

Abstract
Human plasma high density lipoprotein-cholesterol (HDL-C) concentrations are a negative risk factor for atherosclerosis-linked cardiovascular disease (ACVD). Pharmacological attempts to reduce ACVD by increasing plasma HDL-C have been disappointing so that recent research has shifted from HDL-quantity to HDL-quality, i.e., functional vs. dysfunctional HDL. HDL has varying degrees of dysfunction reflected in impaired reverse cholesterol transport (RCT). In the context of atheroprotection, RCT occurs by two mechanisms: One is the well-known trans-hepatic pathway comprising macrophage free cholesterol (FC) efflux, which produces early forms of FC-rich nascent (n)HDL. Lecithin:cholesterol acyltransferase converts HDL-FC to HDL-CE while converting nHDL from a disc to a mature spherical HDL, which transfers its CE to the hepatic HDL receptor, scavenger receptor B1 (SR-B1) for uptake, conversion to bile salts, or transfer to the intestine for excretion. Although widely cited, current evidence suggests that this is a minor pathway and that most HDL- and nHDL-FC rapidly transfer directly to the liver independent of LCAT activity. A small fraction of plasma HDL-FC enters the trans-intestinal efflux pathway comprising direct FC transfer to the intestine. SR-B1−/− mice, which have impaired trans-hepatic FC transport, are characterized by high plasma levels of a dysfunctional FC-rich HDL that increases plasma FC bioavailability in a way that produces whole body hypercholesterolemia and multiple pathologies. The design of future therapeutic strategies to improve RCT will have to be formulated in the context of these dual RCT mechanisms and the role of FC bioavailability.
Keywords: reverse cholesterol transport, HDL biogenesis, atherogenesis, ATP-binding cassette transporter A1, lipoprotein receptors, cholesterol bioavailability
Subject Codes: Lipids and cholesterol, metabolism, atherosclerosis, cell biology/structural biology
The Reverse Cholesterol Transport (RCT) Hypothesis
Atherosclerotic cardiovascular disease (ACVD), a major cause of mortality and morbidity in the United States,(3) is characterized by cholesterol accumulation within the subendothelial space of the arterial wall. Discovery of the reaction of lecithin:cholesterol acyltransferase (LCAT), which esterifies free cholesterol (FC), mainly on high density lipoprotein (HDL), provoked the hypothesis that LCAT is part of a broader cholesterol transport process that sequesters cholesterol as cholesteryl ester (CE) in HDL thereby preventing accumulation of cytotoxic FC levels in peripheral tissues, which cannot catabolize FC.(2,4,5) This process (Figure 1), reverse cholesterol transport (RCT), was hypothesized before the discovery of cellular cholesterol transporters and HDL receptors. In the first RCT step, interaction of apo AI with the cellular lipid transporter ATP-binding cassette subfamily A, member 1 (ABCA1),(6,7) forms nascent HDL (nHDL), which contains mainly apo AI, free cholesterol (FC), and phospholipid (PL);(8,9) though less studied, a related lipid transporter, ABCG1, mediates FC efflux to HDL.(10) In the context of atheroprotection, the first step occurs on macrophages within the subendothelial space of the arterial wall. The second step, nHDL esterification by plasma LCAT converts discoidal nHDL to a mature spherical HDL with a central CE core. In the final step—cholesterol disposal—hepatic scavenger receptor class B member 1 (SR-B1), selectively extracts HDL-lipids (11) for disposal in the bile by a nibbling mechanism in which HDL-apos are excluded from uptake; HDL is reduced to a remnant while releasing lipid-free apo AI.(12) Although cited to near-consensus,(13,14) this trans-hepatic RCT model has not been fully validated.
Figure 1.
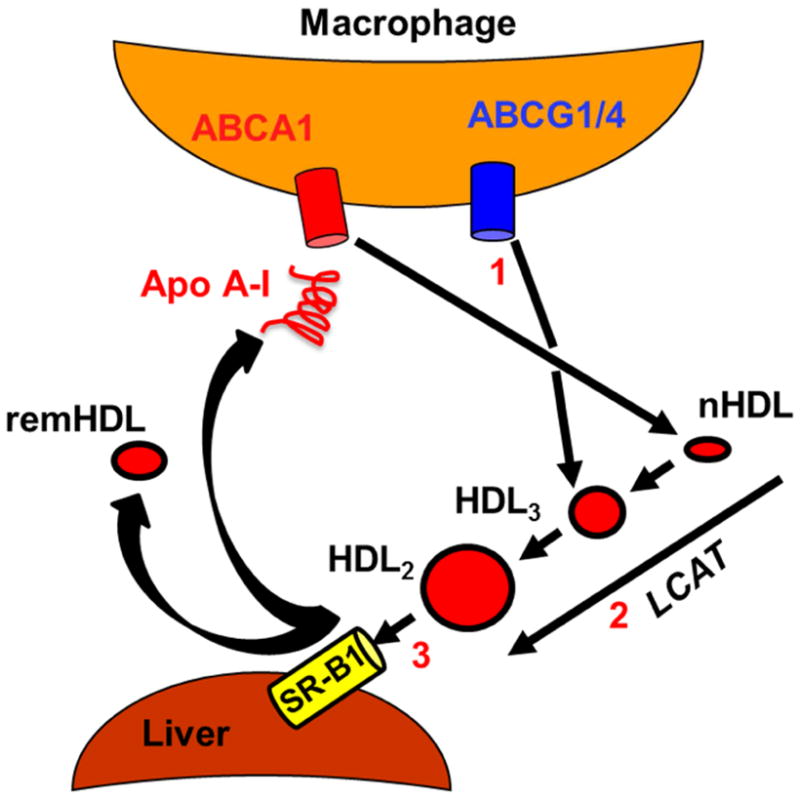
Traditional Model of RCT in the Context of Atheroprotection (Adapted from Glomset and Ross).(2) 1) FC efflux from macrophages initiates the formation of discoidal nHDL, which contains FC, PL, and apo AI. 2) LCAT catalyzes the conversion of FC to CE, which forms a central core within spherical HDL. 3) SR-B1 selectively extracts lipids, especially FC and CE, from the mature HDL particle leaving an apo-rich remnant HDL (remHDL) particle and lipid-free apo AI that returns to another RCT cycle.
