis
Raw reads were aligned to hg19 or mm9 using bwa (version 0.7.2-r351). The resultant sam files were converted to bam with samtools (version 0.1.18 (r982:295)). MACS2 (version 2.0.10.20131216) was used to call peaks on the bam files. The bedGraph files containing signal per million reads produced from MACS2 were converted to bigwig files with ucsctool kit (315). ChIP-seq signals were extracted with bwtool (version 1.0) from bigwig files and visualized in R.
Repeat annotation was downloaded from UCSC for hg19 and mm9, only ERVs were used for downstream analysis. To select ERVs, ERV families originated in Homo sapiens or Mus musculus were downloaded from Repbase (http://www.girinst.org/repbase/). A peak catalogue consisting of all possible peak intervals in ChIP-seq (histones and LSD1) was produced and ERVs were filtered with this catalogue. ChIP-seq signals were extracted with bwtool (version 1.0) from bigwig files and then visualized in R.
RNA-seq analysis
Raw reads were aligned to hg19 or mm9 using STAR (v2.4.2a) with the parameter “quantMode” set as “GeneCounts” to produce count table for each gene. Differential gene analysis was performed on gene raw counts in R with edgeR package (v3.18.1) from bioconductor. Read count table was filtered so that genes with at least one count across conditions were kept. The negative binomial generalized log-linear model was used in differential analysis. A FDR cut-off of 0.05 was used to determine significantly differentially expressed genes. The R package gProfileR (v0.6.4) was used to perform gene enrichment analysis on differential genes. Gene set enrichment analysis (GSEA) was performed with R package clusterProfiler (v3.4.4).
The function analyzeRepeats.pl from Homer (http://homer.salk.edu/homer/) software was used to get raw counts for repeats from RNA-seq data. Differential expression for repeats was performed with edgeR the same way as for genes.
TCGA data analysis
For the association of LSD1 expression with survival, patient vital status (dead and alive) was used as a surrogate end-point and patients dichotomized by LSD1 expression. The proportional hazards (PH) assumption was tested using the cox.zph function in the R Survival package (v2.41-3) with 0.1 as cutoff. A log-rank test was used instead if the PH assumption failed.
For analyzing LSD1 expression level versus T cell infiltration in each tumor, the total expression of CD8α (log2 counts per million) was used to assess the infiltration of cytotoxic T-lymphocytes, and correlations were computed versus LSD1 expression.
For analyzing LSD1 expression level versus IFN signature, genes were extracted from ‘virus’ and ‘interferon’ related terms of the biological pathways enriched in up-regulated genes upon LSD1 KD in MCF7 cells. These genes were further filtered for those that are up-regulated in KD condition to obtain a refined IFN signature gene list. Gene-wise z-score was calculated in each tumor type and the sum of genes in the refined IFN signature gene list was used to characterize IFN signature. Pearson’s correlation was used to evaluate the correlation of LSD1 level (z-score) and IFN signature.
For human SKCM data, patients were divided into tertiles based on LSD1 expression, and then the second and third tertiles were combined into one group (named LSD1-int/high) due to a lack of observable difference in survival curves between them. The online DAVID (https://david.ncifcrf.gov/summary.jsp) bioinformatics resources were used to analyze the differentially expressed genes under the category of GOTERM_BP_DIRECT.
DATA AND SOFTWARE AVAILABILITY
Data resources
The raw and processed data of ChIP-seq and RNA-seq are deposited at the Gene Expression Omnibus (GEO) under the subseries entry GSE112230.
Supplementary Material
(A) The RT-qPCR analysis of transcripts of selected ERVs, IFNs and ISGs in MCF-7 cells treated with or without 2 µM GSK-LSD1 for 5~6 days.
(B) The validation of shRNA-mediated LSD1 KD in MCF-7 cells by immunoblot.
(C) The RT-qPCR analysis of transcripts of selected ERVs, IFNs and ISGs in MCF-7 cells transduced with shRNA against scramble (sh-Ctrl) or LSD1 (sh-LSD1).
(D) IFN-β secretion in LSD1 KD MCF-7 cells measured by ELISA. n.d., not detected.
(E–G) Immunoblot of LSD1 (E) and RT-qPCR analysis of ERV (F) and IFN (G) expression in MCF-7 cells with indicated manipulations.
(H) Immunoblot of DNMT proteins in control and LSD1 KD MCF-7 cells.
(I) 5-methylcytosine content in genomic DNA of control and LSD1 KD MCF-7 cells quantified by HPLC-MS analysis.
(J and K) The expression of selected ERVs, IFNs and ISGs in T47D (J) and 293T cells (K) transduced with shRNA against scramble or LSD1 was measured by RTqPCR.
