

Abstract
It is widely accepted that central and effector memory CD4+ T cells originate from naïve T cells after they have encountered their cognate antigen in the setting of appropriate co-stimulation. However, if this were true the diversity of T cell receptor (TCR) sequences within the naïve T cell compartment should be far greater than that of the memory T cell compartment, which is not supported by TCR sequencing data. Here we demonstrate that aged mice with far fewer naïve T cells, respond to the model antigen, hen eggwhite lysozyme (HEL), by utilizing the same TCR sequence as their younger counterparts. CD4+ T cell repertoire analysis of highly purified T cell populations from naïve animals revealed that the HEL-specific clones displayed effector and central “memory” cell surface phenotypes even prior to having encountered their cognate antigen. Furthermore, HEL-inexperienced CD4+ T cells were found to reside within the naïve, regulatory, central memory, and effector memory T cell populations at similar frequencies and the majority of the CD4+ T cells within the regulatory and memory populations were unexpanded. These findings support a new paradigm for CD4+ T cell maturation in which a specific clone can undergo a differentiation process to exhibit a “memory” or regulatory phenotype without having undergone a clonal expansion event. It also demonstrates that a foreign-specific T cell is just as likely to reside within the regulatory T cell compartment as it would the naïve compartment, arguing against the specificity of the regulatory T cell compartment being skewed towards self-reactive T cell clones. Finally, we demonstrate that the same set of foreign and autoreactive CD4+ T cell clones are repetitively generated throughout adulthood. The latter observation argues against T cell-depleting strategies or autologous stem cell transplantation as therapies for autoimmunity-as the immune system has the ability to regenerate pathogenic clones.
Keywords: Next generation sequencing, T cell repertoire analysis, CD4 T cell, T regulatory cells, memory T cells, virtual memory, autoimmunity, driver T cells, Experimental Autoimmune Encephalomyelitis, hematopoietic stem cell transplantation
Graphical Abstract
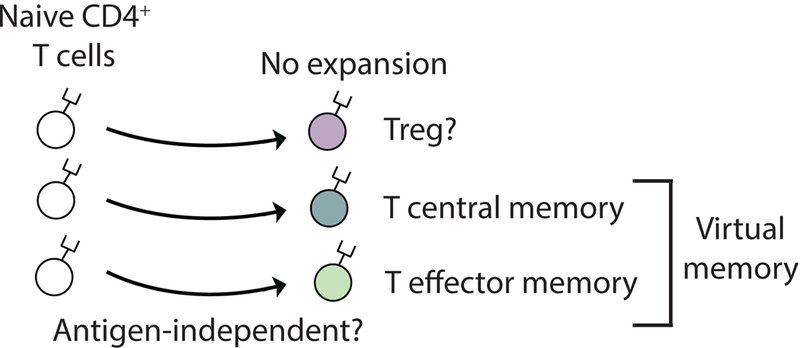
1. Introduction
In the presence of appropriate co-stimulation, naïve T cells respond to their cognate antigen by expanding and differentiating into effector and memory T cells [1]. Central to this theory is the belief that unexpanded antigen-inexperienced CD4+ T cells reside preferentially within the “naïve” T cell repertoire. However, this has never been formally verified, as it is difficult to study individual antigen- inexperienced CD4+ T cell clones due to their extremely low precursor frequencies. Although measurements of T cell receptor excision circles (TREC) clearly demonstrate that the naïve T cell population contains recent thymic emigrants [2, 3], this does not exclude the possibility that antigen-inexperienced T cells may reside in other T cell compartments as well. It is now apparent that the naïve T cell compartment is comprised of a heterogeneous population of T cells, as evident by the existence of subpopulations of naïve T cells that differ with respect to T cell receptor excision circles (TRECs) and cell surface markers such as CD31 [3]. One interpretation of these findings is that some members of the naïve T cell compartment differentiate, albeit slowly, without forming T cell blasts. If this were true, it would also have implications for autoimmunity. The theory of molecular mimicry states that autoimmunity can arise when a naïve T cell bearing a cross-reactive T cell receptor (TCR) foreign and self-directed) becomes activated to a microbial or viral pathogen. However, if naïve T cells are continuously differentiating in the absence of a clonal expansion event it is also possible for autoimmunity to arise from crossreactivity directly within the T cell memory pool, which in theory would require less of a “danger” signal. Herein, we use T cell repertoire analysis to investigate the ultimate fate of the antigen-inexperienced T cell.
The adaptive immune system is reliant upon a diverse set of antigen-specific receptors (T cell receptor [TCR] and B cell receptor [BCR]) to effectively respond to microbial and viral pathogens. Within the β chain of the TCR, the most diverse region is the complementarity determining region 3 (CDR3), which is generated by genetic recombination of variable (V), diversity (D), and joining (J) segments. In total, V(D)J joining has the potential to generate more than 1013 unique CDR3 amino acid sequences [4], although the actual number expressed is likely much less [5]. By determining the relative frequencies of CDR3 lengths (i.e. the “T cell CDR3 spectratype”), or actual CDR3 sequence frequencies using next generation sequencing, one can detect antigen-specific clonotypic T cell expansions from within a large background population of unexpanded T cells [6, 7]. Such analyses have demonstrated that mice of the same inbred strain often expand identical T cells in response to a particular viral exposure or antigenic challenge [8]. Of the endogenous T cell expansions identified by T cell repertoire analysis, perhaps the most well- characterized is the BALB/c I-Ed-restricted HEL-specific Vβ8.2Jβ1.5 CD4+ T cell clonotype (CDR3=CASGTGNNQAPL) [8–12], which responds to its cognate antigen with an IFN-γ-dominated cytokine response [9].
