

Abstract
Receptor protein-tyrosine phosphatase RPTPσ has important functions in modulating neural development and regeneration. Compelling evidence suggests that both heparan sulfate (HS) and chondroitin sulfate (CS) glycosaminoglycans (GAGs) bind to a series of Lys residues located in the first Ig domain of RPTPσ. However, HS promotes and CS inhibits axonal growth. Mutation of these Lys residues abolished binding and signal transduction of RPTPσ to CS, whereas HS binding was reduced, and signaling persisted. This activity was mediated through novel heparin-binding sites identified in the juxtamembrane region. Although different functional outcomes of HS and CS have been previously attributed to the differential oligomeric state of RPTPσ upon GAG binding, we found that RPTPσ was clustered by both heparin and CS GAG rich in 4,6-O-disulfated disaccharide units. We propose an additional mechanism by which RPTPσ distinguishes between HS and CS through these novel binding sites.
Keywords: proteoglycan, heparan sulfate, chondroitin sulfate, cell signaling, glycosaminoglycan, receptor tyrosine phosphatase, RPTPσ
Introduction
Proteoglycans in the extracellular matrix play central roles in maintaining tissue architecture and modulating cell signaling. Emerging evidence suggests that proteoglycans themselves may be independent signaling molecules, binding directly to transmembrane receptors to exert distinct biological effects. In the nervous system, the glycosaminoglycan (GAG)3 chains of proteoglycans bind to members of the type IIa receptor protein-tyrosine phosphatases (RPTPσ/RPTPδ/LAR) as well as reticulon 4 receptors (1–6). Curiously, the binding of RPTPσ to chondroitin sulfate proteoglycans (CSPGs) impedes the growth of axons, whereas binding to heparan sulfate proteoglycans (HSPGs) promotes axonal growth (7). How the binding of CS and HS GAG chains causes opposite actions through binding to RPTPσ is a matter of intense interest.
Members of this subfamily (RPTPs) are composed of two cytoplasmic phosphatase domains and extracellular immunoglobulin (Ig) domains followed by fibronectin type III (FNIII) repeats, similar to the neural cell adhesion molecule (NCAM) family of cell adhesion molecules (Fig. S1). Previous mutagenesis and structural studies have shown that the GAG-binding site for both HS and CS lies in the first Ig domain of RPTPσ and comprises an extended positively charged surface of basic residues (Lys- and Arg-loop) (1, 7, 8). In this study, through the use of full-length RPTPσ, we report that heparin, a close structural relative of HS, up-regulates Tyr phosphorylation within cells independently of this common GAG-binding site of RPTPσ. This led us to identify novel heparin-binding sites between the fourth FNIII domain and the transmembrane domain that are involved in the regulation of intracellular Tyr phosphorylation.
Coles and co-workers (7, 9) reported that HS binding to the common GAG-binding site on RPTPσ induced clustering of the extracellular region of RPTPσ, whereas CS binding did not. They thus proposed an “RPTPσ-dependent proteoglycan switch” where the opposing effects of HS and CS GAGs are attributed to the differential oligomeric state of RPTPσ. Here, we provide evidence that CS GAGs rich in 4,6-O-disulfated disaccharide units known to impede axonal growth (10) can induce RPTPσ clustering. Our observations have allowed us to propose an additional mechanism by which RPTPσ differentiates between HS and CS to modulate axonal growth.
Results
We investigated whether complex formation between full-length RPTPσ and GAGs impacts the receptor's enzymatic activity under physiological conditions. Because HS promotes neuronal outgrowth, we first examined whether the binding of heparin, a close structural relative of HS, to RPTPσ altered intracellular phosphorylation. To increase baseline Tyr phosphorylation in cells, chick v-Src was cotransfected into 293 cells together with RPTPσ and its variants. When 293 cells expressing WT RPTPσ were treated with heparin, the level of Tyr phosphorylation was transiently up-regulated in a dose-dependent manner (Fig. 1, A–C). This up-regulation was abolished by a phosphatase-inactivating mutation of RPTPσ (D1110E) in the cytoplasmic domain, indicating the importance of phosphatase activity of the molecule (Fig. S3). Of particular interest is that the disruption of the Lys-loop within the first Ig domain (ΔLys mutant; K68A/K69A/K71A/K72A) did not abolish the up-regulation of Tyr phosphorylation by heparin (Fig. 1). Although this Lys-loop has been reported to be a sole GAG-binding site in RPTPσ, this finding strongly suggests the presence of another heparin-binding domain(s) in the extracellular portion of RPTPσ.
Figure 1.

Altered Tyr phosphorylation by the addition of heparin. 293 cells expressing GFP (lanes 1), RPTPσ (lanes 2), and RPTPσ ΔLys (lanes 3) were treated with heparin (1 mg/ml) for the indicated times (A) and different concentrations of heparin for 5 (B) and 30 min (C). Proteins were separated by SDS-PAGE followed by immunoblotting with anti-phosphotyrosine antibody. D, cells were treated with heparin, CS-E, and DS (1 mg/ml) for 5 min. Similar expression levels of recombinant proteins were confirmed with anti-His6 tag antibody (Fig. S2). Cont, control; TM, transmembrane.
To biochemically analyze the interaction between RPTPσ and GAGs, the extracellular domains (ECDs) of RPTPσ were expressed as a fusion protein with alkaline phosphatase (AP) and solid-phase binding assays were performed with immobilized heparin, CS, and DS GAGs. (disaccharide composition analyses of GAGs used in this study are shown in Table S1). Fig. 2 shows the binding curves of AP fusion proteins, and the calculated dissociation constant (Kd) and maximum number of binding sites (Bmax) are summarized in Table 1. Scatchard plot analysis of RPTPσ binding data revealed a Kd of 0.760 ± 0.178 nm for heparin, consistent with a previously reported value (1). Among CS/DS GAGs analyzed, only CS-E and DS showed binding to RPTPσ with the typical nanomolar range of Kd values for biologically relevant ligand–receptor interactions. This indicates that RPTPσ distinguishes the sulfation pattern of CS GAGs. Although the disruption of the Lys-loop eliminated the binding of RPTPσ to CS-E and DS, this ΔLys mutant maintained the ability to bind to heparin. Thus, heparin-induced up-regulation of cytoplasmic phosphorylation via RPTPσ with the ΔLys mutation (Fig. 1A) is likely through binding to a region of the ECD other than the Lys-loop. Consistent with the CS/DS binding data, CS/DS-induced up-regulation of Tyr phosphorylation was reduced dramatically by the ΔLys mutation in RPTPσ (Fig. 1D).
