

Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disease affecting over 5 million individuals worldwide. The exact molecular events underlying PD pathogenesis are still not clearly known. Glia maturation factor (GMF), a neuroinflammatory protein in the brain plays an important role in the pathogenesis of PD. Mitochondrial dysfunctions and oxidative stress trigger apoptosis leading to dopaminergic neuronal degeneration in PD. Peroxisome proliferator activated receptor-gamma coactivator-1 alpha (PGC-1α or PPARGC-α) acts as a transcriptional co-regulator of mitochondrial biogenesis and energy metabolism by controlling oxidative phosphorylation, antioxidant activity and autophagy. In this study, we found that incubation of immortalized rat dopaminergic (N27) neurons with GMF influences the expression of peroxisome PGC-1α and increases oxidative stress, mitochondrial dysfunction and apoptotic cell death. We show that incubation with GMF reduces the expression of PGC-1α with concomitant decreases in the mitochondrial complexes. Besides, there is increased oxidative stress and depolarization of mitochondrial membrane potential (MMP) in these cells. Further, GMF reduces tyrosine hydroxylase (TH) expression and shifts Bax/Bcl-2 expression resulting in release of cytochrome-c, and increased activations of effector caspases expressions. Transmission electron microscopy analyses revealed alteration in the mitochondrial architecture. Our results show that GMF acts as an important upstream regulator of PGC-1α in promoting dopaminergic neuronal death through its effect on oxidative stress mediated apoptosis. Our current data suggest that GMF is a critical risk factor for PD and suggest that it could be explored as a potential therapeutic target to inhibit PD progression.
Keywords: Apoptosis, mitochondrial, dysfunctions, neurodegeneration, oxidative stress, Parkinson’s disease
Graphical Abstract
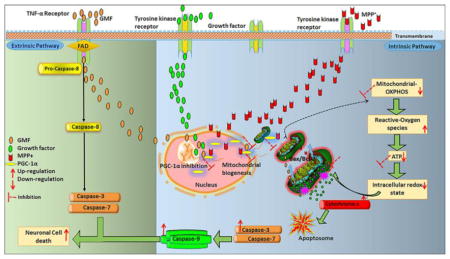
Introduction
Parkinson’s disease (PD), the second most common age dependent chronic neurodegenerative disease after Alzheimer’s disease (AD), arises due to continuous depletion of type-A9-dopaminergic neurons in nigro-striatal pathway. The resultant well-known motor decline symptoms are collectively termed as TRAP comprising Tremor, Rigidity Akinesia and Postural instability in patients [1,2]. Though the exact cause of dopaminergic loss in PD is unknown, various genetic and environmental factors and abnormalities in protein processing, oxidative stress, mitochondrial dysfunction, excito-toxicity, inflammation, immune regulation, and other mechanisms have been reported to contribute to the pathogenesis of PD [3–6].
Mitochondria strongly determines the cellular fate of dopaminergic neurons in either contributing to neuroprotection or neuronal depletion under various intracellular stress conditions [7]. Modulations in mitochondrial physiology directly triggers oxidative stress by increasing reactive oxygen species (ROS). Increased accumulation of intracellular ROS, one of the major factors to induce dopaminergic neuronal loss impairs the mitochondrial function [8]. Neurotoxic downstream regulation of ROS leads to disruption of mitochondrial permeability, causing translocation of Bax/Bcl-2 expressions, increased intracellular cytochrome-c level and subsequent activation of caspase-8, a prime participant in apoptosis signaling cascades that in turn leads to apoptotic cell loss [9].
There are at least two pathways responsible for neuropathological progression in dopaminergic neuronal cells: (a) misfolded protein formation, aggregation and aberrant proteolytic processing and (b) mitochondrial dysfunction with oxidative/nitrosative flux [10,6]. Therefore, inhibition of mitochondrial dysfunctions and oxidative stress mediated apoptotic cascade represent the prime targets to halt the progression of PD. Elevated intracellular oxidative flux, mitochondrial release of cytochrome-c activate other “pro-apoptotic mediators,” such as apoptosis-initiating factor (AIF) into the cytoplasm that triggers a cascade of events, culminating in neuronal death [11,12]. However, little is known about factors that regulate mitochondrial biogenesis and respiration in the dopaminergic neurons of vertebrates, under various physiological and pathological conditions.
Intracellular factors have the ability to reduce oxidative flux by modulating the mitochondrial biogenesis and respiration and are considered as novel therapeutic targets for age dependent chronic neurodegenerative diseases, especially for PD. Recent evidences from meta-analysis implicate transcriptional regulators in neurodegenerative diseases that function by regulating mitochondrial dysfunction [13,14]. Approximately, 1000 mitochondrial proteins are encoded by nuclear transcriptional regulatory genes which control mitochondrial membrane potential and oxidative fluxes, ATP production, mitochondrial fission and fusion [15]. Peroxisome proliferator activated receptor gamma coactivator-1 (PGC-1) family of transcriptional co-activators consists of PGC-1α, PGC-1β and PGC-1 related coactivator (PRC) that enhance the activities of the nuclear respiratory factors NRF1 and NRF2 and induce transactivation of many genes encoding mitochondrial biogenesis-specific proteins involved in the respiratory chain, mitochondrial DNA transcription/replication and protein import/assembly [16–18]. PGC-1α is a master regulator of cellular metabolism, controlling the expression of nuclear regulatory genes implicated in mitochondrial biogenesis, respiration and resistance to oxidative stress [19]. PGC-1α quantitatively and qualitatively neutralizes both the regulation of mitochondrial biogenesis and adenosine triphosphate (ATP) mediated increase in the expression of ROS detoxifying enzymes [20]. PGC-1α and its family of networks regulate a wide array of mitochondrial metabolic functions, enabling them to act as global orchestrators of remodeling of intracellular energy and metabolism.
Glia maturation factor (GMF), a highly conserved neuro-inflammatory acidic protein in the brain that was previously discovered, isolated, sequenced and cloned in our laboratory [21–23]. GMF contains no leader sequence normally found in secreted proteins and is confined to the cells except after cell damage. The intracellular localization of GMF leaves room for the possibility that GMF might have a dual function: an intracellular function in intact cells and an extracellular activity when released after injury. Over the last several years, the intracellular role of GMF has begun to surface. First, we reported that recombinant GMF can be phosphorylated by at least four serine/threonine protein kinases (PKA, PKC, CKII and RSK), all of which are members of signal transduction chains. We next reported that endogenous GMF in cultured cells is rapidly phosphorylated upon external stimulation such as by phorbol ester [24]. We also found that in a test-tube assay, PKA-phosphorylated GMF stimulates the p38 MAP kinase pathway, implicating GMF in the regulation of signal transduction. However, under certain stress conditions such as oxidative stress GMF could be overexpressed and exert its toxic effect on neuronal cells [25]. Additionally, our previous studies have successfully demonstrated shown that GMF expression is implicated in the pathogenesis of AD, PD and MS [26,27]. Further, we have shown that there are increased levels of GMF in the brains of human subjects with AD, PD, Multiple Sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE) [27–29]. Moreover, overexpression of astrocytic-GMF leads to depletion of primary oligodendrocyte myelin genesis in the brain, by interactions between astrocytes, microglia, and oligodendrocytes. These above mentioned findings suggest that GMF-initiates cytotoxicity and neuroinflammatory pathways in neurodegenerative diseases [23]. Our unpublished data show that GMF binds to astrocytes and C6 glioma cells with a high affinity (Kd of 1 × 10-9 M). The binding is saturable, reversible, and is competed by GMF but not by other proteins. Moreover, it has been reported by Ito et al [30] that GMF interacts with the glial cell membrane and their data showed the presence of a cell surface receptor for GMF on glioblasts and the reduction of the thiol groups on GMF promotes binding to its receptor. Also, our recent understanding would support a non- clathrin- mediated, caveolar endocytosis as has been proposed for toxins and growth factors [31] that may be regulated by syntaxin. GMF has been shown to modulate beta-1 integrin recycling [32] to the plasma membrane and binds syntaxin. Our transfection studies showed that overexpression of GMF in brain cells leads to the activation of the stress-related transduction cascades involving p38 MAP kinase and NF-kB. Because of its association with p38 and NF-kB, we postulate that intracellular GMF is involved in stress-induced signal transduction.
Rat mesenchymal dopaminergic cells (1RB3AN27-abbreviated as N27 cells) are directly derived from the mesencephalon region, which is affected in PD. We hypothesized that GMF interferes with PGC-1α expression and oxidative flux, which in turn results in mitochondrial dysfunction-mediated apoptotic cell death. Here we show that GMF treatment induces oxidative stress mediated apoptotic cascade by inhibiting PGC-1α expression in N27 dopaminergic cells. We propose that GMF-mediated regulation of PGC-1α in mitochondrial dynamics determines dopaminergic neuronal survival in neuropathological conditions and therefore could serve as a potential therapeutic target to inhibit PD progression.
Material and Methods
Glia Maturation Factor
GMF was recombinant human protein produced in Escherichia coli as described earlier [21].
Rat Dopaminergic Neuronal (N27) cell Culture
Rat mesencephalic dopaminergic N27 cells were grown in RPMI-1640 (GIBCO, Life Technologies, Grand Island, NY) medium supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO), 1% L-glutamine, penicillin (10 U/ml) and streptomycin (10 U/ml; GIBCO). The cells were seeded at a density of 0.5×106 in a 75-cm2 tissue culture flask (Corning, New York, NY) and incubated at 37°C under saturating humidity in 5% CO2/95% air [33,34]. The doubling time of N27 cells was ~26 h.
Incubation of N27 cells with GMF and MPP+
N27 cells were grown as mentioned above to confluency. Cells were incubated for up to 24 h with 300 μM of MPP+ (dissolved in Dulbecco’s phosphate-buffered saline (DPBS), Life technologies), an active metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [35] or were stimulated with different concentrations of GMF (50, 100 and 200 ng/ml). Post GMF/MPP+ treatment, cells were trypsinized and collected for glutathione peroxidase (GPx), superoxide dismutase (SOD) and ROS assays. Cell lysates were prepared for Western blotting and apoptotic markers expression analysis. Protein concentration of the cell lysates was determined using the bicinchoninic acid assay (BCA) protein assay kit (Thermo Scientific, Waltham, MA) as per the manufacturer’s instructions.
MTT Reduction assay of neuronal viability
The cell viability 3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (MTT) assay was performed with slight modifications of the methods as previously described [36–38]. The viable cells with active mitochondria reduce the colorless tetrazolium salt MTT, producing solid blue water insoluble formazan crystals. MTT was dissolved at a concentration of 5 mg/ml in phosphate buffered saline (PBS) to perform cell viability assay. Exactly 2 h prior to the end of the experiment, the MTT solution (50 μl per well) was added to 24-well plates and the plates were returned to the incubator. Following the 2 h incubation period, the medium was decanted and the formazan precipitates were solubilized with acid isopropanol (0.04 – 0.1 N HCl in absolute isopropanol). The absorbance was measured on a microplate reader (Molecular Devices; Sunnyvale, CA) at a wavelength of 570 nm with background subtraction at 630 nm. The absorbance of the untreated cultures was set as 100%.