Spontaneous Lipid Transfer
Plasma lipids transfer among lipoproteins and cells by several mechanisms. Spontaneous transfer, which occurs without the aid of transfer proteins, occurs on a physiologically meaningful time scale for FC, PL, and non-esterified fatty acids (FA). Halftimes (t1/2) for FC transfer from HDL3 and low density lipoproteins (LDL) to lipoprotein and membrane surfaces are respectively 3 and 45 min.(15) FA transfer halftimes vary according to chain-length and unsaturation. Each methylene unit and double bond decrease and increase respectively the transfer t1/2 by a factor of ~4.(16) The effects of chain length on FA transfer rates are profound. The t1/2 for palmitic acid (16-carbons) transfer is ~3 ms; adding ten methylene units, which gives hexacosanoic acid, increases t1/2 to 3 hours. Hexacosanoic acid is associated with adrenoleukodystrophy, an inherited disorder of FA metabolism in which hexacosanoic acid accumulates in tissues due to slow transfer to peroxisomes for degradation.(17) PL transfer rates also vary similarly with acyl chain composition. The t1/2 for PL and sterol transfer increases with increasing particle size, HDL < LDL < VLDL.(15,18) Given that they are a major component of nHDL,(1,8,9) the fate of nHDL-PL in the RCT pathway is determined in part by acyl chain composition and nHDL size.
Plasma HDL-Modifying Activities
HDL is modified by several plasma enzymes, transfer proteins, and cell surface receptors—cholesteryl ester transfer (CETP),(19) and phospholipid transfer proteins (PLTP),(20) LCAT,(21) endothelial(22) and hepatic lipases,(23) and hepatic SR-B1.(12) CETP transfers CE, triglyceride (TG) and PL among lipoproteins;(24) its most medically relevant activity is exchange of LDL- and HDL-CE for VLDL-TG, which in the context of hypertriglyceridemia gives rise to TG-rich LDL and HDL,(25) which are subsequently reduced to small, dense LDL and HDL3.(25) The in vivo t1/2 of CE transfer activity is on the order of 4–6 h(26) and varies according to ratios and plasma concentrations of lipoproteins, especially VLDL, which is a major acceptor of HDL-CE under hypertriglyceridemic conditions.(25) The main target of PLTP, discovered as a protein that transfers PL among lipoproteins and erythrocytes,(27) is HDL;(28) PLTP also mediates slow fusion of HDL with the concomitant release of apo AI.(20,28–30) Whether the apo AI is lipid-free or just lipid-poor has not been established with certainty; that apo AI binds to lipid surfaces favors the model in which apo AI is released as a lipid-free protein. LCAT transfers the SN-2 acyl chain of phosphatidylcholine to FC and in the absence of FC, LCAT has phospholipase A2 activity; both activities are activated by apo AI.(31) This reaction is important because it converts FC, which has high bioavailabililty, i.e., transfers among lipoprotein and membrane surfaces on a time frame of minutes, to CE, which is transferred by CETP with t1/2 ~5 h.(26) Early reports showed that cellular extraction of HDL-CE by SR-B1 occurs without HDL-protein uptake.(32) SR-B1 also extracts FC, PL, and TG albeit at different rates compared to CE (=1)—1.6, 0.2, and 0.7 respectively,(33) and gradually “nibbles” HDL to smaller and smaller remnants on a time frame of hours.(12) Most HDL-modifying activities,(19–21,23,28) including SR-B1,(12) release lipid-free apo AI. The release of lipid-free apo AI is due to its intrinsic instability.(34–37) The similar energetics for apo AI release from HDL and exchange among HDL (38) suggests that apo AI desorption is the rate-limiting step in HDL disruption. Given that lipid-free apo AI is not readily detected in freshly isolated plasma we surmise that lipid-free apo AI formed in vivo either reassociates with HDL or is rapidly relipidated by cell surface ABCA1 for additional nHDL production.
HDL Metabolism
Although most discussions of HDL metabolism are framed within the current model for trans-hepatic RCT (Figure 1), some studies have shed doubt on its validity: HDL-[3H]FC clearance and hepatic uptake in mice is rapid, t1/2 = 3.2 min, and is even shorter, t1/2 = 1.82 min, in mice over expressing SR-B1;(39). Only ~2% of plasma FC is esterified over 3 min, thus most FC plasma clearance occurs without the esterification step that is implicated in the conventional trans-hepatic RCT model. Considering that human metabolism is slower than that of mice, plasma HDL[3H]FC clearance kinetics in humans is similar (Table 1); most plasma HDL-FC is cleared with t1/2 ~8 min, during which <5% of FC is converted to CE;(40–42) plasma HDL-[3H]FC transfer to bile reaches its maximum value within 40 min,(43) a time frame in which little HDL-FC esterification occurs.
Table 1.
Kinetics of Clearance of HDL and nHDL*
| Human Plasma Lifetime | Mouse Plasma Lifetime | |||
|---|---|---|---|---|
| t1/2 | FCR | t1/2 | FCR | |
| HDL-apo AI | 5.8 d (83) | 0.12/d | 5.8 h (84) | 0.12/h |
| HDL-FC | 7–8 min (40) | 0.09/min | 3.2 min (39) | 0.22/min |
| HDL-CE | 0.92 d (85) | 0.75/d | 3.3–3.5 h(40,42,43,85) | 0.21/h |
| nHDL-FC | - | 5.2 min (1) | 0.13/min | |
| nHDL-PL | - | 2.0 min (1) | 0.35/min | |
| nHDL-apo AI | - | 7.7 h (1) | 0.09/h | |
Fractional catabolic rate (FCR) = ln2/t1/2.