Figure 2.

Solid-phase binding of RPTPσ and RPTPσ Ig1FN3 to immobilized GAGs. The indicated concentrations of AP fusion proteins were incubated with immobilized GAGs, and bound AP activity was measured. Kd and Bmax were calculated using Scatchard plot analysis (inset). Error bars represent S.D.
Table 1.
Kd and Bmax of AP fusion proteins with ECD and Ig1FN3 for the binding to GAGs
Data were expressed as mean ± S.D. n.b. indicates no saturable binding was detected.
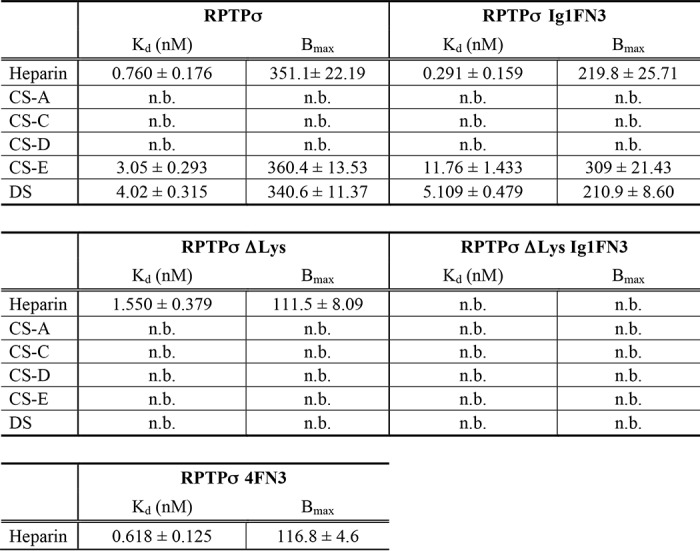
To examine the possible contribution of FNIII-containing domains to heparin binding, we generated deletion mutants consisting of three Ig domains and three FNIII domains (Ig1FN3) and performed solid-phase binding assays. Although removal of the fourth FNIII-containing domain from RPTPσ did not affect the Kd for heparin, we found a substantial reduction of Bmax for heparin (Table 1). When the ΔLys mutation was introduced to the first Ig domain (RPTPσ ΔLys Ig1FN3), its ability to bind heparin was completely eliminated. Furthermore, the fourth FNIII-containing domain along with the juxtamembrane domain (RPTPσ 4FN3) alone was found to bind to heparin independently of the three Ig domains with a Kd similar to that of the WT protein. The fact that the sum of Bmax for RPTPσ Ig1FN3 and RPTPσ 4FN3 (or RPTPσ ΔLys) is nearly equal to Bmax of WT RPTPσ supports the notion that there are multiple heparin-binding domains with a similar Kd (Table 1). In contrast, the Lys-loop in the first Ig domain appeared to be the major binding site for CS-E and DS because RPTPσ ΔLys demonstrated no binding ability to these GAGs. Notably, there is some contribution of the fourth FNIII-containing domain of RPTPσ to the binding to CS-E and DS because the deletion of the fourth FNIII-containing domain affected the Kd for CS-E and Bmax for DS.
To further characterize the second binding domain, a series of deletion mutants were prepared and subjected to affinity chromatography to check their binding ability to heparin (Fig. 3). Ten microliters of 10–20 nm AP fusion proteins were loaded onto the column and eluted with a linear gradient of NaCl (150–2000 mm) under the same buffer as used for solid-phase binding assays. Although AP alone was not recognized by the column at all, both AP fusion proteins with the ECDs of RPTPσ and RPTPσ Ig1FN3 were retained and eluted from the column at NaCl concentrations of 1334 and 1260 mm, respectively. The protein with the ΔLys mutation in RPTPσ was eluted earlier from the column than the WT (964 mm NaCl), and the ΔLys mutation in RPTPσ Ig1FN3 totally abolished its binding, comparable with solid-phase binding data. Although RPTPσ 4FN3 bound and was eluted from the column at 1112 mm as expected, RPTPσ with the ΔLys mutation without the fourth FNIII (RPTPσ ΔLys Δ4FN3) was still retained by the column and eluted at a concentration of NaCl (964 mm) similar to that for RPTPσ 4FN3. This finding strongly suggests that a novel binding site resides within the flanking regions of the fourth FNIII domain. None of the fragments that lack this flanking region (RPTPσ Ig1FN4, RPTPσ ΔLys Ig1FN4, and RPTPσ 4FN3 AL) or this flanking region alone (RPTPσ FLNK) were secreted into the media even in the presence of the signal peptide, but these recombinant proteins were detected in the cell lysates (data not shown).
Figure 3.

Heparin-affinity chromatography of AP fusion proteins. Purified AP fusion proteins were loaded onto a heparin column and eluted with a linear gradient of NaCl (150–2000 mm; dotted line). AP activity of each fraction was measured.
To identify this novel heparin-binding site(s) within the flanking region of the fourth FNIII domain, “heparin protection” assays were performed (11). Amino acid residues potentially involved in the interaction with heparin were labeled by NHS-biotin after acetylation of nonessential lysine/arginine residues exposed to the surface of the molecule. Table S2 shows the modified peptides identified within the region. The lysine/arginine residues selected as potential heparin-binding sites were then mutated to alanine individually, and AP fusion proteins with these mutations were subjected to heparin-affinity chromatography. Additionally, a stretch of positively charged amino acids in the region (residues 762–781) were replaced with alanine to elaborate the possible involvement of the stretch of basic amino acid residues. None of the peptides were recovered from this stretch in LC-MS analysis possibly because of hydrophilic amino acids. Among those tested, mutations within the positive charge stretch (R762A/R763A/R766A/H767A/R769A; ΔRRRHR mutation) and H809A/R810A (ΔHR) together with the ΔLys mutation in the first Ig domain altered the elution profile of the heparin-affinity column (Fig. 4). RPTPσ ΔLys ΔRRRHR and RPTPσ ΔLys ΔHR were retained and eluted earlier than WT at 890 and 964 mm NaCl, respectively. Finally, all three mutations together (RPTPσ ΔLys ΔRRRHR ΔHR) eliminated the ability to bind to the heparin column. Thus, we concluded that these three sites are involved in heparin binding.
Figure 4.

Heparin-affinity chromatography of AP fusion proteins. Purified AP fusion proteins were loaded onto a heparin column and eluted with a linear gradient of NaCl (150–2000 mm; dotted line). AP activity of each fraction was measured.
