

Abstract
BACKGROUND
Neuroendocrine tumors (NETs) can secrete bioactive amines into the bloodstream causing carcinoid syndrome (CS), with symptoms including flushing and diarrhea. However, CS frequency in the NET population has never been rigorously evaluated, nor has its relationship to presenting characteristics. This analysis assessed the proportion of NET patients with CS and associated clinical factors.
METHODS
We identified patients diagnosed 4/2000–12/2011 from the SEER-Medicare database, excluding those with pancreatic tumors or small cell/large cell lung cancer, as well as those without complete data. The incidence of patients with at least two claims of flushing (782·62), diarrhea (564·5, 787·91), or carcinoid syndrome (259·2) during the three months before and after NET diagnosis was assessed. We compared demographic and clinical characteristics between patients with and without CS using chi-squared tests. We used the Cochran-Armitage trend test to identify trends in CS incidence and cox regression to assess the relationship between CS and survival.
FINDINGS
Among 9,512 NET patients, 1,922 (19%) had CS. The proportion of NET patients with CS reported increased from 10·8% (50 of 465 patients) in 2000 to 18·6% (159 of 854 patients) in 2011 (p<0·0001). Patients with CS more likely had regional/distant than localized disease, grade 1/2 than grade 3/4 histology, and small bowel or cecal primary tumors rather than lung or colon. Female (p=0·00030) and non-Hispanic white patients (and p < 0·0001) more likely reported CS. CS was associated with shorter survival (HR 1·1, p = 0·017). Age and patient location were not correlated with the incidence of CS. Use of octreotide was more common in patients with CS, while use of chemotherapy and radiation were used more frequently in patients without CS and use of surgery was not significantly associated with the presence of CS at diagnosis.
INTERPRETATION
This population-based analysis reveals that CS is significantly associated with grade, stage, primary site, and shorter survival. An improved understanding of the heterogeneity of presenting syndromes among patients with NETs may permit more tailored evaluation and management, and enables future research regarding the impact of CS control on patient survival.
FUNDING
Supported by Ipsen, which sponsored purchasing SEER-Medicare data and funded analytical support. The company was not involved in data collection, analysis, or interpretation. This study was not directly funded by NIH.
Introduction
Neuroendocrine tumors (NETs) are relatively rare malignancies that can develop from a diffuse network of neuroendocrine cells throughout the body. Their incidence is increasing, with the most recent Surveillance, Epidemiology, and End Results (SEER) data showing a 5-fold increase from 1973 to 2004, with an annual incidence of 5·25/100,000 in the U.S. in 2004. This increase is likely multifactorial and related to improvements in detection and diagnosis, as well as a true rise in incidence.1 Given the indolent nature of well-differentiated NETs, many patients live over 5 years with metastatic disease, and must endure symptoms for significant periods of time.
Extrapancreatic NETs are historically known as carcinoids due to their “carcinoma-like” histology. These NETs can secrete bioactive amines causing carcinoid syndrome (CS).2 These symptoms include wheezing, skin flushing, diarrhea, and fibrotic valvular heart disease.3 Consistent with observations that serotonin is a product of the enterochromaffin cells4 considered the nonmalignant counterparts of gastrointestinal NETs, serotonin secretion is associated with CS in NET patients.5 Similarly, urinary excretion of the downstream serotonin metabolite, 5-hyrdoxyindolacetic acid, is elevated in patients with CS, more so than serum serotonin, which fluctuates significantly.6 The hormone secretion and associated symptoms can be reduced, but not eliminated, in most patients with somatostatin analogues such as octreotide.7–9
However, the frequency of CS among patients with NETs has not been systematically evaluated. While prior studies have attempted to estimate this frequency, the wide range identified (3–74%) suggests methodological limitations and the need to establish a more accurate incidence rate in the broader NET patient population.10–13 Indeed, prior analyses were small retrospective studies from single centers or relied on surveys, with potential ascertainment bias.
While the reported frequency of CS among NET patients has been inconsistent, the impact of CS on patient quality of life is clear.2,14–16 NET patients with CS demonstrate marked impairments in multiple areas including fatigue, general health, and physical function, compared both to the general population15,16 and to NET patients without overt CS,2 using metrics such as the PROMIS-29 and SF-36. Additionally, smaller studies have suggested that potentially due to subclinical niacin deficiency from tumor consumption of tryptophan to produce serotonin,6,17,18 even NET patients on therapeutic somatostatin analogues have modest subjective and objective cognitive impairments.19,20 Given the importance of understanding the symptom burden of this growing patient population, this study used SEER-Medicare, a large population-based database, to examine trends in the incidence of CS and associated symptoms. Demographic and clinical characteristics were compared between patients with and without CS to determine factors associated with having NETs and CS. Treatment selection and patient outcomes were also evaluated.
Materials and Methods
Data Source
The data source used for this study is the SEER registry linked with Medicare claims data. The SEER program of the National Cancer Institute (NCI) provides information on cancer statistics including tumor characteristics, patient demographics, and cause of death information for persons diagnosed with cancer in the United States. The SEER registries cover approximately 28% of the U.S. population and, when linked to Medicare claims data, the SEER-Medicare data provides detailed patient information such as neighborhood socioeconomic status (SES), comorbidities, and the usage of health care resources. Of note, the SEER registry uses 4 grades based on extraction of pathology reports without central review. Therefore, in cases where pathology reports did not explicitly describe tumors as WHO grade 1, 2, or 3, extraction based on morphologic description was performed, with SEER grade I including tumors classified as “well differentiated,” grade II including tumors classified as “moderately differentiated,” grade III including tumors classified as “poorly differentiated,” and grade IV including tumors classified as “undifferentiated,” or “anaplastic.” SEER grade III or IV cancers would be “grade 3 neuroendocrine carcinomas” in the 2010 WHO nomenclature, which is primarily based on division rate rather than histologic appearance.21 The SEER-Medicare database has been widely used is considered to be representative of the older U.S. population.22
Study Cohort
The study cohort consisted of 9,512 cancer patients over age 65 who were diagnosed with NETs between April 1, 2000 and December 31, 2011. NETs were identified by International Classification of Diseases for Oncology, 3rd Edition (ICD-O-3) codes: 8240, 8241, 8242, 8243, 8244, 8245, 8246, and 8249 (gastrointestinal and pulmonary carcinoids, and neuroendocrine carcinomas). Pancreatic NET patients were excluded due to overlapping symptoms with CS, as well as the lack of serotonin-producing cells in the normal pancreas. Patients less than 65 years old at the time of diagnosis were excluded, as they make up a minority of patients enrolled in Medicare are defined by specific qualifying conditions, therefore making these patients less representative of the overall population. We also required continuous Medicare Parts A and B enrollment (providing coverage for hospital and physician services) and no Health Maintenance Organization (HMO) enrollment during the time period between three months before and three months after the NET diagnosis to ensure consistent data availability and the completeness of medical claims used to identify CS.