nHDL Metabolism
nHDL obtained by incubating apo AI ABCA1-expressing cells has a higher FC content than HDL (60 vs. 10 mol%, where mol% FC = 100% x moles FC/(moles FC + moles PL)),(1,8,22,44,45) likely reflecting the importance of ABCA1 in reducing cellular cholesterol burden. Although nHDL-[3H]FC, [14C]PL, and [125I]apo AI transfer to different lipoproteins at different rates in vitro, at equilibrium their distribution reflects their natural occurrence within plasma lipoproteins.(1) In mice, the rates of transfer of plasma nHDL- and HDL-FC are similar, t1/2 ~5 min and ~2 min respectively; for apo AI t1/2 ~460 min.(39) During the ~5 min required for hepatic nHDL-FC uptake, only 2% of plasma FC was converted to CE. Thus, like human and mouse HDL-FC, nHDL-FC is cleared at a rate that is two orders of magnitude faster than that of apo AI and over a time interval in which little FC is esterified—LCAT plays a minor role in nHDL-FC clearance. The rates of HDL- and nHDL-FC clearance are comparable to those for spontaneous desorption and transfer of HDL-FC to LDL, 2–4 min.(15) This mechanism may in part underlie the rapid hepatic FC uptake. However, overexpression of SR-B1 in mice increases the rate of hepatic uptake of plasma HDL-FC.(39) Collectively, these data support a revised RCT model (Figure 2) in which nHDL-FC transfers to HDL after which it is cleared via hepatic SR-B1. However, contributions of direct FC transfer from nHDL spontaneously or via SR-B1 cannot be excluded.
Figure 2.

Revised RCT Model. ABCA1-expressing cells extrude FC and PL via the interaction of apo AI with ABCA1 giving nHDL (1). nHDL-apo AI and some nHDL-FC and PL rapidly transfer to HDL, t1/2 < 2 min (2); concurrently some nHDL-FC and PL transfer to LDL (3). Over the same time frame, nHDL- and HDL-FC and PL transfer mainly to the liver (5, 6, 7) while some nHDL-apo AI is recycled to ABCA1 (8). Over time, FC and PL equilibrate among erythrocytes (4), extra-hepatic tissues, and lipoproteins. nHDL- and HDL-FC and PL accretion in the liver occurs via spontaneous transfer and SR-B1 (5, 6, 7), the latter being promoted by PLTP (6). Modified from Xu et al. (1).
In mice, plasma nHDL-PL transfers to the liver with t1/2 ~2 min. This is unexpected given that spontaneous PL transfer halftimes are >1 hour.(15,18) Other mechanisms may be involved. One is SR-B1-mediated uptake although the rate of PL uptake is only 10% of FC uptake in SR-B1-expressing cells.(33) Another is direct transfer to cells at the site of contact between the plasma membrane and nHDL. Although PLTP increases PL transfer from nHDL to human Huh7 hepatocytes, a model cell line of human hepatocytes,(1) the increase (+100%) is still too small to account for the rapid in vivo uptake. Lastly, phospholipolytic activities convert PL to FA and lysoPL, which spontaneously transfer between lipoprotein and membrane surfaces with t1/2 ~ 3 msec.(16) Current data support a model (Figure 2) in which nHDL-PL transfers to the liver by spontaneous and PLTP-mediated mechanisms that likely involve SR-B1.(1)
Metabolic Segregation of Trans-Hepatic RCT
The plasma clearance kinetics of HDL- and nHDL-FC, PL, and apo AI have similar trends in mice and humans (Table 1): FC and PL disappear within a few minutes whereas apo AI persists for hours and days for mouse and man respectively. During the 5.8 d required for apo AI clearance from human plasma, CE is cleared 6 times and FC is cleared ~10,000 times. Following their initial rapid hepatic uptake, nHDL-FC and PL distribute over all major tissue sites, especially erythrocytes, while most apo AI remains in the plasma compartment.(1) The initial RCT step is cellular FC efflux to apo AI or HDL from whence it transfers to other lipoproteins and ultimately equilibrates with a total body FC pool which is in a stationary but dynamic state: Stationary because over time there is little change in the plasma concentrations of HDL-apo AI, FC, TG, CE, and PL; dynamic because HDL FC and PL rapidly enter and leave the plasma compartment and exchange with other lipoproteins and tissues, albeit at different rates. Most apo AI associates with HDL but is transferred to the lipid-poor (free?) form by several plasma activities(12,19–21,23,46) making it available for additional cycles of tissue-cholesterol efflux.
SR-B1−/− Mice: An Extreme Model of Dysfunctional HDL and Lipotoxic FC Bioavailability
SR-B1−/− mice have aberrant platelet and erythrocyte morphology and function, and susceptibility to diet-induced atherosclerosis, despite having a high plasma HDL-C concentration;(47,48) moreover, female SR-B1−/− mice are infertile.(49) The cause of these pathophysiologies has been attributed to a dysfunctional HDL for which a molecular mechanism has not been identified. Current evidence suggests that the defect is high FC bioavailability. Compared to WT mice, SR-B1−/− mice have ~2-fold higher plasma HDL levels, (50,51) and more HDL surface as FC (60 vs. 15 mol%);(22) these two attributes are expected to increase HDL-FC bioavailability. A high mol% HDL-FC alone is not atherogenic. However, giving SR-B1−/− mice a high fat, high FC diet increases plasma HDL concentrations and, while maintaining a high mol% HDL-FC, induces atherosclerosis. Thus, even though chow-fed SR-B1−/− mice have several serious metabolic abnormalities, atherogenic HDL in SR-B1−/− mice requires both high mol% FC and high plasma HDL concentration, which would be expected to increase FC bioavailability in a way that increases FC transfer to all cells in contact with plasma. The increased HDL-FC bioavailability overwhelms whole-body cholesterol clearance capacity thereby producing a pathological state in erythrocytes, platelets, the arterial wall, and the ovaries (of female mice). Some dysfunctionality can be reduced by lowering plasma HDL levels and/or mol% FC. In SR-B1−/− mice, probucol, which reduces plasma HDL levels and HDL-FC content,(52) rescues fertility and prevents atherosclerosis.(49,53) Compared to a control diet, dietary β-cyclodextrins, which remove FC from membranes(54) and lower plasma cholesterol (−35%),(55) reduce atherosclerotic plaque size and FC burden in a mouse model of atherosclerosis, the apo E−/− mouse on a high fat, high cholesterol diet.(56)
Other studies in humans are supportive but not axiomatic. A high HDL-cholesterol/PL ratio is atherogenic; patients with the highest ratio of HDL-C/particle had increased carotid atheroprogression compared with those with the lowest ratios, and increase in plaque area over time was greater in those with the highest vs. lowest HDL-C/particle ratio.(57) Without invoking the concept of FC bioavailability, the authors averred that the combination of cholesterol content (mol% FC) and particle number (HDL concentration) better predicted HDL atherogenicity than either parameter taken alone. Similar studies(58,59) are supportive but inconclusive because they did not distinguish between HDL-FC and CE so that FC bioavailability could not be surmised. There are notable exceptions:(60) For example, patients with familial hypoalphalipoproteinemia or familial apo AI/CIII deficiency, both with very low HDL levels, present with the onset of coronary artery disease at ages <40 and <50 years respectively. Tangier patients, who also have low HDL-C, present with coronary artery disease at age <60 years. Thus, FC bioavailability does not explain everything and other unknown HDL attributes or comorbidities are likely atherogenic.