Coles et al. (7) reported heparin-induced clustering of RPTP type IIa members. The fact that RPTPσ binds to CS-E as well as heparin and DS led us to investigate GAG-induced clustering of RPTPσ. Purified RPTPσ Ig1FN3-AP fusion protein was incubated with or without GAGs, and the GAG-protein mixture was separated by size-exclusion chromatography followed by measurement of AP activity (Fig. 5). Incubation with heparin induced a shift toward a multimer of RPTPσ Ig1FN3 (Fig. 5A). The measured molecular masses of RPTPσ IgFN3–AP were 210 kDa without heparin and 400 kDa with heparin. This trend continued as RPTPσ IgFN3–AP was incubated with increasing concentrations of heparin: as the concentration of heparin was increased, we observed a further shift in the peak, which is consistent with the finding by Coles et al. (7). Heparin did not alter the retention time of AP (Fig. 5C) or of the inactive form, RPTPσ ΔLys IgFN3–AP (data not shown). Because we did not find any interaction of RPTPσ IgFN3 with CS-A, CS-C, and CS-D in the solid-phase binding assays, it is reasonable that neither CS-A, CS-C, or CS-D induced clustering of the protein (data not shown). However, incubation with CS-E induced an earlier elution of RPTPσ IgFN3–AP from the column as compared with the protein alone with a retention time equivalent to that of the heparin–RPTPσ complex. This is distinct from the finding by Coles et al. (7) showing that only heparin derivatives induced the clustering of RPTPσ and CS did not. In contrast to heparin, CS-E did not induce further multimerization of RPTPσ IgFN3–AP with an increased concentration of CS-E. A similar clustering was observed when full-length RPTPσ–AP was analyzed (Fig. 5B). Although DS was a good binding partner for RPTPσ in solid-phase assays, DS did not promote the multimerization of RPTPσ Ig1FN3.
Figure 5.
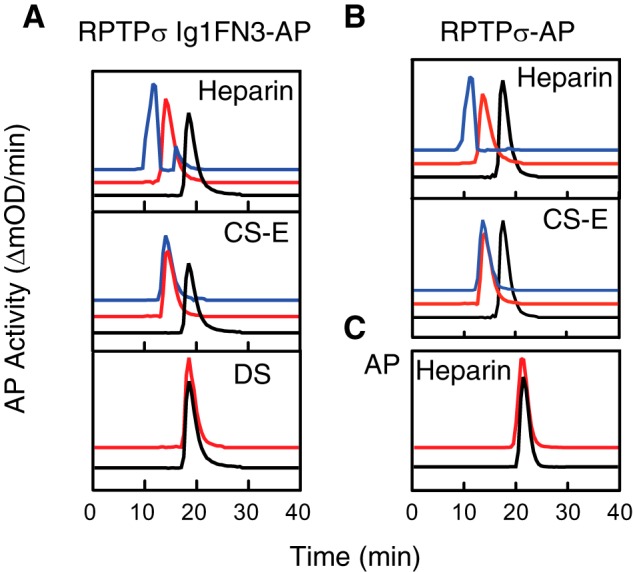
GAG-induced clustering of AP fusion protein. Purified AP fusion proteins were incubated with (red) and without (black) a 4-fold molar excess of GAGs and separated by size-exclusion chromatography. A, RPTPσ Ig1FN3–AP; B, RPTPσ–AP; C, AP. Blue lines indicate incubation with a 60-fold molar excess of GAGs. The molecular mass of the complex between the recombinant protein and a 60-fold molar excess of heparin was not calculated because of the earlier retention time of thyroglobulin (670 kDa) gel-filtration standard protein (Bio-Rad).
To further strengthen the observation of clustering induced by heparin and CS-E, we took a different approach (“sandwich ELISA”) using RPTPσ–Fc and RPTPσ–AP fusion proteins (Fig. 6). RPTPσ–Fc was first immobilized to protein A–precoated plates followed by incubation with GAGs for 1.5 h at which time RPTPσ–AP was added. Without addition of GAGs, we did not detect AP activity, indicating no homophilic binding of RPTPσ. When heparin was added, RPTPσ–AP bound to heparin captured by the immobilized RPTPσ–Fc, showing the binding of multiple RPTPσ molecules to heparin. Although the addition of CS-A, CS-C, and CS-D did not induce the binding of RPTPσ–AP, CS-E permitted the binding of RPTPσ–AP, demonstrating the potential to capture multiple RPTPσ molecules on a CS-E GAG chain. DS did not permit RPTPσ–AP binding, consistent with the data from size-exclusion chromatography (Fig. 5). To exclude the possibility that longer GAG chains confer multivalent binding, the distribution of GAG chain sizes was analyzed by size-exclusion chromatography (Fig. S4). It is clear that there is no correlation between the capacity to cluster RPTPσ and the length of individual GAG chains.
Figure 6.
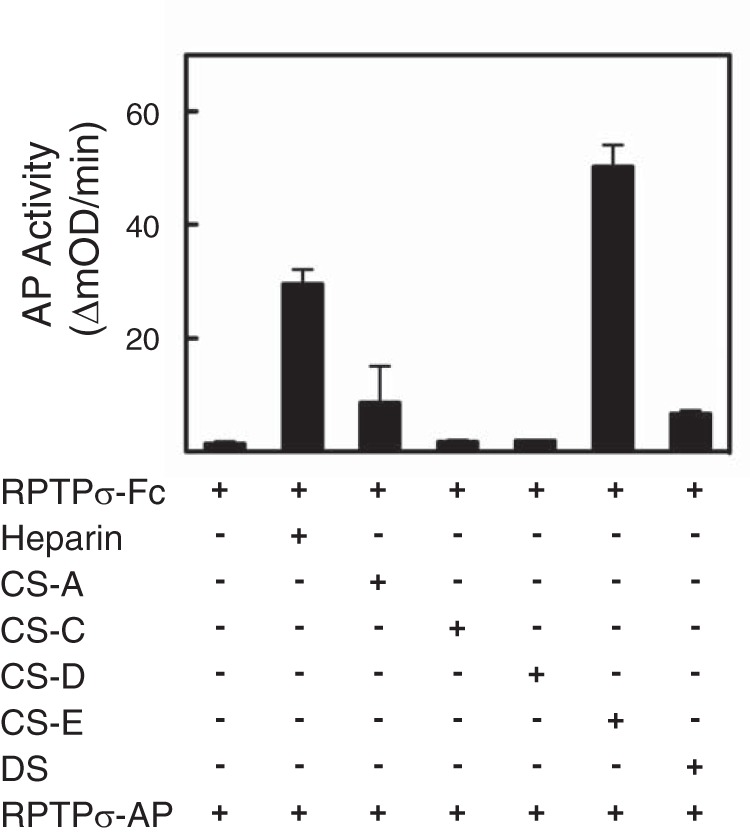
Heparin and CS-E have the potential to support the binding of multiple RPTPσ. After immobilization of RPTPσ–Fc to protein A–precoated plates, GAGs were incubated followed by incubation with RPTPσ–AP. Bound AP activity was measured. Error bars represent S.D.
