

Abstract
Signaling activated by binding of the CXC motif chemokine ligand 12 (CXCL12) to its cognate G protein–coupled receptor (GPCR), chemokine CXC motif receptor 4 (CXCR4), is linked to metastatic disease. However, the mechanisms governing CXCR4 signaling remain poorly understood. Here, we show that endocytosis and early endosome antigen 1 (EEA1), which is part of the endosome fusion machinery, are required for CXCL12-mediated AKT Ser/Thr kinase (Akt) signaling selective for certain Akt substrates. Pharmacological inhibition of endocytosis partially attenuated CXCL12-induced phosphorylation of Akt, but not phosphorylation of ERK-1/2. Similarly, phosphorylation of Akt, but not ERK-1/2, stimulated by CXCL13, the cognate ligand for the chemokine receptor CXCR5, was also attenuated by inhibited endocytosis. Furthermore, siRNA-mediated depletion of the Rab5-effector EEA1, but not of adaptor protein, phosphotyrosine-interacting with PH domain and leucine zipper 1 (APPL1), partially attenuated Akt, but not ERK-1/2, phosphorylation promoted by CXCR4. Attenuation of Akt phosphorylation through inhibition of endocytosis or EEA1 depletion was associated with reduced signaling to Akt substrate forkhead box O1/3a but not the Akt substrates TSC complex subunit 2 or glycogen synthase kinase 3β. This suggested that endocytosis and endosomes govern discrete aspects of CXCR4- or CXCR5-mediated Akt signaling. Consistent with this hypothesis, depletion of EEA1 reduced the ability of CXCL12 to attenuate apoptosis in suspended, but not adherent, HeLa cells. Our results suggest a mechanism whereby compartmentalized chemokine-mediated Akt signaling from endosomes suppresses the cancer-related process known as anoikis. Targeting this signaling pathway may help inhibit metastatic cancer involving receptors such as CXCR4.
Keywords: C-X-C chemokine receptor type 4 (CXCR-4), chemokine, G protein-coupled receptor (GPCR), Akt PKB, FOXO, apoptosis, anoikis, endosome, endocytosis
Introduction
Signaling by the chemokine receptor CXC motif receptor 4 (CXCR4)2 instigated by binding to its cognate ligand, CXCL12, plays an important role in cancer progression (1–3). Upon activation by CXCL12, CXCR4 signals to a variety of intracellular signaling pathways, including the Akt signaling pathway (3, 4). This pathway has been linked to CXCR4-mediated cell survival, especially in the context of metastatic breast cancer (5). Akt is a serine/threonine protein kinase that phosphorylates diverse proteins to regulate many cellular functions (6, 7). Akt acts on several factors relevant to antiapoptosis and on transcription factors that regulate gene expression that promotes a cell survival phenotype (7). However, the mechanisms by which Akt signaling is activated by cell signaling receptors remain poorly understood.
CXCR4 couples with heterotrimeric guanine nucleotide–binding protein Gαi and the associated Gβγ heterodimer to initiate Akt signaling from the inner leaflet of the plasma membrane (8). Similar to other GPCRs, the released Gβγ directly activates phosphoinositide 3-kinase (PI3K), leading to the production of phosphatidylinositol(3,4,5)-trisphosphate (PIP3, also known as PtdIns(3,4,5)P3) at the plasma membrane (9). This results in the recruitment of 3-phosphoinositide–dependent protein kinase-1 (PDK1) and Akt to PIP3 at the plasma membrane via their respective pleckstrin-homology domains, culminating in PDK1 phosphorylation of Thr308 on Akt (Akt1). For full activation, Akt is also phosphorylated at Ser473 by mTORC2. The mechanisms governing temporal and spatial activation of Akt remain poorly understood.
An emerging concept in GPCR signaling is that, in addition to the plasma membrane, GPCRs can signal from an intracellular site generally via either G proteins or β-arrestins (10, 11). In addition to the plasma membrane, β-adrenergic receptor 2 can couple with its cognate heterotrimeric G protein at the surface of endosomes to instigate ligand-dependent cAMP signaling (12, 13). The protease-activated receptor 2 can couple with β-arrestin–dependent formation of multimeric signaling complexes at the surface of endosomes to mediate extracellular signal–regulated kinase 1 and 2 (ERK-1/2) signaling (14). An interesting aspect of signaling from the surface of endosomes is that it is qualitatively distinct from the signaling that is initiated at the cell surface (12).
GPCR-instigated Akt signaling has also been linked to the surface of endosomes (15, 16), but whether this applies broadly to other GPCRs, such as chemokine receptors, remains unknown. Here, we provide evidence that endocytosis and endosomal protein early endosome antigen 1 (EEA1), but not adaptor protein, phosphotyrosine-interacting with PH domain and leucine zipper 1 (APPL1), are required for CXCR4- and CXCR5-promoted Akt activation and signaling. Endocytosis or EEA1 specifies chemokine-instigated phosphorylation of certain Akt substrates but not others. Importantly, EEA1 is required for chemokine-instigated suppression of apoptosis in detached cells but not in adherent cells. Our data are consistent with endosomes specifying context-specific chemokine signaling that is relevant to suppression of the cancer-related process known as anoikis or anchorage-independent cell death.
Results
Role of endocytosis in CXCR4-mediated Akt activation
CXCL12 binding to its cognate receptor, CXCR4, activates many signaling pathways, including the ERK-1/2 and Akt signaling pathways (1, 2, 4). Previously, we reported that CXCR4-mediated activation of ERK-1/2 does not require endocytosis (17). However, whether endocytosis is required for activation of Akt remains an open question. To address this, HeLa cells were treated with dynasore, an inhibitor of dynamin, a GTPase that is required for endocytosis (18) and is often used to inhibit GPCR endocytosis (19, 20). Dynasore treatment of HeLa cells attenuated CXCL12 internalization of CXCR4 (Fig. 1A). To examine whether dynasore treatment impacted CXCR4-mediated activation of Akt, HeLa cells were serum-starved for 3 h and then treated with 10 nm CXCL12 or vehicle for 5 min. Akt activation was assessed by immunoblotting to detect phosphorylation of Akt at serine residue 473 using a phosphospecific antibody as we have described previously (8). Dynasore treatment significantly attenuated CXCL12-instigated phosphorylation of Akt at Ser473 (pAkt-Ser473) by ∼50% compared with control (Fig. 1, B and quantified in C). Dynasore treatment did not impact ERK-1/2 phosphorylation (Fig. 1, B and quantified in D), indicating that dynasore does not globally impact signaling and confirming our previous results that endocytosis is not required for CXCR4-mediated ERK-1/2 activation (17). These data suggest that CXCR4-mediated activation of Akt, in part, requires endocytosis.
Figure 1.