Although T cell repertoire analysis conducted by next generation sequencing can produce millions of DNA reads [7], it traditionally does not possess the sensitivity to reproducibly detect a particular unexpanded, antigen-inexperienced T cell clone from within an individual’s total T cell pool. This limitation is due to a variety of reasons, including the extremely low frequency at which a particular unexpanded T cell clone exists within an individual, the difficulty in amplifying low- abundant sequences, and the sequencing errors inherent to the next-generation parallel sequencing platforms. Herein, we use a protocol that enables high- resolution focused sequencing of the T cell repertoire to answer a simple but fundamental question: where do antigen-inexperienced CD4+ T cells reside?
Our specific hypothesis was that antigen-inexperienced T cells can reside outside of the naïve T cell compartment. This was based upon the unexpectedly high degree of CDR3 diversity observed within the regulatory and memory T cell populations [13]. Our most striking finding is that unexpanded antigen- inexperienced T cells reside at relatively similar frequencies in all of the archetypical CD4+ T cell compartments (naïve, regulatory, effector memory and central memory). In essence, we contend that some of the differentiated memory and regulatory CD4+ T cells can also be “naïve” with respect to their history of antigen recognition. These results are a radical departure from conventional immunological theory and have implications to the field of autoimmunity.
2. Materials and Methods
2.1. Mice
Female BALB/c, OT-II TCR transgenic and B10.PL mice were obtained from The Jackson Laboratory. Female BALB/c 18-month-old mice were obtained from the National Institutes of Aging. Mice were maintained under pathogen-free conditions. Care of all experimental animals was in accordance with institutional and National Institutes of Health guidelines.
2.2. Flow cytometry analysis and cell sorting
Mouse whole blood was treated with Ammonium Chloride Tris (ACT) lysis buffer. The Peripheral Blood Mononuclear Cell (PBMCs) were treated with Fc-bloock (BD) on ice for 15 minutes and stained with PE labeled anti-CD3e (BD Biosciences), APC- labeled anti-CD4 (Clone: RM4–5) (BD Biosciences), and FITC labeled anti-CD8 (BD Biosciences), for 30 min on ice. After washing these samples two times, they were then analyzed on a BD FACSCalibur flow cytometer.
Splenocytes isolated from 12-to-14-week-old unimmunized BALB/c mice were were treated with Fc-block (BD) on ice for 15 minutes and stained with anti-CD25 PE (BD Biosciences), anti-CD4 Pacific Blue (BD Biosciences), anti-CD62L FITC (BD Biosciences), anti-CD44 Alexa700 (BD Biosciences), and CD127PE-Cy7 (BD Biosciences) or an appropriate isotype control antibody. Cells were then sorted into the following populations: CD4+ CD25low CD62L+ CD44low for naïve; CD4+ CD25 med/low CD62L+ CD44high central memory; CD4+ CD25med/low CD62L- CD44high effector memory; and CD4+CD25highCD127low/- Treg cells using a FACSAria™ II flow cytometer system (Becton Dickinson). The purity of sorted cells was confirmed by reanalysis of sorted cells. Data were analyzed with FlowJo software (Tree Star Inc.). Sorted cell subsets were used for in vitro culture or stabilized with RNAlater (Ambion) for subsequent RNA isolation and library preparation.
2.3. Generation of DCs and DC-stimulated T-cell culture.
DCs were generated in vitro from bone marrow cells. BM cells were cultured in complete RPMI containing 10% heat-inactivated FBS in the presence of 20 ng/ml rm GM-CSF (Peprotech) for 7 days. On day 4 fresh medium containing 20 ng/ml rmGM- CSF and 50 ng/ml rm IL-4 (Peprotech) was added. Nonadherent CD11c+ DCs were sorted using anti-CD11c-coated magnetic beads, according to the manufacturer’s directions (Miltenyi Biotec, Auburn, CA, USA), on Day 7. The absence of T cells in CD11c+ BMDCs preparations was confirmed by a Vβ8.2 -Jβ1.5 TCR specific PCR. To evaluate HEL responses in vitro BMDCs at responder:stimulator cell ratio 1 : 10 were co-cultured with or without sorted; CD4+CD25 med/lowCD62L+/−CD44high memory T cells in presence or absence of 10 μg /ml HEL. After 96 hr cells were harvested and stabilized with RNAlater (Ambion) for subsequent RNA isolation and library preparations.
2.4. HEL Immunization and in vitro recall assay.
Female BALB/c mice were immunized subcutaneously with 10μg of HEL (Sigma) or PBS (Gibco) emulsified in CFA (DIFCO) or IFA. Draining lymph nodes were harvested and single-cell suspensions were prepared in petri dishes containing DME (GIBCO). Large debris was removed by decanting, followed by two washes in DME. LN cells were cultured with 10 μg /ml HEL, 20ng/ml of PPD, 10 μg /ml of Mus musculus lysozyme 2 (ML2 ) peptide RAWVAWRAHCQ, 10 μg /ml of Mus musculus lysozyme 1 (ML1) peptide RAWVAWRTQCQ or PBS. Peptides were purchased from A&A Lab (San Diego).
Cells were suspended at a concentration of 5 × 106 cells per milliliter in chemically defined, HL-1 serum-free medium (Lonza) supplemented with streptomycin and penicillin (Gibco). 24-well plates were used for culture. Incubation conditions maintained at 5% CO2 and 37°C. After 72 hr cell culture supernatants and cells were collected.