(L) Volcano and M-A plots of differentially expressed genes in LSD1 KD versus control MCF-7 cells analyzed by RNA-seq. Dots in red represent significantly increased genes (log2(FC-KD/Ctrl) > 1 and FDR < 0.05) and dots in blue represent significantly decreased genes (log2(FC-KD/Ctrl) < −1 and FDR < 0.05) in LSD1 KD versus control cells.
(M) A heatmap for differential transcript expression of repetitive elements between LSD1 KD and WT control MCF-7 cells.
(N) LSD1 and H3K4me2 ChIP-seq signals at genomic loci of HERV-E subfamily in control and LSD1 KD MCF-7 cells.
(O) The assessment of both sense and antisense transcripts of selected ERVs using strand specific primers for reverse transcription and PCR amplification. An asterisk indicates non-specific bands.
(P) The RT-qPCR analysis of the expression of IFNs and ISGs in MCF-7 cells transduced with pHAGE-EGFP or pHAGE-HERV-(K+E).
The RT-qPCR data were normalized to GAPDH and presented as fold changes of gene expression in the test sample compared to the control. Error bars represent SEM from three experiments (A, C, G and I) or two experiments (F and P), or represent SD between triplicates (D and J) or duplicates (K) in one of two experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant, as determined by unpaired t-test.
(A) Total RNA extract from control or LSD1 KD MCF-7 cells was digested with mock or RNase A under high salt condition (350 mM NaCl) followed by a second round of RNA extraction with TRIzol. The RNA transcripts of selected retrotransposons were measured by RT-qPCR with GAPDH as an internal control. The ratios of (retrotransposon/GAPDH)RNase-A/(retrotransposon/GAPDH)mock were calculated as enrichment folds.
(B) The RT-qPCR analysis of selected retrotransposon transcripts captured by J2 antibody in pulldown assay. n.d., not detected.
(C) A heatmap showing the expression of nucleic acid receptors in control and LSD1 KD MCF-7 cells analyzed by RNA-seq.
(D) KD validation of protein expression in MCF-7 cells transduced with indicated shRNAs by immunoblot analysis.
(E) Immunoblot analysis of ISG15 expression in MCF-7 cells transduced with indicated shRNAs.
(F and G) Immunoblot of MAVS protein (F) and RT-qPCR analysis of transcripts of selected ERVs, IFNs and ISGs (G) in MCF-7 cells transduced with shRNA against scramble, LSD1 or LSD1 plus MAVS.
(H) MCF-7 cells (control, cGAS KD and STING KD) were transfected with 5 µg fragmented genomic DNA (gDNA) from mammalian cells or mock for 5 hours, and the expression of IFNs and ISGs was analyzed by RT-qPCR.
(I and J) Immunoblots of cGAS and STING proteins (I) and RT-qPCR analysis of transcripts of selected ERVs, IFNs and ISGs (J) in MCF-7 cells transduced with indicated shRNAs.
The RT-qPCR data were normalized to GAPDH and presented as fold changes of gene expression in the test sample compared to the control (G, H and J). Error bars represent SEM from three experiments (A and J), or represent SD between duplicates (B, G and H) in one of two experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant, as determined by unpaired t-test.
(A) Immunoblot of Drosha protein in control and LSD1 KD MCF-7 cells.
(B–E) The transcripts of selected IFNs and ISGs were analyzed by RT-qPCR (B and D), and the KD of DICER and TRBP2 was validated by immunoblot (C and E) in MCF-7 cells transduced with indicated shRNAs.
(F) Immunoblot analysis of DICER, AGO2 and TRBP2 in MCF-7 cells transduced with indicated shRNAs.
(G) The measurement of RNA level of AGO1-4, DICER and TRBP2 by RT-qPCR in control and LSD1 KD MCF-7 cells.
(H) The physical interaction between LSD1 and TRBP2 was examined by co-immunoprecipitation assay using whole cell lysates (WCL) of MCF-7 cells stably expressing FH-TRBP2 or empty vector (−).
(I) Cellular fraction into WCL, cytoplasmic extract (CytoE) or nuclear extract (NE), and immunoblot analysis of DICER, AGO2, TRBP2 and LSD1 in wild type MCF-7 cells.
(J and K) Purified FH-AGO2 from MCF-7 cells treated by LSD1 KD or GSK-LSD1 was visualized by silver staining (J), and analyzed by mass spectrometry, with a representative MS/MS spectrum showing mono-methyl K726 in LSD1 inhibited samples (K).
(L) Dot blot detecting the reactivity of K726me1-specific antibody against un-, monoor di-methylated AGO2 peptides.