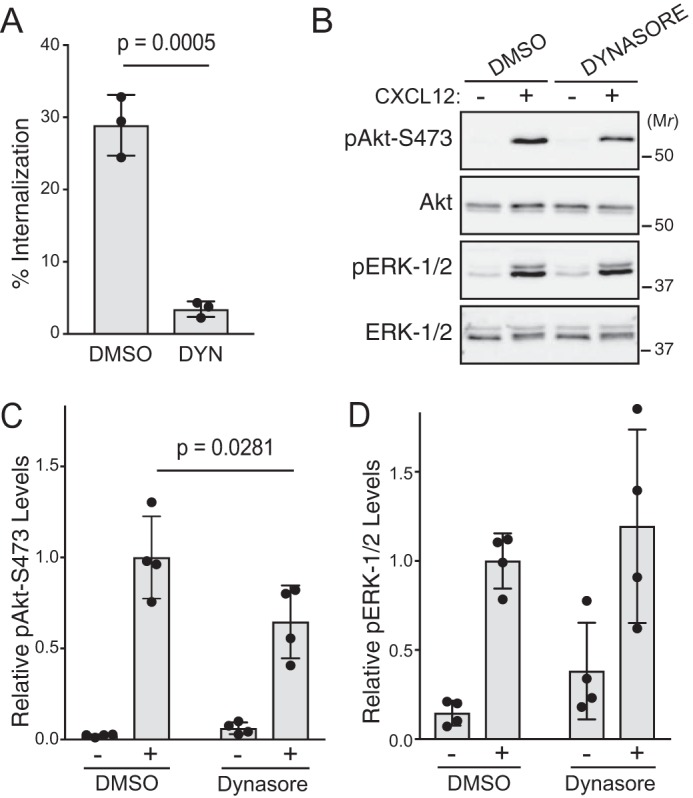
Role of endocytosis in CXCL12-instigated Akt phosphorylation. A, CXCR4 internalization was quantified by ELISA. HeLa cells transfected with HA-CXCR4 were treated with 80 μm dynasore or DMSO for 30 min followed by 10 nm CXCL12 or vehicle for 5 min. Bars represent the average percentage of CXCL12-induced receptor internalization from three independent experiments. Error bars represent the S.D. Data were analyzed by Student's unpaired t test. p value is indicated. B, immunoblot analysis of Akt and ERK-1/2 phosphorylation. Serum-starved HeLa cells were treated with 80 μm dynasore or DMSO for 30–60 min followed by 10 nm CXCL12 or vehicle for 5 min. Whole-cell lysates were analyzed by immunoblotting for the indicated proteins. Representative immunoblots from four independent experiments are shown. C and D, immunoblots were analyzed by densitometry. Bars represent the mean of the relative levels of pAkt-Ser473 (C) or pERK-1/2 (D) to DMSO- and CXCL12-treated samples. Error bars represent the S.D. Data were analyzed by two-way ANOVA followed by Tukey's multiple comparison test. p value is indicated.
To determine how broadly applicable the requirement for endocytosis is in chemokine receptor–instigated Akt activation, we examined CXCL13, the sole cognate ligand for the chemokine receptor CXCR5 (3). This receptor is expressed endogenously in HeLa cells (21), and treatment of HeLa cells with increasing doses of CXCL13 robustly promoted phosphorylation of Akt and ERK-1/2 (Fig. 2, A and quantified in B). Similar to CXCR4, CXCR5 couples with a pertussis toxin–sensitive heterotrimeric G protein to phosphorylate Akt or ERK-1/2 (Fig. 2, C and quantified in D and E, respectively). HeLa cells also express the CXC chemokine receptors CXCR7 (also known as ACKR3) and CXCR3 (21); however, treatment of HeLa cells with CXCL11 or CXCL10, ligands for these receptors, respectively, did not significantly promote phosphorylation of Akt (Fig. 2, F and quantified in G).
Figure 2.

Chemokine-mediated signaling in HeLa cells. A, immunoblot analysis of CXCL13-induced phosphorylation of Akt and ERK-1/2 in HeLa cells. Serum-starved cells were treated with increasing doses of CXCL13 (10–1000 nm), 10 nm CXCL12, or vehicle for 5 min. Whole-cell lysates were analyzed by immunoblotting for the indicated proteins. Representative immunoblots from three independent experiments are shown. B, immunoblots were analyzed by densitometry. Bars represent the mean of the relative levels of pAkt-Ser473 or pERK-1/2 to CXCL12 treated samples. Error bars represent the S.D. Data were analyzed by two-way ANOVA followed by Tukey's multiple comparison test. C, CXCL13-instigated phosphorylation of Akt and ERK-1/2 is pertussis toxin–sensitive. HeLa cells were serum-starved and treated with or without 50 ng/ml pertussis toxin for 7 h followed by 10 nm CXCL12 or 100 nm CXCL13. Whole-cell lysates were analyzed by immunoblotting for the indicated proteins. Representative immunoblots from three independent experiments are shown. D and E, immunoblots were analyzed by densitometry. Bars represent the mean of the relative levels of pAkt-Ser473 (D) or pERK-1/2 (E) to CXCL12-treated samples. Error bars represent the S.D. F, the effect of chemokines CXCL11 and CXCL10 in HeLa cells on phosphorylation of Akt or ERK-1/2. Serum-starved cells were treated with vehicle or increasing doses (10–1000 nm) of CXCL11, CXCL10, or 10 nm CXCL12 for 5 min. Whole-cell lysates were analyzed by immunoblotting for the indicated proteins. Representative immunoblots from three independent experiments are shown. Panels are separated from each other to remove irrelevant intervening bands but are from the same exposure. G, immunoblots were analyzed by densitometry. Bars represent the mean of the relative levels of pAkt-Ser473 to CXCL12-treated samples. Error bars represent the S.D.
We focused on CXCL13 signaling via CXCR5 for further experiments. Similar to CXCL12 (Fig. 1A), dynasore treatment of HeLa cells significantly attenuated CXCL13 internalization of CXCR5 (Fig. 3A) and phosphorylation of Akt, but not ERK-1/2, by CXCL13 (Fig. 3, B and quantified in C and D, respectively). To determine whether the requirement for endocytosis was restricted to HeLa cells, we examined chemokine-mediated phosphorylation of Akt in WEHI-231 cells, a B-lymphoma cell line that expresses CXCR4 and CXCR5 (22). Dynasore treatment of WEHI-231 cells significantly attenuated CXCL12- or CXCL13-instigated phosphorylation of Akt (Fig. 4, A and quantified in B). These data suggest that endocytosis-mediated Akt activation may be broadly applicable to chemokine receptors and cell types.
Figure 3.

Role of endocytosis in CXCL12- or CXCL13-instigated Akt signaling. A, CXCR5 internalization was quantified by ELISA. HeLa cells transfected with HA-CXCR5 were treated with 80 μm dynasore or DMSO for 30 min followed by 100 nm CXCL13 or vehicle for 5 min. Bars represent the average percentage of CXCL13-induced receptor internalization from three independent experiments. Error bars represent the S.D. Data were analyzed by a Student's unpaired t test. p value is indicated. B, CXCL12- or CXCL13-instigated Akt signaling was analyzed by immunoblotting. HeLa cells were treated with 80 μm dynasore or DMSO for 30 min followed by stimulation with 10 nm CXCL12 or 100 nm CXCL13 for 5 min at 37 °C. Equal amounts of whole-cell lysates were analyzed by immunoblotting for the phosphorylated or total forms of the indicated proteins. C–G, immunoblots were analyzed by densitometry. Bars represent the mean of the relative levels of pAkt-Ser473 (C), pERK-1/2 (D), pFOXO1/3a (E), pGSK3β (F), or pTSC2 (G) to DMSO- and CXCL12-treated samples. Error bars represent the S.D. Significance was determined by two-way ANOVA followed by a Newman-Keuls (C) or Tukey's (E) multiple comparison test. Asterisks in C indicate significance between DMSO-CXCL12 versus dynasore-CXCL12 (*) and DMSO-CXCL13 versus dynasore-CXCL13 (**). p values are indicated in E.
Figure 4.