2.5. Bone marrow transplantation.
Bone marrow cell (BMC) suspensions were prepared by gently releasing cells from mice backbones, femurs, and tibiae into PBS with a mortar and pestle and filtered through a cell strainer (BD Biosciences San Jose, CA) to remove particulates. Red blood cells were removed by ACT lysis buffer. To deplete T cells BM cells were incubated with anti-Thy1.2 (30H12) antibody (1–2 mg/ 106 cells) for 30 min on ice and anti-rabbit complement (CEDARLANE, Ontario, Canada) and the cells were placed in an incubator set at 37°C set and 5% CO2 for 45 min. Afterwards cells were washed in PBS, and adjusted to 5×106 cells/ml and mice were injected with 106 cells in 0.2 mL of PBS.
Recipient (BALB/c) mice received total body γ-irradiation (850 cGy/mouse). After irradiation, recipient mice were depleted of CD4+ T cells by injection of anti-CD4 (GK1.5, 300 mg/mouse). 1 day after CD4+ T cell depletion mice were injected with T cell-depleted BM cells (106/0.2ml /mouse) via their tail vein. Third complementarity region CDR3 size spectratyping was used as a tool for monitoring T-cell repertoire reconstitution after T-cell-depleted bone marrow transplantation. Anti-CD4 (GK1.5) and anti-Thy1.2 (30H12) were obtained from ATCC.
2.6. Induction of EAE (Experimental Autoimmune Encephalomyelitis).
To induce EAE, B10.PL mice were injected subcutaneously with a CFA emulstion containing 100 μ Ac1–9 and 200 μ of Mycobacterium tuberculosis H37Ra (Difco) divided among 3 sites on their backs. 1 and 3 day after the immunization, the mice were injected intraperitoneally with 100 ng of pertussis toxin (List Biological Laboratory) in PBS. Disease score was measured as follows: 1, tail paralysis; 2, hind limb weakness; 3, hind limb paralysis; 4, hind and forelimb paralysis; 5, moribund or death.
2.7. RNA Isolation and cDNA synthesis.
Total RNA was isolated using the Qiagen RNEasy Plus Kit (Qiagen) and treated with RNA-free DNase I (Ambion) to remove genomic DNA. RNA concentrations were quantified using a Qubit® Fluorometer and the Quant-iT RNA assay kit (Invitrogen). cDNA was synthesized by reverse transcription using the SuperScript® III First- Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer’s instructions.
2.8. TCR CDR3 spectrotyping.
TCR CDR3 spectrotyping analysis was performed as has been described in detail elsewhere [10]. Briefly, from each cDNA, PCR reactions were performed using Vβprimers, a common Cβ primer and Platinum® Taq DNA Polymerase. Vβ primers and a fluorescent-conjugated (6-FAM) Jβ primers were used for second round PCR reaction. PCR products were then analyzed on an ABI 3100 Prism Genetic (Applied Biosystems, Carlsbad, CA) and Peak Scanner Software v1.0 (Applied Biosystems).
2.9. DNA capillary sequencing.
Sequencing was performed as has been described in details previously [31]. Briefly, cDNA was amplified with specific primers and Phusion™ Hot Start High- Fidelity DNA Polymerase (Finnzyme F-540). After adding deoxyadenosine (A) to the 3’ ends of PCR products, PCR products were TA ligated and cloned into PCR 2.1- TOPO vector using the PCR 2.1-TOPO TA Cloning Kit (Invitrogen) according to the manufacturer’s instructions. cDNA-containing plasmid DNA was extracted using QIAprep 96 Turbo Miniprep Kit (QIAGEN) and sequenced with M13 primers.
2.10. CDR3 definition.
CDR3P length was defined as all bases between the second cysteine of TRBV and the phenylalanine in the TRBJ segment motif FGXG according International ImMunoGeneTics [IMGT] nomenclature. Gene names of Vβ and Jβ are according to the Immunogenetics (IMGT) gene name nomenclature for T cell Receptor [32] of mice.
2.11. Amplification of complementarity-determining region 3 (CDR3) of Vβ8.2 -Jβ1.5 TCR
Reverse transcription was performed using the TCR Cp-specific primer MCβ12RT (5’GGAGACCTTGGGTGGAGTCA3’) and the Superscript® III First-Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer’s instructions. Quantitative real-time PCR was performed for each sample to identify the minimum number of PCR cycles needed to ideally amplify TCR β without over amplification. qRT-PCR was carried out with the primer pairs MVβ8.2 (TGGCTACCCCCTCTCAGACATCA) and Mjβ1.5 (GAGTCCCCTCTCCAAAAAGCG) and used the Platinum® Taq DNA Polymerase HiFi kit (Invitrogen) and EvaGreen® Dye (Biotium). PCR cycle: 94°C for 2 min, 94°C 30 sec, 60°C 20 sec, 68°C 30 sec for 40 cycles, followed by a final extension step of 68°C for 5 min.
Once the optimal number of PCR cycles was established, final PCR was carried out with primer pairs MVB8.2 and MJb1.5, and the Platinum® Taq DNA Polymerase HiFi kit (Invitrogen). All amplification reactions were assayed in at least 10 separate tubes to minimize PCR amplification bias. The reaction products were purified using the MinElute PCR Purification Kit (Qiagen) and quantified using a Qubit® Fluorometer and Quant-iT dsDNA BR Assay Kit (Invitrogen).
2.12. Library preparation and sequencing.
High-throughput sequencing was done on an Illumina HiSeq 2000 platform. All libraries from unimmunized animals were prepared using TruSeq DNA Sample Preparation Kit v2 (Illumina Inc.) according to the manufacturer’s instructions (gelmethod version of TruSeq DNA Sample Prep v2 Low Throughput protocol] with minor modifications. Briefly 300–500 ng of PCR fragment was used for each library. MinElute PCR Purification Kit (Qiagen] was used to purify DNA after the end- repaired step. E-gel SizeSelect 2% gel (Invitrogen] was used to purify adaptor ligated products. Enriched library were purified using Agencourt® AMPure® Kit (Beckman Coulter, Inc]. Enriched library were quantified using a Qubit Fluorometer and the Quant-iT HS DNA assay kit (Invitrogen)
All amplicon libraries from immunized animals or stimulated cells were prepared using Nextera XT Index kit (Illumina Inc.] according to the manufacturer’s instructions for “ 16S Metagenomic Sequencing Library Preparation” protocol with minor modifications.