Identification of CS
As previously described,23 we identified the presence of CS by International Classification of Disease 9th Revision (ICD-9) codes: flushing (782·62), diarrhea (564·5, 787·91), and carcinoid syndrome (259·2). We defined patients to have CS if they had at least two claims with any of the above mentioned ICD-9 codes during a six month time window between three months before and three months after the NET diagnosis. We therefore tightened the case ascertainment criterion for CS, increasing the specificity for an accurate diagnosis, but sacrificing sensitivity, potentially permitting the relatively rare patients who develop CS subsequently to go undetected. This concession was necessary to avoid confounding from coding for diarrhea or CS that arises as a complication of therapies, such as bowel resections or steatorrhea from somatostatin analogue therapy.
Demographic and clinical factors
The demographic information included age [65–69, 70–74, 75–79, ≥80], gender [male vs. female], race/ethnicity [non-Hispanic white, non-Hispanic black, Hispanics or all others], region [Northeast, West, Midwest, South], and urban/rural status [metropolitan vs. non-metropolitan]. Clinical factors included tumor grade, stage, and primary site, as well as treatment variables such as octreotide, chemotherapy, radiation, and surgery.
Statistical Analysis
We used the Cochran-Armitage trend test to evaluate the trend in CS. This method was chosen over parametric regression to avoid the assumption that the change over time could be described with a simple function. We used the chi-square test to compare demographic and clinical characteristics between patients with and without CS, as well as the frequency with which interventions were performed in patients with and without CS and the proportion of patients experiencing CS by grade, stage, and primary site. Multivariate Cox regression analysis was used to identify prognostic variables, with plots of log of the cumulative hazards function versus log of survival time employed to ensure that the proportional hazards assumption was satisfied (Supplementary Figure 1, pages 2–14). Multivariate logistic regression was performed to determine the use of specific interventions in patients with versus without CS while controlling for other explanatory variables. Median survival was estimated using the Kaplan-Meier method.
All statistical analyses were conducted in SAS 9·3 (SAS Institute, Cary NC). The Institutional Review Board at The University of Texas MD Anderson Cancer Center exempted this study for approval because all patients were de-identified.
Role of the funding source
The sponsor had no involvement in the study design, data collection, analysis, or interpretation, and was furthermore not involved in the writing of the report. DMH, CS, AD, YX, YC, SZ, YTS, and JCY all had access to all raw data. The corresponding author had full access to all of the data and the final responsibility to submit for publication.
Results
Overall frequency of reported CS
Figure 1 depicts the selection criteria by which the NET cohort was generated, resulting in 9,512 patients for analysis. The proportion of newly diagnosed NET patients also diagnosed with CS or its component symptoms was assessed for each year between 2000 and 2011 (Figure 2). Among the 9,512 patients diagnosed with NETs over this period, 1,784 (18·8%) manifested CS in the six months surrounding the diagnosis of NET. The proportion of patients diagnosed with CS increased over this time period from 10·8% (50 of 465 patients) in 2000 to 18·6% (159 of 854) in 2011 (Figure 1, p<0·0001 by Cochran-Armitage test).
Figure 1.
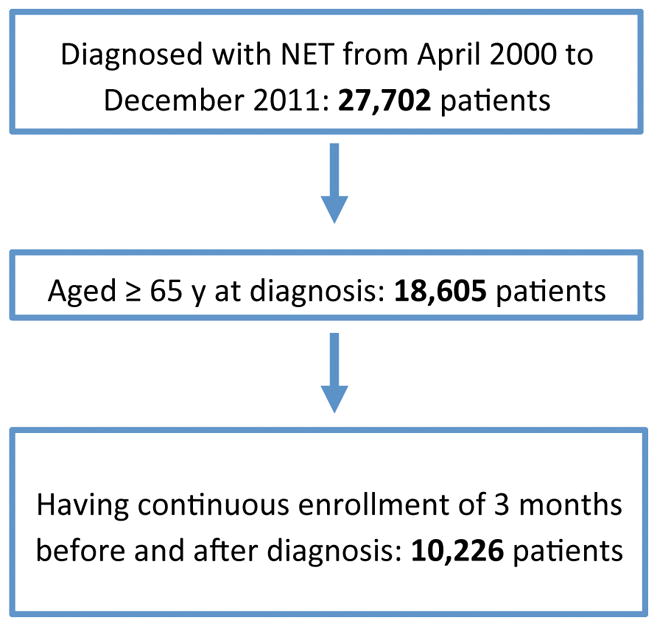
Creation of a NET cohort using SEER-Medicare data from 2000–2011.
Figure 2. Incidence of total NETs and carcinoid syndrome, 2000–2011.

Upper Panel. Incidence of total diagnosed NETs and carcinoid syndrome. Lower Panel. Percentage of patients with incident NETs diagnosed with carcinoid syndrome.
CS and demographics
Table 1 contains patient characteristics that were evaluated for association with the diagnosis of CS. The percentage of patients with CS as compared to those patients without did not differ significantly with respect to age at diagnosis, geographic region, or metropolitan vs. rural status. Interestingly, female patients were more likely to have CS diagnosed (1091 of 5449 [20%] compared to 695 of 4063 [17·1%] for males; p=0·00030). Lastly, race/ethnicity was associated with a significant difference in the reported incidence of CS. CS occurred in 1483 (19·8%) of 7501 non-Hispanic white, 166 (16·3%) of 1016 non-Hispanic black, and 137 (13·8%) of 995 Hispanic or other race cases. These differences were highly statistically significant (p<0·0001).
Table 1. Patient characteristics and treatments associated with carcinoid syndrome.