One question that arises here is whether these novel heparin-binding sites are involved in the altered phosphorylation upon heparin binding. 293 cells expressing RPTPσ ΔLys ΔRRRHR ΔHR were treated with heparin, and intracellular phosphorylation was examined (Fig. 7). Although heparin induced a quick up-regulation of Tyr phosphorylation through WT RPTPσ but not the phosphatase-deficient mutant, we observed a very high level of baseline Tyr phosphorylation without heparin in cells expressing RPTPσ ΔLys ΔRRRHR ΔHR. Addition of heparin did not alter the level of phosphorylation. This implies that elimination of all heparin-binding sites on RPTPσ caused the dysregulation of Tyr phosphorylation within the cells.
Figure 7.
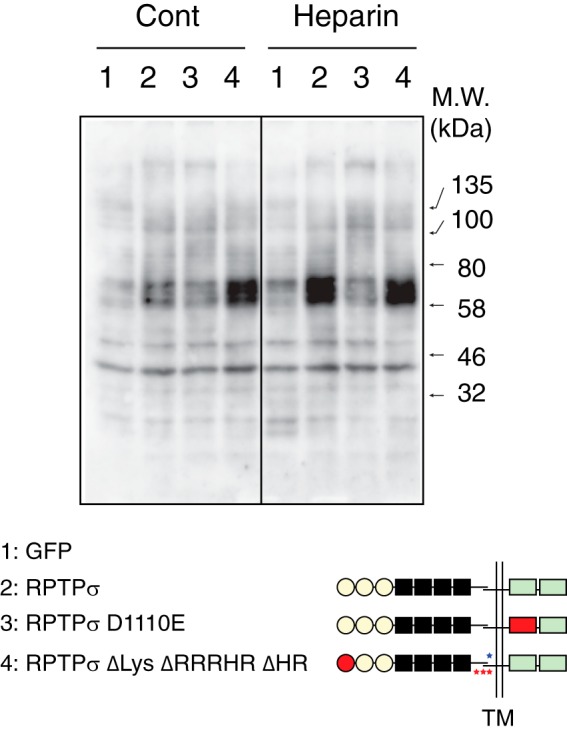
Dysregulated Tyr phosphorylation by disruption of all heparin-binding sites on RPTPσ. 293 cells expressing GFP (lanes 1), RPTPσ (lanes 2), RPTPσ D1110E (lanes 3), and RPTPσ ΔLys ΔRRRHR ΔHR (lanes 4) were treated with heparin for 5 min, and Tyr phosphorylation was examined by immunoblotting. Similar expression levels of recombinant proteins were confirmed with anti-His6 tag antibody (Fig. S2). Cont, control; TM, transmembrane.
Based on the identification of these novel heparin-binding sites in RPTPσ, we propose a model for differential interactions of HS and CS GAG chains with RPTPσ (Fig. 8). One complication in proving this model is that endogenous HS chains may occupy the receptor, thereby decreasing the response to exogenous GAGs. To test this, we expressed RPTPσ in 293 cells and then treated the cells with heparinase III to reduce cell-surface HS before adding heparin. We found that this treatment vastly increased the sensitivity to heparin, suggesting the involvement of endogenous HS in signaling through RPTPσ (Fig. S5).
Figure 8.
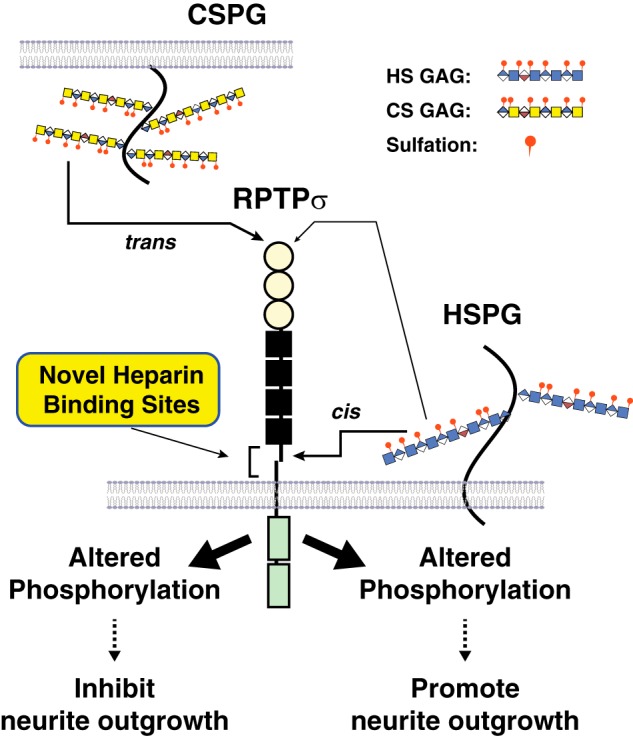
Model illustrating HS and CS binding to RPTPσ. HSPGs on the same cell interact in cis with RPTPσ through the novel binding sites near the transmembrane domain and regulate the enzymatic activity of RPTPσ, resulting in the altered Tyr phosphorylation of certain sets of proteins and the recognition of HS as a permissive cue. When CS/DS GAGs are presented in trans, binding occurs through the first Ig domain far from the transmembrane domain and triggers the changes in phosphorylation of distinct sets of proteins within the cells, resulting in the recognition of CS/DS as an inhibitory cue by RPTPσ.
Discussion
The interactions of proteoglycan GAG chains with type IIa tyrosine phosphatases are of considerable importance during brain development and in regenerative neurobiology. Structural analyses have indicated that the GAG-binding site in the first Ig domain mediates these interactions (7). This has led to the conundrum that both HS and CS bind to this site but produce opposite actions. We now provide evidence for additional binding sites, specific to HS, in the juxtamembrane domain.