Transintestinal Cholesterol Excretion (TICE) and FC Bioavailability
Early studies indicated FC transfer to the feces occurs independent of bile;(61,62) FC appears in the feces of dogs with total bile diversion.(63) Studies in mice with deleted ABCG5/ABCG8, the hepatic FC transporter that transfers FC into bile, revealed the underlying mechanism.(64) These mice have biliary cholesterol secretion rates near nil but high fecal neutral sterol levels.(65) Moreover, fecal sterol excretion of Cyp7A1−/− mice is two-times that of WT mice.(66) These findings implicate an alternative route for FC excretion, direct TICE.
The molecular steps associated with TICE are not known. In the context of FC bioavailability there are two likely mechanisms. One is that TICE is mediated by cholesterol transporters located at the plasma membranes at various interfaces between the plasma compartment and the intestine. One likely candidate, SR-B1, which mediates bidirectional FC transport,(67) has been studied.(68) Although intestinal SR-B1 levels correlate with the TICE rates, TICE was actually higher in SR-B1−/− vs. WT mice, perhaps due to the diversion of FC from hepatic disposal to plasma. The other mechanism is spontaneous transfer from plasma lipoproteins to multiple tissue sites including intestine. The FC transfer halftime from HDL to lipoprotein and membrane surfaces is ~5 min.(15) FC translocation across membranes is faster, <1 sec.(69–71) Thus, TICE might begin with FC transfer from lipoproteins to membranes in contact with the plasma compartment followed by diffusion to numerous phospholipid-containing loci between plasma and the intestine. To have net transfer to excretion, there must be an irreversible mechanism that sequesters FC. Given that intestinal PL content is a positive regulator of TICE and that PL are the cholesterophilic component(72,73) of bile, a high concentration of intestinal bile-phospholipid would support net RCT via TICE. Dietary modifications that maintain a high bile-phospholipid content might stimulate TICE, thereby enhancing whole body FC disposal. Ezetimibe, which blocks intestinal FC absorption, doubles the flux of plasma-derived FC into fecal neutral sterols and increases total fecal neutral sterol excretion, plasma CE clearance, and plasma de novo-synthesized cholesterol.(74) Ezetimibe also increases plasma-to-feces FC transfer and elimination via TICE 4-fold.(74) Thus, ezetimibe could also maintain a positive FC gradient between the intestinal lumen and the rest of the body. However, in the context of statin co-therapy, the effect of ezetimibe on the reduction of major vascular events was not different from that of an equivalent reduction in the LDL-C concentration by a higher statin dose.(75) A therapeutic role for ezetimibe via TICE might be verified if its effects on CVD event reduction could be inversely correlated with plasma FC, HDL-FC, and LDL-FC concentrations.
Conclusions
RCT comprises both trans-hepatic and trans-intestinal pathways in which nHDL- and HDL-C are transported as FC. Although HDL-CE is also cleared by trans-hepatic RCT, this is a minor pathway for HDL in humans and mice and for nHDL in mice. Based on current knowledge, trans-intestinal RCT is likely mediated by spontaneous transfer whereas trans-hepatic RCT is mediated by spontaneous and SR-B1-mediated transfer. In vitro spontaneous PL transfer and in vivo PL clearance rates decrease with increasing acyl chain length suggesting that PL clearance is mediated, in part, by spontaneous transfer. nHDL-apo AI transfers solely to HDL in vivo and several plasma activities elicit the release of lipid-free apo AI from an unstable HDL for additional RCT cycles. Thus, nHDL-FC, PL, and apo AI are metabolically segregated and do not enter a common HDL pathway. While CETP and PLTP modify HDL composition in vitro, in vivo they are minor contributors to trans-hepatic RCT. Deletion of the HDL receptor, SR-B1, is associated with dysfunctional HDL and multiple pathologies for which the underlying cause is excess FC bioavailability. Although current evidence suggests that a high mol% HDL-FC and a high plasma HDL concentration likely contribute to HDL dysfunction, this rule is not axiomatic, suggesting there are other determinants of HDL dysfunctionality. Independent of HDL concentration and mol% HDL-FC, HDL dysfunctionality can be due to myeloperoxidase-mediated apo AI oxidation,(76) a high content of triglyceride,(77) or apo CIII content,(78) and paraoxanase-1-,(79) or apo M-deficiency.(80) One or more of these HDL qualities are found among patients with type 2 diabetes, hypertriglyceridemia,(81) and HIV-positive status after receiving anti-retroviral therapy.(82) Future therapeutic strategies for managing atherogenic HDL should consider the roles of both the trans-hepatic and trans-intestinal RCT pathways. In summary, we propose that the focus of cholesterol clearance, and RCT, should shift to FC, and homeostasis of FC bioavailability, rather than the current focus on HDL-total cholesterol and CE.