Number (percent) of patients each group identified as manifesting carcinoid syndrome. For treatment selections below double line, percentage reflects proportion of patients with/without carcinoid syndrome for whom the treatment was selected.
| Covariates | With Syndrome | Without Syndrome | P |
|---|---|---|---|
| Age | 0.652 | ||
| <70 | 498 (18.52%) | 2191 (81.48%) | |
| 70–74 | 451 (18.29%) | 2015 (81.71%) | |
| 75–79 | 391 (18.74%) | 1696 (81.26%) | |
| >=80 | 446 (19.65%) | 1824 (80.35%) | |
| Gender | 0.0003 | ||
| male | 695 (17.11%) | 3368 (82.89%) | |
| female | 1091 (20.02%) | 4358 (79.98%) | |
| Race | <.0001 | ||
| Non-Hispanic White | 1483 (19.77%) | 6018 (80.23%) | |
| Non-Hispanic Black | 166 (16.34%) | 850 (83.66%) | |
| Hispanic or Others | 137 (13.77%) | 858 (86.23%) | |
| Stage | <.0001 | ||
| In Situ | Masked* | Masked* | |
| Localized | 473 (11.93%) | 3492 (88.07%) | |
| Regional | 397 (21.95%) | 1412 (78.05%) | |
| Distant | 515 (24.87%) | 1556 (75.13%) | |
| Unstaged or Unknown | 397 (24.02%) | 1256 (75.98%) | |
| Histological Grade | <.0001 | ||
| G1 | 1302 (22.55%) | 4472 (77.45%) | |
| G2 | 114 (17.51%) | 537 (82.49%) | |
| G3 | 77 (8.20%) | 862 (91.80%) | |
| G4 | 18 (5.14%) | 332 (94.86%) | |
| Unknown | 237 (15.29%) | 1313 (84.71%) | |
| Mixed Histology | 38 (15.32%) | 210 (84.68%) | |
| Region | 0.054 | ||
| Midwest | 214 (18.18%) | 963 (81.82%) | |
| Northeast | 408 (20.96%) | 1539 (79.04%) | |
| South | 453 (18.13%) | 2045 (81.87%) | |
| West | 711 (18.28%) | 3179 (81.72%) | |
| Site | <.0001 | ||
| APP | 29 (16.67%) | 145 (83.33%) | |
| CEC | 96 (32.21%) | 202 (67.79%) | |
| COL | 170 (11.47%) | 1312 (88.53%) | |
| LUN | 229 (7.63%) | 2773 (92.37%) | |
| OTH | 541 (24.08%) | 1706 (75.92%) | |
| SMB | 717 (32.43%) | 1494 (67.57%) | |
| Urban/Rural Status | 0.534 | ||
| metropolitan | 1518 (18.88%) | 6521 (81.12%) | |
| non-metropolitan | 268 (18.19%) | 1205 (81.81%) | |
|
| |||
| Octreotide Treatment | <.0001 | ||
| Yes | 465 (82.45%) | 99 (17.55%) | |
| No | 1321 (14.76%) | 7627 (85.24%) | |
| Chemotherapy | <.0001 | ||
| Yes | 284 (15.05%) | 1603 (84.95%) | |
| No | 1502 (19.70%) | 6123 (80.30%) | |
| Radiotherapy | <.0001 | ||
| Yes | 84 (8.79%) | 872 (91.21%) | |
| No | 1702 (19.89%) | 6854 (80.11%) | |
| Surgery | 0.226 | ||
| Yes | 992 (18.35%) | 4413 (81.65%) | |
| No | 794 (19.33%) | 3313 (80.67%) | |
Masked per SEER Medicare user agreement for confidentiality.
APP: Appendix; CEC: Cecum; COL: Colon or rectum; LUN: Lung, bronchus, larynx, trachea or other respiratory organ; OTH: Other; SMB: Duodenum, jejunum, ileum.
CS and NET grade
The proportion of patients exhibiting symptoms of CS was assessed in relation to tumor grade. Well-differentiated tumors of grade 1 or 2 histology were associated with CS in 22·6% (1302 of 5774) and 17·5% (114 of 651) of cases, respectively (Table 1). Poorly differentiated tumors of grade 3 or 4 histology were associated with CS in 8·2% (77 of 939) and 5·1% (18 of 350) of cases, respectively. This difference was highly statistically significant (p<0·0001).
CS and NET stage
Using SEER staging, the proportion of NETs manifesting CS was evaluated in relation to extent of disease. Localized NETs were associated with CS in 11·9% (473 of 3965) of cases, whereas regionally advanced and distant metastatic NETs were associated with carcinoid syndrome in 22% (397 of 1809) and 24·9% (515 of 2071) of cases, respectively. This difference was highly statistically significant (p<0·0001).
CS and NET primary site
Consistent with prior SEER analyses, the most frequently observed primary sites for NET were respiratory organs, small bowel, and colorectal. CS was closely associated with primary site (p<0·0001). CS was present in 717 (32·4%) of 2211 small bowel and 170 (32·2%) of 1482 cecal NETs. Conversely, CS was present in 229 (7·6%) of 3002 pulmonary NETs, 170 (11·5%) of 1482 colon NETs, and 29 (16·7%) of 174 appendix NETs.
Integrating grade, stage, and primary site into one analysis yielded consistent data with each of these component analyses. This analysis (Table 2) reveals that well-differentiated tumors produced CS with greater frequency in more advanced disease, within each primary site grouping (p<0·0001).
Table 2. Frequency of carcinoid syndrome in patients with well-differentiated NETs.
Proportion of patients with carcinoid syndrome with a histologic diagnosis of G1–2 NET, grouped by primary site and SEER stage.
| localized | regional | distant | |
|---|---|---|---|
| Appendix | Masked* | Masked* | Masked* |
| Cecum | Masked* | 38/113 (33.63%) | 28/54 (51.85%) |
| Colon | 78/945 (8.25%) | 14/54 (25.93%) | 26/75 (34.67%) |
| Larynx, bronchus, lung, trachea and other respiratory organs | 83/1044 (7.95%) | 19/239 (7.95%) | 30/196 (15.31%) |
| Others | 102/604 (16.89%) | 16/62 (25.81%) | 52/100 (52%) |
| Small intestine | 155/817 (18.97%) | 248/670 (37.01%) | 242/436 (55.5%) |
Masked per SEER Medicare user agreement for confidentiality.