Briefly the 1st stage PCR was performed with KAPA HiFi HotStart ReadyMix (KAPA Biosystems) and the primer pairs designed according protocol guidelines:
1st stage PCR Forward Primer: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTGGCTACCCCCTCTCAGACATCA 3’
1st stage PCR Reverse Primer: 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGAGTCCCCTCTCCAAAAAGCG 3’
1st stage PCR products were purified using the Agencourt® AMPure® Kit (Beckman Coulter, Inc].
Illumina sequencing adapters and dual - index barcodes were added in subsequent 2nd stage PCR using Nextera XT indices and HotStart ReadyMix (KAPA Biosystems]. Indexed library were purified using Agencourt® AMPure® Kit (Beckman Coulter, Inc], quantified using a Qubit Fluorometer and the Quant-iT HS DNA assay kit (Invitrogen). Further library validation, polling and sequencing was performed by BGI Americas.
Sequencing was performed in 150-bp paired-end mode with Illumina HiSeq 2000 chemistry. Raw sequencing reads were processed for FASTQ conversion and demultiplexing by BGI Americas using CASAVA 1.8.2. Sequences were then processed as described in Fig. 2A using an in-house developed software.
Fig. 2. In unimmunized animals HEL-specific T cells are present in the naive, regulatory, effector memory, and central memory T cell compartments.
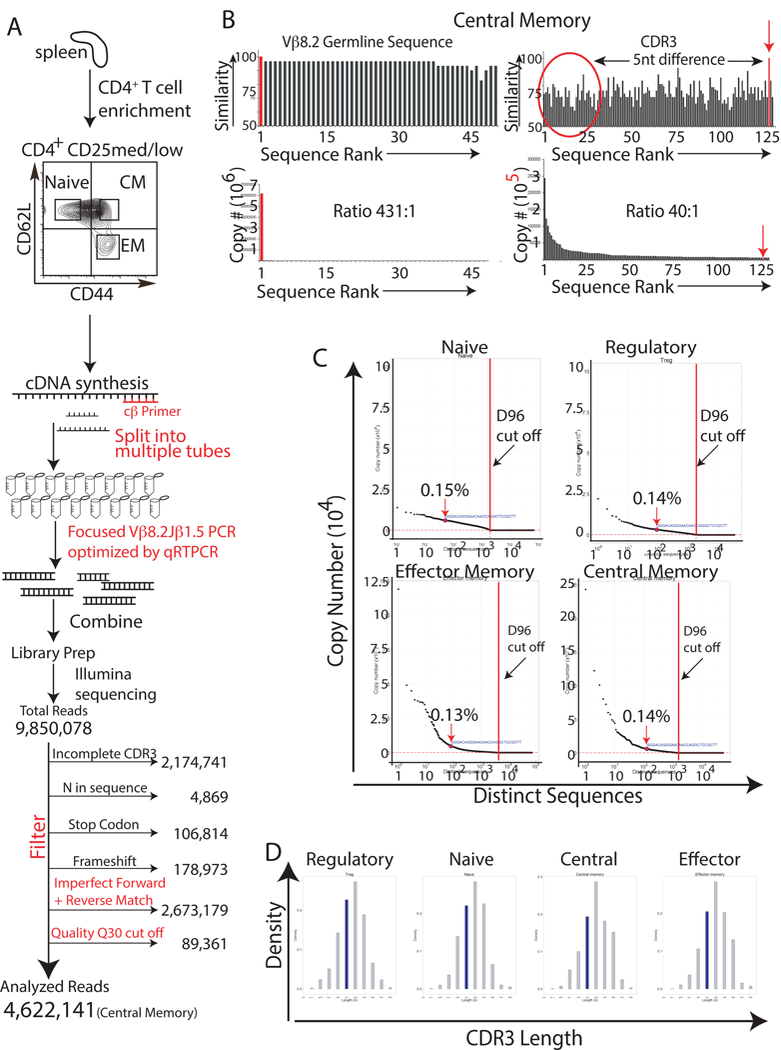
(A) Splenocytes from antigen-naive, 12-to-14-week-old BALB/c mice were FACS sorted to isolate naive, regulatory, effector memory, and central memory T cells using antibodies specific to CD4, CD25, CD127, CD44, and CD62L. RNA was then harvested from the isolated T cells and cDNA was synthesized using a TCR-specific primer to the constant region of the TCR β gene. To minimize amplification bias, the cDNA was then split into multiple PCR reaction tubes. Primers to Vβ8.2 and Jβ1.5 and high high-fidelity Taq DNA polymerase were then used to amplify the CDR3 region of just the Vβ8.2Jβ1.5 subpopulation of T cells. To minimize amplification bias, the number of PCR cyles was optimized by real-time PCR to ensure that PCR reactions were stopped during the linear phase of the amplification. Amplified products were used to generate sequencing libraries. 9,850,078 total reads were obtained from the central memory T cell library. These sequences were then filtered to remove sequences with incomplete CDR3 regions, N’s, and frameshifts. Sequences were also removed if they did not meet a Phred quality score cut-off of 30, or if their forward and reverse sequences did not match perfectly. The remaining 4,622,141 (central memory) CDR3 sequences were then analyzed to determine the frequency of the HEL-specific sequence. The same process was repeated for the other T cell subpopulations. Results are representative of three independent experiments. (B) The germline Vβ8.2 sequence, upstream of the CDR3 region, was used to determine the frequency of sequencing/amplification errors. Similarity scores for the different reads, and the read’s copy number are represented graphically against sequence rank order; the reads were ranked based upon their copy number with “1” being the most abundant read. Results are representative of three independent experiments. (C) Graphs of copy number vs. distinct CDR3 sequence revealed that the HEL- specific Vβ8.2Jβ1.5 CDR3 sequence was present within the naive, regulatory, effector memory, and central memory T cell populations and that the sequence was not expanded when compared with other CDR3 sequences. D96 cut off values(red dot lines) were calculated [5] to identify CDR3 sequences that resided at unacceptably low frequencies, i.e. those that had an increased possibility of originating from amplification or sequencing errors. Results are representative of three independent experiments. (D) In silico spectratyping of CDR3 lengths revealed Gaussian distributions for the naive, regulatory, central memory, and effector memory Vβ8.2Jβ1.5 spectra. Results are representative of at lest three independent experiments.