Collectively, our studies and those of others have led to a revised RCT model as follows:
nHDL-FC and human and mouse HDL-FC are cleared faster than apo AI and HDL-CE.
FC esterification by LCAT plays a minor role in RCT.
In mice, plasma nHDL-PL is rapidly transferred to the liver via a PLTP-dependent mechanism that involves both spontaneous and SR-B1-mediated paths.
This revised RCT model provokes new mechanistic questions about the role of dysfunctional HDL in impaired RCT.
Is high plasma FC bioavailability a marker of CVD risk; if so, how may this be lowered therapeutically?
Mol% HDL-FC and plasma HDL levels may contribute to dysfunctional HDL; do these attributes appear in the plasma HDL of CVD patients?
Sequestration of FC in the intestine is expected to support the TICE pathway; are there dietary or pharmacological means of TICE enhancement that reduce plasma HDL-FC and does reduction of plasma total cholesterol also reduce mol% HDL-FC?
Addressing these and other questions as they emerge will refine the model of RCT in ways that will better guide the design of new therapies for atheroprotection.
Highlights.
nHDL-FC and human and mouse HDL-FC are cleared faster than apo AI and HDL-CE.
FC esterification by LCAT plays a minor role in RCT.
Mouse plasma nHDL-PL rapidly transfers to the liver via a PLTP-dependent mechanism.
Mol% HDL-FC and plasma HDL levels may contribute to dysfunctional HDL.
High plasma FC bioavailability may be a marker of CVD risk.
Acknowledgments
Funding: Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Numbers HL056865 (CR and HJP), the Bass Foundation (AMG, Fort Worth TX) and Houston Methodist Foundation (Houston TX) funding from Charif Souki, Patrick Studdert and the Jerold B. Katz family.
We thank Drs. W. Sean Davidson and Nicholas Lyssenko for reading and critiquing our paper prior to submission.
Abbreviations
- ABCA1
ATP-binding cassette subfamily A, member 1
- ACVD
Atherosclerotic cardiovascular disease
- Apo
Apolipoprotein
- SR-B1
Scavenger Receptor Class B, Type 1
- RCT
reverse cholesterol transport
- LCAT
lecithin:cholesterol acyltransferase
- FC
Free cholesterol
- CE
Cholesteryl ester
- nHDL
nascent high density lipoproteins
- LDL
Low density lipoproteins
- FA
Fatty acid
- PL
Phospholipid
- CYP7A1
Cholesterol-7-α-hydroxylase
- CETP
Cholesteryl ester transfer protein
- PLTP
Phospholipid transfer protein
- TG
Triglyceride
- WT
Wild type
- Tg
transgenic
Footnotes
Disclosures: BKG, CR, and HJP have nothing to disclose. AMG is on the Board of Directors with Aegerion Pharmaceuticals, Arisaph Pharmaceuticals, and Esperion Therapeutics, a member of the Data Safety Monitoring Board for Ionis Pharmaceuticals, and received consulting fees from Kowa Pharmaceuticals, Merck and Co, Pfizer, and Vatera Capital. HJP prepared an initial draft and conceptualized the paper. CR and BKG critiqued and edited subsequent drafts. AMG provided critiques on the metabolic and medical aspects of the review. All authors have approved the final article as submitted.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xu B, Gillard BK, Gotto AM, Jr, Rosales C, Pownall HJ. ABCA1-Derived Nascent High-Density Lipoprotein-Apolipoprotein AI and Lipids Metabolically Segregate. Arterioscler Thromb Vasc Biol. 2017;37:2260–2270. doi: 10.1161/ATVBAHA.117.310290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ross R, Glomset JA. Atherosclerosis and the arterial smooth muscle cell: Proliferation of smooth muscle is a key event in the genesis of the lesions of atherosclerosis. Science. 1973;180:1332–9. doi: 10.1126/science.180.4093.1332. [DOI] [PubMed] [Google Scholar]
- 3.Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glomset JA. The plasma lecithins:cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–67. [PubMed] [Google Scholar]
- 5.Glomset JA, Wright JL. Some Properties of a Cholesterol Esterifying Enzyme in Human Plasma. Biochim Biophys Acta. 1964;89:266–76. doi: 10.1016/0926-6569(64)90215-9. [DOI] [PubMed] [Google Scholar]
- 6.Brooks-Wilson A, Marcil M, Clee SM, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22:336–45. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 7.Bodzioch M, Orso E, Klucken J, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22:347–51. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 8.Lyssenko NN, Nickel M, Tang C, Phillips MC. Factors controlling nascent high-density lipoprotein particle heterogeneity: ATP-binding cassette transporter A1 activity and cell lipid and apolipoprotein AI availability. FASEB J. 2013;27:2880–92. doi: 10.1096/fj.12-216564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sorci-Thomas MG, Owen JS, Fulp B, et al. Nascent high density lipoproteins formed by ABCA1 resemble lipid rafts and are structurally organized by three apoA-I monomers. J Lipid Res. 2012;53:1890–909. doi: 10.1194/jlr.M026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci U S A. 2004;101:9774–9. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–20. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 12.Gillard BK, Bassett GR, Gotto AM, Jr, Rosales C, Pownall HJ. Scavenger receptor B1 (SR-B1) profoundly excludes high density lipoprotein (HDL) apolipoprotein AII as it nibbles HDL-cholesteryl ester. J Biol Chem. 2017;292:8864–8873. doi: 10.1074/jbc.M117.781963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toth PP, Barter PJ, Rosenson RS, et al. High-density lipoproteins: a consensus statement from the National Lipid Association. J Clin Lipidol. 2013;7:484–525. doi: 10.1016/j.jacl.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Rosenson RS, Brewer HB, Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–19. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lund-Katz S, Hammerschlag B, Phillips MC. Kinetics and mechanism of free cholesterol exchange between human serum high- and low-density lipoproteins. Biochemistry. 1982;21:2964–9. doi: 10.1021/bi00541a025. [DOI] [PubMed] [Google Scholar]