Association with survival
Consistent with prior analyses, Cox regression revealed grade, stage, and primary site to be significantly associated with overall survival. With a median of 5·5 years (95% CI 5·3 – 5·7, IQR 1·4- not reached) of follow-up, 1012 of 1786 patients with CS died, as did 4209 of the 7726 patients without CS. CS was independently associated with shorter survival (Figure 3a), with median overall survival of CS patients being 5·6 years (95% CI 5·4–5·9) and median overall survival for patients without CS being 5 years (95% CI 4·5–5·4), (HR 1·1 [95% CI 1·02–1·2, p=0·017]). To further explore this observation, we evaluated the prognostic importance of CS in the subset of patients with metastatic grade 1–2 small bowel NET (Figure 3b). With a median of 5·3 years (95% CI, 4·8 – 6·4, IQR 2·4 – not reached) of follow-up, 144 of 242 patients with CS died, as did 103 of 194 patients without CS. The difference in overall survival was even more striking, with a median overall survival of 7·1 years (95% CI 5·2–8·1) in patients without CS and 4·7 years (95% CI 4–5·4) in patients with CS (p=0·013), with similar differences in disease-specific survival (p=0·0060, data not shown). These survival durations are consistent with the median overall survival of 56 months for the complete population of patients with metastatic small bowel NETs previously observed.1
Figure 3. Overall survival of patients diagnosed with NET, 2000–2011.


Overall survival of the entire NET cohort (a) and specifically for the subgroup of patients with metastatic grade 1–2 small bowel NETs (b).
Treatment selection
While the use of therapy over longer periods of time is limited by significant data loss, we explored the treatments employed in the three months following diagnosis. Uncontrolled comparison of treatment use (Table 1) suggests more frequent octreotide use and surgery in patients with CS, and less frequent radiation and chemotherapy. In a multivariate logistic regression analysis controlling for grade, stage, and primary site, patients with CS received significantly different treatments than patients without CS during this time period (Table 4). Use of the FDA-approved somatostatin analogue for CS control, octreotide LAR, was instituted within three months of diagnosis in more patients with CS (OR 18·5, 95% CI 14·5–23·7, p < 0·0001). Chemotherapy use was more common in CS patients (OR 1·31, 95% CI 1·09–1·57, p=0·0030), while primary tumor resection was less frequently performed (OR 0·82, 95% CI 0·71–0·95, p=0·0090), and radiotherapy (including both external beam radiotherapy and selective internal radiotherapy) trended toward less frequent use among patients with CS (OR 0·79, 95% CI 0·61–1·02, p=0·073).
Table 4. Treatment utilization in NET patients with and without carcinoid syndrome.
The use of surgery, radiotherapy, chemotherapy, and octreotide was assessed during the 3 months following NET diagnosis using multivariate logistic regression analysis incorporating other potential explanatory variables.
| Table 4a. Treatment use in the 3 months following NET diagnosis (Octreotide and Chemotherapy)
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Octreotide | Chemotherapy | |||||||
|
|
|
|||||||
| OR | 95% OR CI | P | OR | 95% OR CI | P | |||
|
|
|
|||||||
| Lower Limit | Upper Limit | Lower Limit | Upper Limit | |||||
| Age | ||||||||
| 65–69 | 1 | 1 | ||||||
| 70–74 | 0.954 | 0.72 | 1.263 | 0.5593 | 1.029 | 0.865 | 1.223 | <.0001 |
| 75–79 | 0.888 | 0.661 | 1.192 | 0.8343 | 0.862 | 0.718 | 1.034 | 0.1997 |
| >=80 | 0.793 | 0.593 | 1.061 | 0.1559 | 0.461 | 0.382 | 0.557 | <.0001 |
| Gender | ||||||||
| Male | 1 | 1 | ||||||
| Female | 0.893 | 0.724 | 1.101 | 0.2881 | 0.829 | 0.728 | 0.944 | 0.0046 |
| Race | ||||||||
| Non-Hispanic White | 1 | 1 | ||||||
| Non-Hispanic Black | 0.777 | 0.534 | 1.131 | 0.9127 | 0.717 | 0.564 | 0.913 | 0.0113 |
| Hispanic or Others | 0.576 | 0.372 | 0.892 | 0.0718 | 1.004 | 0.799 | 1.263 | 0.1817 |
| Histological Stage | ||||||||
| Localized | 1 | 1 | ||||||
| Regional | 2.168 | 1.382 | 3.403 | 0.0002 | 3.163 | 2.562 | 3.904 | 0.0194 |
| Distant | 14.22 | 9.792 | 20.651 | <.0001 | 6.777 | 5.567 | 8.252 | <.0001 |
| Unstaged or Unknown | 5.119 | 3.477 | 7.535 | 0.0012 | 2.583 | 2.042 | 3.268 | 0.4647 |
| Grade | ||||||||
| G1 | 1 | 1 | ||||||
| G2 | 1.342 | 0.885 | 2.034 | <.0001 | 2.871 | 2.209 | 3.732 | <.0001 |
| G3 | 0.167 | 0.081 | 0.343 | 0.0182 | 13.242 | 10.78 | 16.267 | <.0001 |
| G4 | 0.122 | 0.029 | 0.518 | 0.0685 | 18.073 | 13.544 | 24.115 | <.0001 |
| Mixed | 1.087 | 0.824 | 1.434 | <.0001 | 8.779 | 7.28 | 10.588 | <.0001 |
| Unknown | 0.11 | 0.02 | 0.601 | 0.0882 | 6.974 | 4.373 | 11.123 | 0.3879 |
| Carcinoid Syndrome | ||||||||
| Without Syndrome | 1 | 1 | ||||||
| With Syndrome | 18.538 | 14.484 | 23.727 | <.0001 | 1.312 | 1.096 | 1.57 | 0.0031 |
| Region | ||||||||
| Midwest | 1 | 1 | ||||||
| Northeast | 1.027 | 0.708 | 1.491 | 0.9778 | 1.144 | 0.895 | 1.462 | 0.3778 |
| South | 1.046 | 0.728 | 1.503 | 0.83 | 1.142 | 0.904 | 1.441 | 0.3669 |
| West | 1.024 | 0.727 | 1.443 | 0.9993 | 1.044 | 0.834 | 1.307 | 0.522 |