LDH Release Assay of Neuronal Cytotoxicity
The amount of lactate dehydrogenase (LDH) released into the culture medium upon cell lysis was measured by the conversion of a tetrazolium salt into red formazan product according to manufacturer’s instructions (Cayman Chemical, Ann Arbor, MI.; LDH kit No: 601170). The absorbance, proportional to the lysed cells was measured at 490 nm. The amount of LDH released by the cells in the presence of 1 % Triton X-100 was considered as maximal absorbance [38,39].
Oxidative Stress Markers: Determination of Oxidants, Antioxidants and Reactive Oxygen Species (ROS)
N27 cells (1×106 cells/flask in 8 ml medium) were seeded in a six well tissue culture plate (1×105cells/ml), followed by incubation with GMF and/MPP+. After the incubation period, the cells were collected and centrifuged at 2000 rpm for 10 min at 4°C. Cells were lysed at 4 °C with radio immunoprecipitation assay (RIPA) cell lysis buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, 1 mM EDTA, 1 % NP-40, 0.5 % deoxycholate) containing protease inhibitors (Roche Diagnostics, Indianapolis, IN) and phosphatase inhibitor cocktails (Cell Signaling Technology, Danvers, MA). The cell lysate was used for further biochemical analysis.
Cellular GPx (kit No. 703102) and SOD (kit No. 706002) were determined according to the kit manufacturer’s instructions (Cayman Chemical). The activity of GPx was calculated as micromoles of GSH formed per milligram protein, using a molar extinction coefficient of 13.6×103 M−1 cm−1. The SOD results were expressed as U1/micromoles of SOD released per milligram protein.
The levels of intracellular ROS was measured according to manufacturer’s instruction (Abcam, Cambridge, MA; kit No: 113851) using a cell-permeant fluorogenic - probe, 2, 7-diacetyl dichlorofluorescein (DCFH-DA). Inside the cell, the dye is deacetylated by cellular esterases, then oxidized by ROS and converted into fluorescent dichlorofluorescein (DCF). After the experimental period, ROS formation was determined by fluorescence microscopy [38,40].
Determination of Apoptosis by Dual Staining
The dual staining with Acridine orange (AO) and ethidium bromide (EtBr) for DNA was performed to visualize condensed chromatin of dead apoptotic cells as reported previously [41,42]. Dual stained cells were viewed under a fluorescence microscope (ELWD 0.3, Nikon Inc. Japan). The number of cells showing features of apoptosis was counted as percentage of the total number of cells present in the field.
Determination of Mitochondrial Membrane Potential (MMP or ΔΨm)
The alterations in ΔΨm in different treatment groups was observed microscopically, using the fluorescent dye rhodamine-123 (Rh-123), as previously described [42,43]. Briefly, after incubation with compounds for 24 h, 1 μL of fluorescent dye Rh-123 (5mmol/l) was added to the cells and incubated for 15 min at 37°C. The cells were then washed with PBS, observed under fluorescence microscope by using blue filter (450–490 nm) (Olympus BX60 fluorescence microscope). Polarized mitochondria emit orange red fluorescence and depolarized mitochondria emit green fluorescence.
Determination of Intracellular Adenosine Triphosphate (ATP) Levels
N27 cells were grow in T25 cm2 cell culture flasks (1×106). Briefly, after the incubation period with the compounds the cells were collected and washed with cold PBS. Cell lysates were prepared from treated cells and the samples were used to measure intracellular ATP by using the ATP assay kit (Cat No. ab83355; Abcam) according to the manufacturer’s instructions [44].
Determination of Mitochondrial Morphology
The mitochondrial morphology was determined in N27 cells after the incubation with or without GMF. The cells were stained with MitoTracker green CMXRos (Molecular probes, Life technologies, Eugene, OR; Cat No: M7512) according to the manufacturer’s instructions and imaged by using fluorescent microscope [45].
Western Blotting
Briefly, N27 cells were seeded in T25 cm2 cell culture flasks and allowed to grow to required confluency (about 65% confluency). Then the cells were incubated with GMF and MPP+ for 24 h and harvested by trypsinization and washed with PBS. Cells were lysed with RIPA cell lysis buffer containing protease inhibitors and phosphatase inhibitor cocktails. Protein concentration was measured in the lysates as previously mentioned. Lysates were loaded into 2–14% NuPAGE Tris-Glycine gradient gel (Invitrogen, Life technologies, Carlsbad, CA). An equal amount of protein (ranging from 20–35 μg) loaded per lane along with a lane containing pre-stained protein markers (Invitrogen). The separated proteins were blotted onto PVDF/nitrocellulose membrane by wet protein transfer system (Life technologies). After blocking with 5% nonfat milk/bovine serum albumin in Tris-Buffered saline-tween20 (TBS-T) for 1 h, the membranes were then incubated with various antibodies: anti-PGC-α (Cat No. ab106814), mitochondrial complexes (anti-ATP5A; Cat No. ab108327, anti-UQCRC2; Cat No. ab14745), anti-SDHB(Cat No. ab14714), anti-NDUFB8 (Cat No. ab110242), anti-cytochrome c (Cat No. ab110325), anti-caspase 8 (Cat No. ab32397), anti-Bax (Cat No. ab32503; purchased from abcam, Inc), anti-Bcl-2 (Cat No. sc-492; SCBT, Dallas, TX),, anti-caspase 3 (Cat No.CST 9662), anti-caspase 7 (Cat No. CST12827), and anti-caspase 9 (Cat No. CST 9508; all purchased from Cell Signaling Tech), and β-actin (Cat No. A1978; Sigma). The following dilutions were used for PGC-α (1:500), mitochondrial complexes (ATP5A (1:1000), NDUFB8 (1:800), SDHB (1:3000), UQCRC-2 (1:000) and MTCO1 (1:500), Bcl-2 and Bax (1:500), cytochrome-c, caspase 3 and 9 (1:600), and β-actin (1:2000) diluted in 5% BSA in TBS-T or PBS-T. After the primary antibodies incubation, the membranes were incubated with appropriate secondary antibodies at a concentration of 1:2000 dilution. Then, the membranes were washed with Tris-buffered saline with 0.05% Tween-20 (TBST) thrice for 5 min interval. The bands were visualized by treating the membranes with ECL prime western blotting detection reagent (Cat No. RPN2232; Amersham, GE healthcare Life Sciences, Pittsburgh, PA) or SuperSignal West Pico PLUS Substrate (Cat No. 34580; ThermoScientific). Blots were stripped and re-probed for β-actin (Sigma) as a loading control. Densitometry was performed by using ChemiDoc-It2 Imaging System (UVP LLC, Upland, CA) analysis software.
Immunocytochemistry
N27 cells were cultivated on glass coverslips pre-coated with Poly-D-Lysine (Millipore, Billerica, MA). The cells were fixed with 4% paraformaldehyde for 1 h and permeabilized by incubation in PBS/0.1% Triton X-100 (PBST) for 15 min. Then the cells were rinsed three times in PBS, blocked with 5% normal goat serum (NGS) for 30 min and finally incubated for 2 h at room temperature with anti-PGC-1α (1:500 dilution) and tyrosine hydroxylase (TH; 1:1000) primary antibody. The cells were then incubated with goat anti-rabbit IgG conjugated with green fluorescent dye Alexa Fluor 488 secondary antibody (cat no. A-11001, ThermoFisher scientific) for 1 h at room temperature. The coverslips were stained with VECTASHIELD Antifade Mounting Medium with DAPI (Vector laboratories) to stain DNA. Finally, the coverslips were mounted with Vinol (Sigma) onto microscope slides and cells were examined under a Nikon fluorescent microscope.
Confocal imaging was performed on a Leica TCP SP8 laser scanning confocal microscope with a 405-nm diode laser and tunable super continuum white light laser using 63X oil immersion objective (Molecular cytology core facility, University of Missouri). Briefly, after the treatment with GMF/MPP+, N27 cells were incubated with fixative (4% paraformaldehyde) for 1 h at 4 °C and permeabilized by incubation in PBST for 15 min. Then the cells were rinsed three times in PBS, blocked with 5% normal goat serum (NGS) for 30 min and finally incubated for 2 h at room temperature with anti-PGC-1α (1:500 dilution). PGC-1α (green) was visualized with goat anti-rabbit IgG conjugated with green fluorescent dye Alexa Fluor 488. After PGC-1α immunostaining cells were incubated with MitoTracker Red CMXRos (1mM) for 20 min and finally counterstained with VECTASHIELD Antifade Mounting Medium with DAPI. The following excitation/emission band-pass wavelengths were used: 405/420–480 nm (DAPI), 495/505–550 nm (Alexa Fluor 488) and 570/580–630 nm (Alexa Fluor 568).
Transmission Electron Microscopy
For transmission electron microscopy, cells were grown on gold-coated sapphire discs (3 mm; Wohlwend GmbH, Switzerland). The cells were cryo-immobilized by high-pressure freezing using the Wohlwend HPF Compact 02 and freeze substituted in acetone containing 0.1% uranyl acetate, 0.5% osmium tetroxide, 0.1% imidazole, and 4% of water. Freeze substitution was performed in a metal block warming up from liquid nitrogen to room temperature over a period of 2.5 h. After embedding the samples in Epon/Araldite, 75nm thin sections were prepared and mounted on formvar/carbon coated copper grids for transmission electron microscopy. Thick and ultrathin sections were generated using a Leica UC6 ultra microtome (Leeds Precision Instruments, Minneapolis, MN). Samples were imaged with a JEOL 1400 transmission electron microscope at an acceleration voltage of 80 kV (Electron Microscopy core facility, University of Missouri, Columbia).
Statistical Analysis
The results were analyzed using GraphPad InStat 3 statistical software. Mean ± SD was calculated and analyzed using One-way Analysis of Variance (ANOVA) followed by Tukey-Kramer multiple comparison tests to determine statistically significant differences between the groups. Results were expressed as mean ± SD for four experiments in each group. p values < 0.05 were considered significant.