When HS and CS-E/DS were bound to RPTPσ, the level of intracellular phosphorylation was altered. An intact Lys-loop in the first Ig domain was essential for these changes in response to CS-E/DS. However, HS-induced up-regulation of phosphorylation was independent of the Lys-loop. Biochemical analyses revealed that although CS-E and DS bind to the Lys-loop within the typical nanomolar range of Kd values heparin binds to multiple domains, including the Lys-loop, with a subnanomolar range of Kd values. Two novel heparin-specific binding sites were identified within the juxtamembrane domain, and the disruption of all three sites (RPTPσ ΔLys ΔRRRHR ΔHR) abolished the ability to bind heparin.
If binding of the GAGs to the juxtamembrane domain alters phosphatase activity, the intracellular phosphorylation state of cells expressing RPTPσ ΔLys ΔRRRHR ΔHR should be similar to that of WT RPTPσ without heparin, and heparin should not induce an up-regulation of phosphorylation. However, the steady-state level of phosphorylation was very high in cells expressing PPTPσ ΔLys ΔRRRHR ΔHR, and heparin treatment did not alter this situation, which is surprising. Thus, we speculate that the juxtamembrane domain, including the novel heparin-binding sites, regulates the intracellular phosphorylation state coordinated by RPTPσ (Fig. 8). In this model, HSPGs on the same cell surface interact in cis with RPTPσ through the novel binding sites near the transmembrane domain and regulate the enzymatic activity of RPTPσ. Native HSPGs in the glycocalyx may limit the complex formation with RPTPσ (12), resulting in the regulated cytoplasmic phosphorylation state. This notion is supported by the fact that heparinase III treatment increased the sensitivity to exogenous heparin (Fig. S5). When HS GAGs with specific sulfation patterns interact with RPTPσ through this region, certain sets of protein phosphorylation are altered in the cells, and the ligands work as permissive cues. When CS/DS GAGs are presented in trans, the binding is through the first Ig domain far from the transmembrane domain and triggers the changes in phosphorylation of distinct sets of proteins within the cells, resulting in the recognition of CS/DS as inhibitory cues by RPTPσ. A flexible architecture of the RPTPσ ECD (RPTPσ Ig1FN3 amino acids 1–602) is reported to be an important factor for trans-synaptic complex formation (12). The current study demonstrates a role for the juxtamembrane region (amino acids 672–847) in the signaling through HS. It is evident that interdomain interaction (7, 12) is present within RPTPσ Ig1FN3; thus, it is intriguing how the flexibility of the entire ECD is modified by the presence of the juxtamembrane domain.
We focused on the alteration of intracellular Tyr phosphorylation induced by GAG binding. It is not clear whether these changes are derived from direct actions of RPTPσ phosphatase or indirectly, but phosphatase activity in the first cytoplasmic domain is required for intracellular signaling by heparin. There are several reports showing altered phosphorylation of endogenous cytoplasmic proteins through RPTPs (13, 14). However, it is still possible that the intracellular domain of RPTPσ functions as a scaffold protein. A deficient mutant of the Drosophila type IIa RPTP dLAR can be rescued by a catalytically inactive receptor but not by a dLAR mutant lacking the second inactive phosphatase domain (15). To fully understand multiple ligand interactions and signaling pathways leading to distinct functional outcomes, additional effort is needed to clarify endogenous substrates for RPTPs as well as nonsubstrates associating with the cytoplasmic region of the molecule.
Several biological functions of type IIa RPTPs are attributed to the induction of reciprocal higher-order clustering of the receptors after ligand binding (7, 12, 14). A differential oligomeric state of RPTPs triggered by divergent ligands is an attractive model underlying distinct functional outcomes. We found that both heparin and CS-E induced the clustering of the RPTPσ ECD independently of the novel heparin-binding sites. It remains to be elucidated whether polyvalent binding of RPTPσ to GAGs or binding of oligomers of RPTPσ to GAGs is induced. Heparin promotes and CS-E inhibits axonal growth (7, 10). An intriguing clue is that heparin induces the clustering of RPTPσ to a higher degree than does CS-E. The fact that DS, another GAG that inhibits axonal growth similarly to CS-E, did not induce the clustering of RPTPσ ECD indicates that GAG-induced altered phosphorylation under physiological conditions is likely to be dissociated from differential clustering by GAGs.
The opposite outcomes from HS and CS binding to RPTPσ could imply totally separate mechanisms, not a simple explanation such as differential phosphorylation signaling. For example, the idea that the degree of oligomerization of RPTPσ may be different in response to particular ligands is still feasible for GAG signaling. As mentioned above, axonal behavior is a series of complex events, and GAG-induced altered phosphorylation may not be directly linked to axonal behavior of neurons. Instead, a higher degree of oligomerization of RPTPσ induced by permissive cues could trigger the growth-promotional pathways by more efficiently recruiting specific molecules favorable for axonal growth; a lower degree or no clustering by repellent cues may not reach the threshold to trigger such pathways, and other mechanisms such as phosphorylation could lead to different outcomes, resulting in the proteoglycan switch. Involvement of phosphorylation in CSPG's inhibitory effect is supported by the fact that small compounds that directly inhibit RPTPσ phosphatase activity overcame neurite outgrowth inhibition by CSPG using PC12 cells (16) and that CSPG induces specific phosphorylation changes within cells (17). The role of RPTPσ clustering and downstream signaling in general by HS and CS is a matter for future investigations.
Among CS GAGs, only CS-E exhibited high-affinity binding sites for RPTPσ. It is striking that CS-A and CS-C, the major components of CS GAGs in vivo, did not support the binding, consistent with our previous finding for RPTPσ (6, 18). These data emphasize the importance of appreciating the sulfation pattern and sources of CS in interpreting biological data (19). CS-E is not a major component of CS GAGs derived from natural sources. Because the Lys- and Arg-loops located in the first Ig domain are responsible for heparin binding (1, 7), negative charges derived from 4,6-O-disulfated disaccharides in the CS-E chain are likely to be critical for the interaction with RPTPσ. However, mechanisms by which RPTPσ distinguish the sulfation patterns of CS GAGs must exist because CS-D, rich in 2,6-O-disulfated disaccharides, did not bind to RPTPσ despite having a similar negative charge as CS-E. Although DS binds with high affinity to RPTPσ, DS, in contrast to HS and CS-E, did not induce clustering of RPTPσ. This reinforces the sharp differences between closely related GAG chains. The information for suitable length of DS GAG required for the binding is not available at the moment. Many different functions of DS have been found and proposed during development and in disease (20). Additional studies will be required to recognize the importance of the interaction between RPTPσ and DS.