2.13. PCR for OT-II mice.
Reverse transcription was performed using the iScript™ Advanced cDNA Synthesis Kit for RT-qPCR (Bio-Rad) according to the manufacturer’s instructions. PCR was performed using Platinum® Taq DNA Polymerase HiFi kit (Invitrogen) and either MVβ5.1 and MJβ2.4 primer pairs, or the Mouse RT2 qPCR Primer Assay for the housekeeping gene Tbp (Qiagen). PCR cycle: 94°C for 3min, 94°C for 10 sec, 55°C for 30 sec, 68°C for 30 sec for 30 cycles, followed by a final extension step of 68°C for 5 min. PCR products were visualized on an E-Gel® SizeSelect™ 2% agarose gel (Invitrogen) with the SYBR® Safe DNA Gel Stain (Invitrogen).
2.14. ELISA assaays.
ELISA assay was performed using the Mouse IFN-γ ELISA Kit (Sigma) following the manufacturer’s instructions. Cell culture supernatants were collected and cells were pelleted by centrifugation. Cell-free supernatants were frozen until they were analyzed. Each sample was analyzed in duplicate and IFN-γ concentration in each sample was estimated from an IFN-γ standard curve.
2.15. Data access
The sequences reported in this paper have been deposited in the National Center for Biotechnology Information Sequence Read Archive, www.ncbi.nlm.nih.gov/sra (SRA accession numbers:SRP049789, SRP049700; NCBI BioProject numbers: PRJNA266900, PRJNA266905).
2.16. Statistics
All bar graphs represent mean + standard deviation (SD). Statistical significance was determined using Student t-test. P values less than 0.05 were considered significant. *p < 0.05; **p < 0.01; ***p < 0.001.
3. Results
3.1. Young and aged animals mount similar CD4+ T cell responses to HEL.
As animals age, they become progressively deprived of naïve T cells, as defined by their surface expression of CD25, CD44, and CD62L (med/low, low, and high, respectively). For instance, 18-month-old BALB/c mice are almost entirely devoid of naïve T cells whereas these cells are plentiful in their 7-week-old counterparts (Fig. 1A). Given the discrepancy in the number of naïve T cells between young and aged animals, we sought to compare their T cell responses to a never-before-seen antigen, HEL. As previously reported, young BALB/c mice expanded Vβ8.2Jβ1.5 T cells in response to HEL priming (Fig. 1B). The HEL-specificity of the Vβ8.2Jβ1.5 expansion is evident by its presence in an antigen-specific manner when HEL was used for in vivo priming and in vitro recall; i.e. it was not expanded when lymphocytes were in vitro culture with medium alone (Fig. 1B) or with the control antigen, purified protein derivative of Mycobacterium tuberculosis (PPD) (Fig. S1D). Furthermore, DNA capillary sequencing of the expansion produced the characteristic HEL-specific CDR3-sequence TGTGCCAGCGGGACAGGGAACAACCAGGCTCCGCTT (Table S1), known to be expressed by HEL-specific T cell clones [8]. Like their 7-week-old counterparts, 18- month-old mice also responded to HEL priming by expanding the same HEL-specific T cell clone (Fig. 1B), the sequence of which was also confirmed by DNA capillary sequencing (Table S1). Consistent with prior studies [9], the presence of the Vβ8.2Jβ1.5 HEL-specific expansion correlated with increased IFN-γ production in response to in vitro culture with HEL (Fig. 1C). From these results, we hypothesized that antigen-inexperienced CD4+ T cells may reside in T cell compartments other than the population traditionally labeled as “naïve,” as the clone was detected in older mice that do not have many naïve T cells. We next set out to directly test this hypothesis.
Fig. 1. Aged and young mice expand similar sets of T cells to a HEL.