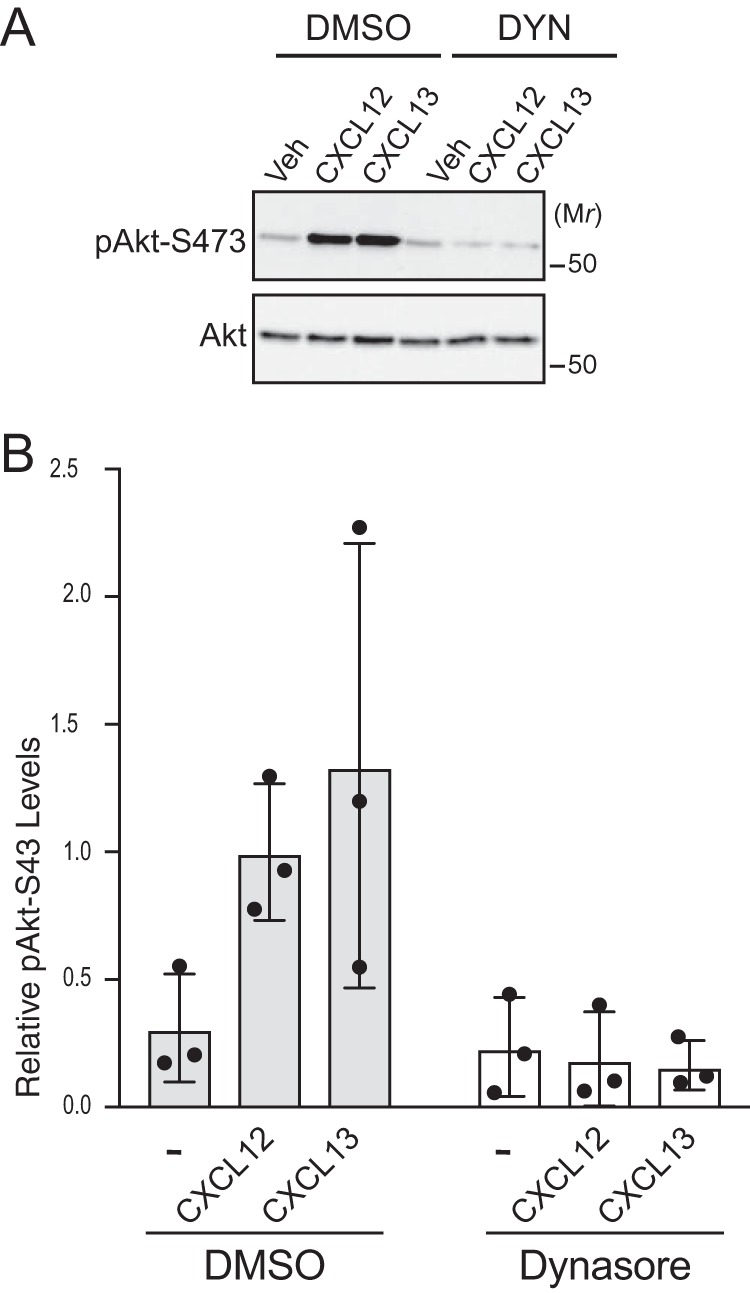
Role of endocytosis in CXCL12- or CXCL13-instigated Akt phosphorylation in WEHI-231 cells. A, CXCL12- or CXCL13-instigated Akt phosphorylation in WEHI-231 cells. Serum-starved WEHI-231 cells were treated with 80 μm dynasore (DYN) or DMSO for 30 min and then 10 nm CXCL12, 100 nm CXCL13, or vehicle (Veh) for 5 min at 37 °C. Whole-cell lysates were analyzed by immunoblotting for pAkt-Ser473 and Akt. B, immunoblots were analyzed by densitometry. Bars represent the mean from three independent experiments relative to cells treated with DMSO and CXCL12. Error bars represent the S.D.
Chemokine receptor signaling to forkhead box O1/3a (FoxO1/3a) transcription factors is impaired by dynasore treatment
Once activated, Akt can phosphorylate many proteins involved in discrete processes (6, 7). To examine whether endocytosis impacts Akt signaling, we examined the phosphorylation status of Akt substrates FoxO1/3a, glycogen synthase kinase 3β (GSK3β), and tuberous sclerosis complex 2 (TSC2) (7). Akt phosphorylates FoxO1/3a at Thr24/32, GSK3β at Ser9, and TSC2 at Thr1462 (7). Dynasore treatment significantly attenuated CXCL12- or CXCL13-instigated phosphorylation of FoxO1/3a but not TSC2 or GSK3β (Fig. 3, B and quantified in E, F, and G, respectively). Taken together, these data suggest that endocytosis specifies Akt substrate phosphorylation downstream of chemokine receptor signaling.
Role of APPL1 and EEA1 in CXCR4-mediated Akt signaling
Given that endocytosis is required for CXCR4-mediated Akt signaling, we next addressed whether endosomes are also required. Rab5-positive endosomes containing the Rab5-effector protein APPL1 have been linked to Akt signaling (23–25). APPL1 interacts with certain signaling receptors, suggesting that APPL1 endosomes may provide a platform for the assembly of signaling complexes (23–27). Depletion of APPL1 with two discrete siRNAs significantly attenuated insulin-instigated phosphorylation of Akt at Ser473 (Fig. 5, A and quantified in B), consistent with a role of APPL1 in Akt signaling (25). In contrast, depletion of APPL1 did not impact CXCL12- or epidermal growth factor (EGF)-instigated phosphorylation of Akt (Fig. 5, A and quantified in B). These results reveal that APPL1 is not required for CXCR4-mediated Akt activation.
Figure 5.
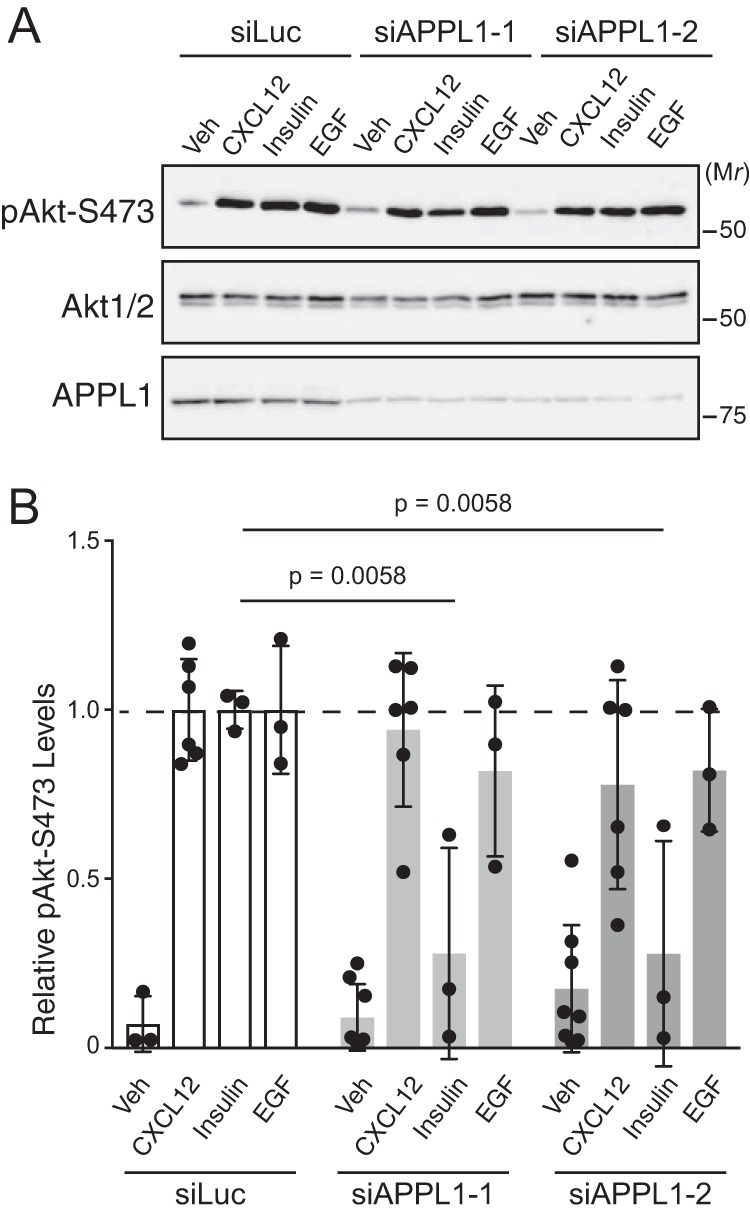
Role of APPL1 in CXCL12-instigated Akt phosphorylation. A, CXCL12-, insulin-, and EGF-instigated Akt phosphorylation was examined in cells depleted of APPL1. Serum-starved HeLa cells transfected with control siRNA (siLuc) or two distinct siRNAs against APPL1 (siAPPL1-1 and siAPPL1-2) were treated with CXCL12 (10 nm), insulin (50 nm), or EGF (100 ng/ml) for 5 min at 37 °C. Whole-cell lysates were analyzed by immunoblotting for pAkt-Ser473, total Akt-1/2, and APPL1. B, immunoblots were analyzed by densitometry. Bars represent the mean of the relative levels of pAkt-Ser473 to siLuc- and CXCL12-treated cells. Error bars represent the S.D. Data were analyzed by two-way ANOVA followed by Tukey's multiple comparison test. The p values for insulin-treated cells are indicated. CXCL12- or EGF-treated samples were not significant. Veh, vehicle.