Results
GMF Treatment Causes Neuronal Loss Brought about by Oxidative Stress in N27 Cells
To determine whether or not GMF treatment induces oxidative stress, N27 cells were incubated with different concentrations of GMF (50, 100 and 200 ng/ml) for 24 h. The results with LDH release (Fig. 1) shows that GMF treatment caused significant cell death but does not seem to cause cell necrosis. Also, neuronal injury determined by MTT reduction showed a concentration-dependent increase (Fig. 1), demonstrating that while GMF is not highly cytotoxic it does inhibit the mitochondrial activity of the cells. Based on this result, a dose of 100 ng/ml of GMF was chosen for all further studies. GMF significantly decreased the activities of the antioxidant enzymes; GPx and SOD (Fig. 2A and 2B) and increased ROS accumulation (Fig. 3) as compared to the control cells (*p<0.05). The formation of intracellular ROS was measured in terms of the green fluorescence of DCF. GMF significantly increased the green fluorescence intensity in comparison to control cells as shown in Fig. 3. Additionally, N27 cells incubated with both GMF and MPP+(300μM) significantly elevated the activities of GPx and SOD, and produced increased levels of ROS (*p<0.05). These findings demonstrate that GMF treatment induced intracellular oxidative flux that led to changes in cell viability.
Fig. 1.

Effect of GMF on dopaminergic N27 cells viability and proliferation. The dose (0, 50, 100 and 200 ng/ml) dependent effect of GMF toxicity was determined by MTT reduction and LDH release. Different doses of GMF significantly reduced cell viability, and increased LDH release of N27 cells when incubated for 24 h. Values are presented as mean ± SD of four experiments in each group. The maximum of above 50% inhibition concentration value was found to be 100 ng/ml. *indicates significance compared to control cells.
Fig. 2.

GMF potentiates the neurotoxicity through oxidative stress indices in N27 cells. Incubation of N27 cells with GMF (100 ng/ml) for 24 h significantly reduced the activities of GPx (A) and SOD (B) as compared control cells. Incubation of N27 cells with both GMF and MPP+ (300 μm) significantly downregulates the antioxidant status by reduce GPx, and SOD activities when compared with cells incubated only with GMF or MPP+. Values are given as mean ± SD. of four experiments in each group. SOD units-enzyme concentration required for 50% inhibition of nitro blue tetrazolium reduction in 1 min. GPx units-micrograms of glutathione consumed/minute. *p< 0.05 compared to control, and *p< 0.05 compared to GMF treated group.
Fig. 3.
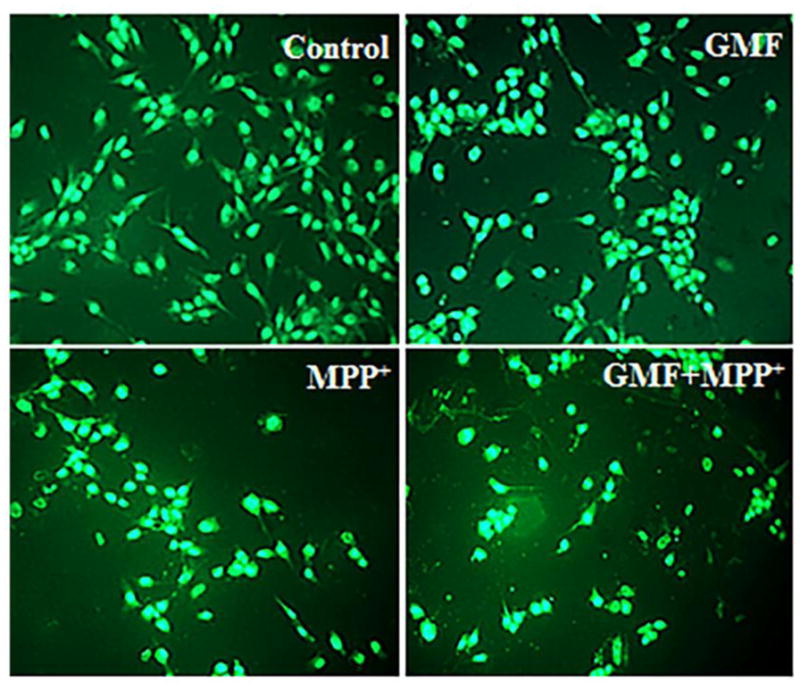
Effect of GMF on ROS expression in dopaminergic N27 cells. N27 cells were seeded (3×106) in 96 well plate and incubated with GMF (100 ng/ml) and/or MPP+ (300 μm) for 24 h. After the incubation period cells washed with PBS and stained with DCFDA green flourescent dye for 35 mins. Microphotographs showing the toxic putative effect of GMF induced ROS generation (green flourescence) by DCFDA staining (200X). The optimum dose of GMF treatment significantly increased the levels of ROS (similar to MPP+ treated cells) as compared to control cells. Incubation of cells with both GMF and MPP+ drastically increased ROS generation compared with cells incubated with only GMF or MPP+. Values are given as mean ± SD. of four experiments in each group. *p< 0.05 compared to control, and *p< 0.05 compared to GMF treated group.
GMF Increased the Apoptotic Cell Death
To address the question, whether GMF and MPP+ treatment leads to increased apoptotic cell death, N27 cells were co-stained with ethidium bromide/acridine orange (EtBr/AO). EtBr/AO stained cells revealed the characteristic morphology of apoptosis in N27 cells. The bright uniform green color nuclei represents viable cells while the red and orange nuclei represented dead cells due to apoptosis (Fig. 4a and b). The control cells showed bright uniform green nuclei and normal morphology. In contrast, cells exposed to GMF showed orange luminescent apoptotic body formation and the number of apoptotic cells increased (*p<0.05) in GMF and MPP+ treated cells when compared with the control cells.
Fig. 4.

Effect of GMF on apoptotic morphological changes in dopaminergic N27 cells. N27 cells were seeded (3×106) in 96 well plate and incubated with GMF (100 ng/ml) and/or MPP+ (300 μm) for 24 h. After the incubation period the cells werewashed with PBS and stained with EtBr/AO fluorescent dye for 15 mins. Images were taken using fluorescence microscope at 200X. Photomicrographs show that GMF exposure increased apoptotic cell death of dopaminergic cells (A). The percentage of viable cells were measured after termination of incubation period (B). The bright green color indicate control cells, orange color indicates the apoptotic cell death (B). *p< 0.05 compared to control, and *p< 0.05 compared to GMF treated group.
GMF Induced Depolarization of Mitochondrial Membrane Potential (ΔΨm)
To determine whether or not GMF induces depolarization of mitochondrial membrane potential, N27 cells exposed to GMF/MPP+ were monitored for any changes in mitochondrial membrane potential. Incubation of N27 cells with GMF/MPP+ induced changes in the mitochondrial membrane potential in term of red/green fluorescence ratio as detected with Rhodamine-123 (Fig. 5a and b). Incubation of N27 cells with GMF alone showed an increase in the average green fluorescence similar to that seen with MPP+ when compared with control cells. Additionally, N27 cells when incubated with both GMF and MPP+ further increased (*p<0.05) the green flourescence, indicating a highly depolarized state of the mitochondria compared to the other treated cells.
Fig. 5.

Effect of GMF on mitochondrial membrane potential (MMP) depolarization in N27 dopaminergic cells. N27 cells were seeded (3×106) in 96 well plate and incubated with GMF (100 ng/ml) and/or MPP+ (300 μm) for 24 h. After the incubation period cells were washed with PBS and stained with the fluorescent dye rhodamine 123 (Rh-123) for 15 mins. Images were taken using fluorescence microscopy at 200X. The photomicrographs show the alteration in MMP induced by GMF in N27 cells after 24 h treatment (A). Expression of GMF significantly decreased MMP (similar to MPP+ treated cells) as compared with control cells (B). The values are given as mean ± SD. of four experiments in each group. *p< 0.05 compared to control, and *p< 0.05 compared to GMF treated group.
GMF Depletes Cellular ATP Levels
To understand the potential molecular mechanism underlying GMF-induced apoptosis we measured intracellular ATP levels. N27 cells were exposed to either GMF or MPP+ or a combination of both GMF and MPP+ and the cellular ATP content determined. N27 cells exposed to GMF or MPP+ significantly depleted the intracellular ATP levels when compared to untreated cells (*p<0.05), as shown in Fig. 6. Incubation with both GMF and MPP+ simultaneously further reduced the ATP levels. There were no significant differences in the ATP levels between GMF or MPP+ treated cells.
Fig. 6.

Effect of GMF on intracellular ATP levels in dopaminergic N27 cells. N27 cells were seeded (1×106) in T25 cell culture flask and incubated with GMF (100 ng/ml) and/or MPP+ (300 μm) for 24 h. After incubation period cells harvested and washed with PBS. The Cell lysates were prepared and intracellular ATP levels determined as per manufacture’s instruction. GMF significantly reduced intracellular ATPlevel (similar to MPP+ treated cells) as compared with control cells. Values are given as mean ± SD. of four experiments in each group. *p< 0.05 compared to control, and *p< 0.05 compared to GMF treated group.
Functional Mitochondria shown by Mito-tracker Staining
To determine the functional mitochondrial populations or abundance in GMF and MPP+ treated cells we treated N27 cells with mitochondrial-specific fluorescent dye MitoTracker Red CMXRos to label mitochondria and DNA specific Hoechst staining. Incubation of N27 cells with GMF or MPP+ showed that the primary organelle affected by this treatment was mitochondria (as shown in Fig. 7). In the photomicrograph red color indicates the positive mitochondrial abundance and accumulation and blue color in Fig. 7 indicates the DNA. GMF completely damaged the mitochondrial morphology when compared to the controls, similar to MPP+ treated cells demonstrating that the effect of GMF targeted the mitochondrial functions.
Fig. 7.