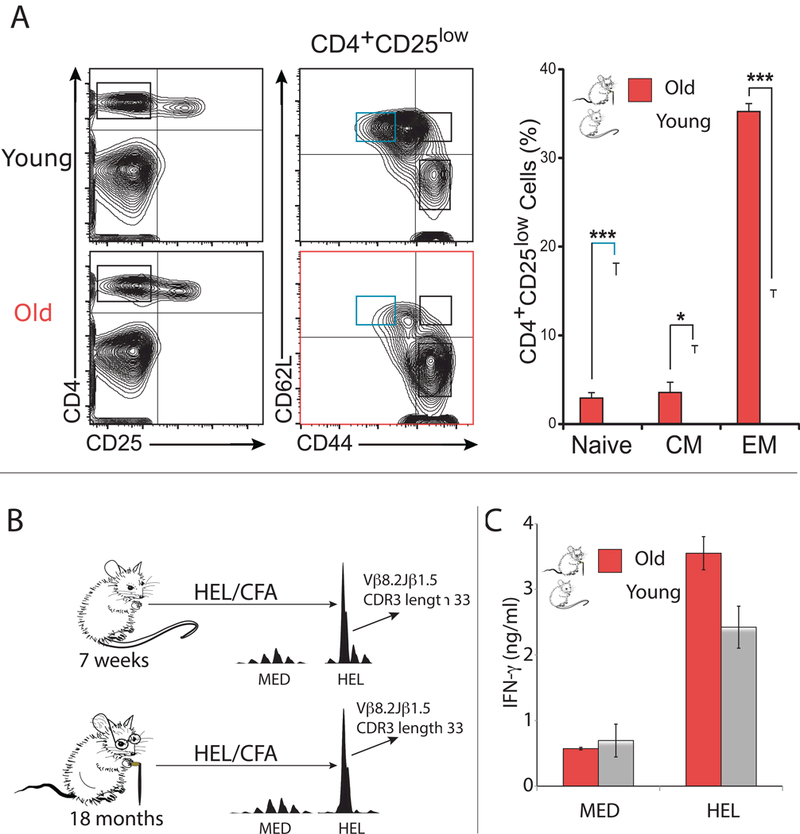
(A) Flow cytometry of mice splenocytes demonstrates that the CD4+, CD25med/low, CD44high, CD62L- T effector memory population is dramatically expanded in aged (18-month-old) BALB/c mice (p<0.001 student’s t-test), while their CD4+, CD25 med/low, CD44low, CD62L+ naïve T cell population is statistically significantly diminished (p<0.001 student’s t-test). (B) Aged (18-month-old) and young (7-week- old) BALB/c mice were immunized subcutaneously with HEL emulsified in CFA. Nine days later, draining lymph nodes were extracted and incubated with HEL or serum-free medium alone. After 72h incubation TCR CDR3 spectrotyping analysis of TCR specific for HELwas performed. Results are representative of three independent experiments. In cultures incubated with HEL, there was a considerable expansion of Vβ8.2Jβ1.5 T cells with a CDR3 length of 36 nucleotide. (C) Aged (18- month-old) and young (7-week-old) BALB/c mice were immunized subcutaneously with HEL emulsified in CFA. Lymph nodes were harvested 9 days later and primary cultures were incubated with HEL or serum-free medium alone. ELISA was performed for IFN-γ detection on the 72-hour supernatants.
3.2. Antigen-inexperienced T cells reside at similar frequencies within the naïve, regulatory, central memory and effector memory T cell compartments.
To determine which T cell compartment(s) contain antigen-inexperienced HEL- specific T cells in unimmunized BALB/c mice, RNA was extracted from FACS- isolated pure populations of central and effector memory T cells (18-month-old mice); and regulatory, naïve, central and effector memory T cells (12-week-oldmice). The entire preparation of RNA was then used to make cDNA. Using multiple reaction tubes the entire pool of cDNA was then used for a focused amplification of just the Vβ8.2Jβ1.5 T cell repertoire (Fig. 2A, S2A, S2B). Following next-generation parallel sequencing of the Vβ8.2Jβ1.5 repertoire, an in-house developed software was used to analyze the raw data in order to extract CDR3 sequences and discard reads that did not satisfy a set of stringent inclusion/exclusion criteria, outlined in Fig. 2A. Specifically, sequences were eliminated if: 1) they contained a frame shift, incomplete CDR3 sequences, or an N in their sequence; 2) their forward and reverse reads did not align perfectly; and 3) if they contained one or more base reads with a Phred Quality Score less than 30. The latter was enforced to insure a base call accuracy of 99.9%. Finally, a D96 cutoff value (explained in reference [5]) was calculated for each set of TCR sequences obtained from the young mice. Sequences that did not meet this cutoff criterion were ignored (i.e. those to the right of the red dotted lines in Fig. 2C). In silico spectratyping of the remaining sequences revealed that T cell populations isolated from unimmunized animals had TCR CDR3 lengths that followed a Gaussian distribution, indicating that large T cell expansions were not present (Fig. 2D and S3A). This was true for all isolated T cell populations (regulatory, naïve, central and effector memory).
Next, the individual reads were examined to determine which of the T cell compartment(s) housed antigen-inexperienced HEL-specific CD4+ T cell clones. Since HEL was a never-before-seen antigen for these animals, we predicted that some of the HEL-specific T cells would reside within the naïve T cell compartment. Indeed, in young mice 0.15% of the Vβ8.2Jβ1.5 CDR3 sequences within the naïve T cell compartment were HEL-specific (Fig. 2C). However, this sequence was also present within the regulatory (0.14%), central memory (0.14%), and effector memory (0.13%) compartments. These results were repeated in 3 independent sorting experiments, all of which yielded similar results (Fig. S1A). The HEL-specific CD4+ clone was also detected at very low frequency within the effector and central memory T cell compartments of the 18-month-old mice (Fig. S3B). In all cases the clone was detected in an unexpanded state, as indicated by its relatively low “sequence rank” (Fig. 2B and C, Fig. S3B, and Fig. S4). To demonstrate the difference in “sequence rank” between antigen-inexperienced T cells and those that have encountered their cognate antigen, BALB/c mice were transiently exposed to HEL:IFA (injected and then removed by surgical excision) or PBS:IFA at four weeks of life. The mice were then sacrificed at 14 weeks and T cell repertoire analysis was conducted on splenic T cells. Fig. 3A demonstrates that a transient exposure to HEL early in life resulted in a 9-fold increase in the frequency of HEL-specific T cell clone, shifting its sequence rank to the left, indicating prior antigen exposure.