The RT-qPCR data were normalized to GAPDH and presented as fold changes of gene expression in the test sample compared to the control (B, D and G). Error bars represent SD between duplicates in one experiment (B and D), or represent SEM from two experiments (G). *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant, as determined by unpaired t-test.
(A–C) The RNA expression of selected retrotransposons, IFNs and ISGs in control and LSD1 KO LLC (A), D4m.3A (B) and B16 (C) cells analyzed by RT-qPCR.
(D) The assessment of dsRNA enrichment in control and LSD1 KO D4m.3A cells by RNase A digestion under 350 mM NaCl condition and RT-qPCR analysis.
(E–H) B16 cells with indicated manipulations by shRNA-mediated KD (E and F) or CRISPR/Cas9-mediated KO (G and H) were seeded in 6-well plates (500 cells per well) in triplicates and allowed to grow for 6 days before staining with crystal violet. Colony areas were quantified and compared to sh-Ctrl (F) or scramble (H).
(I) Total RNA extract from scramble, LSD1 KO or LSD1/MDA5 DKO B16 cells was dotted on Hybond N+ membranes in replicates, visualized by methylene blue staining and immunoblotted with J2 antibody.
(J) The RT-qPCR analysis of selected retrotransposons, IFNs and ISGs in control and MDA5 KO B16 cells.
(K and L) B16 cells (scramble, LSD1 KO and MDA5 KO) were seeded in 12-well plates in quadruplicates (200 cells per well) and allowed to grow for 6 days before staining with crystal violet. Colony areas were quantified and compared to scramble (L).
(M) The RT-qPCR analysis of IFNs and ISGs in control, LSD1KO and LSD1/TLR3 DKO B16 cells.
(N and O) B16 cells (scramble, LSD1 KO, LSD1/IFN-β DKO and IFN-β KO) were seeded in 12-well plates in triplicates (200 cells per well) and allowed to grow for 6 days before staining with crystal violet. Colony areas were quantified and compared to scramble (O).
(P) The RT-qPCR analysis of selected retrotransposons, IFNs and ISGs in control, LSD1 KO and LSD1/IFN-β DKO B16 cells.
The RT-qPCR data were normalized to GAPDH and presented as fold changes of gene expression in the test sample compared to the control (A, B, C, J, M and P). Error bars represent SEM from two experiments (A and P) or three experiments (B, C and D), or represent SD between duplicates (J and M), triplicates (F, H and O) or quadruplicates (L) in one of two experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant, as determined by unpaired t-test.
(A) Sequencing results of genomic Lsd1 exon targeted by gRNA5 (highlighted sequences) in corresponding LLC clones and in alignment with reference sequences.
(B) Immunoblot of LSD1 in CRISPR/Cas9-modified LLC clones.
(C) Sequencing results of genomic Lsd1 exons targeted by two gRNAs in corresponding D4m.3A clones and in alignment with reference sequences.
(D) Immunoblot of LSD1 in CRISPR/Cas9-modified D4m.3A clones.
(E) Sequencing results of genomic Lsd1, Mda5, Ifnar1, Ifnb and Tlr3 exons targeted by gRNAs in corresponding B16 clones and in alignment with reference sequences.
(F and G) Immunoblot of LSD1 with two antibodies in multiple B16 clones transfected with different gRNAs targeting Lsd1.
(H and I) Immunoblot of LSD1, MDA5 (H) and IFNAR1 (I) in CRISPR/Cas9-modified B16 cells as indicated. Arrow indicates IFNAR1 bands.
(J) ELISA detection of secreted IFN-β in scramble, IFN-β KO, LSD1/IFN-β DKO B16 cells transfected by 2 µg poly(I:C) for 23 hours.
(A) Flow cytometry analysis of T cells (CD4, CD8 and Treg) in draining lymph nodes (dLNs) of B16 tumor-bearing immunocompetent mice (n=5 for scramble, n=5 for LSD1 KO and n=6 for LSD1/MDA5 DKO).
(B and C) M-A plots of differentially expressed genes analyzed by RNA-seq using GFP-labeled B16 tumor cells (n=3 per group of scramble, LSD1 KO and LSD1/MDA5 DKO) isolated from tumor-bearing immunocompetent mice, referred as ex vivo cells hereafter. Dots in red represent significantly increased genes (log2(FC) > 1 and FDR < 0.05) and dots in blue represent significantly decreased genes (log2(FC) < −1 and FDR < 0.05) in LSD1 KO versus scramble cells (left plot) or LSD1/MDA5 DKO versus scramble cells (right plot).
(C) Differential expression (FDR < 0.05) of ERVs between scramble and LSD1 KO ex vivo B16 cells shown in a heatmap.