| Site | ||||||||
| LUN | 1 | 1 | ||||||
| APP | 3.508 | 0.927 | 13.282 | 0.3664 | 0.372 | 0.201 | 0.688 | 0.1804 |
| CEC | 1.586 | 0.858 | 2.932 | 0.2376 | 0.401 | 0.275 | 0.585 | 0.0943 |
| COL | 1.473 | 0.852 | 2.546 | 0.0981 | 0.564 | 0.444 | 0.717 | 0.4943 |
| OTH | 5.524 | 3.722 | 8.198 | <.0001 | 0.89 | 0.739 | 1.071 | <.0001 |
| SMB | 2.075 | 1.413 | 3.049 | 0.859 | 0.277 | 0.215 | 0.355 | <.0001 |
| Urban-Rural Status | ||||||||
| Non-Metropolitan | 1 | 1 | ||||||
| Metropolitan | 1.112 | 0.819 | 1.51 | 0.4945 | 1.062 | 0.878 | 1.285 | 0.5365 |
| Table 4b Treatment use in the 3 months following NET diagnosis (Radiotherapy and Surgery)
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Radiotherapy | Resection of primary tumors | |||||||
|
|
|
|||||||
| OR | 95% OR CI | P | OR | 95% OR CI | P | |||
|
|
|
|||||||
| Lower Limit | Upper Limit | Lower Limit | Upper Limit | |||||
| Age | ||||||||
| 65–69 | 1 | 1 | ||||||
| 70–74 | 1.066 | 0.865 | 1.315 | 0.3877 | 0.902 | 0.778 | 1.046 | 0.0025 |
| 75–79 | 0.969 | 0.776 | 1.21 | 0.5747 | 0.734 | 0.628 | 0.857 | 0.2083 |
| >=80 | 0.998 | 0.802 | 1.243 | 0.8917 | 0.563 | 0.484 | 0.656 | <.0001 |
| Gender | ||||||||
| Male | 1 | 1 | ||||||
| Female | 1.041 | 0.89 | 1.217 | 0.6141 | 1.197 | 1.07 | 1.338 | 0.0016 |
| Race | ||||||||
| Non-Hispanic White | 1 | 1 | ||||||
| Non-Hispanic Black | 1.066 | 0.808 | 1.405 | 0.2679 | 0.577 | 0.481 | 0.693 | 0.0004 |
| Hispanic or Others | 0.807 | 0.603 | 1.078 | 0.1213 | 0.674 | 0.56 | 0.812 | 0.2337 |
| Histological Stage | ||||||||
| Localized | 1 | 1 | ||||||
| Regional | 3.418 | 2.631 | 4.441 | <.0001 | 1.11 | 0.943 | 1.308 | <.0001 |
| Distant | 3.176 | 2.474 | 4.076 | <.0001 | 0.181 | 0.155 | 0.211 | <.0001 |
| Unstaged or Unknown | 2.039 | 1.508 | 2.757 | 0.5062 | 0.048 | 0.04 | 0.058 | <.0001 |
| Grade | ||||||||
| G1 | 1 | 1 | ||||||
| G2 | 2.022 | 1.417 | 2.887 | 0.0002 | 1.558 | 1.253 | 1.937 | <.0001 |
| G3 | 6.334 | 4.89 | 8.205 | <.0001 | 0.701 | 0.583 | 0.843 | 0.2954 |
| G4 | 6.341 | 4.572 | 8.794 | <.0001 | 0.637 | 0.481 | 0.843 | 0.1296 |
| Mixed | 5.843 | 4.575 | 7.462 | <.0001 | 0.202 | 0.166 | 0.245 | <.0001 |
| Unknown | 3.838 | 1.838 | 8.013 | 0.7595 | 1.44 | 0.867 | 2.391 | 0.0035 |
| Carcinoid Syndrome | ||||||||
| Without Syndrome | 1 | 1 | ||||||
| With Syndrome | 0.789 | 0.609 | 1.021 | 0.0718 | 0.82 | 0.707 | 0.952 | 0.0092 |
| Region | ||||||||
| Midwest | 1 | 1 | ||||||
| Northeast | 0.903 | 0.674 | 1.21 | 0.3207 | 1.174 | 0.961 | 1.433 | 0.0029 |
| South | 1.054 | 0.801 | 1.386 | 0.2806 | 0.866 | 0.718 | 1.045 | 0.0051 |
| West | 0.952 | 0.729 | 1.242 | 0.6979 | 0.978 | 0.816 | 1.173 | 0.6499 |
| Site | ||||||||
| LUN | 1 | 1 | ||||||
| APP | 0.026 | 0.003 | 0.208 | 0.037 | 7.981 | 3.932 | 16.2 | 0.007 |
| CEC | 0.057 | 0.021 | 0.155 | 0.02 | 10.407 | 7.051 | 15.359 | <.0001 |
| COL | 0.298 | 0.215 | 0.412 | 0.012 | 3.898 | 3.265 | 4.653 | 0.3383 |
| OTH | 0.55 | 0.443 | 0.682 | <.0001 | 1.253 | 1.075 | 1.46 | <.0001 |
| SMB | 0.082 | 0.05 | 0.135 | 0.015 | 5.063 | 4.282 | 5.986 | <.0001 |
| Urban-Rural Status | ||||||||
| Non-Metropolitan | 1 | 1 | ||||||
| Metropolitan | 1.094 | 0.87 | 1.375 | 0.4433 | 1.031 | 0.88 | 1.208 | 0.7048 |
Discussion
In this review of approximately 10,000 incident cases of NETs in patients 65 years of age and older diagnosed over more than a decade, the aggregate proportion of patients manifesting CS was 18·8%, with a range from 7·6% to 32·4% in lung and small bowel NETs, respectively. To the best of our knowledge, this work represents the first comprehensive epidemiologic analysis of the incidence of carcinoid syndrome among newly diagnosed patients with NETs. Since nearly 1 in every 5 patients with NETs has CS, ameliorating the symptoms should be an important aspect of patient care. Additionally, our data regarding worse survival among patients with CS suggest that controlling hormonal secretion may be of even more vital importance than previously appreciated. Previous studies on CS used small samples from referral centers to estimate that CS is present in between 3–74% in NET patients at diagnosis. This large range of functional NET percentages needed refinement, and the 19% incidence rate identified in this study is much more precise. The proportion of patients being diagnosed with CS increased over the assessed time period, potentially reflecting increasing diagnostic accuracy among clinicians as the awareness and prevalence of NETs has risen. While prior work has established the incidence of NETs to be increasing overall, the incidence has not risen substantially between 2003–2011 in the specific population included in our dataset. This lack of continued increase could be related to changes in the true occurrence of invasive cancer or achievement of maximum saturation with imaging tests that detect tumors in this population. Furthermore, our analysis revealed increased incidence rates of CS diagnosis in females and in non-Hispanic whites, which could be related to improved awareness and diagnosis in these populations.