Our discovery of new heparin-binding sites near the transmembrane domain of RPTPσ provides new insights into GAG signaling. Novel roles of type IIa RPTP family members have been demonstrated as presynaptic proteins that interact with multiple postsynaptic partners to mediate synapse organization (14, 21–25). Heparin is reported to compete with postsynaptic binding partners for presynaptic RPTPs, resulting in reduced synaptogenesis in vitro. Considering therapeutic approaches for neuronal regeneration in the mammalian central nervous system, CSPGs in the glial scar are presented in trans to RPTPσ on neurons. Thus, the juxtamembrane domain of RPTPσ presents a potential target. In addition to blocking the effects of negative guidance cues, small compounds or HS mimetics specific for these novel binding sites could promote neuronal regeneration without reducing synaptogenesis.
Experimental procedures
DNA constructs and preparation of recombinant proteins
Full-length RPRPσ (accession number NM_001252456) was amplified and subcloned into NheI/XhoI sites of pAPTAG5 (GenHunter), which generates full-length RPRPσ with a c-Myc epitope and a His6 tag at the C terminus. The RPRPσ ECD (amino acids 1–847) and Ig1FN3 (amino acids 1–602) were amplified and subcloned into NheI/HindIII sites of pAPTAG5, which generates alkaline phosphatase fusion proteins with a c-Myc epitope and a His6 tag at their C termini. The series of RPTPσ fragments with mutations used in this study are listed in Table S3, and their schematic structures are shown in Fig. S1. The entire ECD of RPRPσ (amino acids 1–847) was also subcloned into EcoRI/EcoRV sites of pFUSE-hIgG1-Fc1 (InvivoGen) to express as an Fc fusion protein.
HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum and antibiotics. Cells cultured in T160 flasks were transiently transfected with 20 μg of AP/Fc fusion protein DNA plasmids with Avalanche®-Omni Transfection Reagent (EZ Biosystems) according to the manufacturer's protocol. Conditioned media were collected 72–96 h after transfection. Secreted recombinant proteins were purified by immobilized metal-affinity chromatography for AP fusion proteins (Profinity IMAC nickel-charged resin, Bio-Rad) and by Protein G-Sepharose 4 Fast Flow for Fc fusion proteins (GE Healthcare). Before the elution of the recombinant proteins, both columns were thoroughly washed with 1.5 m NaCl. Purified proteins were extensively dialyzed against HBS (10 mm Hepes, 150 mm NaCl, pH 7.3).
Preparation of GAGs for solid-phase binding assays
Heparin (porcine intestinal mucosa, H-3393), CS-A (bovine trachea, C-8529), CS-C (shark cartilage, C-4384), and dermatan sulfate (porcine intestinal mucosa, C-3788) were purchased from Sigma. CS-D (shark cartilage, 400676-1) and CS-E (squid cartilage, 400678-1) were obtained from Seikagaku (Japan). GAGs were biotinylated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and EZ-Link sulfo-NHS-LC-biotin (Thermo Scientific) according to a published procedure (26). The final concentration of GAGs was determined using a modification of the carbazole reaction (27). The degree of biotinylation in CS GAGs was monitored with 4′-hydroxyazobenzene-2-carboxylic acid (HABA) reagent (Thermo Scientific), and the modified GAGs with comparable degrees of biotinylation were used in this study.
Solid-phase binding assays
Solid-phase binding assays were performed in an ELISA format (6). Briefly, biotinylated GAGs were immobilized to Immulon 4 plates (Thermo Scientific) precoated with streptavidin (Life Technologies). AP fusion proteins were then incubated for 2 h at 4 °C, and AP activity of the bound fraction was measured in a microplate reader (SpectraMax190, Molecular Devices) at 37 °C under kinetics mode with BluePhos substrate (KPL). Data were expressed as Δmilli-OD/min. Kd and Bmax were calculated by Prism6 (GraphPad).
Sandwich ELISA
RPTPσ–Fc (50–100 nm) was incubated with Immulon 4 plates that were precoated with protein A (Life Technologies). One microgram of GAGs was incubated for 1.5 h at 4 °C followed by incubation with RPTPσ–AP (2 nm) for 1.5 h at 4 °C. After the rinse, the bound AP activity was measured as described under “Solid-phase binding assays.” All binding procedures were performed in HBS containing 0.1% Tween 20.
Heparin-affinity chromatography
Affinity chromatography was performed with a HiTrap Heparin HP column (1 ml, GE Healthcare Life Sciences) using HPLC (Hitachi L-7100) that was equilibrated with 10 mm Hepes buffer, pH 7.3, containing 150 mm NaCl and 0.1% Tween 20. Ten microliters of 10–20 nm AP fusion proteins were loaded onto the column and eluted with a linear gradient of NaCl (150–2000 mm) for 25 min in the same buffer at a flow rate of 0.3 ml/min. Eluted proteins were detected by the measurement of AP activity as described above.
Heparin protection assay
Potential heparin-binding sites were screened according to the published method with minor modification (11). Purified GFP–RPTPσ from 293 cells (3 nmol) was captured by HiTrap heparin beads. After acetylation of Lys and Arg residues exposed to the protein surface with sulfo-NHS-acetate, the modified protein was released from the heparin beads and subjected to treatment with NHS-biotin for selective labeling of Lys and Arg residues involved in the interaction with heparin. After digestion with chymotrypsin, biotinylated peptides were enriched by PierceTM Streptavidin Magnetic Beads, and eluted peptides were analyzed on an Eksigent NanoLC system connected to an Orbitrap Elite mass spectrometer (Thermo Scientific) equipped with a nanoelectrospray ion source. Peptides were loaded onto a peptide trap cartridge (Agilent Technologies) at a flow rate of 6 μl/min. The trapped peptides were fractionated with a reverse-phase PicoFrit column (New Objective, Woburn, MA) using a linear gradient of 5–35% acetonitrile in 0.1% formic acid. The gradient time was 45 min at a flow rate of 0.25 μl/min. Precursor mass spectra (MS1) were acquired in the Orbitrap at 60,000 resolution, and product mass spectra (MS2) were acquired with the ion trap.
GAG-induced clustering of AP fusion proteins
Purified AP fusion proteins were incubated with or without GAGs for 30 min at 4 °C. GAG–protein complexes were separated in HBS containing 0.1% Tween 20 with size-exclusion chromatography (TSK BioAssist G3SWxl, 7.8 × 300 mm, Tosoh Corp.) attached to a Shimadzu UFLC system at a flow rate of 0.5 ml/min. The AP activity of each fraction was measured as described above.