(D and E) LSD1 and H3K4me2 ChIP-seq signals at genomic loci of 74 ERV subfamilies (D) and representative RLTR13D3 subfamily (E) in ex vivo scramble and LSD1 KO B16 cells.
(F and G) GSEA analysis for inflammatory response (F) and positive regulation of cell proliferation (G) in ex vivo LSD1 KO versus scramble B16 cells.
(H) Tumor growth of immunocompetent and TCRα KO mice inoculated with 500k scramble or LSD1 KO D4m.3A cells.
(I) CD4+ and CD8+ TILs in transplanted D4m.3A tumors (n=9 for scramble and n=10 for LSD1 KO) in immunocompetent mice were analyzed by flow cytometry.
(J and K) Flow cytometry of MHC-1 (J) and PD-L1 (K) on the cell surface of D4m.3A tumor cells (n=9 for scramble and n=10 for LSD1 KO) isolated from tumor-bearing immunocompetent mice.
*p < 0.05, **p<0.01, ****p<0.0001, ns, not significant, as determined by unpaired t-test.
(A) The frequencies of mutation, amplification and deletion of LSD1 across a panel of cancer types were analyzed using cBioPortal. Only cancer types with at least one case of altered LSD1 were shown in the chart.
(B) The survival curves of LSD1-low (red) and LSD1-high (blue) patient groups dichotomously divided by LSD1 median in two cancer types ACC and SARC using TCGA dataset.
(C) The survival curves of LSD1-low (first tertile in red, n=113) and LSD1-int/high (second and third tertiles in blue, n=210) SKCM patient groups divided based on LSD1 expression using TCGA dataset.
(D) A plot of top 10 GO terms based on p value generated by DAVID functional annotation of differentially expressed genes (FC > 1.5 or FC< 0.67, and FDR < 0.05) in LSD1-low group versus LSD1-int/high group of SKCM (increased genes – red, decreased genes – blue).
(E and F) Comparison of CD8α (E) and GzmB (F) expression between the LSD1-low group (red) and the LSD1-int/high group (blue) of SKCM patients.
(G) A proposed model depicting how LSD1 regulates tumor responses to host immunity and checkpoint blockade therapy.
Table S1. DNA oligo sequences, Related to STAR Methods.
Highlights.
ERV induction and RISC reduction activate dsRNA-IFN pathway upon LSD1 inhibition
LSD1 loss in tumor cells stimulates anti-tumor T cell immunity
LSD1 ablation enhances tumor immunogenicity and T cell infiltration
LSD1 inhibition overcomes resistance to anti-PD-1 therapy in a mouse melanoma model
Acknowledgments
We thank Cheryl Arrowsmith for kindly providing the chemical compounds used in our screen; Hank Qi, Joan Brugge, David Fisher and Glenn Dranoff for sharing cell lines, and Santa Cruz Biotechnology for providing antibodies. We thank Richard Gregory, Carl Novina, Wolf Reik and all the Shi lab members for discussion and suggestions. This work was supported by a grant from the National Institutes of Health to Y.S. (R01 CA118487 and R35 CA210104) and funds from Boston Children’s Hospital. Y.S. is an American Cancer Society Research Professor. This work was also supported by grants from the Evergrande Center for Immunologic Diseases and Ludwig Center at Harvard Medical School to A.H.S., as well as trainings grant from the National Institutes of Health (T32CA207021 to M.W.L.).
Y.S. is a cofounder of Constellation Pharmaceuticals, Inc. and Athelas Pharmaceuticals, Inc and a member of the scientific advisory boards. A.H.S. has served as a paid consultant for Novartis, is a member of scientific advisory boards for Surface Oncology, SQZ Biotech, Adaptimmune and Elstar, and has patents on the PD-1 pathway licensed by Roche and Novartis. G.J.F. has patents/pending royalties on the PD-1 pathway from Roche, Merck, Bristol-Myers-Squibb, EMD-Serono, Boehringer-Ingelheim, Dako, AstraZeneca, and Novartis. G.J.F. has served on advisory boards for Roche, Bristol-Myers-Squibb, Xios and Quiet.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
W.S. and Y.S. conceived the project; W.S., M.W.L., A.H.S. and Y.S. designed experiments; W.S. performed and analyzed all in vitro cell experiments under the supervision of Y.S.; M.W.L. performed and analyzed all in vivo mouse experiments with assistance from T.H.N. under the supervision of A.H.S.; S.C., A.C., J.R.C., Y.L., H.Y., E.M.V.A., D.D.D.C. and H.H.H. analyzed ChIP-seq, RNA-seq and public TCGA data; G.J.F. contributed to mouse anti-PD-1 experiments; H.C. helped with 5mdC HLPC-MS analysis; P.H.H. performed protein MS analysis; W.S. and Y.S. wrote the manuscript with input from M.W.L. and A.H.S.