Interestingly, while this study confirms that CS is associated with advanced disease, it revealed a surprisingly high incidence of CS in patients with localized and regional disease, particularly among patients with small bowel NET, with CS in 19% and 37% of patients with local and regional disease, respectively. These data suggest that unappreciated hepatic metastases or CS in locoregional disease may more common than previously reported.24–26 Additionally, we report a surprisingly high frequency of CS among patients with colorectal NET, likely reflecting the imprecision of this classification, which bridges midgut and hindgut, each of which would be expected a priori to have different frequencies of CS. Finally, lung NETs demonstrated a surprisingly low frequency of CS diagnosis given that secreted products bypass liver clearance, perhaps reflecting atypical syndromic presentations.27
In addition to these fundamental observations about the frequency of CS and patient demographics from a large population of NET patients, several important correlations between CS and clinical tumor characteristics also became evident. Consistent with long-held clinical impressions in the neuroendocrine field, CS was more common in well-differentiated tumors arising from midgut organs, and in patients with higher disease burden, likely from the release of serotonin into the post-hepatic circulation. While these relationships have long been suspected, this study presents the first large-scale population analysis to support them.
Additionally, this analysis reveals differences in treatment patterns among patients with and without CS. Patients with CS were more likely to receive octreotide or chemotherapy in the three months following diagnosis, whereas patients without CS were more likely to undergo primary tumor resection. Radiotherapy trended toward less frequent use in patients with CS, likely because external beam radiotherapy is recommended for some patients with high-grade neuroendocrine carcinomas who less commonly manifest CS, whereas selective internal radiotherapy is commonly used to reduce the symptom burden of CS in patients with well-differentiated tumors. With the exception of radiotherapy, which is used commonly for patients with high-grade neuroendocrine carcinomas, more aggressive therapy was used for patients with CS. These findings are somewhat unexpected, given that CS was most frequently observed in chemo-insensitive small bowel NETs, but appear to be influenced significantly by the use of chemotherapy in lung NETs, particularly of intermediate grade, which is recommended by some specialty societies.28
Perhaps most intriguingly, this study reveals the first evidence to our knowledge that CS is associated with overall survival, irrespective of known clinical prognostic variables such as grade, stage, and primary site. While the relative magnitude of the difference is modest in the overall population, the absolute difference is more substantial, given the long clinical course of these tumors. Particularly striking is the absolute magnitude of the difference in patients with metastatic well-differentiated small bowel NETs, which was over two years at the median. While the presence of CS may reflect unmeasured differences in tumor burden, it remains possible that CS reflects a biological difference in tumor aggression. The impact of CS control independent of tumor control on patient survival similarly remains an open question.
There are several limitations to our dataset that must be considered in its interpretation. Given the reliance on Medicare claims data, this analysis is restricted to older adults, which may limit its generalizability. As prior data showed a median age at NET diagnosis of 63, this analysis should capture a substantial proportion of patients diagnosed with NET, but there are some patient subpopulations, namely appendiceal and thymic NET, with younger median ages at diagnosis (48 and 56, respectively) that may lead to relative underrepresentation in this dataset.1 As the comorbidity frequency among older patients often differs from that of younger patients, there might be competing causes in this population for diarrhea and flushing which could, in some cases, lead to misdiagnosis of CS. Therefore, it remains possible that results would better reflect the general population if we had data for a broader group of patients. Additionally, these data are limited by the quality of coding on which the analysis is based. Any errors in diagnosis or coding could negatively impact the accuracy of our analysis, and information regarding liver function, which impacts clearance of tumor-secreted bioactive amines, or quantitation of tumor burden, impacting the amount of hormone secretion, is similarly unavailable from claims data. Furthermore, while carcinoid heart disease has a diagnostic code in ICD10, there was no such specific code in ICD9, making it impossible to measure carcinoid heart disease as an endpoint or cause of death. Coding accuracy is of particular interest with respect to SEER vs. WHO grading, as extraction of grade from pathologic reports adds the category of “grade IV” neuroendocrine cancers, which are not a part of the standard WHO nomenclature. As has previously been performed1,23, we collapsed SEER grade III and IV cancers into one category for these analyses.
Quantitation of tumor hormone secretion on a patient level is also absent. Given that both sensitivity and specificity for hormonal syndromes would be limited with the coding query we performed, pancreatic NETs were excluded from this analysis. We therefore have not estimated the frequency of overt functional syndromes in that patient population.
Carcinoid syndrome remains a cause of significant symptom burden for patients with NETs and impacts patient quality of life. This work also suggests that it may impact overall survival, and is associated with use of costly chemotherapy and somatostatin analogues in the three months following diagnosis. As prior work has demonstrated, the incidence of these tumors continues to rise. Given recent improvements in NET treatment, a greater number of patients live with NETs for longer periods of time. Therefore, understanding the incidence of CS is critical toward efforts to design studies to test interventions that may improve the lives of affected patients. Furthermore, appreciating the true frequency of CS is necessary to estimate the burden of disease and appreciate the need for innovations in this field, so that resources can be appropriately dedicated to relieving the symptom burden of NET patients.
Supplementary Material
Table 3. Cox Regression Model of Overall Survival in Patients diagnosed with NET, 2000–2011.
Multivariate Cox regression including carcinoid syndrome diagnosis and known prognostic variables was performed to evaluate association with overall survival. APP: Appendix; CEC: Cecum; COL: Colon or rectum; LUN: Lung, bronchus, larynx, trachea and other respiratory organs; OTH: Other; SMB: Duodenum, jejunum, ileum.