A second class of Rab5-positive endosomes contains the Rab5-effector EEA1 (28). EEA1-positive endosomes have been previously linked to GPCR-mediated Akt signaling (15). Depletion of EEA1 with two discrete siRNAs significantly attenuated CXCL12-instigated phosphorylation of Akt at Ser473 (Fig. 6, A and quantified in B). Phosphorylation of ERK-1/2 was not impacted by EEA1 depletion (Fig. 6, A and quantified in D), indicating that there was not a global defect in signaling. Furthermore, depleting EEA1 attenuated CXCL12-instigated signaling to Akt substrate FoxO1/3a but not GSK3β or TSC2 (Fig. 6, A and quantified in C and D, respectively). Together these data suggest that EEA1-positive, but not APPL1-positive, endosomes specify discrete Akt substrate phosphorylation downstream of CXCR4 signaling.
Figure 6.

Role of EEA1 in CXCL12-instigated Akt phosphorylation. A, CXCL12-instigated Akt phosphorylation was examined in cells depleted of EEA1. Serum-starved HeLa cells transfected with control siRNA (siLuc) or two distinct siRNAs against EEA1 (siEEA1-1 and siEEA1-2) were treated with 10 nm CXCL12 for 5 min at 37 °C. Whole-cell lysates were analyzed by immunoblotting for the phosphorylated or total forms of the indicated proteins. B–D, immunoblots were analyzed by densitometry. Bars represent the mean relative levels of pAkt-Ser473 (B), pFoxO1/3a (C), or pTSC2, pGSK3β, and pERK-1/2 (D) to siLuc- and CXCL12-treated samples. Error bars represent the S.D. Data were analyzed by two-way ANOVA followed by Tukey's multiple comparison test. p values are indicated.
EEA1 is required for CXCR4-mediated suppression of apoptosis in detached, but not adherent, cells
Akt promotes cell survival by suppressing apoptosis via directly phosphorylating and inactivating proapoptotic proteins (7). CXCR4-mediated Akt signaling has been linked to cell survival, which is key to its role in metastatic disease (29). This is especially relevant when tumor cells detach and transit the bloodstream to distant sites. Typically, upon detachment, cells undergo anchorage-independent cell death or apoptosis known as anoikis (30). Because EEA1 is required for Akt signaling, we tested whether EEA1 is required for suppression of apoptosis by CXCR4 in detached or adherent cells. In control siRNA cells that were maintained in an adherent setting, CXCL12 reduced PARP cleavage, a hallmark of apoptosis. In detached cells, cleaved PARP was somewhat elevated compared with adherent cells, suggesting greater apoptosis. Importantly, the ability of CXCL12 to reduce PARP cleavage was similar or not statistically significant between adherent and detached cells (Fig. 7, A and quantified in B). In EEA1-depleted cells, basal levels of cleaved PARP were higher than in control cells, suggesting active apoptosis. Importantly, CXCL12 significantly reduced PARP cleavage in adherent cells but not in detached cells (Fig. 7, A and quantified in B). To further confirm these findings, in parallel samples we quantified caspase-3/7 activity using a luminescence assay. In control siRNA cells, the ability of CXCL12 to reduce caspase-3/7 activity was not significantly different between adherent and detached cells (Fig. 7C). In contrast, EEA1 depletion significantly attenuated the ability of CXCL12 to reduce caspase-3/7 activity in detached cells but not in adherent cells (Fig. 7C). These results are consistent with EEA1 being required for CXCR4-mediated suppression of apoptosis.
Figure 7.

Role of EEA1 in CXCR4-mediated suppression of apoptosis. A, examination of CXCL12-promoted suppression of apoptosis in HeLa cells depleted of EEA1. Cells transfected with control siRNA (siLuc) or siRNAs against EEA1 (siEEA1-1 or siEEA1-2) were left adherent on the culture dish or left detached by seeding onto poly(2-hydroxyethyl methacrylate dishes where cells were unable to adhere. Adherent (Ad) or detached (Det) cells were treated with 10 nm CXCL12 for 2–5 h at 37 °C. Equal amounts of cleared lysates were analyzed by immunoblotting for PARP. Representative immunoblots are shown from four independent experiments. B, immunoblots were analyzed by densitometry. Bars represent the mean of cleaved PARP levels relative to siLuc- and vehicle-treated samples. Error bars represent the S.D. Data were analyzed by two-way ANOVA, followed by Tukey's multiple comparison test. p values are indicated. C, parallel samples from A were examined for caspase-3/7 activity as described under “Experimental procedures.” Bars represent the mean -fold change in caspase-3/7 activity with siLuc-transfected cells that were left adherent to the culture dish and treated with vehicle. Error bars represent the S.D. Single points were removed from the analysis if greater than one S.D. from the mean. Data were analyzed by one-way ANOVA followed by Tukey's multiple comparison test. The p values are indicated. n.s., not significant.
Discussion
Our study provides mechanistic insight into which chemokine receptors activate Akt signaling. Our data reveal that CXCR4-mediated Akt activation and signaling require endocytosis and the Rab5-effector EEA1 but not APPL1, suggesting a role for EEA1-positive endosomes in Akt signaling. This is likely broadly applicable to other chemokine receptors because CXCL13, the cognate ligand for the chemokine receptor CXCR5, also requires endocytosis for Akt signaling. This is not cell type–dependent because, in addition to HeLa cells, endocytosis is also required for CXCR4- or CXCR5-mediated Akt activation in WEHI-231 cells. CXCL12 time-dependent stimulation indicates that endocytosis impacts the efficiency of Akt phosphorylation, rather than the kinetics, but does not impact ERK-1/2 phosphorylation (Fig. S1). Further, our data suggest that Akt signaling from EEA1-positive endosomes specifies Akt substrate specificity. Furthermore, and importantly, EEA1-positive endosomes are likely required for CXCL12-instigated suppression of anoikis, a type of anchorage-independent cell death. Our study provides evidence that GPCRs can drive Akt signaling with qualitatively distinct functional consequences depending on the compartment from which Akt signaling occurs.