- 16.Massey JB, Bick DH, Pownall HJ. Spontaneous transfer of monoacyl amphiphiles between lipid and protein surfaces. Biophys J. 1997;72:1732–43. doi: 10.1016/S0006-3495(97)78819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho JK, Moser H, Kishimoto Y, Hamilton JA. Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J Clin Invest. 1995;96:1455–63. doi: 10.1172/JCI118182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pownall HJ, Bick DL, Massey JB. Spontaneous phospholipid transfer: development of a quantitative model. Biochemistry. 1991;30:5696–700. doi: 10.1021/bi00237a009. [DOI] [PubMed] [Google Scholar]
- 19.Rye KA, Hime NJ, Barter PJ. Evidence that cholesteryl ester transfer protein-mediated reductions in reconstituted high density lipoprotein size involve particle fusion. J Biol Chem. 1997;272:3953–60. doi: 10.1074/jbc.272.7.3953. [DOI] [PubMed] [Google Scholar]
- 20.Settasatian N, Duong M, Curtiss LK, et al. The mechanism of the remodeling of high density lipoproteins by phospholipid transfer protein. J Biol Chem. 2001;276:26898–905. doi: 10.1074/jbc.M010708200. [DOI] [PubMed] [Google Scholar]
- 21.Liang HQ, Rye KA, Barter PJ. Remodelling of reconstituted high density lipoproteins by lecithin: cholesterol acyltransferase. J Lipid Res. 1996;37:1962–70. [PubMed] [Google Scholar]
- 22.Ma K, Forte T, Otvos JD, Chan L. Differential additive effects of endothelial lipase and scavenger receptor-class B type I on high-density lipoprotein metabolism in knockout mouse models. Arterioscler Thromb Vasc Biol. 2005;25:149–54. doi: 10.1161/01.ATV.0000150414.89591.6a. [DOI] [PubMed] [Google Scholar]
- 23.Kee P, Rye KA, Taylor JL, Barrett PH, Barter PJ. Metabolism of apoA-I as lipid-free protein or as component of discoidal and spherical reconstituted HDLs: studies in wild-type and hepatic lipase transgenic rabbits. Arterioscler Thromb Vasc Biol. 2002;22:1912–7. doi: 10.1161/01.atv.0000038485.94020.7f. [DOI] [PubMed] [Google Scholar]
- 24.Bruce C, Tall AR. Cholesteryl ester transfer proteins, reverse cholesterol transport, and atherosclerosis. Curr Opin Lipidol. 1995;6:306–11. doi: 10.1097/00041433-199510000-00010. [DOI] [PubMed] [Google Scholar]
- 25.Pownall HJ, Brauchi D, Kilinc C, et al. Correlation of serum triglyceride and its reduction by omega-3 fatty acids with lipid transfer activity and the neutral lipid compositions of high-density and low-density lipoproteins. Atherosclerosis. 1999;143:285–97. doi: 10.1016/s0021-9150(98)00301-3. [DOI] [PubMed] [Google Scholar]
- 26.Barter PJ, Jones ME. Rate of exchange of esterified cholesterol between human plasma low and high density lipoproteins. Atherosclerosis. 1979;34:67–74. doi: 10.1016/0021-9150(79)90107-2. [DOI] [PubMed] [Google Scholar]
- 27.Brewster ME, Ihm J, Brainard JR, Harmony JA. Transfer of phosphatidylcholine facilitated by a component of human plasma. Biochim Biophys Acta. 1978;529:147–59. doi: 10.1016/0005-2760(78)90113-3. [DOI] [PubMed] [Google Scholar]
- 28.Rao R, Albers JJ, Wolfbauer G, Pownall HJ. Molecular and macromolecular specificity of human plasma phospholipid transfer protein. Biochemistry. 1997;36:3645–53. doi: 10.1021/bi962776b. [DOI] [PubMed] [Google Scholar]
- 29.Lusa S, Jauhiainen M, Metso J, Somerharju P, Ehnholm C. The mechanism of human plasma phospholipid transfer protein-induced enlargement of high-density lipoprotein particles: evidence for particle fusion. Biochem J. 1996;313( Pt 1):275–82. doi: 10.1042/bj3130275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rye KA, Jauhiainen M, Barter PJ, Ehnholm C. Triglyceride-enrichment of high density lipoproteins enhances their remodelling by phospholipid transfer protein. J Lipid Res. 1998;39:613–22. [PubMed] [Google Scholar]
- 31.Aron L, Jones S, Fielding CJ. Human plasma lecithin-cholesterol acyltransferase. Characterization of cofactor-dependent phospholipase activity. J Biol Chem. 1978;253:7220–6. [PubMed] [Google Scholar]
- 32.Glass C, Pittman RC, Weinstein DB, Steinberg D. Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc Natl Acad Sci U S A. 1983;80:5435–9. doi: 10.1073/pnas.80.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thuahnai ST, Lund-Katz S, Williams DL, Phillips MC. Scavenger receptor class B, type I-mediated uptake of various lipids into cells. Influence of the nature of the donor particle interaction with the receptor. J Biol Chem. 2001;276:43801–8. doi: 10.1074/jbc.M106695200. [DOI] [PubMed] [Google Scholar]
- 34.Kunitake ST, Kane JP. Factors affecting the integrity of high density lipoproteins in the ultracentrifuge. J Lipid Res. 1982;23:936–40. [PubMed] [Google Scholar]
- 35.Mehta R, Gantz DL, Gursky O. Human plasma high-density lipoproteins are stabilized by kinetic factors. J Mol Biol. 2003;328:183–92. doi: 10.1016/s0022-2836(03)00155-4. [DOI] [PubMed] [Google Scholar]
- 36.Pownall HJ, Hosken BD, Gillard BK, Higgins CL, Lin HY, Massey JB. Speciation of human plasma high-density lipoprotein (HDL): HDL stability and apolipoprotein A-I partitioning. Biochemistry. 2007;46:7449–59. doi: 10.1021/bi700496w. [DOI] [PubMed] [Google Scholar]
- 37.Pownall HJ. Remodeling of human plasma lipoproteins by detergent perturbation. Biochemistry. 2005;44:9714–22. doi: 10.1021/bi050729q. [DOI] [PubMed] [Google Scholar]