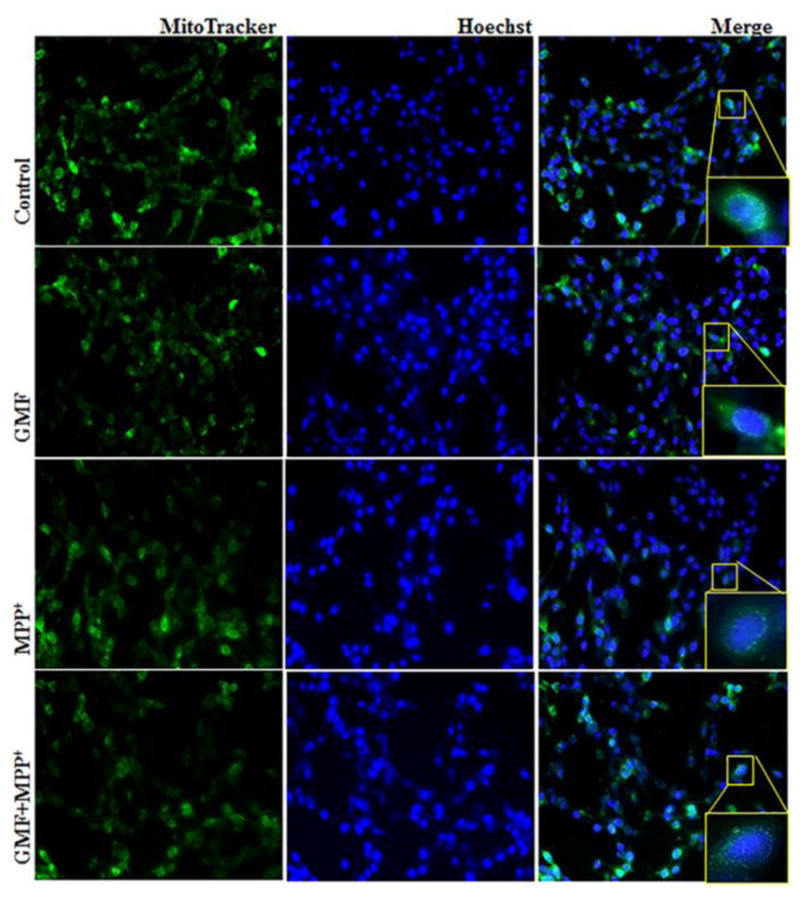
Effect of GMF exposure on intracellular mitochondrial morphology detected by MitoTracker Red CMXRos fluorescent staining method. N27 cells were seeded (3×106) in 6 well plate and incubated with GMF (100 ng/ml) and/or MPP+ (300 μm) for 24 h under standard conditions. After incubation period cells washed with PBS. 1 mM of MitoTracker Red CMXRos and 16 mM Hoechst fluorescent dye were prepared and incubated with cells for 20 mins. Exposure of GMF to dopaminergic N27 cells directly affects the cytoplasmic mitochondria mass and induced mitochondrial morphological changes compared with control cells. GMF treatment reduced the MitoTracker Red CMXRos fluorescent stained mitochondria mass as compared with control cells. Images were taken using fluorescence microscopy at 200X.
GMF Interferes with the Mitochondrial PGC-1α Expression
PGC-1α is a key regulator of mitochondrial biogenesis and activity. To find out whether GMF affects the PGC-1α protein expression; N27 cells, were incubated with GMF (100ng/ml) for 24 h and subsequently lysed and subjected to electrophoresis and western blotting. Figure 8 shows the PGC-1α expression after GMF or MPP+ treatments. It is clear that both treatments lead to decreased expression of PGC-1α which was also reduced after the combined treatment.
Fig. 8.

Effect of GMF on PGC-1α expression. N27 cells were seeded in T25 cell culture flask and incubated with GMF (100 ng/ml) and MPP+ (300 μm) for 24 h under standard conditions. After the incubation period cells were washed with PBS and cell lysates were prepared from these cells for western blot studies. GMF treatment significantly reduced expression of PGC-1α expression as seen by immunoblotting as compared with control cells (A). β-actin was used as an internal protein control to show equal protein loading (B). Western blot bands were quantified and the values are expressed as arbitrary units and given as mean ± SD of four experiments in each group. *p< 0.05 compared to control
The apoptotic effect brought about by GMF or MPP+ is mediated through their action on mitochondrial functions as evident from results detailed above with generation of ROS and with depolarization of mitochondrial membrane potential. The depletion of cellular ATP production and interference with mitochondrial oxidative phosphorylation brought about by GMF and MPP+ points to their effects on mitochondrial biogenesis that could be under the control of the transcription coactivator PGC-1α. The action of GMF in the suppression of the mitochondrial biogenesis as demonstrated in the reduced expression of PGC-1α level was studied by immunocytochmical-confocal imaging method. Fig. 9 shows that GMF alone treatment significantly reduced PGC-1α and positive mitochondrial morphology expression as compared to the control cells. The combined treatment of GMF and MPP+ drastically reduced PGC-1α expression when compared with the other treated groups. These findings indicates that GMF directly interacted with mitochondrial biogenesis by restricting the expression of PGC-1α (Fig. 8 and 9).
Fig. 9.
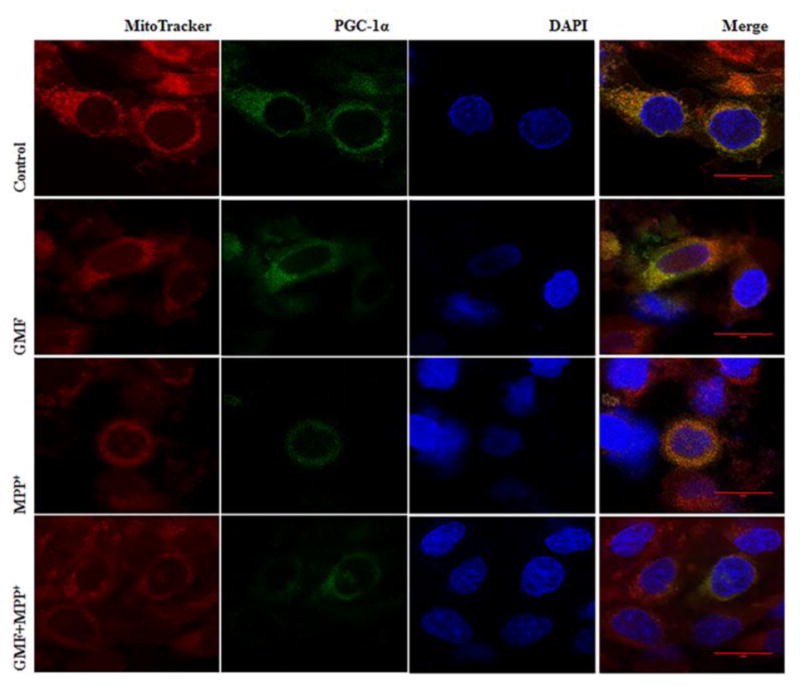
Influence of GMF on PGC-1α immunocytochemical expression in rat dopaminergic N27 cells. N27 cells were incubated in the presence or absence of GMF (100 ng/ml) or MPP+ and immunostained for PGC-1α, and further stained with MitoTracker Red CMXRos finally counterstained with DAPI. Representative images show that exposure of N27 cells to GMF significantly reduced PGC-1α expression, and reduced positive mitochondrial staining (like to MPP+ treated cells) as compared to control cells. Incubation of N27 cells with both GMF and MPP+ simultaneously significantly reduced PGC-1α expression as compared with other group. Images were taken using Leica TCP SP8 laser scanning confocal microscope with a 405-nm diode laser and tunable super continuum white light laser using 63X oil immersion objective.
GMF Interferes withMitochondrial Oxidative Phosphorylation (OXPHOS) Complex Proteins
The Effect on the Levels of Mitochondrial OXPHOS proteins
We determined the effect of GMF treatment on the levels of mitochondrial OXPHOS complexes (C1-NDUFB8, C2-SDHB C3-UQCRC2 and C5-ATP5A) in N27 cells. The effect of GMF on levels of the mitochondrial OXPHOS protein complexes in N27 cells, as shown in Fig. 10. Treatment with only GMF significantly affected the levels of the mitochondrial OXPHOS complexes (I, II, III and V) and the action was similar to MPP+ treated cells. However, treatment with GMF together with MPP+ inhibited the mitochondrial OXPHOS complex proteins expression to a greater degree. This finding emphasizes that GMF induced cellular oxidative stress is brought about through its action on the mitochondria and it mediates its neuronal insults by inhibition of the OXPHOS protein complexes.
Fig. 10.

Effect of GMF induced mitochondrial OXPHOS expression in dopaminergic N27 cells. N27 cells were seeded in T25 cell culture flask with appropriate concentration and incubated with GMF (100 ng/ml) and/or MPP+ (300 μm) for 24 h under standard conditions. After incubation period cells washed with PBS and cell lysates were prepared from these cells used to detect the mitochondrial OXPHOS complexes expressions by western blot (A). GMF incubation significantly reduced mitochondrial complexes (NDUFB8, SDHB, UQCRC and ATP5A) as compared with control cells (B). Protein expressions were quantified by using β-actin as an internal control and values are given as mean ± S.D. of four experiments in each group. *p< 0.05 compared to control, and *p< 0.05 compared to GMF treated group.
GMF Treatment Predominantly Increased the Cytosolic Cytochrome-c
To examine the effect of GMF on mitochondrial permeability we analyzed the release of cytochrome-c in N27 cells on both the cytosolic and mitochondrial fractions by western blotting. Many studies provide evidence that the action of neurotoxins is brought about by its effect on mitochondrial functions mediating the release of number of proapoptotic factors like cytochrome-c. The release of cytochrome-c with or without treatment of GMF is as shown in Fig. 11A. Treatment with GMF or MPP+ significantly increased cytosolic release of cytochrome-c and reduced in mitochondrial cytochrome-c when compared to the controls, while the combined treatment of GMF and MPP+ increased the cytochrome-c expression significantly (*p<0.05). In the treated cells, the fold cytosolic expression of cytochrome-c indicates that the increased colloidal osmotic pressure in outer mitochondria and swelling in inner cristae ultimately led to the release of cytochrome-c from outer membrane by GMF or MPP+ toxicity.
Fig. 11.
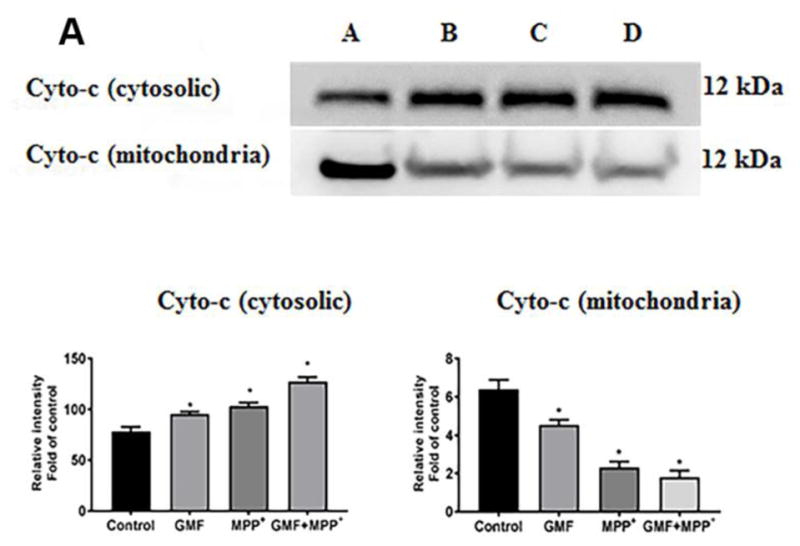
Influences of GMF on the apoptotic and anti-apoptotic markers expression. Cells were treatedin the presence orabsence of GMF (100 ng/ml) and/or MPP+ (300 μm) for 24 h. After the incubation period cell lysates from these cells are used to detect the apoptotic and anti-apoptotic markers expressions by western blot Exposure of N27 cells to GMF significantly increased the cytosolic cytochrome-c (A), increased Bax, reduced Bcl2 (B) and increased caspases 3, 7, 8 and 9; (C) expression when compared with control cells. The band density was quantified by densitometry. β-actin was used as internal standard to normalize the intensity of the protein expressions. Values are given as mean ± SD. of four experiments in each group. *p< 0.05 compared to control, and *p< 0.05 compared to GMF treated group.