DECLARATION OF INTERESTS
All other authors declare no competing interests.
Supplemental information includes seven figures and one table.
References
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrens RV, Andrews S, Spensberger D, Santos F, Dean W, Gould P, Sharif J, Olova N, Chandra T, Koseki H, et al. An endosiRNA-Based Repression Mechanism Counteracts Transposon Activation during Global DNA Demethylation in Embryonic Stem Cells. Cell Stem Cell. 2017;21:694–703. e697. doi: 10.1016/j.stem.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes E, Shi Y. Diverse epigenetic mechanisms of human disease. Annu Rev Genet. 2014;48:237–268. doi: 10.1146/annurev-genet-120213-092518. [DOI] [PubMed] [Google Scholar]
- Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17:1142–1149. doi: 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- Chen S, Lee LF, Fisher TS, Jessen B, Elliott M, Evering W, Logronio K, Tu GH, Tsaparikos K, Li X, et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res. 2015;3:149–160. doi: 10.1158/2326-6066.CIR-14-0118. [DOI] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2015;162:974–986. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Feschotte C, Gilbert C. Endogenous viruses: insights into viral evolution and impact on host biology. Nat Rev Genet. 2012;13:283–296. doi: 10.1038/nrg3199. [DOI] [PubMed] [Google Scholar]
- Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357 doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KT, Li L, Chu Y, Janowski BA, Corey DR. RNAi factors are present and active in human cell nuclei. Cell reports. 2014;6:211–221. doi: 10.1016/j.celrep.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, Carter R, Awad W, Neale G, Thomas PG, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell. 2017;170:142–157. e119. doi: 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini A, Minucci S. A comprehensive review of lysine-specific demethylase 1 and its roles in cancer. Epigenomics. 2017;9:1123–1142. doi: 10.2217/epi-2017-0022. [DOI] [PubMed] [Google Scholar]
- Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, Mullins DW, Brinckerhoff CE. Multiple murine BRaf(V600E) melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res. 2014;27:495–501. doi: 10.1111/pcmr.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, Sharpe AH. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017;214:895–904. doi: 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassiotis G, Stoye JP. Immune responses to endogenous retroelements: taking the bad with the good. Nat Rev Immunol. 2016;16:207–219. doi: 10.1038/nri.2016.27. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nature immunology. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell. 2015;162:1242–1256. doi: 10.1016/j.cell.2015.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH, Ohlfest JR, Hu P, Epstein AL. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother. 2013;36:477–489. doi: 10.1097/01.cji.0000436722.46675.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Leung DC, Lorincz MC. Silencing of endogenous retroviruses: when and why do histone marks predominate? Trends Biochem Sci. 2012;37:127–133. doi: 10.1016/j.tibs.2011.11.006. [DOI] [PubMed] [Google Scholar]
- Li Y, Basavappa M, Lu J, Dong S, Cronkite DA, Prior JT, Reinecker HC, Hertzog P, Han Y, Li WX, et al. Induction and suppression of antiviral RNA interference by influenza A virus in mammalian cells. Nat Microbiol. 2016;2:16250. doi: 10.1038/nmicrobiol.2016.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, Sharpe AH. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33:2706–2716. doi: 10.1002/eji.200324228. [DOI] [PubMed] [Google Scholar]
- Macfarlan TS, Gifford WD, Agarwal S, Driscoll S, Lettieri K, Wang J, Andrews SE, Franco L, Rosenfeld MG, Ren B, et al. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev. 2011;25:594–607. doi: 10.1101/gad.2008511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell. 2015;28:57–69. doi: 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Mosammaparast N, Kim H, Laurent B, Zhao Y, Lim HJ, Majid MC, Dango S, Luo Y, Hempel K, Sowa ME, et al. The histone demethylase LSD1/KDM1A promotes the DNA damage response. J Cell Biol. 2013;203:457–470. doi: 10.1083/jcb.201302092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Lai EC. Endogenous small interfering RNAs in animals. Nat Rev Mol Cell Biol. 2008;9:673–678. doi: 10.1038/nrm2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16:131–144. doi: 10.1038/nrc.2016.14. [DOI] [PubMed] [Google Scholar]