- 38.Handa D, Kimura H, Oka T, et al. Kinetic and thermodynamic analyses of spontaneous exchange between high-density lipoprotein-bound and lipid-free apolipoprotein A-I. Biochemistry. 2015;54:1123–31. doi: 10.1021/bi501345j. [DOI] [PubMed] [Google Scholar]
- 39.Ji Y, Wang N, Ramakrishnan R, et al. Hepatic scavenger receptor BI promotes rapid clearance of high density lipoprotein free cholesterol and its transport into bile. J Biol Chem. 1999;274:33398–402. doi: 10.1074/jbc.274.47.33398. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz CC, Berman M, Vlahcevic ZR, Swell L. Multicompartmental analysis of cholesterol metabolism in man. Quantitative kinetic evaluation of precursor sources and turnover of high density lipoprotein cholesterol esters. J Clin Invest. 1982;70:863–76. doi: 10.1172/JCI110683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwartz CC, Vlahcevic ZR, Berman M, Meadows JG, Nisman RM, Swell L. Central role of high density lipoprotein in plasma free cholesterol metabolism. J Clin Invest. 1982;70:105–16. doi: 10.1172/JCI110582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwartz CC, VandenBroek JM, Cooper PS. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J Lipid Res. 2004;45:1594–607. doi: 10.1194/jlr.M300511-JLR200. [DOI] [PubMed] [Google Scholar]
- 43.Schwartz CC, Halloran LG, Vlahcevic ZR, Gregory DH, Swell L. Preferential utilization of free cholesterol from high-density lipoproteins for biliary cholesterol secretion in man. Science. 1978;200:62–4. doi: 10.1126/science.204996. [DOI] [PubMed] [Google Scholar]
- 44.Havel RJ, Goldstein JL, Brown MS. Lipoproteins and Lipid Transport. In: Bondy PE, Rosenberg LE, editors. Metabolic Control of Disease. Philadelphia: Saunders Publishing; 1980. pp. 393–493. [Google Scholar]
- 45.Van Eck M, Twisk J, Hoekstra M, et al. Differential effects of scavenger receptor BI deficiency on lipid metabolism in cells of the arterial wall and in the liver. J Biol Chem. 2003;278:23699–705. doi: 10.1074/jbc.M211233200. [DOI] [PubMed] [Google Scholar]
- 46.Gillard BK, Courtney HS, Massey JB, Pownall HJ. Serum opacity factor unmasks human plasma high-density lipoprotein instability via selective delipidation and apolipoprotein A-I desorption. Biochemistry. 2007;46:12968–78. doi: 10.1021/bi701525w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holm TM, Braun A, Trigatti BL, et al. Failure of red blood cell maturation in mice with defects in the high-density lipoprotein receptor SR-BI. Blood. 2002;99:1817–24. doi: 10.1182/blood.v99.5.1817. [DOI] [PubMed] [Google Scholar]
- 48.Dole VS, Matuskova J, Vasile E, et al. Thrombocytopenia and platelet abnormalities in high-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:1111–6. doi: 10.1161/ATVBAHA.108.162347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miettinen HE, Rayburn H, Krieger M. Abnormal lipoprotein metabolism and reversible female infertility in HDL receptor (SR-BI)-deficient mice. J Clin Invest. 2001;108:1717–22. doi: 10.1172/JCI13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A. 1997;94:12610–5. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trigatti B, Rayburn H, Vinals M, et al. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci U S A. 1999;96:9322–7. doi: 10.1073/pnas.96.16.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rinninger F, Wang N, Ramakrishnan R, Jiang XC, Tall AR. Probucol enhances selective uptake of HDL-associated cholesteryl esters in vitro by a scavenger receptor B-I-dependent mechanism. Arterioscler Thromb Vasc Biol. 1999;19:1325–32. doi: 10.1161/01.atv.19.5.1325. [DOI] [PubMed] [Google Scholar]
- 53.Braun A, Zhang S, Miettinen HE, et al. Probucol prevents early coronary heart disease and death in the high-density lipoprotein receptor SR-BI/apolipoprotein E double knockout mouse. Proc Natl Acad Sci U S A. 2003;100:7283–8. doi: 10.1073/pnas.1237725100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kilsdonk EP, Yancey PG, Stoudt GW, et al. Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem. 1995;270:17250–6. doi: 10.1074/jbc.270.29.17250. [DOI] [PubMed] [Google Scholar]
- 55.Riottot M, Olivier P, Huet A, et al. Hypolipidemic effects of beta-cyclodextrin in the hamster and in the genetically hypercholesterolemic Rico rat. Lipids. 1993;28:181–8. doi: 10.1007/BF02536637. [DOI] [PubMed] [Google Scholar]
- 56.Zimmer S, Grebe A, Bakke SS, et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med. 2016;8:333ra50. doi: 10.1126/scitranslmed.aad6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qi Y, Fan J, Liu J, et al. Cholesterol-overloaded HDL particles are independently associated with progression of carotid atherosclerosis in a cardiovascular disease-free population: a community-based cohort study. J Am Coll Cardiol. 2015;65:355–363. doi: 10.1016/j.jacc.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 58.Mora S, Glynn RJ, Ridker PM. High-density lipoprotein cholesterol, size, particle number, and residual vascular risk after potent statin therapy. Circulation. 2013;128:1189–97. doi: 10.1161/CIRCULATIONAHA.113.002671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mackey RH, Greenland P, Goff DC, Jr, Lloyd-Jones D, Sibley CT, Mora S. High-density lipoprotein cholesterol and particle concentrations, carotid atherosclerosis, and coronary events: MESA (multi-ethnic study of atherosclerosis) J Am Coll Cardiol. 2012;60:508–16. doi: 10.1016/j.jacc.2012.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schaefer EJ. Clinical, biochemical, and genetic features in familial disorders of high density lipoprotein deficiency. Arteriosclerosis. 1984;4:303–22. doi: 10.1161/01.atv.4.4.303. [DOI] [PubMed] [Google Scholar]
- 61.Miettinen TA, Proia A, McNamara DJ. Origins of fecal neutral steroids in rats. J Lipid Res. 1981;22:485–95. [PubMed] [Google Scholar]