Effect of GMF on Bax/Bcl-2 Ratio Detected by Western Blotting
Understanding the apoptotic effects mediated by the action of GMF or MPP+ would require following the effects on expression of known pro-apoptotic molecule Bax in relation to the anti-apoptotic molecule Bcl-2. We determined the expression of Bax and Bcl2 expression in N27 cells by western blotting after treatment with GMF or MPP+. Cells treated with GMF significantly elevated expression of the proapoptotic protein Bax and depleted the expression of the antiapoptotic protein Bcl-2. Similar results were obtained with the MPP+ treated cells as compared with untreated control cells. Cells treated with both GMF and MPP+ significantly elevated Bax expression and reduced Bcl-2 expression significantly when compared to the untreated cells (*p<0.05; Fig. 11B). This finding confirms the microscopic data that GMF treatment directly influences the apoptotic pathway.
GMF induces the expression and activation of caspases
During the apoptosis execution pathway, cytochrome-c from mitochondria is released into the cytosol, which then activates effector caspases. The caspases act as key executioners for apoptosis resulting in neuronal death. To further determine, whether GMF treatment initiates the apoptotic cell death in dopaminergic N27 cells we performed electrophoresis followed by electro-transfer and immunoblotting as shown in Fig. 11C. The results of the present study showed that GMF treatment induced a significant increase in caspases expression pattern (caspase-3, -7, -8 and -9) and increased activation of caspase 8 brought about by the increased release of cytosolic cytochrome-c which was similar in the MPP+ treated cells. This trend was significantly elevated in combined incubation with both GMF and MPP+ when compared to treatment with only GMF or MPP+ treated cells (*p<0.05; Fig 11C).
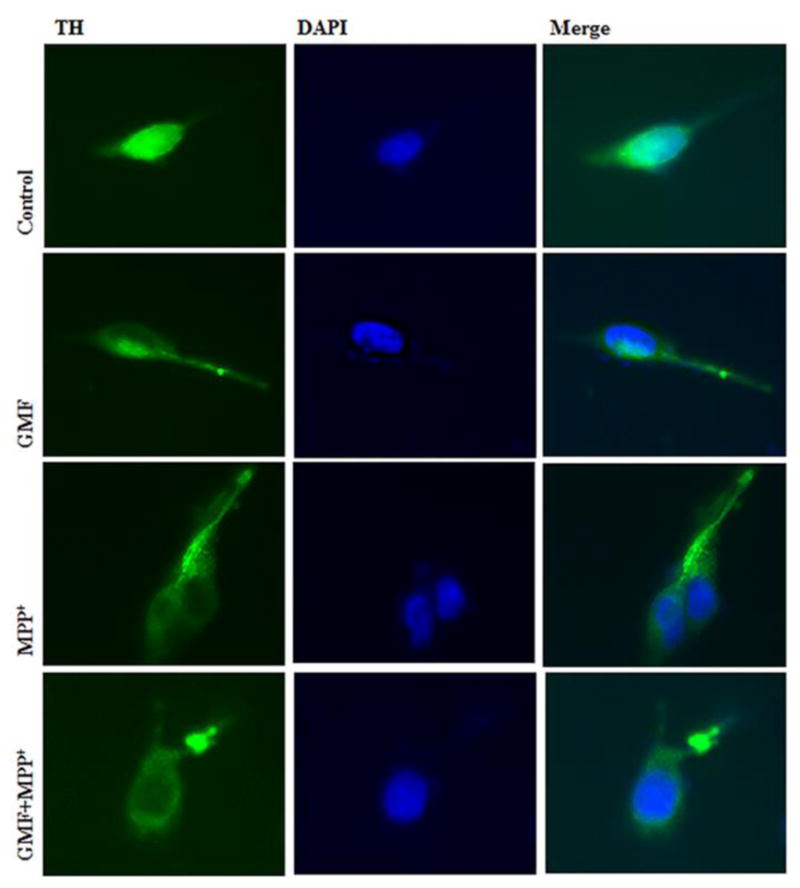
GMF Treatment Reduces Immunocytochemical Expression of TH in N27 Cells
To determine the toxic effects of GMF exposure on N27 dopaminergic cells we studied the TH expression by immunocytochemical method. Fig. 12 shows that phenotypic TH expression was significantly reduced in cells exposed to GMF as compared with control cells. N27 cells simultaneously incubated with GMF and MPP+ significantly reduced TH protein expression as compared with other groups. No significant changes were observed in untreated control cells. That the GMF/MPP+ treatment reduces dopaminergic specific marker TH protein expression indicates that both stimulants regulates in progression.
Fig. 12.

Effects of GMF on TH immunocytochemical expression in rat dopaminergic N27 cells. N27 cells were incubated in the presence or absence of GMF (100 ng/ml) or MPP+ and double- stained with TH and DAPI. Exposure to GMF showed moderate e TH positive expression as compared with untreated control cells. Incubation of N27 cells with both GMF and MPP+ significantly reduced TH expression as compared with other group. Images were taken using fluorescence microscopy at 200X.
GMF Induces Ultrastructural changes in the mitochondria and chromatin detected by transmission electron microscope
We used transmission electron microscope (TEM) to detect GMF induced intracellular morphological changes to mitochondria and the nucleus in dopaminergic N27 cells. Ultrastructural changes in the intracellular cytoplasmic region are depicted in Fig. 13. N27 cells exposed to GMF for 24 h significantly altered mitochondrial integrity, decreased mitochondrial biogenesis hence impaired mitochondrial respiration and brought about chromatin condensation and nuclear fragmentation (all hallmark of apoptotic cell death) compared with untreated control cells. TEM analysis showed decreased numbers of mitochondria with altered cristae. No significant changes were observed in untreated control cells. TEM results clearly showed that GMF induces apoptotic cell death and it is mediated by changes in mitochondrial homeostasis.
Fig. 13.
Influences of GMF on the ultrastructural changes (mitochondrial integrity and chromatin condensation) in rat dopaminergic N27 cells. Representative transmission electron microscopy (TEM) images analysis of N27 dopaminergic cells exposed to GMF (100 ng/ml) for 24 h. (A and B) untreated control N27 dopaminergic cells; (C, D, E and F) exposed with GMF for 24 h. Exposure toGMF altered the mitochondrial integrity by inhibiting mitochondrial biogenesis and by chromatin condensations as shown in representative images. The figure shows morphology of the mitochondrial structure and the damaged organelle structure (white colored arrows indicate mitochondrial biogenesis; yellow colored arrows indicate mitochondria and red color arrows indicate chromatin condensation) scale bar 1 and 2 μm.
Discussion
Parkinson’s disease is (PD) currently considered as a mitochondrial dysfunction and oxidative stress mediated, chronic, age dependent, neurodegenerative disease that mainly targets dopaminergic system in the aged brain and leads to motor impairments. The loss of dopaminergic neurons in the SN and its consequences is the widely accepted fate in PD [46]. We used N27 cells because they primarily replicate the key features of mammalian nigral dopaminergic neurons [47]. N27 cells are considered better in vitro cell culture tools to explore dopaminergic neurotoxicity, because they represent a homogenous population of dopaminergic TH positive cells with functional characteristics, including cellular signaling, similar to dopaminergic neurons [48,49]. In the present study, we report that GMF inhibits mitochondrial biogenesis with progressive depletion of dopaminergic neurons by three different aspects: (1) induce intracellular oxidative stress and depletion of intracellular ATP (2) induce the release the apoptotic mediators from mitochondrial pores and execute intrinsic cell death pathway and 3) inhibit PGC-1α expression.
Approximately 50% of dopaminergic neuronal population in the nigral region degenerate in the initial stages of PD and the remaining 50% of dopaminergic neurons are affected by the intracellular stress event. Even in the present N27 cellular model it was important to employ concentrations of toxins that would recreate situations seen in early stages of PD [50,51]. In our present study results obtained from the MTT assay indicated that GMF (100 ng/ml) significantly affected N27 cell viability (Fig. 1), which is consistent with previous studies demonstrating that exposure of cells to neurotoxic insults results in a mixed population of cells, in which some are healthy, some are no longer viable, and some are dead [52,53].
Dopaminergic neuronal system requires high amount of mitochondrial activity to generate a large amount of ATP during the action potential generation and excitatory signal transduction in mammalian brain [54]. Normally, during ATP production, excessive ROS; superoxide and hydroxyl radicals that are produced in the cytoplasm from mitochondria are cleaved into O2 and H2O by intracellular redox enzymes such as SOD and GPx [55–57]. It has been reported that neurons are more prone to oxidative flux, because 20% of the total oxygen is consumed by the brain than any other organ in the body; also it is made up of more sensitive amino acids and has its own anti-oxidant defense enzymes [58]. Especially, the substantia nigra of pars compacta (SNpc) region is more prone to intracellular insults, because it operates under pro-oxidative state in comparison to any other part of the brain. Moreover, the SNpc region has a high metabolic rate combined with high content of easily oxidizable species including dopamine (DA), DA-derived ROS, neuromelanin, polyunsaturated fatty acids, iron and a low content of antioxidants (glutathione in particular). All oxidative species, render the SN region to be highly vulnerable to peroxynitrite and sulfite [58,59].
Fluctuation in intracellular redox state and stress produced by damaged mitochondrial electron transport chain is considered to be a typical trigger underlying the pathogenesis of mitochondrial dysfunction-related neurological diseases. MPP+, a classical metabolite of MPTP, interferes with the mitochondrial respiration by inhibiting mitochondrial complexes and impairing ATP synthesis. Impaired or reduced complexes activity was found in mitochondria derived from the platelets of PD patients [60]. Perusal of current literature suggests that mitochondrial metabolism is also responsible for the generation and accumulation of ROS in the intracellular region. ATP synthesis or inhibition in electron transport chain complexes activity leads to depletion of intracellular ATP stores and abnormal ROS accumulation [61,62]. Maintenance of intracellular ATP levels by the process of oxidative phosphorylation is a critical feature for all eukaryotes to respond to extra and intra cellular requirements. Reduced expression of PGC-1α severely challenges the maintenance of cytoplasmic ATP levels, since deficiencies in incomplete OXPHOS complexes functions lead to energy deprivation and cell death [63]. Both MPP+ and GMF have been implicated in the pathogenesis of PD. Earlier studies from this laboratory [64,65] have shown that both affect glial cells similarly by releasing chemokines and cytokines. MPP+ has been shown to be an inhibitor of mitochondrial complex I. In the present work, we show that GMF treatment affects expression of mitochondrial complexes very similar to MPP+ treatment and thus would have similar function in blocking complex I (Fig. 10A). However, it is worth noting that downregulation of GMF by GMF-specific shRNA protected dopaminergic (DA) neurons from MPP+ induced neurotoxicity [38]. Ongoing studies in our laboratory have shown that intracellular GMF translocates to the mitochondria in an activation dependent manner and thus the effects of GMF on the mitochondria are planned for future publication. The present study (Fig 8 and 9) show that GMF interfered with PGC-1α expression causing the diminished expression of OXPHOS complexes, which in turn resulted in depletion of intracellular ATP stores, similar to MPP+ treated cells. A similar function for PGC-1a has been shown in earlier studies [63].