Detection of altered Tyr phosphorylation induced by GAG
293 cells cultured in collagen-coated 12-well plates were cotransfected with pGFPTAG5, pTAG5-RPTPσ, or its mutants (0.9 μg/well) together with pLNCX chick v-Src (a gift from Joan Brugge, Addgene plasmid number 14578, 0.1 μg/well). The media were replaced with DMEM without serum 30 h after transfection. Sixteen hours later, transfected cells were treated with GAGs for the indicated time. Cell lysates were prepared with 2× SDS sample buffer, and protein concentration was measured by a BCA protein assay kit (Thermo Fisher Scientific). Cell lysates were separated by 10% SDS-PAGE under reducing conditions followed by transfer to polyvinylidene difluoride membranes. After blocking, His6-tagged proteins were probed with rabbit Anti-6X His tag® antibody (Abcam, ab9108), and phosphorylated Tyr was probed with phospho-Tyr mouse mAb (Cell Signaling Technology, 9416). Bound antibodies were visualized with a myECLTM Imager (Thermo Fisher) using LumiGLO® Peroxidase Chemiluminescent Substrate kit (KPL). When polyvinylidene difluoride membranes were reprobed, bound antibodies were removed by RestoreTM Western Blotting Stripping Buffer (Thermo Fisher) following the manufacturer's protocol and reprobed with mouse anti-β-actin antibody (Sigma, A5316). To examine the effect of endogenous HSPGs on the cell surface, transfected cells as mentioned above were treated with 1 unit/ml heparinase III (Sigma, H8891) for 8 h in the absence of serum. After the rinse, cells were treated with heparin for 5 min, and cell lysates were collected to analyze tyrosine phosphorylation.
GAG analysis
CS GAGs were digested with chondroitinase ABC (Sigma, C3667), and the resulting disaccharides were labeled with the fluorescent tag 2-aminobenzamide, and disaccharide composition was analyzed with HPLC (28). Heparin composition analysis was performed by the University of California San Diego Glycotechnology Core. Size of GAGs used in this study was examined in 150 mm ammonium bicarbonate by size-exclusion chromatography (Superdex 200, 10 × 300 mm, GE Healthcare) attached to a Shimadzu UFLC system at a flow rate of 0.3 ml/min. The amount of uronic acid in each fraction was measured by the carbazole method (27).
Author contributions
Y. K., A. A. M., P. Y., N. J. B., and R. J. data curation; Y. K. and H. M. G. supervision; Y. K. investigation; Y. K. methodology; Y. K. writing-original draft; Y. K. project administration; Y. K. and H. M. G. writing-review and editing; P. Y. resources.
Supplementary Material
Acknowledgments
We thank K. Yagyu and S. Higashi for technical assistance. We thank Dr. Y. Takagi and C. Pearson for helpful discussion. Mass spectrometry was done in the National Heart, Lung, and Blood Institute Proteomics Core Facility (Drs. Marjan Gucek and Yong Chen).
This work was supported by the Division of Intramural Research, NHLBI, National Institutes of Health, and the Mizutani Foundation for Glycoscience. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5 and Tables S1–S3.
- GAG
- glycosaminoglycan
- HS
- heparan sulfate
- CS
- chondroitin sulfate
- CSPG
- chondroitin sulfate proteoglycan
- HSPG
- heparan sulfate proteoglycan
- RPTP
- receptor protein-tyrosine phosphatase
- FNIII
- fibronectin type III repeat
- ECD
- extracellular domain
- AP
- alkaline phosphatase
- Kd
- dissociation constant
- Bmax
- maximum number of binding sites.
References
- 1. Aricescu A. R., McKinnell I. W., Halfter W., and Stoker A. W. (2002) Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase σ. Mol. Cell. Biol. 22, 1881–1892 10.1128/MCB.22.6.1881-1892.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shen Y., Tenney A. P., Busch S. A., Horn K. P., Cuascut F. X., Liu K., He Z., Silver J., and Flanagan J. G. (2009) PTPσ is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 326, 592–596 10.1126/science.1178310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fox A. N., and Zinn K. (2005) The heparan sulfate proteoglycan syndecan is an in vivo ligand for the Drosophila LAR receptor tyrosine phosphatase. Curr. Biol. 15, 1701–1711 10.1016/j.cub.2005.08.035 [DOI] [PubMed] [Google Scholar]
- 4. Johnson K. G., Tenney A. P., Ghose A., Duckworth A. M., Higashi M. E., Parfitt K., Marcu O., Heslip T. R., Marsh J. L., Schwarz T. L., Flanagan J. G., and Van Vactor D. (2006) The HSPGs Syndecan and Dallylike bind the receptor phosphatase LAR and exert distinct effects on synaptic development. Neuron 49, 517–531 10.1016/j.neuron.2006.01.026 [DOI] [PubMed] [Google Scholar]
- 5. Fisher D., Xing B., Dill J., Li H., Hoang H. H., Zhao Z., Yang X. L., Bachoo R., Cannon S., Longo F. M., Sheng M., Silver J., and Li S. (2011) Leukocyte common antigen-related phosphatase is a functional receptor for chondroitin sulfate proteoglycan axon growth inhibitors. J. Neurosci. 31, 14051–14066 10.1523/JNEUROSCI.1737-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dickendesher T. L., Baldwin K. T., Mironova Y. A., Koriyama Y., Raiker S. J., Askew K. L., Wood A., Geoffroy C. G., Zheng B., Liepmann C. D., Katagiri Y., Benowitz L. I., Geller H. M., and Giger R. J. (2012) NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nat. Neurosci. 15, 703–712 10.1038/nn.3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coles C. H., Shen Y., Tenney A. P., Siebold C., Sutton G. C., Lu W., Gallagher J. T., Jones E. Y., Flanagan J. G., and Aricescu A. R. (2011) Proteoglycan-specific molecular switch for RPTPσ clustering and neuronal extension. Science 332, 484–488 10.1126/science.1200840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Biersmith B. H., Hammel M., Geisbrecht E. R., and Bouyain S. (2011) The immunoglobulin-like domains 1 and 2 of the protein tyrosine phosphatase LAR adopt an unusual horseshoe-like conformation. J. Mol. Biol. 408, 616–627 10.1016/j.jmb.2011.