| Entire Cohort | ||||
|---|---|---|---|---|
|
| ||||
| HR | 95% HR CI | P | ||
|
| ||||
| Lower Limit | Upper Limit | |||
| Age | ||||
| 65–69 | 1 | |||
| 70–74 | 1.213 | 1.118 | 1.316 | <.0001 |
| 75–79 | 1.426 | 1.313 | 1.548 | <.0001 |
| >=80 | 2.246 | 2.077 | 2.428 | <.0001 |
| Gender | ||||
| Male | 1 | |||
| Female | 0.779 | 0.736 | 0.824 | <.0001 |
| Race | ||||
| Non-Hispanic White | 1 | |||
| Non-Hispanic Black | 1.175 | 1.07 | 1.291 | 0.0008 |
| Hispanic or Others | 0.947 | 0.855 | 1.049 | 0.2964 |
| Histological Stage | ||||
| Localized | 1 | |||
| Regional | 1.527 | 1.397 | 1.668 | <.0001 |
| Distant | 2.769 | 2.543 | 3.016 | <.0001 |
| Unstaged or Unknown | 1.265 | 1.146 | 1.397 | <.0001 |
| Grade | ||||
| G1 | 1 | |||
| G2 | 1.383 | 1.228 | 1.557 | <.0001 |
| G3 | 2.692 | 2.441 | 2.968 | <.0001 |
| G4 | 2.344 | 2.043 | 2.688 | <.0001 |
| Mixed | 2.04 | 1.872 | 2.222 | <.0001 |
| Unknown | 1.497 | 1.173 | 1.911 | 0.0012 |
| Carcinoid Syndrome | ||||
| Without Syndrome | 1 | |||
| With Syndrome | 1.102 | 1.016 | 1.194 | 0.0186 |
| Chemotherapy | ||||
| No | 1 | |||
| Yes | 1.386 | 1.285 | 1.495 | <.0001 |
| Radiotherapy | ||||
| No | 1 | |||
| Yes | 1.189 | 1.091 | 1.294 | <.0001 |
| Resection | ||||
| No | 1 | |||
| Yes | 0.607 | 0.564 | 0.655 | <.0001 |
| Octreotide | ||||
| No | 1 | |||
| Yes | 1.119 | 0.993 | 1.26 | 0.0643 |
| Region | ||||
| Midwest | 1 | |||
| Northeast | 0.882 | 0.798 | 0.976 | 0.0147 |
| South | 0.999 | 0.909 | 1.097 | 0.9766 |
| West | 0.9 | 0.822 | 0.986 | 0.023 |
| Site | ||||
| LUN | 1 | |||
| APP | 0.931 | 0.682 | 1.271 | 0.654 |
| CEC | 0.928 | 0.784 | 1.097 | 0.3797 |
| COL | 0.823 | 0.742 | 0.914 | 0.0003 |
| OTH | 1.156 | 1.066 | 1.253 | 0.0005 |
| SMB | 0.746 | 0.679 | 0.821 | <.0001 |
| Urban-Rural Status | ||||
| Non-Metropolitan | 1 | |||
| Metropolitan | 0.964 | 0.891 | 1.044 | 0.3661 |
Research in Context.
Evidence before this study
A Pubmed search of MeSH terms “Malignant Carcinoid Syndrome/epidemiology” and “Carcinoid Tumor” revealed 44 publications from 1/1/2000–10/15/2016. Two included over 150 patients. One study included 2,001 patients and the other included 3,379 pNET patients and 8,088 GI-NET patients. Carcinoid syndrome was estimated to occur in 3·2% of patients in the larger study using retrospective questionnaires and in 21·4% of patients in the other study.
Added value of this study
This study used prospective databases to identify approximately 10,000 extrapancreatic NET patients and used claims data to identify those patients suffering from carcinoid syndrome. It is the largest, most rigorous analysis of the epidemiology of carcinoid syndrome and associated clinicopathological factors.
Implications of all the available evidence
Nearly 20% of NET patients have carcinoid syndrome, which is associated with grade, stage, and primary tumor site. Carcinoid syndrome is associated with survival. This work highlights the prognostic importance of prioritizing the diagnosis of carcinoid syndrome in patients with NETs, and raises potential future research questions regarding the survival benefit of carcinoid syndrome control.
Acknowledgments
This study used the linked SEER-Medicare database. The interpretation and reporting of these data are the sole responsibility of the authors. The authors acknowledge the efforts of the Applied Research Program, NCI; the Office of Research, Development and Information, CMS; Information Management Services (IMS), Inc.; and the Surveillance, Epidemiology, and End Results (SEER) Program tumor registries in the creation of the SEER-Medicare database.
Footnotes
Authors’ Contributions
DMH contributed conception and design, data analysis and interpretation, manuscript writing, and final manuscript approval. CS contributed conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, and final manuscript approval. AD contributed conception and design, manuscript writing, and final manuscript approval. YX contributed conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, and final manuscript approval. YC contributed data analysis and interpretation, manuscript writing, and final manuscript approval. SZ contributed data analysis and interpretation, manuscript writing, and final manuscript approval. YTS contributed conception and design, data analysis and interpretation, manuscript writing, and final manuscript approval. JCY contributed conception and design, data analysis and interpretation, manuscript writing, and final manuscript approval.
Declaration of interests
Dr. Halperin reports grants from NCI, during the conduct of the study. Dr. Shen has nothing to disclose. Dr. Dasari has nothing to disclose. Dr. Xu has nothing to disclose. Ms. Chu has nothing to disclose. Dr. Zhou has nothing to disclose. Dr. Shih has nothing to disclose. Dr. Yao reports grants from NCI, during the conduct of the study.
Previously presented at ASCO Annual Meeting in Chicago, IL, June 2016.