Mitochondrion by itself can acts as a primary source of ROS production in most of the tissues and cells. It has been estimated that over 0.2% – 2% of O2 consumed is converted into O2•- by mitochondrial electrons transport chain (ETC), during ATP synthesis [61]. The vast majority of basal cellular ROS (estimated at approximately 90%) are generated from the inhibition of mitochondrial complex chain reactions. This could be attributed to its quick reactive nature resulting in progressive distress in dopaminergic neuronal cells [66,67]. Accumulated ROS reacts with the nearest nucleic acids, proteins, lipids and the organelles resulting in dopaminergic neuronal loss. In the present study, incubation of cells for 24 h with GMF increased the ROS accumulation. The resultant fluctuation in the activities of GPx and SOD causes the oxidative flux. The resultant decrease in the activities of GPx and SOD lead to a reduced intracellular redox state (Fig. 2A and B). As per our hypothesis, GMF directly inhibited the mitochondrial complexes, increased ROS production and altered the intracellular redox pool similar to MPP+ treated cells evident from earlier findings [38,68,69].
Several studies have demonstrated that oxidative stress and mitochondrial dysfunction plays an important role in the development of PD pathology [70]. However, biogenesis of mitochondria and the production of energy in the form of ATP from the mitochondria are different during normal and diseased conditions [20]. The PGC-1α, a nodal transcriptional coactivator factor, plays a central role in mitochondrial biogenesis [71,18]. Recently, it was reported that PGC-1α had the ability to re-modulate the individual functions and compositions of the cells in normal and as well as in diseased condition [20]. Another study [72] reported that PGC-1α regulated mitochondrial biogenesis and respiration by the NRFs and mitochondrial uncoupled proteins during energy deprivation [73]. Reduced expression of PGC-1α led to reduced nuclear encoded subunits, which was closely associated with the mitochondrial respiratory complex and subsequently increased dopaminergic cell death [74]. It was reported that PGC-1α-null mice were more prone to neurodegeneration in the striatum and different layers of the mid brain and the cortex [75], with similar type of findings were reported in PD and Huntington disease [76,19,77]. Due to the multiple roles played in the mitochondrial biogenesis, PGC-1α has been suggested to serve as a neuroprotective agent against various intra and extra cellular stimuli or xenobiotic stimuli-induced neurodegeneration. It was reported that PGC-1α null mice were more sensitive to MPTP-induced cytoplasmic oxidative flux toxicity than the control mice [19]. Increased expression of PGC-1α significantly ameliorated the mitochondrial mediated anti-oxidant deficiency in glial cells resulting in PGC-1α mediated cellular protection [78–81]. Our present study also confirmed that incubation with GMF significantly reduced the PGC-1α expression which was similar with MPP+ treated cells as compared with untreated control N27 dopaminergic neuronal cells (as shown in Fig. 9).
Downregulation of PGC-1α directly reduced the number of mitochondria and the mitochondrial membrane potential, thereby leading to oxidative stress mediated apoptotic dopaminergic neuronal depletion. Earlier, it was reported that increased mitochondrial depolarization by the inhibition of mitochondrial complexes activity in presence of paraquate or 6-OHDA caused dopaminergic neuronal vulnerability and resultant dysregulation of Bax/Bcl-2 expression [82–84]. The present results showed that N27 dopaminergic neuronal cells incubated with GMF significantly increased ROS concentration and mitochondrial membrane depolarization, similar to MPP+ treated cells when compared with the control cells (Fig. 3, 5 and 11B).
Oxidative stress mediated apoptosis is currently considered as an important mode of dopaminergic cell death in PD [85]. The ROS-induced alteration or shift in the ratio between Bax/Bcl-2 expressions ultimately lead to initiation of intrinsic apoptotic cell death pathways [86,87,85]. We have previously demonstrated that overexpression of GMF in N18 neuroblastoma cells directly induced glycogen synthase kinase 3β (GSK-3β) activation mediated caspase-dependent apoptosis and neuronal loss [37]. Furthermore, overexpression of GMF directly enhanced the caspases dependent apoptotic intrinsic pathway by reduced intracellular redox state [88,37]. Extracellular secretory nerve factor might induce a typical apoptotic neuronal cell death in the invitro model of PD [89]. Previous reports found that over expression of GMF resulted in an increase in the levels of SOD and LDH while reducing the activity of GPx, ultimately causing apoptotic cell loss [88,90]. Our results in addition to corroborating the above findings shows that GMF aggravates and extends anti-oxidant flux resulting in apoptotic cell death (Figs. 1, 3, 4 and 5) by the activation of apoptotic markers and leads to chromatin condensation and cell death (Figs. 11A, B and C and Fig. 13).
When the outer mitochondrial membrane is breached, the intra cellular pro-apoptogenic factors such as cytochrome-c located and released from inter-membrane space of the mitochondria [91] causes an activation of caspases such as caspase -6, -7, -8 and-9 during pathological conditions [92,93]. In the present study, we examined the mechanistic involvement of GMF in apoptotic pathway mediating neuronal death. In this regard, apoptotic and anti-apoptotic markers expression in N27 cells were also assessed. The present study demonstrates that exposure of GMF to the dopaminergic cells significantly down regulated Bcl-2 expression and upregulated Bax expression similar to MPP+ treated cells (as shown in Fig. 11B). The present study clearly demonstrates that incubation of N27 cells with GMF significantly increased oxidative stress, impaired mitochondrial morphology and mitochondrial biogenesis by inhibiting PGC-1α expression. Further, dysfunction of mitochondria lead to shifts in Bax/Bcl-2 and initiated cytochrome-c release, which subsequently activated the expression of caspases resulting in apoptotic dopaminergic N27 neuronal cell death (Figs. 8, 9, 10 and 11). Our previous findings showed that absence of GMF significantly alleviated the TH positive dopaminergic neuronal density in PD brain [68]. Immunocytochemistry results shows that N27 cells express TH protein, which is reduced after GMF exposure. Further, our results confirm that N27 cells selectively express TH protein [94,95]. The chromatin condensation leading to nuclear fragmentation is a hallmark of apoptotic cell death. In our present study ultrastructural changes detected by TEM demonstrates that exposure of N27 cells to GMF significantly induced both chromatin condensation and mitochondrial damage as shown in Fig. 13. GMF significantly elevates the expression of apoptotic proteases resulting in mitochondrial dysfunction, therby leading to dopaminergic cell death.
Conclusions
GMF is a brain specific abundant, biologically active neuro-inflammatory factor, however, its precise role in mitochondrial biogenesis as well as mitochondrial dysfunction especially. Our present findings indicate that GMF directly or indirectly inhibits the expression of PGC-1α, and results in the inhibition of mitochondrial biogenesis during oxidative conditions. GMF-induces inhibition of mitochondrial complexes and triggers an intra-cellular oxidative flux, causing the shift in Bax/Bcl-2 expression. This in turn leads to the release of cytochrome-c, activation of caspases triggering apoptotic cell death. In the current study, MPP+ a well-studied neurotoxin was also included alongside with GMF. We conclude that GMF causes mitochondrial dependent redox flux that leads to apoptotic cell death in dopaminergic neurons. Future studies aimed at understanding the mechanism of action of GMF to inhibit PD pathogenesis are underway in the laboratory.
Fig. 14.

Overall mechanism of action of GMF in N27 dopaminergic neuronal cell death through apoptotic intrinsic pathway by inhibition of PGC-1α expression. Inhibition of PGC-1α expression leads to depletion of intracellularATP stores, and causes oxidative flux further leads to shift in Bax and Bcl2 expression with subsequent release of cytochrome-c. Released cytochrome-c binds with Apaf (apoptosis activating factor) to form the apoptosome, which activates effector caspases expression and activation. This further leads to apoptotic dopaminergic neuronal death. Red colored arrows indicates increase or decrease in the intracellular markers expression.
Acknowledgments
This work was supported by Veterans Affairs Merit Award I01BX002477 and National Institutes of Health Grants NS073670 and AG048205 to AZ. We would like to thank Dr. Alexander Jurkevich, Associate Director of Molecular Cytology core, University of Missouri, Columbia-MO for his help in preparation and validation of confocal images.
Footnotes
Conflict of Interest
The authors declare that there are no conflicts of interest.