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doody K. M., Stanford S. M., Sacchetti C., Svensson M. N., Coles C. H., Mitakidis N., Kiosses W. B., Bartok B., Fos C., Cory E., Sah R. L., Liu-Bryan R., Boyle D. L., Arnett H. A., Mustelin T., et al. (2015) Targeting phosphatase-dependent proteoglycan switch for rheumatoid arthritis therapy. Sci. Transl. Med. 7, 288ra276 10.1126/scitranslmed.aaa4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brown J. M., Xia J., Zhuang B., Cho K. S., Rogers C. J., Gama C. I., Rawat M., Tully S. E., Uetani N., Mason D. E., Tremblay M. L., Peters E. C., Habuchi O., Chen D. F., and Hsieh-Wilson L. C. (2012) A sulfated carbohydrate epitope inhibits axon regeneration after injury. Proc. Natl. Acad. Sci. U.S.A. 109, 4768–4773 10.1073/pnas.1121318109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ori A., Free P., Courty J., Wilkinson M. C., and Fernig D. G. (2009) Identification of heparin-binding sites in proteins by selective labeling. Mol. Cell. Proteomics 8, 2256–2265 10.1074/mcp.M900031-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coles C. H., Mitakidis N., Zhang P., Elegheert J., Lu W., Stoker A. W., Nakagawa T., Craig A. M., Jones E. Y., and Aricescu A. R. (2014) Structural basis for extracellular cis and trans RPTPσ signal competition in synaptogenesis. Nat. Commun. 5, 5209 10.1038/ncomms6209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ohtake Y., Wong D., Abdul-Muneer P. M., Selzer M. E., and Li S. (2016) Two PTP receptors mediate CSPG inhibition by convergent and divergent signaling pathways in neurons. Sci. Rep. 6, 37152 10.1038/srep37152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Won S. Y., Kim C. Y., Kim D., Ko J., Um J. W., Lee S. B., Buck M., Kim E., Heo W. D., Lee J. O., and Kim H. M. (2017) LAR-RPTP clustering is modulated by competitive binding between synaptic adhesion partners and heparan sulfate. Front. Mol. Neurosci. 10, 327 10.3389/fnmol.2017.00327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krueger N. X., Reddy R. S., Johnson K., Bateman J., Kaufmann N., Scalice D., Van Vactor D., and Saito H. (2003) Functions of the ectodomain and cytoplasmic tyrosine phosphatase domains of receptor protein tyrosine phosphatase Dlar in vivo. Mol. Cell. Biol. 23, 6909–6921 10.1128/MCB.23.19.6909-6921.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee H. S., Ku B., Park T. H., Park H., Choi J. K., Chang K. T., Kim C. H., Ryu S. E., and Kim S. J. (2016) Identification of novel protein tyrosine phosphatase σ inhibitors promoting neurite extension. Bioorg. Med. Chem. Lett. 26, 87–93 10.1016/j.bmcl.2015.11.026 [DOI] [PubMed] [Google Scholar]
- 17. Yu P., Pisitkun T., Wang G., Wang R., Katagiri Y., Gucek M., Knepper M. A., and Geller H. M. (2013) Global analysis of neuronal phosphoproteome regulation by chondroitin sulfate proteoglycans. PLoS One 8, e59285 10.1371/journal.pone.0059285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yi J. H., Katagiri Y., Yu P., Lourie J., Bangayan N. J., Symes A. J., and Geller H. M. (2014) Receptor protein tyrosine phosphatase σ binds to neurons in the adult mouse brain. Exp. Neurol. 255, 12–18 10.1016/j.expneurol.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu P., Pearson C. S., and Geller H. M. (2018) Flexible roles for proteoglycan sulfation and receptor signaling. Trends Neurosci. 41, 47–61 10.1016/j.tins.2017.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thelin M. A., Bartolini B., Axelsson J., Gustafsson R., Tykesson E., Pera E., Oldberg Å., Maccarana M., and Malmstrom A. (2013) Biological functions of iduronic acid in chondroitin/dermatan sulfate. FEBS J. 280, 2431–2446 10.1111/febs.12214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takahashi H., Katayama K., Sohya K., Miyamoto H., Prasad T., Matsumoto Y., Ota M., Yasuda H., Tsumoto T., Aruga J., and Craig A. M. (2012) Selective control of inhibitory synapse development by Slitrk3-PTPδ trans-synaptic interaction. Nat. Neurosci. 15, 389–398 10.1038/nn.3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takahashi H., and Craig A. M. (2013) Protein tyrosine phosphatases PTPδ, PTPσ, and LAR: presynaptic hubs for synapse organization. Trends Neurosci. 36, 522–534 10.1016/j.tins.2013.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Woo J., Kwon S. K., Choi S., Kim S., Lee J. R., Dunah A. W., Sheng M., and Kim E. (2009) Trans-synaptic adhesion between NGL-3 and LAR regulates the formation of excitatory synapses. Nat. Neurosci. 12, 428–437 10.1038/nn.2279 [DOI] [PubMed] [Google Scholar]
- 24. Kwon S. K., Woo J., Kim S. Y., Kim H., and Kim E. (2010) Trans-synaptic adhesions between netrin-G ligand-3 (NGL-3) and receptor tyrosine phosphatases LAR, protein-tyrosine phosphatase δ (PTPδ), and PTPσ via specific domains regulate excitatory synapse formation. J. Biol. Chem. 285, 13966–13978 10.1074/jbc.M109.061127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takahashi H., Arstikaitis P., Prasad T., Bartlett T. E., Wang Y. T., Murphy T. H., and Craig A. M. (2011) Postsynaptic TrkC and presynaptic PTPσ function as a bidirectional excitatory synaptic organizing complex. Neuron 69, 287–303 10.1016/j.neuron.2010.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Briani C., Berger J. S., and Latov N. (1998) Antibodies to chondroitin sulfate C: a new detection assay and correlations with neurological diseases. J. Neuroimmunol. 84, 117–121 10.1016/S0165-5728(97)00209-9 [DOI] [PubMed] [Google Scholar]
- 27. Bitter T., and Muir H. M. (1962) A modified uronic acid carbazole reaction. Anal. Biochem. 4, 330–334 10.1016/0003-2697(62)90095-7 [DOI] [PubMed] [Google Scholar]
- 28. Wang H., Katagiri Y., McCann T. E., Unsworth E., Goldsmith P., Yu Z. X., Tan F., Santiago L., Mills E. M., Wang Y., Symes A. J., and Geller H. M. (2008) Chondroitin-4-sulfation negatively regulates axonal guidance and growth. J. Cell Sci. 121, 3083–3091 10.1242/jcs.032649 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.