Fig 3. Frequency of the characteristic HEL-specific CD4+ T cell clone in antigen- naive and immunized mice.
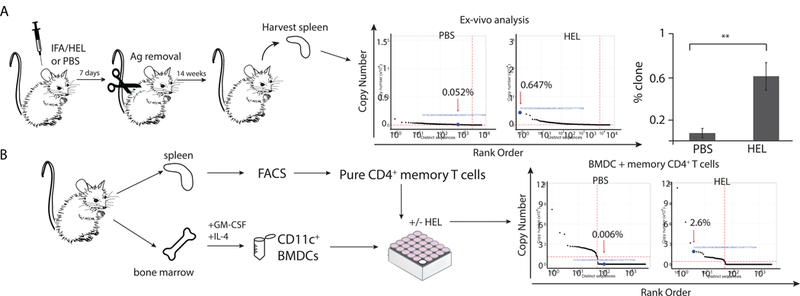
(A) 4 week old BALB/c mice were injected with PBS or HEL emulsified in IFA and four day later removed by surgical excision. The mice were then sacrificed at 12 weeks and for focused Vβ8.2Jβ1.5 TCR next generation repertoire analysis was conducted on splenic T cells. Significance calculated by Student’s t-test. Graphs of copy number vs. distinct nucleotide CDR3 sequence are shown. Red arrow indicates location of HEL-specific Vβ8.2Jβ1.5 CDR3 sequence(CDR3=CASGTGNNQAPL). Results are representative of at lest three independent experiments. (B) Spleen cells from unimmunized 12 week old BALB/c mice were harvested and stained with antibodies specific to CD4, CD25, CD44, CD127, and CD62L. Lymphocytes were then FACS sorted to isolate pure populations of memory (CD4+, CD25med/low, CD44 high, CD62L−/+) T cells. Isolated memory CD4+ T cells were cocultured for 96 hr with in vitro generated BMDCs in presence or absence of HEL. RNA was extracted for focused Vβ8.2Jβ1.5 TCR next generation repertoire analysis. We were unable to generate focused Vβ8.2Jβ1 TCR libraries from BMDCs cultured alone in presence or absence of HEL. These confirms absence of T cell in our BMDCs preparations. Resulting reads were then analyzed using an in-house developed software. Graphs of copy number vs. distinct nucleotide CDR3 sequence are shown. Red arrow indicates location of HEL-specific Vβ8.2Jβ1.5 CDR3 sequence(CDR3=CASGTGNNQAPL).
3.3. Validation of the HEL-specific CDR3 sequence.
Although high fidelity Taq polymerase was used in the PCR reactions, and a set of stringent inclusion/exclusion criteria were employed to remove suboptimal reads (Fig. 2A), the possibility of the HEL-specific TCR sequence appearing from either an amplification error or a sequencing error had to be entertained, as next-generation sequencing platforms are prone to read errors. The software we developed in-house to process the raw next-generation parallel sequencing data extracted CDR3 sequences in part by discarding all reads that did not align perfectly with the CDR3- flanking germline sequences for Vβ8.2 and Jβ 1.5 (Fig. S2C). A stringent set of inclusion/exclusion criteria was then applied to eliminate sequences containing potential errors within the CDR3 region (Fig. 2A). For samples obtained from young mice, D96 cut off values were also calculated for each run [explained in reference [5]. Errors located within the germline Vβ region (i.e. outside of the CDR3 sequence) were then used as standards for sequencing error rate analysis. This analysis revealed that the vast majority of the observed errors were single nucleotide substitutions, which have been previously reported as the most common errors encountered in next-generation parallel sequencing of rearranged TCR genes [14]. For the central memory population, the observed error rate was 1.57% (i.e. 1.57% of the reads contained at least one error within the sequenced germline Vβ8.2 region, 30 residues total). The “similarity score” between the sequences containing these error(s) and the germline Vβ8.2 sequence was uniformly high (greater than 90% for the most common Vβ8.2 sequences with errors) (Fig. 2B). This finding also held true for all other T cell populations examined (regulatory, naïve, and effector memory) (Fig S4). In contrast, there was little similarity between the HEL-specific CDR3 DNA sequence and the most common CDR3 sequences within the central memory population (Fig. 2B). The ratio between the most common central memory Vβ8.2Jβ1.5 CDR3 sequence and the HEL-specific CDR3 sequence was also relatively low (40.3 to 1)(Fig. 2B). It is therefore highly unlikely that the observed HEL-specific CDR3 sequence originated as a result of a sequencing or amplification error. Firstly, the HEL-specific sequence does not show strong similarity to any of the most common CDR3 sequences found within the pool of central memory T cells (Fig. 2B). If the HEL-specific sequence had indeed resulted from a sequencing error, presumably one of the more common CDR3 sequences would have served as its template. Secondly, the HEL-specific sequence was detected in every purified T cell population, a finding that was consistent in all biological replicates for these experiments (Fig. S1A).
Using the highest observed error rate for single nucleotide substitutions within the CDR3-upstream germline region (Fig. S2D), we were able to approximate the probability that our HEL-specific CDR3 sequence was a result of a sequencing or amplification error. For the purpose of this calculation we made three assumptions. 1) Error rates within and outside of the CDR3 region were equivalent. 2) If the HEL- specific CDR3 sequence arose as a byproduct of a sequencing error, one of the non- HEL-specific TCR CDR3 sequences must have served as its template. 3) Only errors resulting in nucleotide substitutions (i.e. not insertions or deletions) could have given rise to the HEL-specific CDR3 sequence. Allowing for these assumptions, we aligned the entire Vβ8.2Jβ1.5 repertoire to the HEL-specific TCR sequence, and then calculated the probability that our observed HEL-specific sequence was generated from another TCR sequence of equal length. By this calculation, the probability that the HEL-specific sequence arose from an error in sequencing was infinitesimally small: 1 × 10-18.