- Qi HH, Ongusaha PP, Myllyharju J, Cheng D, Pakkanen O, Shi Y, Lee SW, Peng J, Shi Y. Prolyl 4-hydroxylation regulates Argonaute 2 stability. Nature. 2008;455:421–424. doi: 10.1038/nature07186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell. 2015;162:961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U, Lapaquette P, Andrieux A, Faure G, Dejean A. Sumoylation of human argonaute 2 at lysine-402 regulates its stability. PLoS One. 2014;9:e102957. doi: 10.1371/journal.pone.0102957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nature reviews Immunology. 2018;18:153–167. doi: 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, Wenzel A, Hicks J, Ballew M, Stone M, et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell. 2017;171:1284–1300. e1221. doi: 10.1016/j.cell.2017.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- White E, Schlackow M, Kamieniarz-Gdula K, Proudfoot NJ, Gullerova M. Human nuclear Dicer restricts the deleterious accumulation of endogenous double-stranded RNA. Nat Struct Mol Biol. 2014;21:552–559. doi: 10.1038/nsmb.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesen JL, Tomasi TB. Dicer is regulated by cellular stresses and interferons. Mol Immunol. 2009;46:1222–1228. doi: 10.1016/j.molimm.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lu F, Wang J, Yin F, Xu Z, Qi D, Wu X, Cao Y, Liang W, Liu Y, et al. Pluripotent stem cell protein Sox2 confers sensitivity to LSD1 inhibition in cancer cells. Cell Rep. 2013;5:445–457. doi: 10.1016/j.celrep.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv324. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) The RT-qPCR analysis of transcripts of selected ERVs, IFNs and ISGs in MCF-7 cells treated with or without 2 µM GSK-LSD1 for 5~6 days.
(B) The validation of shRNA-mediated LSD1 KD in MCF-7 cells by immunoblot.
(C) The RT-qPCR analysis of transcripts of selected ERVs, IFNs and ISGs in MCF-7 cells transduced with shRNA against scramble (sh-Ctrl) or LSD1 (sh-LSD1).
(D) IFN-β secretion in LSD1 KD MCF-7 cells measured by ELISA. n.d., not detected.
(E–G) Immunoblot of LSD1 (E) and RT-qPCR analysis of ERV (F) and IFN (G) expression in MCF-7 cells with indicated manipulations.
(H) Immunoblot of DNMT proteins in control and LSD1 KD MCF-7 cells.
(I) 5-methylcytosine content in genomic DNA of control and LSD1 KD MCF-7 cells quantified by HPLC-MS analysis.
(J and K) The expression of selected ERVs, IFNs and ISGs in T47D (J) and 293T cells (K) transduced with shRNA against scramble or LSD1 was measured by RTqPCR.
(L) Volcano and M-A plots of differentially expressed genes in LSD1 KD versus control MCF-7 cells analyzed by RNA-seq. Dots in red represent significantly increased genes (log2(FC-KD/Ctrl) > 1 and FDR < 0.05) and dots in blue represent significantly decreased genes (log2(FC-KD/Ctrl) < −1 and FDR < 0.05) in LSD1 KD versus control cells.
(M) A heatmap for differential transcript expression of repetitive elements between LSD1 KD and WT control MCF-7 cells.
(N) LSD1 and H3K4me2 ChIP-seq signals at genomic loci of HERV-E subfamily in control and LSD1 KD MCF-7 cells.
(O) The assessment of both sense and antisense transcripts of selected ERVs using strand specific primers for reverse transcription and PCR amplification. An asterisk indicates non-specific bands.
(P) The RT-qPCR analysis of the expression of IFNs and ISGs in MCF-7 cells transduced with pHAGE-EGFP or pHAGE-HERV-(K+E).
The RT-qPCR data were normalized to GAPDH and presented as fold changes of gene expression in the test sample compared to the control. Error bars represent SEM from three experiments (A, C, G and I) or two experiments (F and P), or represent SD between triplicates (D and J) or duplicates (K) in one of two experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant, as determined by unpaired t-test.
(A) Total RNA extract from control or LSD1 KD MCF-7 cells was digested with mock or RNase A under high salt condition (350 mM NaCl) followed by a second round of RNA extraction with TRIzol. The RNA transcripts of selected retrotransposons were measured by RT-qPCR with GAPDH as an internal control. The ratios of (retrotransposon/GAPDH)RNase-A/(retrotransposon/GAPDH)mock were calculated as enrichment folds.
(B) The RT-qPCR analysis of selected retrotransposon transcripts captured by J2 antibody in pulldown assay. n.d., not detected.
(C) A heatmap showing the expression of nucleic acid receptors in control and LSD1 KD MCF-7 cells analyzed by RNA-seq.
(D) KD validation of protein expression in MCF-7 cells transduced with indicated shRNAs by immunoblot analysis.
(E) Immunoblot analysis of ISG15 expression in MCF-7 cells transduced with indicated shRNAs.