References
- 1.Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–72. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 2.Beaumont JL, Cella D, Phan AT, Choi S, Liu Z, Yao JC. Comparison of health-related quality of life in patients with neuroendocrine tumors with quality of life in the general US population. Pancreas. 2012;41(3):461–6. doi: 10.1097/MPA.0b013e3182328045. [DOI] [PubMed] [Google Scholar]
- 3.Davis Z, Moertel CG, McIlrath DC. The malignant carcinoid syndrome. Surgery, gynecology & obstetrics. 1973;137(4):637–44. [PubMed] [Google Scholar]
- 4.Erspamer V, Asero B. Identification of enteramine, the specific hormone of the enterochromaffin cell system, as 5-hydroxytryptamine. Nature. 1952;169(4306):800–1. doi: 10.1038/169800b0. [DOI] [PubMed] [Google Scholar]
- 5.Lembeck F. 5-Hydroxytryptamine in a Carcinoid Tumour. Nature. 1953;172:910–1. [Google Scholar]
- 6.Udenfriend S, Weissbach H, Sjoerdsma A. Studies on tryptophan and serotonin in patients with malignant carcinoid. Science (New York, NY) 1956;123(3199):669. doi: 10.1126/science.123.3199.669. [DOI] [PubMed] [Google Scholar]
- 7.Kvols LK, Martin JK, Marsh HM, Moertel CG. Rapid reversal of carcinoid crisis with a somatostatin analogue. N Engl J Med. 1985;313(19):1229–30. doi: 10.1056/NEJM198511073131915. [DOI] [PubMed] [Google Scholar]
- 8.Kvols LK, Moertel CG, O’Connell MJ, Schutt AJ, Rubin J, Hahn RG. Treatment of the malignant carcinoid syndrome. Evaluation of a long-acting somatostatin analogue. N Engl J Med. 1986;315(11):663–6. doi: 10.1056/NEJM198609113151102. [DOI] [PubMed] [Google Scholar]
- 9.Rubin J, Ajani J, Schirmer W, et al. Octreotide acetate long-acting formulation versus open-label subcutaneous octreotide acetate in malignant carcinoid syndrome. J Clin Oncol. 1999;17(2):600–6. doi: 10.1200/JCO.1999.17.2.600. [DOI] [PubMed] [Google Scholar]
- 10.Janson ET, Holmberg L, Stridsberg M, et al. Carcinoid tumors: analysis of prognostic factors and survival in 301 patients from a referral center. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 1997;8(7):685–90. doi: 10.1023/a:1008215730767. [DOI] [PubMed] [Google Scholar]
- 11.Landerholm K, Falkmer S, Jarhult J. Epidemiology of small bowel carcinoids in a defined population. World J Surg. 2010;34(7):1500–5. doi: 10.1007/s00268-010-0519-z. [DOI] [PubMed] [Google Scholar]
- 12.Ito T, Igarashi H, Nakamura K, et al. Epidemiological trends of pancreatic and gastrointestinal neuroendocrine tumors in Japan: a nationwide survey analysis. J Gastroenterol. 2015;50(1):58–64. doi: 10.1007/s00535-014-0934-2. [DOI] [PubMed] [Google Scholar]
- 13.Polikarpova SB, Lubimova NV, Ogereliev AS, Britvin TA, Davidov MI. Clinical and biochemical aspects of the carcinoid syndrome in neuroendocrine tumors of the abdominal and retroperitoneal organs and its impact for the disease prognosis. Bulletin of experimental biology and medicine. 2009;148(5):803–6. doi: 10.1007/s10517-010-0821-7. [DOI] [PubMed] [Google Scholar]
- 14.Pearman TP, Beaumont JL, Cella D, Neary MP, Yao J. Health-related quality of life in patients with neuroendocrine tumors: an investigation of treatment type, disease status, and symptom burden. Support Care Cancer. 2016 doi: 10.1007/s00520-016-3189-z. [DOI] [PubMed] [Google Scholar]
- 15.Frojd C, Larsson G, Lampic C, von Essen L. Health related quality of life and psychosocial function among patients with carcinoid tumours. A longitudinal, prospective, and comparative study. Health Qual Life Outcomes. 2007;5:18. doi: 10.1186/1477-7525-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haugland T, Vatn MH, Veenstra M, Wahl AK, Natvig GK. Health related quality of life in patients with neuroendocrine tumors compared with the general Norwegian population. Quality of life research: an international journal of quality of life aspects of treatment, care and rehabilitation. 2009;18(6):719–26. doi: 10.1007/s11136-009-9487-x. [DOI] [PubMed] [Google Scholar]
- 17.Fleischmajer R, Hyman AB. Clinical significance of derangements of tryptophan metabolism. A review of pellagra, carcinoid and H disease. Archives of dermatology. 1961;84:563–73. doi: 10.1001/archderm.1961.01580160027003. [DOI] [PubMed] [Google Scholar]
- 18.Green RF. Subclinical pellagra and idiopathic hypogeusia. JAMA: the journal of the American Medical Association. 1971;218(8):1303. [PubMed] [Google Scholar]
- 19.Chambers AJ, Longman RS, Pasieka JL, et al. Impairment of cognitive function reported by patients suffering from carcinoid syndrome. World J Surg. 2010;34(6):1356–60. doi: 10.1007/s00268-010-0404-9. [DOI] [PubMed] [Google Scholar]
- 20.Pasieka JL, Longman RS, Chambers AJ, Rorstad O, Rach-Longman K, Dixon E. Cognitive impairment associated with carcinoid syndrome. Ann Surg. 2014;259(2):355–9. doi: 10.1097/SLA.0b013e318288ff6d. [DOI] [PubMed] [Google Scholar]
- 21.Pasaoglu E, Dursun N, Ozyalvacli G, Hacihasanoglu E, Behzatoglu K, Calay O. Comparison of World Health Organization 2000/2004 and World Health Organization 2010 classifications for gastrointestinal and pancreatic neuroendocrine tumors. Ann Diagn Pathol. 2015;19(2):81–7. doi: 10.1016/j.anndiagpath.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Warren JL, Klabunde CN, Schrag D, Bach PB, Riley GF. Overview of the SEER-Medicare data: content, research applications, and generalizability to the United States elderly population. Medical care. 2002;40(8 Suppl):IV-3–18. doi: 10.1097/01.MLR.0000020942.47004.03. [DOI] [PubMed] [Google Scholar]
- 23.Shen C, Shih YC, Xu Y, Yao JC. Octreotide long-acting repeatable among elderly patients with neuroendocrine tumors: a survival analysis of SEER-Medicare data. Cancer Epidemiol Biomarkers Prev. 2015;24(11):1656–65. doi: 10.1158/1055-9965.EPI-15-0336. [DOI] [PubMed] [Google Scholar]
- 24.Datta S, Williams N, Suortamo S, et al. Carcinoid syndrome from small bowel endocrine carcinoma in the absence of hepatic metastasis. Age Ageing. 2011;40(6):760–2. doi: 10.1093/ageing/afr122. [DOI] [PubMed] [Google Scholar]
- 25.Feldman JM, Jones RS. Carcinoid syndrome from gastrointestinal carcinoids without liver metastasis. Ann Surg. 1982;196(1):33–7. doi: 10.1097/00000658-198207000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haq AU, Yook CR, Hiremath V, Kasimis BS. Carcinoid syndrome in the absence of liver metastasis: a case report and review of literature. Med Pediatr Oncol. 1992;20(3):221–3. doi: 10.1002/mpo.2950200307. [DOI] [PubMed] [Google Scholar]
- 27.Ahlman H, Wangberg B, Nilsson O, et al. Aspects on diagnosis and treatment of the foregut carcinoid syndrome. Scandinavian journal of gastroenterology. 1992;27(6):459–71. doi: 10.3109/00365529209000106. [DOI] [PubMed] [Google Scholar]
- 28.Kalemkerian GP, Loo BW, Jr, Akerley W, et al. Small cell lung cancer, version 2.2017. Journal of the National Comprehensive Cancer Network: JNCCN. 2016 doi: 10.6004/jnccn.2013.0084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.