- 62.Chevallier F. Study of the origin of fecal sterols in the rat by means of radioactive indicators. 1. Demonstration of the secretion of sterols into the intestinal contents. Bull Soc Chim Biol (Paris) 1960;42:623–32. [PubMed] [Google Scholar]
- 63.Pertsemlidis D, Kirchman EH, Ahrens EH., Jr Regulation of cholesterol metabolism in the dog. I. Effects of complete bile diversion and of cholesterol feeding on absorption, synthesis, accumulation, and excretion rates measured during life. J Clin Invest. 1973;52:2353–67. doi: 10.1172/JCI107424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berge KE, Tian H, Graf GA, et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771–5. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 65.Yu L, Li-Hawkins J, Hammer RE, et al. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest. 2002;110:671–80. doi: 10.1172/JCI16001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schwarz M, Russell DW, Dietschy JM, Turley SD. Marked reduction in bile acid synthesis in cholesterol 7alpha-hydroxylase-deficient mice does not lead to diminished tissue cholesterol turnover or to hypercholesterolemia. J Lipid Res. 1998;39:1833–43. [PubMed] [Google Scholar]
- 67.Yancey PG, de la Llera-Moya M, Swarnakar S, et al. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J Biol Chem. 2000;275:36596–604. doi: 10.1074/jbc.M006924200. [DOI] [PubMed] [Google Scholar]
- 68.van der Velde AE, Vrins CL, van den Oever K, et al. Regulation of direct transintestinal cholesterol excretion in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G203–G208. doi: 10.1152/ajpgi.90231.2008. [DOI] [PubMed] [Google Scholar]
- 69.Lange Y, Dolde J, Steck TL. The rate of transmembrane movement of cholesterol in the human erythrocyte. J Biol Chem. 1981;256:5321–3. [PubMed] [Google Scholar]
- 70.Hamilton JA. Fast flip-flop of cholesterol and fatty acids in membranes: implications for membrane transport proteins. Curr Opin Lipidol. 2003;14:263–71. doi: 10.1097/00041433-200306000-00006. [DOI] [PubMed] [Google Scholar]
- 71.Backer JM, Dawidowicz EA. The rapid transmembrane movement of cholesterol in small unilamellar vesicles. Biochim Biophys Acta. 1979;551:260–70. doi: 10.1016/0005-2736(89)90004-7. [DOI] [PubMed] [Google Scholar]
- 72.Niu SL, Litman BJ. Determination of membrane cholesterol partition coefficient using a lipid vesicle-cyclodextrin binary system: effect of phospholipid acyl chain unsaturation and headgroup composition. Biophys J. 2002;83:3408–15. doi: 10.1016/S0006-3495(02)75340-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tchoua U, Gillard BK, Pownall HJ. HDL superphospholipidation enhances key steps in reverse cholesterol transport. Atherosclerosis. 2010;209:430–5. doi: 10.1016/j.atherosclerosis.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davidson MH, Voogt J, Luchoomun J, et al. Inhibition of intestinal cholesterol absorption with ezetimibe increases components of reverse cholesterol transport in humans. Atherosclerosis. 2013;230:322–9. doi: 10.1016/j.atherosclerosis.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 75.Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med. 2015;372:2387–97. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 76.Nicholls SJ, Zheng L, Hazen SL. Formation of dysfunctional high-density lipoprotein by myeloperoxidase. Trends Cardiovasc Med. 2005;15:212–9. doi: 10.1016/j.tcm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 77.Lamarche B, Rashid S, Lewis GF. HDL metabolism in hypertriglyceridemic states: an overview. Clin Chim Acta. 1999;286:145–61. doi: 10.1016/s0009-8981(99)00098-4. [DOI] [PubMed] [Google Scholar]
- 78.Hsieh JY, Chang CT, Huang MT, et al. Biochemical and functional characterization of charge-defined subfractions of high-density lipoprotein from normal adults. Anal Chem. 2013;85:11440–11448. doi: 10.1021/ac402516u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mackness B, Mackness M. Anti-inflammatory properties of paraoxonase-1 in atherosclerosis. Adv Exp Med Biol. 2010;660:143–51. doi: 10.1007/978-1-60761-350-3_13. [DOI] [PubMed] [Google Scholar]
- 80.Wolfrum C, Poy MN, Stoffel M. Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat Med. 2005;11:418–22. doi: 10.1038/nm1211. [DOI] [PubMed] [Google Scholar]
- 81.Vollenweider P, von Eckardstein A, Widmann C. HDLs, diabetes, and metabolic syndrome. Handb Exp Pharmacol. 2015;224:405–21. doi: 10.1007/978-3-319-09665-0_12. [DOI] [PubMed] [Google Scholar]
- 82.Gillard BK, Raya JL, Ruiz-Esponda R, et al. Impaired lipoprotein processing in HIV patients on antiretroviral therapy: aberrant high-density lipoprotein lipids, stability, and function. Arterioscler Thromb Vasc Biol. 2013;33:1714–21. doi: 10.1161/ATVBAHA.113.301538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blum CB, Levy RI, Eisenberg S, Hall M, 3rd, Goebel RH, Berman M. High density lipoprotein metabolism in man. J Clin Invest. 1977;60:795–807. doi: 10.1172/JCI108833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chajek-Shaul T, Hayek T, Walsh A, Breslow JL. Expression of the human apolipoprotein A-I gene in transgenic mice alters high density lipoprotein (HDL) particle size distribution and diminishes selective uptake of HDL cholesteryl esters. Proc Natl Acad Sci U S A. 1991;88:6731–5. doi: 10.1073/pnas.88.15.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nestel PJ, Monger EA. Turnover of plasma esterified cholesterol in normocholesterolemic and hypercholesterolemic subjects and its relation to body build. J Clin Invest. 1967;46:967–74. doi: 10.1172/JCI105603. [DOI] [PMC free article] [PubMed] [Google Scholar]