(F and G) Immunoblot of MAVS protein (F) and RT-qPCR analysis of transcripts of selected ERVs, IFNs and ISGs (G) in MCF-7 cells transduced with shRNA against scramble, LSD1 or LSD1 plus MAVS.
(H) MCF-7 cells (control, cGAS KD and STING KD) were transfected with 5 µg fragmented genomic DNA (gDNA) from mammalian cells or mock for 5 hours, and the expression of IFNs and ISGs was analyzed by RT-qPCR.
(I and J) Immunoblots of cGAS and STING proteins (I) and RT-qPCR analysis of transcripts of selected ERVs, IFNs and ISGs (J) in MCF-7 cells transduced with indicated shRNAs.
The RT-qPCR data were normalized to GAPDH and presented as fold changes of gene expression in the test sample compared to the control (G, H and J). Error bars represent SEM from three experiments (A and J), or represent SD between duplicates (B, G and H) in one of two experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant, as determined by unpaired t-test.
(A) Immunoblot of Drosha protein in control and LSD1 KD MCF-7 cells.
(B–E) The transcripts of selected IFNs and ISGs were analyzed by RT-qPCR (B and D), and the KD of DICER and TRBP2 was validated by immunoblot (C and E) in MCF-7 cells transduced with indicated shRNAs.
(F) Immunoblot analysis of DICER, AGO2 and TRBP2 in MCF-7 cells transduced with indicated shRNAs.
(G) The measurement of RNA level of AGO1-4, DICER and TRBP2 by RT-qPCR in control and LSD1 KD MCF-7 cells.
(H) The physical interaction between LSD1 and TRBP2 was examined by co-immunoprecipitation assay using whole cell lysates (WCL) of MCF-7 cells stably expressing FH-TRBP2 or empty vector (−).
(I) Cellular fraction into WCL, cytoplasmic extract (CytoE) or nuclear extract (NE), and immunoblot analysis of DICER, AGO2, TRBP2 and LSD1 in wild type MCF-7 cells.
(J and K) Purified FH-AGO2 from MCF-7 cells treated by LSD1 KD or GSK-LSD1 was visualized by silver staining (J), and analyzed by mass spectrometry, with a representative MS/MS spectrum showing mono-methyl K726 in LSD1 inhibited samples (K).
(L) Dot blot detecting the reactivity of K726me1-specific antibody against un-, monoor di-methylated AGO2 peptides.
The RT-qPCR data were normalized to GAPDH and presented as fold changes of gene expression in the test sample compared to the control (B, D and G). Error bars represent SD between duplicates in one experiment (B and D), or represent SEM from two experiments (G). *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant, as determined by unpaired t-test.
(A–C) The RNA expression of selected retrotransposons, IFNs and ISGs in control and LSD1 KO LLC (A), D4m.3A (B) and B16 (C) cells analyzed by RT-qPCR.
(D) The assessment of dsRNA enrichment in control and LSD1 KO D4m.3A cells by RNase A digestion under 350 mM NaCl condition and RT-qPCR analysis.
(E–H) B16 cells with indicated manipulations by shRNA-mediated KD (E and F) or CRISPR/Cas9-mediated KO (G and H) were seeded in 6-well plates (500 cells per well) in triplicates and allowed to grow for 6 days before staining with crystal violet. Colony areas were quantified and compared to sh-Ctrl (F) or scramble (H).
(I) Total RNA extract from scramble, LSD1 KO or LSD1/MDA5 DKO B16 cells was dotted on Hybond N+ membranes in replicates, visualized by methylene blue staining and immunoblotted with J2 antibody.
(J) The RT-qPCR analysis of selected retrotransposons, IFNs and ISGs in control and MDA5 KO B16 cells.
(K and L) B16 cells (scramble, LSD1 KO and MDA5 KO) were seeded in 12-well plates in quadruplicates (200 cells per well) and allowed to grow for 6 days before staining with crystal violet. Colony areas were quantified and compared to scramble (L).
(M) The RT-qPCR analysis of IFNs and ISGs in control, LSD1KO and LSD1/TLR3 DKO B16 cells.
(N and O) B16 cells (scramble, LSD1 KO, LSD1/IFN-β DKO and IFN-β KO) were seeded in 12-well plates in triplicates (200 cells per well) and allowed to grow for 6 days before staining with crystal violet. Colony areas were quantified and compared to scramble (O).
(P) The RT-qPCR analysis of selected retrotransposons, IFNs and ISGs in control, LSD1 KO and LSD1/IFN-β DKO B16 cells.