Our results extend the concept that GPCR signaling requires endocytosis. Blocking endocytosis pharmacologically attenuates CXCR4- and CXCR5-mediated Akt activation and signaling (Figs. 1 and 3). Dynasore attenuated Akt phosphorylation by ∼50%, suggesting that canonical Akt activation at the plasma membrane remains intact when endocytosis is blocked. Although dynasore is known to have off-target effects (31), it efficiently blocks endocytosis, and it was only acutely applied in our experiments (Figs. 1A and 3A), thus likely avoiding any cellular changes that may occur when using RNAi or dominant-negative approaches. Dynasore did not globally impact signaling as ERK-1/2 phosphorylation by CXCR4 or CXCR5 remained intact (Figs. 1 and 3). We have previously shown that endocytosis is not required for ERK-1/2 phosphorylation by CXCR4 but requires the protein caveolin-1, a major component of caveolae or lipid rafts at the plasma membrane (17). Therefore, CXCR4 signaling is highly compartmentalized whereby discrete receptor and G-protein pools likely mediate Akt or ERK-1/2 signaling.
Endosomes are required for Akt activation and signaling. EEA1 is a Rab5 effector and a key player of the endosome fusion machinery; therefore, its depletion likely disrupts endosome dynamics (28, 32), suggesting that an intact endosomal network, rather than a direct effect of EEA1, is required for Akt activation and signaling (Fig. 6). EEA1 depletion did not impact ERK-1/2 phosphorylation (Fig. 6), indicating that there is not a global impact on signaling when EEA1 is depleted and that EEA1-positive endosomes are not required for ERK-1/2 signaling by CXCR4. A requirement for EEA1 in Akt signaling by GPCRs is in line with previous studies. EEA1-positive endosomes have been linked to Akt activation by angiotensin II in vascular smooth muscle cells (15). Another Rab5-effector, APPL1, which is mainly localized to a subpopulation of peripheral endosomes devoid of EEA1 (33), is not involved in CXCR4-mediated Akt activation or signaling (Fig. 5). APPL1-positive endosomes have been implicated in Akt and ERK-1/2 signaling mediated by several cell signaling receptors, such as the insulin receptor (Fig. 5) and the GPCR that binds to lysophosphatidic acid (34). It is likely that APPL1-positive endosomes may be required for Akt signaling of a subset of cell signaling receptors. APPL1 has a related family member called APPL2 (25), which we have not examined in this study and therefore cannot rule out. Our results are consistent with EEA1-positive endosomes being required for Akt activation and signaling mediated by a subset of GPCRs.
EEA1-positive endosomes may serve as an ideal signaling platform for assembling complexes required for activation of Akt signaling. We have yet to directly demonstrate that CXCR4 or CXCR5 located at endosomes directly drives Akt signaling. However, we have previously shown that CXCR4 traffics very efficiently to EEA1-positive endosomes (35–37). Other factors required for Akt phosphorylation have also been found at endosomes. Previously, we have shown that PDK1 localizes to EEA1-positive endosomes (38), and mTORC2 activity and phosphorylation of Akt have been linked to the surface of endosomes (39). We recently reported that ESCRTs, which are typically found at the surface of endosomes, are also required for CXCR4-mediated Akt activation and signaling (8). Because heterotrimeric G proteins are required for CXCR4-mediated Akt signaling, they may also be required to be present on endosomes. There is evidence that Gβγ-mediated PI3K signaling may occur from Rab11-positive recycling endosomes via the lysophosphatidic acid receptor (16), although it is unclear how this localization of PI3K relates to the site of relevant phosphoinositide production. Sphingosine 1-phosphate–mediated Gαi-signaling has been linked to the Golgi, not endosomes, and instigated by an unnatural ligand (40). Whether heterotrimeric G protein Gαi localizes to EEA1-positive endosomes remains unclear. β-Arrestins have also been linked to GPCR-mediated Akt activation from endosomes (41); however, considering that CXCR4- or CXCR5-mediated Akt activation is completely G protein–dependent (Fig. 2C), it is difficult to envision a role for β-arrestins. However, β-arrestin–dependent signaling is complex, and its relationship to G-protein signaling may be more intertwined than previously appreciated (42); therefore, β-arrestins cannot be completely ruled out at this time.
We provide evidence that EEA1-positive endosomes specify Akt substrate specificity. Pharmacological inhibition of endocytosis (Figs. 1 and 3) or depletion of EEA1 (Fig. 6) attenuates CXCR4- or CXCR5-mediated phosphorylation of Akt substrates FoxO1/3a but not TSC2 or GSK3β. Notably, ESCRTs are also required for CXCR4-mediated Akt signaling to FoxO1/3a but not GSK3β or TSC2 (8). As mentioned above, this signaling specificity could relate to localization of Akt activation, but it could also relate to localization of its substrates. GSK3β or TSC2 is likely localized to compartments other than EEA1-positive endosomes (7, 25). FoxO1/3a are members of the FoxO family of transcription factors (43). Akt phosphorylation of FoxO1/3a promotes their association with 14-3-3 phospho-binding proteins, leading to their nuclear export and retention in the cytoplasm (44). How endosomes relate to this canonical manner by which Akt regulates FoxO phosphorylation and localization remains unclear and requires further investigation.
Akt signaling is typically antiapoptotic by directly inhibiting the activity of proapoptotic proteins and by suppressing the expression of proapoptotic genes in part by blocking the function of transcription factors FoxO1 and FoxO3a (7). Our data provide evidence that EEA1 is required for CXCR4-mediated suppression of apoptosis in detached cells but not adherent cells (Fig. 7). This is consistent with the notion that endosomes are required for CXCR4-mediated suppression of the cancer-related process known as anoikis. Our results are somewhat reminiscent of integrin-mediated suppression of anoikis, which requires endocytosis and EEA1-positive endosomes (45). In this example, endosomes provide a platform for noncanonical activation of focal adhesion kinase (FAK) and survival signaling (45). FAK is not required for Akt activation, nor is Akt involved in FAK activation downstream of CXCR4.3 Although endosomes may have a general role in cell signaling to suppress anoikis, the mechanisms we describe here may be unique to GPCRs. It is important to note that in siLuc-treated cells CXCL12-mediated suppression of PARP cleavage or caspase-3/7 activity was similar, yet only moderate in both the adherent and detached settings (Fig. 7). Additional studies with more robust cellular and physiological model systems are needed to examine this further. Another interesting aspect of our findings is that although EEA1 depletion attenuates CXCR4-mediated phosphorylation of Akt and FoxO1/3a (Fig. 5) suppression of apoptosis is still intact in adherent cells (Fig. 7). This is likely because other aspects of CXCR4 signaling are involved in suppressing apoptosis in adherent cells (3). However, this signaling redundancy is lost when cells are kept in suspension, consistent with endosomes selectively specifying Akt signaling that suppresses anoikis. How this signaling relates to nonadherent cells, such as WEHI-231 cells (Fig. 4), or in cancers in which CXCR4 signaling may have opposite effects on anoikis (46, 47) requires further investigation.