References
- 1.Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog Neurobiol. 2001;65(2):135–172. doi: 10.1016/s0301-0082(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 2.Frank C, Pari G, Rossiter JP. Approach to diagnosis of Parkinson disease. Can Fam Physician. 2006;52:862–868. [PMC free article] [PubMed] [Google Scholar]
- 3.Cory-Slechta DA, Thiruchelvam M, Di Monte DA. Letter regarding: “Paraquat: The Red Herring of Parkinson’s Disease Research”. Toxicological Sciences. 2008;103(1):215–216. doi: 10.1093/toxsci/kfm309. [DOI] [PubMed] [Google Scholar]
- 4.Eriksen JL, Wszolek Z, Petrucelli L. MOlecular pathogenesis of parkinson disease. Archives of Neurology. 2005;62(3):353–357. doi: 10.1001/archneur.62.3.353. [DOI] [PubMed] [Google Scholar]
- 5.Kieburtz K, Wunderle KB. Parkinson’s disease: evidence for environmental risk factors. Mov Disord. 2013;28(1):8–13. doi: 10.1002/mds.25150. [DOI] [PubMed] [Google Scholar]
- 6.Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, Soldner F, Sunico CR, Nagar S, Talantova M, Lee B, Lopez K, Nutter A, Shan B, Molokanova E, Zhang Y, Han X, Nakamura T, Masliah E, Yates JR, 3rd, Nakanishi N, Andreyev AY, Okamoto S, Jaenisch R, Ambasudhan R, Lipton SA. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell. 2013;155(6):1351–1364. doi: 10.1016/j.cell.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80(1):315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- 8.Lee SB, Bae IH, Bae YS, Um HD. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem. 2006:281. doi: 10.1074/jbc.M606702200. [DOI] [PubMed] [Google Scholar]
- 9.Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1(2):120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 10.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 11.Hu XM, Zeng FD. Protective effects of sodium beta-aescin on ischemia-reperfusion injury in rat brain. Yao Xue Xue Bao. 2004;39(6):419–423. [PubMed] [Google Scholar]
- 12.Selvakumar GP, Manivasagam T, Rekha KR, Jayaraj RL, Elangovan N. Escin, a novel triterpene, mitigates chronic MPTP/p-induced dopaminergic toxicity by attenuating mitochondrial dysfunction, oxidative stress, and apoptosis. J Mol Neurosci. 2015;55(1):184–197. doi: 10.1007/s12031-014-0303-x. [DOI] [PubMed] [Google Scholar]
- 13.Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res. 2017;39(1):73–82. doi: 10.1080/01616412.2016.1251711. [DOI] [PubMed] [Google Scholar]
- 14.Urrutia PJ, Mena NP, Nunez MT. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol. 2014;5:38. doi: 10.3389/fphar.2014.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng A, Hou Y, Mattson MP. Mitochondria and neuroplasticity. ASN Neuro. 2010;2(5):e00045. doi: 10.1042/AN20100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1(6):361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta. 2002;1576(1–2):1–14. doi: 10.1016/s0167-4781(02)00343-3. [DOI] [PubMed] [Google Scholar]
- 18.Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. 2011;1813(7):1269–1278. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 20.Austin S, St-Pierre J. PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci. 2012;125(Pt 21):4963–4971. doi: 10.1242/jcs.113662. [DOI] [PubMed] [Google Scholar]
- 21.Kaplan R, Zaheer A, Jaye M, Lim R. Molecular cloning and expression of biologically active human glia maturation factor-beta. J Neurochem. 1991;57(2):483–490. doi: 10.1111/j.1471-4159.1991.tb03777.x. [DOI] [PubMed] [Google Scholar]
- 22.Lim R, Zaheer A, Lane WS. Complete amino acid sequence of bovine glia maturation factor beta. Proc Natl Acad Sci U S A. 1990;87(14):5233–5237. doi: 10.1073/pnas.87.14.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaheer A, Fink BD, Lim R. Expression of Glia Maturation Factor β mRNA and Protein in Rat Organs and Cells. Journal of Neurochemistry. 1993;60(3):914–920. doi: 10.1111/j.1471-4159.1993.tb03237.x. [DOI] [PubMed] [Google Scholar]
- 24.Lim R, Zaheer A. Phorbol ester stimulates rapid intracellular phosphorylation of glia maturation factor. Biochem Biophys Res Commun. 1995;211(3):928–934. doi: 10.1006/bbrc.1995.1901. [DOI] [PubMed] [Google Scholar]
- 25.Lim R, Zaheer A. In vitro enhancement of p38 mitogen-activated protein kinase activity by phosphorylated glia maturation factor. J Biol Chem. 1996;271(38):22953–22956. doi: 10.1074/jbc.271.38.22953. [DOI] [PubMed] [Google Scholar]
- 26.Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H, Zaheer S, Iyer SS, Zaheer A. Neuroinflammation Induces Neurodegeneration. J Neurol Neurosurg Spine. 2016;1(1) [PMC free article] [PubMed] [Google Scholar]
- 27.Kempuraj D, Khan MM, Thangavel R, Xiong Z, Yang E, Zaheer A. Glia maturation factor induces interleukin-33 release from astrocytes: implications for neurodegenerative diseases. J Neuroimmune Pharmacol. 2013;8(3):643–650. doi: 10.1007/s11481-013-9439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaheer S, Wu Y, Bassett J, Yang B, Zaheer A. Glia maturation factor regulation of STAT expression: a novel mechanism in experimental autoimmune encephalomyelitis. Neurochem Res. 2007;32(12):2123–2131. doi: 10.1007/s11064-007-9383-0. [DOI] [PubMed] [Google Scholar]
- 29.Zaheer S, Wu Y, Sahu SK, Zaheer A. Suppression of neuro inflammation in experimental autoimmune encephalomyelitis by glia maturation factor antibody. Brain Res. 2011;1373:230–239. doi: 10.1016/j.brainres.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito J, Kato T, Yamakawa Y, Kato H, Sakazaki Y, Lim R, Tanaka R. Interaction of glia maturation factor with the glial cell membrane. Brain Res. 1982;243(2):309–314. doi: 10.1016/0006-8993(82)90254-2. [DOI] [PubMed] [Google Scholar]
- 31.Choudhury A, Marks DL, Proctor KM, Gould GW, Pagano RE. Regulation of caveolar endocytosis by syntaxin 6-dependent delivery of membrane components to the cell surface. Nat Cell Biol. 2006;8(4):317–328. doi: 10.1038/ncb1380. [DOI] [PubMed] [Google Scholar]
- 32.Aerbajinai W, Liu L, Zhu J, Kumkhaek C, Chin K, Rodgers GP. Glia Maturation Factor-gamma Regulates Monocyte Migration through Modulation of beta1-Integrin. J Biol Chem. 2016;291(16):8549–8564. doi: 10.1074/jbc.M115.674200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Afeseh Ngwa H, Kanthasamy A, Anantharam V, Song C, Witte T, Houk R, Kanthasamy AG. Vanadium induces dopaminergic neurotoxicity via protein kinase Cdelta dependent oxidative signaling mechanisms: relevance to etiopathogenesis of Parkinson’s disease. Toxicol Appl Pharmacol. 2009;240(2):273–285. doi: 10.1016/j.taap.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Afeseh Ngwa H, Kanthasamy A, Gu Y, Fang N, Anantharam V, Kanthasamy AG. Manganese nanoparticle activates mitochondrial dependent apoptotic signaling and autophagy in dopaminergic neuronal cells. Toxicol Appl Pharmacol. 2011;256(3):227–240. doi: 10.1016/j.taap.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zawada WM, Banninger GP, Thornton J, Marriott B, Cantu D, Rachubinski AL, Das M, Griffin WS, Jones SM. Generation of reactive oxygen species in 1-methyl-4-phenylpyridinium (MPP+) treated dopaminergic neurons occurs as an NADPH oxidase-dependent two-wave cascade. J Neuroinflammation. 2011;8:129. doi: 10.1186/1742-2094-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119(2):203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 37.Zaheer A, Knight S, Zaheer A, Ahrens M, Sahu SK, Yang B. Glia maturation factor overexpression in neuroblastoma cells activates glycogen synthase kinase-3beta and caspase-3. Brain Res. 2008;1190:206–214. doi: 10.1016/j.brainres.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khan MM, Kempuraj D, Zaheer S, Zaheer A. Glia maturation factor deficiency suppresses 1-methyl-4-phenylpyridinium-induced oxidative stress in astrocytes. J Mol Neurosci. 2014;53(4):590–599. doi: 10.1007/s12031-013-0225-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaheer A, Zaheer S, Sahu SK, Knight S, Khosravi H, Mathur SN, Lim R. A novel role of glia maturation factor: induction of granulocyte-macrophage colony-stimulating factor and pro-inflammatory cytokines. J Neurochem. 2007;101(2):364–376. doi: 10.1111/j.1471-4159.2006.04385.x. [DOI] [PubMed] [Google Scholar]
- 40.Lunov O, Zablotskii V, Churpita O, Chanova E, Sykova E, Dejneka A, Kubinova S. Cell death induced by ozone and various non-thermal plasmas: therapeutic perspectives and limitations. Sci Rep. 2014;4:7129. doi: 10.1038/srep07129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kasibhatla S, Amarante-Mendes GP, Finucane D, Brunner T, Bossy-Wetzel E, Green DR. Acridine Orange/Ethidium Bromide (AO/EB) Staining to Detect Apoptosis. CSH Protoc. 2006;2006(3) doi: 10.1101/pdb.prot4493. [DOI] [PubMed] [Google Scholar]
- 42.Rajendra Prasad N, Karthikeyan A, Karthikeyan S, Reddy BV. Inhibitory effect of caffeic acid on cancer cell proliferation by oxidative mechanism in human HT-1080 fibrosarcoma cell line. Mol Cell Biochem. 2011;349(1–2):11–19. doi: 10.1007/s11010-010-0655-7. [DOI] [PubMed] [Google Scholar]
- 43.Tamilselvam K, Braidy N, Manivasagam T, Essa MM, Prasad NR, Karthikeyan S, Thenmozhi AJ, Selvaraju S, Guillemin GJ. Neuroprotective effects of hesperidin, a plant flavanone, on rotenone-induced oxidative stress and apoptosis in a cellular model for Parkinson’s disease. Oxid Med Cell Longev. 2013;2013:102741. doi: 10.1155/2013/102741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sastre E, Caracuel L, Prieto I, Llevenes P, Aller MA, Arias J, Balfagon G, Blanco-Rivero J. Decompensated liver cirrhosis and neural regulation of mesenteric vascular tone in rats: role of sympathetic, nitrergic and sensory innervations. Sci Rep. 2016;6:31076. doi: 10.1038/srep31076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu S, Zhou Z, Zhang L, Yu Z, Zhang W, Wang Y, Wang X, Li M, Chen Y, Chen C, He M, Zhang G, Zhong M. Exposure to 1800 MHz radiofrequency radiation induces oxidative damage to mitochondrial DNA in primary cultured neurons. Brain Res. 2010;1311:189–196. doi: 10.1016/j.brainres.2009.10.062. [DOI] [PubMed] [Google Scholar]
- 46.Dickson DW. Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med. 2012;2(8) doi: 10.1101/cshperspect.a009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drechsel DA, Liang LP, Patel M. 1-methyl-4-phenylpyridinium-induced alterations of glutathione status in immortalized rat dopaminergic neurons. Toxicol Appl Pharmacol. 2007;220(3):341–348. doi: 10.1016/j.taap.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anantharam A, Diverse-Pierluissi MA. Biochemical approaches to study interaction of calcium channels with RGS12 in primary neuronal cultures. Methods Enzymol. 2002;345:60–70. doi: 10.1016/s0076-6879(02)45007-0. [DOI] [PubMed] [Google Scholar]
- 49.Kaul S, Kanthasamy A, Kitazawa M, Anantharam V, Kanthasamy AG. Caspase-3 dependent proteolytic activation of protein kinase C delta mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: relevance to oxidative stress in dopaminergic degeneration. Eur J Neurosci. 2003;18(6):1387–1401. doi: 10.1046/j.1460-9568.2003.02864.x. [DOI] [PubMed] [Google Scholar]