The RT-qPCR data were normalized to GAPDH and presented as fold changes of gene expression in the test sample compared to the control (A, B, C, J, M and P). Error bars represent SEM from two experiments (A and P) or three experiments (B, C and D), or represent SD between duplicates (J and M), triplicates (F, H and O) or quadruplicates (L) in one of two experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant, as determined by unpaired t-test.
(A) Sequencing results of genomic Lsd1 exon targeted by gRNA5 (highlighted sequences) in corresponding LLC clones and in alignment with reference sequences.
(B) Immunoblot of LSD1 in CRISPR/Cas9-modified LLC clones.
(C) Sequencing results of genomic Lsd1 exons targeted by two gRNAs in corresponding D4m.3A clones and in alignment with reference sequences.
(D) Immunoblot of LSD1 in CRISPR/Cas9-modified D4m.3A clones.
(E) Sequencing results of genomic Lsd1, Mda5, Ifnar1, Ifnb and Tlr3 exons targeted by gRNAs in corresponding B16 clones and in alignment with reference sequences.
(F and G) Immunoblot of LSD1 with two antibodies in multiple B16 clones transfected with different gRNAs targeting Lsd1.
(H and I) Immunoblot of LSD1, MDA5 (H) and IFNAR1 (I) in CRISPR/Cas9-modified B16 cells as indicated. Arrow indicates IFNAR1 bands.
(J) ELISA detection of secreted IFN-β in scramble, IFN-β KO, LSD1/IFN-β DKO B16 cells transfected by 2 µg poly(I:C) for 23 hours.
(A) Flow cytometry analysis of T cells (CD4, CD8 and Treg) in draining lymph nodes (dLNs) of B16 tumor-bearing immunocompetent mice (n=5 for scramble, n=5 for LSD1 KO and n=6 for LSD1/MDA5 DKO).
(B and C) M-A plots of differentially expressed genes analyzed by RNA-seq using GFP-labeled B16 tumor cells (n=3 per group of scramble, LSD1 KO and LSD1/MDA5 DKO) isolated from tumor-bearing immunocompetent mice, referred as ex vivo cells hereafter. Dots in red represent significantly increased genes (log2(FC) > 1 and FDR < 0.05) and dots in blue represent significantly decreased genes (log2(FC) < −1 and FDR < 0.05) in LSD1 KO versus scramble cells (left plot) or LSD1/MDA5 DKO versus scramble cells (right plot).
(C) Differential expression (FDR < 0.05) of ERVs between scramble and LSD1 KO ex vivo B16 cells shown in a heatmap.
(D and E) LSD1 and H3K4me2 ChIP-seq signals at genomic loci of 74 ERV subfamilies (D) and representative RLTR13D3 subfamily (E) in ex vivo scramble and LSD1 KO B16 cells.
(F and G) GSEA analysis for inflammatory response (F) and positive regulation of cell proliferation (G) in ex vivo LSD1 KO versus scramble B16 cells.
(H) Tumor growth of immunocompetent and TCRα KO mice inoculated with 500k scramble or LSD1 KO D4m.3A cells.
(I) CD4+ and CD8+ TILs in transplanted D4m.3A tumors (n=9 for scramble and n=10 for LSD1 KO) in immunocompetent mice were analyzed by flow cytometry.
(J and K) Flow cytometry of MHC-1 (J) and PD-L1 (K) on the cell surface of D4m.3A tumor cells (n=9 for scramble and n=10 for LSD1 KO) isolated from tumor-bearing immunocompetent mice.
*p < 0.05, **p<0.01, ****p<0.0001, ns, not significant, as determined by unpaired t-test.
(A) The frequencies of mutation, amplification and deletion of LSD1 across a panel of cancer types were analyzed using cBioPortal. Only cancer types with at least one case of altered LSD1 were shown in the chart.
(B) The survival curves of LSD1-low (red) and LSD1-high (blue) patient groups dichotomously divided by LSD1 median in two cancer types ACC and SARC using TCGA dataset.
(C) The survival curves of LSD1-low (first tertile in red, n=113) and LSD1-int/high (second and third tertiles in blue, n=210) SKCM patient groups divided based on LSD1 expression using TCGA dataset.
(D) A plot of top 10 GO terms based on p value generated by DAVID functional annotation of differentially expressed genes (FC > 1.5 or FC< 0.67, and FDR < 0.05) in LSD1-low group versus LSD1-int/high group of SKCM (increased genes – red, decreased genes – blue).
(E and F) Comparison of CD8α (E) and GzmB (F) expression between the LSD1-low group (red) and the LSD1-int/high group (blue) of SKCM patients.
(G) A proposed model depicting how LSD1 regulates tumor responses to host immunity and checkpoint blockade therapy.
Table S1. DNA oligo sequences, Related to STAR Methods.