Our study provides evidence that chemokine receptor–mediated Akt signaling occurs in a compartmentalized manner to regulate context-specific aspects of cell physiology. Our data are consistent with chemokine receptor–mediated Akt signaling occurring from the plasma membrane and from the surface of EEA1-positive endosomes. Signaling from endosomes may be selective for signaling to certain Akt substrates (FoxO1/3a) but not others (GSK3β and TSC2), suggesting that Akt signaling from endosomes may lead to different cellular outcomes than signaling from the plasma membrane. Endosomes may be required for suppression of apoptosis, but only in detached cells, not adherent cells. This is consistent with endosomes sensing and specifying (i.e. decoding) proper context-specific signaling. Although this compartmentalized signaling may be a general property of chemokine receptors, it remains to be determined whether it can be broadly applied to other GPCRs. It will be important in the future to further elucidate the mechanisms involved in Akt signaling from endosomes and how it suppresses apoptosis. This may reveal novel aspects of this signaling that can be targeted in cancer linked to GPCRs such as CXCR4.
Experimental procedures
Cell culture, antibodies, and reagents
HeLa (CCL-2) and WEHI-231 (CRL-1701) cells were from the American Type Culture Collection (Manassas, VA). Cells were maintained in minimum essential medium (MEM; Gibco, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich). Antibodies against pAkt-Ser473 (catalogue number 9271), Akt (catalogue number 9272), GSK3β (catalogue number 12456), pGSK3β-Ser9 (catalogue number 5558), pFoxO1/3a-Thr24/Thr32 (catalogue number 9464), FoxO3a (catalogue number 2497), FoxO1 (catalogue number 2880), TSC2 (catalogue number 4308), pTSC2-Thr1462 (catalogue number 3617), PARP (catalogue number 9542), and cleaved caspase-3 (catalogue number 9664) were from Cell Signaling Technology (Danvers, MA). Antibodies against ERK-1/2 (catalogue number M8159) and pERK-1/2 (catalogue number M8159) were from Sigma. The antibody against APPL1 was from Protein Tech (catalogue number 19885), and the antibody against EEA1 was from BD Transduction Laboratories (catalogue number BDB610456). The anti-β-tubulin (catalogue number E7) antibody was from Developmental Studies Hybridoma Bank (Iowa City, IA). Horseradish peroxidase-conjugated secondary antibodies were from Vector Laboratories (Burlingame, CA). CXCL12 (catalogue number PFP001) and CXCL13(1–87) (catalogue number PFP019) were from Protein Foundry (Milwaukee, WI). Insulin (catalogue number 300-191P) was from Gemini Bio-Products (West Sacramento, CA). Dimethyl sulfoxide (DMSO; catalogue number D8418) and EGF were from Sigma. Pertussis toxin (catalogue number 3097) and dynasore (catalogue number 2897) were from Tocris (Minneapolis, MN).
siRNAs and transient transfection
siRNAs directed against control luciferase (catalogue number 51-01-08-22), EEA1 (catalogue numbers HSC.RNAI.N003566.12.1 and HSC.RNAI.N003566.12.4), and APPL1 (catalogue numbers HSC.RNAI.N012096.12.1 and HSC.RNAI.N012096.12.3) were from Integrated DNA Technologies (Coralville, IA). Lipofectamine 3000 (catalogue number L3000008, Life Technologies) transfection reagent was used for transfection of siRNA. HeLa cells were grown in 10-cm or 6-well dishes with 10 nm final siRNA against EEA1 or APPL1 or control luciferase siRN, similarly to what we have described previously (8).
Signaling assay
HeLa cells that were either transfected with siRNA or untransfected were passaged onto 6-well dishes and grown for an additional 24 h to ∼90% confluence. Cells were washed once with warm MEM containing 20 mm HEPES, pH 7.4, and incubated in the same medium for 3 h at 37 °C. Cells transfected with siRNA were treated with 10 nm CXCL12, 100 nm CXCL13, 100 ng/ml EGF, 50 nm insulin, or vehicle (water; 1:1000 dilution) for 5 min at 37 °C. To examine chemokine signaling in HeLa cells, cells were treated with 10 nm CXCL12 and 10–1000 nm CXCL13, CXCL11, or CXCL10. To examine the effect of dynasore, untransfected HeLa or WEHI-231 cells grown in 6-well dishes were preincubated with 80 μm dynasore or DMSO (1:1000 dilution) for 30–60 min at 37 °C before treatment with CXCL12 or CXCL13 for 5 min. HeLa cells were then washed once with cold PBS and scraped off the dish in 300 μl of 2× sample buffer (8% SDS, 10% glycerol, 5% β-mercaptoethanol, 37.5 mm Tris-HCl, pH 6.5, 0.003% bromphenol blue). WEHI-231 cells were transferred to round-bottom centrifuge tubes, centrifuged at 1000 × g for 5 min at 4 °C, washed once in PBS, and centrifuged to pellet cells. Cell pellets were solubilized in 300 μl of 2× sample buffer. Equal amounts were analyzed by 10% SDS-PAGE and immunoblotting with antibodies against phosphorylated and total proteins. The phosphorylation status of various proteins was quantitated by densitometric analysis of similar exposures across multiple experiments using ImageJ software (National Institutes of Health, Bethesda, MD).
Anoikis assay
Cells were plated onto 6-well dishes 24 h after transfection. Twenty-four hours later cells were analyzed in adhesion and detached settings. Cells were washed once with warm MEM containing 20 mm HEPES, pH 7.4, and incubated in the same medium for 1 h at 37 °C. For the adhesion setting, adherent cells were then treated with 10 nm CXCL12 or vehicle and incubated at 37 °C for 2–5 h. For the detached setting, cells were detached from the plate with HyQTase (Hyclone Laboratories, Logan, UT); centrifuged; washed in PBS; resuspended in MEM containing 20 mm HEPES, pH 7.4; and seeded onto 6-well plates pretreated with 10 mg/ml poly(2-hydroxyethyl methacrylate) (catalogue number P3932, Sigma) in 100% ethanol to prevent adhesion. Cells were then treated with 10 nm CXCL12 or vehicle and incubated at 37 °C for 2–5 h. Adherent cells were washed once with cold PBS and harvested in 300 μl of cold radioimmune precipitation assay buffer (50 mm Tris-HCl pH 8.0, 150 mm NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, 10 mm NaF, 10 mm sodium orthovanadate, and protease inhibitors (10 μg/ml each of aprotinin, pepstatin A, and leupeptin; Roche Applied Science)). Detached cells were collected into microcentrifuge tubes and centrifuged at 4 °C at 1000 × g for 5 min. Supernatant was aspirated, and cells were washed twice with cold PBS. Cells were lysed in 300 μl of radioimmune precipitation assay buffer. Lysates were cleared by centrifugation at 14,000 rpm for 20 min at 4 °C using a 5417r Eppendorf microcentrifuge. Protein concentration of cleared lysates was determined using a BCA protein assay kit (Pierce Thermo Fisher). A small aliquot of sample (40 μl) was saved in 5× sample buffer for immunoblotting PARP, cleaved caspase-3, and EEA1. The remaining sample with protein amount ranging from 10 to 58 μg across multiple experiments was used in accordance with Caspase-Glo 3/7 Assay kit (catalogue number G8091, Promega, Madison, WI) to measure the luminescence associated with the amount of caspase activity per sample.