- 50.Hancock DB, Martin ER, Mayhew GM, Stajich JM, Jewett R, Stacy MA, Scott BL, Vance JM, Scott WK. Pesticide exposure and risk of Parkinson’s disease: a family-based case-control study. BMC Neurol. 2008;8:6. doi: 10.1186/1471-2377-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Collier TJ, Kanaan NM, Kordower JH. Ageing as a primary risk factor for Parkinson’s disease: evidence from studies of non-human primates. Nat Rev Neurosci. 2011;12(6):359–366. doi: 10.1038/nrn3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yong-Kee CJ, Sidorova E, Hanif A, Perera G, Nash JE. Mitochondrial dysfunction precedes other sub-cellular abnormalities in an in vitro model linked with cell death in Parkinson’s disease. Neurotox Res. 2012;21(2):185–194. doi: 10.1007/s12640-011-9259-6. [DOI] [PubMed] [Google Scholar]
- 53.Munch G, Gasic-Milenkovic J, Dukic-Stefanovic S, Kuhla B, Heinrich K, Riederer P, Huttunen HJ, Founds H, Sajithlal G. Microglial activation induces cell death, inhibits neurite outgrowth and causes neurite retraction of differentiated neuroblastoma cells. Exp Brain Res. 2003;150(1):1–8. doi: 10.1007/s00221-003-1389-5. [DOI] [PubMed] [Google Scholar]
- 54.Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21(10):1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- 55.Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis. 2013;3(4):461–491. doi: 10.3233/JPD-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hwang O. Role of oxidative stress in Parkinson’s disease. Exp Neurobiol. 2013;22(1):11–17. doi: 10.5607/en.2013.22.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rahman K. Studies on free radicals, antioxidants, and co-factors. Clin Interv Aging. 2007;2(2):219–236. [PMC free article] [PubMed] [Google Scholar]
- 58.Floyd RA. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med. 1999;222(3):236–245. doi: 10.1046/j.1525-1373.1999.d01-140.x. [DOI] [PubMed] [Google Scholar]
- 59.Marshall KA, Reist M, Jenner P, Halliwell B. The neuronal toxicity of sulfite plus peroxynitrite is enhanced by glutathione depletion: implications for Parkinson’s disease. Free Radic Biol Med. 1999;27(5–6):515–520. doi: 10.1016/s0891-5849(99)00094-5. [DOI] [PubMed] [Google Scholar]
- 60.Vila M, Ramonet D, Perier C. Mitochondrial alterations in Parkinson’s disease: new clues. J Neurochem. 2008;107(2):317–328. doi: 10.1111/j.1471-4159.2008.05604.x. [DOI] [PubMed] [Google Scholar]
- 61.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 62.Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292(2):C670–686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 63.Rohas LM, St-Pierre J, Uldry M, Jager S, Handschin C, Spiegelman BM. A fundamental system of cellular energy homeostasis regulated by PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104(19):7933–7938. doi: 10.1073/pnas.0702683104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kempuraj D, Selvakumar GP, Zaheer S, Thangavel R, Ahmed ME, Raikwar S, Govindarajan R, Iyer S, Zaheer A. Cross-Talk between Glia, Neurons and Mast Cells in Neuroinflammation Associated with Parkinson’s Disease. J Neuroimmune Pharmacol. 2017 doi: 10.1007/s11481-017-9766-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kempuraj D, Thangavel R, Yang E, Pattani S, Zaheer S, Santillan DA, Santillan MK, Zaheer A. Dopaminergic Toxin 1-Methyl-4-Phenylpyridinium, Proteins alpha-Synuclein and Glia Maturation Factor Activate Mast Cells and Release Inflammatory Mediators. PLoS One. 2015;10(8):e0135776. doi: 10.1371/journal.pone.0135776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanthasamy A, Jin H, Mehrotra S, Mishra R, Kanthasamy A, Rana A. Novel cell death signaling pathways in neurotoxicity models of dopaminergic degeneration: relevance to oxidative stress and neuroinflammation in Parkinson’s disease. Neurotoxicology. 2010;31(5):555–561. doi: 10.1016/j.neuro.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zawada WM, Banninger GP, Thornton J, Marriott B, Cantu D, Rachubinski AL, Das M, Griffin WST, Jones SM. Generation of reactive oxygen species in 1-methyl-4-phenylpyridinium (MPP+) treated dopaminergic neurons occurs as an NADPH oxidase-dependent two-wave cascade. Journal of Neuroinflammation. 2011;8(1):1–13. doi: 10.1186/1742-2094-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khan MM, Zaheer S, Thangavel R, Patel M, Kempuraj D, Zaheer A. Absence of glia maturation factor protects dopaminergic neurons and improves motor behavior in mouse model of parkinsonism. Neurochem Res. 2015;40(5):980–990. doi: 10.1007/s11064-015-1553-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zaheer A, Yang B, Cao X, Lim R. Decreased copper-zinc superoxide dismutase activity and increased resistance to oxidative stress in glia maturation factor-null astrocytes. Neurochem Res. 2004;29(8):1473–1480. doi: 10.1023/b:nere.0000029558.82943.00. [DOI] [PubMed] [Google Scholar]
- 70.Hastings TG. The role of dopamine oxidation in mitochondrial dysfunction: implications for Parkinson’s disease. J Bioenerg Biomembr. 2009;41(6):469–472. doi: 10.1007/s10863-009-9257-z. [DOI] [PubMed] [Google Scholar]
- 71.Zhu L, Wang Q, Zhang L, Fang Z, Zhao F, Lv Z, Gu Z, Zhang J, Wang J, Zen K, Xiang Y, Wang D, Zhang C-Y. Hypoxia induces PGC-1[alpha] expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. 2010;20(6):676–687. doi: 10.1038/cr.2010.46. doi: http://www.nature.com/cr/journal/v20/n6/suppinfo/cr201046s1.html. [DOI] [PubMed] [Google Scholar]
- 72.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98(1):115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 73.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 74.Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, Grunblatt E, Moran LB, Mandel SA, Riederer P, Miller RM, Federoff HJ, Wullner U, Papapetropoulos S, Youdim MB, Cantuti-Castelvetri I, Young AB, Vance JM, Davis RL, Hedreen JC, Adler CH, Beach TG, Graeber MB, Middleton FA, Rochet JC, Scherzer CR Global PDGEC. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med. 2010;2(52):52ra73. doi: 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma D, Li S, Lucas EK, Cowell RM, Lin JD. Neuronal inactivation of peroxisome proliferator-activated receptor gamma coactivator 1alpha (PGC-1alpha) protects mice from diet-induced obesity and leads to degenerative lesions. J Biol Chem. 2010;285(50):39087–39095. doi: 10.1074/jbc.M110.151688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127(1):59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 77.Tsunemi T, La Spada AR. PGC-1alpha at the intersection of bioenergetics regulation and neuron function: from Huntington’s disease to Parkinson’s disease and beyond. Prog Neurobiol. 2012;97(2):142–151. doi: 10.1016/j.pneurobio.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang H, Kang S-U, Zhang S, Karuppagounder S, Xu J, Lee Y-K, Kang B-G, Lee Y, Zhang J, Pletnikova O, Troncoso JC, Pirooznia S, Andrabi SA, Dawson VL, Dawson TM. Adult Conditional Knockout of PGC-1α Leads to Loss of Dopamine Neurons. eneuro. 2016;3(4) doi: 10.1523/eneuro.0183-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nijland PG, Witte ME, van het Hof B, van der Pol S, Bauer J, Lassmann H, van der Valk P, de Vries HE, van Horssen J. Astroglial PGC-1alpha increases mitochondrial antioxidant capacity and suppresses inflammation: implications for multiple sclerosis. Acta Neuropathol Commun. 2014;2:170. doi: 10.1186/s40478-014-0170-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144(5):689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A, Jiang H, Kang SU, Andrabi SA, Dawson VL, Shin JH, Dawson TM. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci U S A. 2015;112(37):11696–11701. doi: 10.1073/pnas.1500624112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lopert P, Day BJ, Patel M. Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. PLoS One. 2012;7(11):e50683. doi: 10.1371/journal.pone.0050683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lopert P, Patel M. Nicotinamide nucleotide transhydrogenase (Nnt) links the substrate requirement in brain mitochondria for hydrogen peroxide removal to the thioredoxin/peroxiredoxin (Trx/Prx) system. J Biol Chem. 2014;289(22):15611–15620. doi: 10.1074/jbc.M113.533653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mosley RL, Benner EJ, Kadiu I, Thomas M, Boska MD, Hasan K, Laurie C, Gendelman HE. Neuroinflammation, Oxidative Stress and the Pathogenesis of Parkinson’s Disease. Clin Neurosci Res. 2006;6(5):261–281. doi: 10.1016/j.cnr.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F, Yang L, Beal MF, Andersen JK. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease. J Neurosci. 2001;21(24):9519–9528. doi: 10.1523/JNEUROSCI.21-24-09519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barzilai A, Daily D, Zilkha-Falb R, Ziv I, Offen D, Melamed E, Shirvan A. The molecular mechanisms of dopamine toxicity. Adv Neurol. 2003;91:73–82. [PubMed] [Google Scholar]
- 87.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94(4):481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 88.Kaimori JY, Takenaka M, Nakajima H, Hamano T, Horio M, Sugaya T, Ito T, Hori M, Okubo K, Imai E. Induction of glia maturation factor-beta in proximal tubular cells leads to vulnerability to oxidative injury through the p38 pathway and changes in antioxidant enzyme activities. J Biol Chem. 2003;278(35):33519–33527. doi: 10.1074/jbc.M301552200. [DOI] [PubMed] [Google Scholar]
- 89.Offen D, Ziv I, Barzilai A, Gorodin S, Glater E, Hochman A, Melamed E. Dopamine-melanin induces apoptosis in PC12 cells; possible implications for the etiology of Parkinson’s disease. Neurochem Int. 1997;31(2):207–216. doi: 10.1016/s0197-0186(96)00150-7. [DOI] [PubMed] [Google Scholar]
- 90.Lim R, Zaheer A, Kraakevik JA, Darby CJ, Oberley LW. Overexpression of glia maturation factor in C6 cells promotes differentiation and activates superoxide dismutase. Neurochem Res. 1998;23(11):1445–1451. doi: 10.1023/a:1020715126326. [DOI] [PubMed] [Google Scholar]
- 91.Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2(1):63–67. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- 92.Antonsson B, Martinou JC. The Bcl-2 protein family. Exp Cell Res. 2000;256(1):50–57. doi: 10.1006/excr.2000.4839. [DOI] [PubMed] [Google Scholar]
- 93.Ullah I, Ullah N, Naseer MI, Lee HY, Kim MO. Neuroprotection with metformin and thymoquinone against ethanol-induced apoptotic neurodegeneration in prenatal rat cortical neurons. BMC Neurosci. 2012;13:11. doi: 10.1186/1471-2202-13-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gao L, Zhou W, Symmes B, Freed CR. Re-Cloning the N27 Dopamine Cell Line to Improve a Cell Culture Model of Parkinson’s Disease. PLoS One. 2016;11(8):e0160847. doi: 10.1371/journal.pone.0160847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ziegler U, Groscurth P. Morphological features of cell death. News Physiol Sci. 2004;19:124–128. doi: 10.1152/nips.01519.2004. [DOI] [PubMed] [Google Scholar]