Internalization assay
Whole-cell ELISA was used to measure CXCR4 and CXCR5 internalization essentially as we have described previously (36, 48). HeLa cells grown on 10-cm dishes were transiently transfected with HA-CXCR4 (10 μg) or HA-CXCR5 (10 μg) using polyethylenimine. The next day ∼200,000 cells were seeded onto poly-l-lysine–coated 24-well plates. Cells were serum-starved for 1 h and pretreated with DMSO or 80 μm dynasore for 30 min followed by stimulation with 10 nm CXCL12 or 100 nm CXCL13 for 5 min. After fixation with 3.7% paraformaldehyde for 5 min and incubation with Tris-buffered saline (TBS) supplemented with 1% bovine serum albumin (BSA) in TBS for 45 min, cells were incubated with anti-HA mAb (catalogue number 901513, Biolegend) for 1 h at room temperature. Cells were washed and incubated with alkaline phosphatase–conjugated anti-mouse IgG (catalogue number A5153, Sigma) in 1% BSA in TBS for 1 h at room temperature. Cells were washed and incubated in p-nitrophenyl phosphate diluted in diethanolamine buffer (catalogue number 9701861, Bio-Rad). Reactions were stopped by adding 0.4 m NaOH, and an aliquot was used to measure the absorbance at 405 nm. Percent receptor internalization was calculated by subtracting the fraction of absorbance after agonist treatment and vehicle treatment (following background subtraction) from 1 and multiplying by 100.
Statistical analysis
Data are represented as the mean ± S.D. of at least three independent experiments or determinations. All statistical tests were done using GraphPad Prism 7.0c for Mac OS X (GraphPad Software, San Diego). Student's t test was used to compare the difference between two groups, one-way analysis of variance (ANOVA) was used to compare the difference between three or more groups, and two-way ANOVA was used to compare the difference between different groups under different treatment conditions. ANOVA was followed by a Tukey's or Newman–Keuls post hoc test. A probability (p) value of 0.05 was considered significant. Specific values are provided in the figure panels or in the figure legends.
Author contributions
E. J. E., S. A. M., and A. M. data curation; E. J. E., S. A. M., and A. M. formal analysis; E. J. E., S. A. M., and A. M. investigation; E. J. E., S. A. M., and A. M. methodology; E. J. E., S. A. M., and A. M. writing-review and editing; A. M. conceptualization; A. M. funding acquisition; A. M. writing-original draft; A. M. project administration.
Supplementary Material
This work was supported by National Institutes of Health Grants GM106727 and GM122889 (to A. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
E. J. English and A. Marchese, unpublished data.
- CXCR
- chemokine CXC motif receptor
- CXCL
- CXC motif chemokine ligand
- GPCR
- G protein–coupled receptor
- EEA1
- early endosome antigen 1
- ERK-1/2
- extracellular signal–regulated kinases 1 and 2
- APPL1
- adaptor protein, phosphotyrosine-interacting with PH domain and leucine zipper 1
- PH
- pleckstrin homology
- TSC2
- tuberous sclerosis complex 2
- PI3K
- phosphoinositide 3-kinase
- PIP3
- phosphatidylinositol(3,4,5)-trisphosphate
- PDK1
- 3-phosphoinositide–dependent protein kinase-1
- mTORC2
- mTOR complex 2
- pAkt-Ser473
- phosphorylation of Akt at Ser473
- FoxO
- forkhead box O
- GSK3β
- glycogen synthase kinase 3β
- EGF
- epidermal growth factor
- PARP
- poly(ADP-ribose) polymerase
- ESCRT
- endosomal sorting complex required for transport
- FAK
- focal adhesion kinase
- MEM
- minimum essential medium
- TBS
- Tris-buffered saline
- ANOVA
- analysis of variance.
References
- 1. Raman D., Baugher P. J., Thu Y. M., and Richmond A. (2007) Role of chemokines in tumor growth. Cancer Lett. 256, 137–165 10.1016/j.canlet.2007.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balkwill F. (2004) Cancer and the chemokine network. Nat. Rev. Cancer 4, 540–550 10.1038/nrc1388 [DOI] [PubMed] [Google Scholar]
- 3. Lacalle R. A., Blanco R., Carmona-Rodríguez L., Martín-Leal A., Mira E., and Mañes S. (2017) Chemokine receptor signaling and the hallmarks of cancer. Int. Rev. Cell Mol. Biol. 331, 181–244 10.1016/bs.ircmb.2016.09.011 [DOI] [PubMed] [Google Scholar]
- 4. Busillo J. M., and Benovic J. L. (2007) Regulation of CXCR4 signaling. Biochim. Biophys. Acta 1768, 952–963 10.1016/j.bbamem.2006.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang X. H., Jin X., Malladi S., Zou Y., Wen Y. H., Brogi E., Smid M., Foekens J. A., and Massagué J. (2013) Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 154, 1060–1073 10.1016/j.cell.2013.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pearce L. R., Komander D., and Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22 10.1038/nrn2754,10.1038/nrm2822 [DOI] [PubMed] [Google Scholar]
- 7. Manning B. D., and Toker A. (2017) AKT/PKB signaling: navigating the network. Cell 169, 381–405 10.1016/j.cell.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verma R., and Marchese A. (2015) The endosomal sorting complex required for transport pathway mediates chemokine receptor CXCR4-promoted lysosomal degradation of the mammalian target of rapamycin antagonist DEPTOR. J. Biol. Chem. 290, 6810–6824 10.1074/jbc.M114.606699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vanhaesebroeck B., Stephens L., and Hawkins P. (2012) PI3K signalling: the path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 13, 195–203 10.1038/nrm3290 [DOI] [PubMed] [Google Scholar]
- 10. Tsvetanova N. G., Irannejad R., and von Zastrow M. (2015) G protein-coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes. J. Biol. Chem. 290, 6689–6696 10.1074/jbc.R114.617951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shenoy S. K., and Lefkowitz R. J. (2011) β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 10.1016/j.tips.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsvetanova N. G., and von Zastrow M. (2014) Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol. 10, 1061–1065 10.1038/nchembio.1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Irannejad R., Tomshine J. C., Tomshine J. R., Chevalier M., Mahoney J. P., Steyaert J., Rasmussen S. G., Sunahara R. K., El-Samad H., Huang B., and von Zastrow M. (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538 10.1038/nature12000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. DeFea K. A., Zalevsky J., Thoma M. S., Déry O., Mullins R. D., and Bunnett N. W. (2000) β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 148, 1267–1281 10.1083/jcb.148.6.1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nazarewicz R. R., Salazar G., Patrushev N., San Martin A., Hilenski L., Xiong S., and Alexander R. W. (2011) Early endosomal antigen 1 (EEA1) is an obligate scaffold for angiotensin II-induced, PKC-α-dependent Akt activation in endosomes. J. Biol. Chem. 286, 2886–2895 10.1074/jbc.M110.141499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. García-Regalado A., Guzmán-Hernández M. L., Ramírez-Rangel I., Robles-Molina E., Balla T., Vázquez-Prado J., and Reyes-Cruz G. (2008) G protein-coupled receptor-promoted trafficking of Gβ1γ2 leads to AKT activation at endosomes via a mechanism mediated by Gβ1γ2-Rab11a interaction. Mol. Biol. Cell 19, 4188–4200 10.1091/mbc.e07-10-1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malik R., Soh U. J., Trejo J., and Marchese A. (2012) Novel roles for the E3 ubiquitin ligase atrophin-interacting protein 4 and signal transduction adaptor molecule 1 in G protein-coupled receptor signaling. J. Biol. Chem. 287, 9013–9027 10.1074/jbc.M111.336792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schmid S. L. (2017) Reciprocal regulation of signaling and endocytosis: implications for the evolving cancer cell. J. Cell Biol. 216, 2623–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Godbole A., Lyga S., Lohse M. J., and Calebiro D. (2017) Internalized TSH receptors en route to the TGN induce local Gs-protein signaling and gene transcription. Nat. Commun. 8, 443 10.1038/s41467-017-00357-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kotowski S. J., Hopf F. W., Seif T., Bonci A., and von Zastrow M. (2011) Endocytosis promotes rapid dopaminergic signaling. Neuron 71, 278–290 10.1016/j.neuron.2011.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nagaraj N., Wisniewski J. R., Geiger T., Cox J., Kircher M., Kelso J., Pääbo S., and Mann M. (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 7, 548 10.1038/msb.2011.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keppler S. J., Gasparrini F., Burbage M., Aggarwal S., Frederico B., Geha R. S., Way M., Bruckbauer A., and Batista F. D. (2015) Wiskott-Aldrich syndrome interacting protein deficiency uncovers the role of the co-receptor CD19 as a generic hub for PI3 kinase signaling in B cells. Immunity 43, 660–673 10.1016/j.immuni.2015.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin D. C., Quevedo C., Brewer N. E., Bell A., Testa J. R., Grimes M. L., Miller F. D., and Kaplan D. R. (2006) APPL1 associates with TrkA and GIPC1 and is required for nerve growth factor-mediated signal transduction. Mol. Cell. Biol. 26, 8928–8941 10.1128/MCB.00228-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mao X., Kikani C. K., Riojas R. A., Langlais P., Wang L., Ramos F. J., Fang Q., Christ-Roberts C. Y., Hong J. Y., Kim R. Y., Liu F., and Dong L. Q. (2006) APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat. Cell Biol. 8, 516–523 10.1038/ncb1404 [DOI] [PubMed] [Google Scholar]
- 25. Schenck A., Goto-Silva L., Collinet C., Rhinn M., Giner A., Habermann B., Brand M., and Zerial M. (2008) The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell 133, 486–497 10.1016/j.cell.2008.02.044 [DOI] [PubMed] [Google Scholar]
- 26. Thomas R. M., Nechamen C. A., Mazurkiewicz J. E., Ulloa-Aguirre A., and Dias J. A. (2011) The adapter protein APPL1 links FSH receptor to inositol 1,4,5-trisphosphate production and is implicated in intracellular Ca2+ mobilization. Endocrinology 152, 1691–1701 10.1210/en.2010-1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nechamen C. A., Thomas R. M., and Dias J. A. (2007) APPL1, APPL2, Akt2 and FOXO1a interact with FSHR in a potential signaling complex. Mol. Cell. Endocrinol. 260–262, 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Christoforidis S., McBride H. M., Burgoyne R. D., and Zerial M. (1999) The Rab5 effector EEA1 is a core component of endosome docking. Nature 397, 621–625 10.1038/17618 [DOI] [PubMed] [Google Scholar]
- 29. Zhang X. H., Wang Q., Gerald W., Hudis C. A., Norton L., Smid M., Foekens J. A., and Massagué J. (2009) Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 16, 67–78 10.1016/j.ccr.2009.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frisch S. M., and Francis H. (1994) Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 124, 619–626 10.1083/jcb.124.4.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park R. J., Shen H., Liu L., Liu X., Ferguson S. M., and De Camilli P. (2013) Dynamin triple knockout cells reveal off target effects of commonly used dynamin inhibitors. J. Cell Sci. 126, 5305–5312 10.1242/jcs.138578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Villasenor R., Nonaka H., Del Conte-Zerial P., Kalaidzidis Y., and Zerial M. (2015) Regulation of EGFR signal transduction by analogue-to-digital conversion in endosomes. eLife 4, 06156 10.7554/eLife.06156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zoncu R., Perera R. M., Balkin D. M., Pirruccello M., Toomre D., and De Camilli P. (2009) A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell 136, 1110–1121 10.1016/j.cell.2009.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Varsano T., Taupin V., Guo L., Baterina O. Y. Jr, and Farquhar M. G. (2012) The PDZ protein GIPC regulates trafficking of the LPA1 receptor from APPL signaling endosomes and attenuates the cell's response to LPA. PLoS One 7, e49227 10.1371/journal.pone.0049227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marchese A., Raiborg C., Santini F., Keen J. H., Stenmark H., and Benovic J. L. (2003) The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell 5, 709–722 10.1016/S1534-5807(03)00321-6 [DOI] [PubMed] [Google Scholar]
- 36. Bhandari D., Trejo J., Benovic J. L., and Marchese A. (2007) Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atrophin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4. J. Biol. Chem. 282, 36971–36979 10.1074/jbc.M705085200 [DOI] [PubMed] [Google Scholar]
- 37. Malik R., and Marchese A. (2010) Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol. Biol. Cell 21, 2529–2541 10.1091/mbc.e10-02-0169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Slagsvold T., Marchese A., Brech A., and Stenmark H. (2006) CISK attenuates degradation of the chemokine receptor CXCR4 via the ubiquitin ligase AIP4. EMBO J. 25, 3738–3749 10.1038/sj.emboj.7601267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ebner M., Sinkovics B., Szczygieł M., Ribeiro D. W., and Yudushkin I. (2017) Localization of mTORC2 activity inside cells. J. Cell Biol. 216, 343–353 10.1083/jcb.201610060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mullershausen F., Zecri F., Cetin C., Billich A., Guerini D., and Seuwen K. (2009) Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat. Chem. Biol. 5, 428–434 10.1038/nchembio.173 [DOI] [PubMed] [Google Scholar]
- 41. Smith T. H., Li J. G., Dores M. R., and Trejo J. (2017) Protease-activated receptor-4 and purinergic receptor P2Y12 dimerize, co-internalize, and activate Akt signaling via endosomal recruitment of β-arrestin. J. Biol. Chem. 292, 13867–13878 10.1074/jbc.M117.782359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jean-Alphonse F. G., Wehbi V. L., Chen J., Noda M., Taboas J. M., Xiao K., and Vilardaga J. P. (2017) β2-Adrenergic receptor control of endosomal PTH receptor signaling via Gβγ. Nat. Chem. Biol. 13, 259–261 10.1038/nchembio.2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tran H., Brunet A., Griffith E. C., and Greenberg M. E. (2003) The many forks in FOXO's road. Sci. STKE 2003, RE5 10.1126/stke.2003.172.re5 [DOI] [PubMed] [Google Scholar]
- 44. Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., and Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 10.1016/S0092-8674(00)80595-4 [DOI] [PubMed] [Google Scholar]
- 45. Alanko J., Mai A., Jacquemet G., Schauer K., Kaukonen R., Saari M., Goud B., and Ivaska J. (2015) Integrin endosomal signalling suppresses anoikis. Nat. Cell Biol. 17, 1412–1421 10.1038/ncb3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kochetkova M., Kumar S., and McColl S. R. (2009) Chemokine receptors CXCR4 and CCR7 promote metastasis by preventing anoikis in cancer cells. Cell Death Differ. 16, 664–673 10.1038/cdd.2008.190 [DOI] [PubMed] [Google Scholar]
- 47. Drury L. J., Wendt M. K., and Dwinell M. B. (2010) CXCL12 chemokine expression and secretion regulates colorectal carcinoma cell anoikis through Bim-mediated intrinsic apoptosis. PLoS One 5, e12895 10.1371/journal.pone.0012895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marchese A., and Benovic J. L. (2001) Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem. 276, 45509–45512 10.1074/jbc.C100527200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.