Following completion of our analysis, an open source program, MiTCR, designed to generate error-corrected datasets of TCR CDR3 sequences, was then used to validate our findings [15]. The analysis pipeline of this program uses three logical blocks. First it extracts CDR3 sequences. It then “builds” clonotypes by using high-quality CDR3 reads as “core clonotypes,” to which low quality reads are mapped. Low quality reads may also be dropped depending on the circumstances. The final logic block eliminates sequencing and amplification errors by co-clustering core clonotypes that contain mismatched nucleotides. Although the strategy used by this software was entirely different from that employed by our in-house software, the results were essentially the same; the HEL specific sequence was detected in the naïve, regulatory, central memory, and effector memory T cell populations at similar frequencies (Fig. S1B).
3.4. Validation of the FACS-isolated T cell populations.
In addition to sequencing error, it was also possible that the detected HEL-specific TCR sequence came about due to cross-contamination during T cell purification. To rule out this possibility, a variety of sorting controls were conducted to ensure the accuracy of our FACS protocol. The results from these experiments allowed us to eliminate this possibility, see supplemental materials (Fig. S5, S6, S7).
3.5. Potential for cross-recognition with “self”.
It has been demonstrated that “memory-like” CD8+ T cells can arise from cross-recognition with “self” peptide-MHC complexes [16]. Although the relatively low “sequence rank” of the HEL-specific CD4+ T cell clone makes it unlikely that it acquired its memory phenotype through antigen recognition and expansion, we felt it prudent to search for cross-reactivity to “self”. It has been well established that the Vβ8.2Jβ1.5 HEL-specific CD4+ T cell clone investigated here responds to immunodominant determinant of HEL p:106–116 [8]. Given that the mouse genome codes for two different lysozyme proteins, the mouse analogs to HEL, we sought to characterize the ability of the Vβ8.2Jβ1.5 HEL-specific CD4+ T cell clone to cross- recognize mouse lysozyme (ML). Thus, ML peptides homologous to HELp:106–116, ML1 and ML2, were synthesized and used for in vitro cross-recognition assays (Fig. S1C, D). In the first experiment, BALB/c mice were immunized with PBS emulsified in CFA. Primary lymphocytes were harvested on day 9 and split into parallel cultures that were incubated with either medium alone, HEL, ML1, ML2, or PPD. Only the cultures incubated with HEL demonstrated a significant in vitro expansion of the Vβ8.2Jβ1.5 HEL-specific T cell clones (Fig. S1C). In a related experiment mice were immunized with HEL emulsified in CFA. This resulted in an in vivo expansion of the Vβ8.2Jβ1.5 HEL-specific T cell clone, which expanded to occupy approximately 15% of the Vβ8.2Jβ1.5 T cell repertoire (Fig. S1D). However, in vitro culture of the HEL-primed lymphocytes with ML1, ML2, or PPD failed to expand this clone further. Only incubation with HEL resulted in further T cell proliferation (Fig. S1D). From these experiments we can conclude that the Vβ8.2Jβ1.5 HEL-specific T cell clone does not expand to mouse lysozyme, which shares significant sequence homology with HEL.
3.6. Responsiveness of Vβ8.2Jβ1.5 HEL-specific antigen-inexperienced T cells to HEL.
The above experiments assume that T cells expressing the Vβ8.2Jβ1.5-encoded CASGTGNNQAPL β chain are responsive to HEL. However, although the CASGTGNNQAPL sequence is clearly seen in HEL-specific T cell clones, it is possible that some CASGTGNNQAPL β chain T cells are NOT HEL-specific due to an their a chain. Thus, to prove that at least some of the Vp8.2JP1.5-encoded CASGTGNNQAPL T cells present in the memory pool were indeed HEL-specific we sought to validate their ability to expand in response to HEL. To demonstrate this, a highly purified population a CD4+ CD25 med/low CD62L+/− CD44high memory T cells were FACS isolated from HEL-naïve BALB/c mice and cultured in the presence and absence of HEL together with bone marrow derived CD11c+ dendritic cells, serving as antigen presenting cells for the in vitro assay. In vitro culture with HEL for four days expanded the Vβ8.2Jβ1.5-encoded CASGTGNNQAPL clone more than 400 fold (Fig. 3B). This expansion was not seen in control cultures, solidifying the existence of unexpanded HEL-specific and responsive T cells in the memory CD4+T cell compartment.
3.7. Young and aged animals are able to regenerate dominant TCR sequences following treatment with T cell-depleting antibodies.
In experimental animal models it has been well characterized that a few T cell clones can “drive” an autoreactive immune response 14,34. In these experimental systems T cell-directed, including clone-specific therapeutic strategies, can abrogate the pathogenic immune response.36 We thus sought to determine if the immune system is able to recover a dominant T cell expansion following antibody-mediated T cell depletion. Using an anti-CD4 T cell depleting monoclonal antibody, young (7 weeks) and aged (18 months) BALB/c mice were rendered temporarily devoid of CD4+ T cells (Fig. 4A). The depletion lasted for 3-weeks, after which the CD4+ T cell population recovered spontaneously (Fig 4A). Following T cell recovery, mice were then immunized with an HEL:CFA (Complete Freund’s Adjuvant) emulsion and draining lymph nodes were harvested 9 days later. T cell repertoire analysis of the draining lymph node cells revealed that following CD4+ T cell depletion and recovery young and aged mice were able to regenerate a characteristic HEL-specific Vβ8.2Jβ1.5 T cell clone (Fig. 4B). Thus, even in the presence of thymic atrophy, the immune system is able to regenerate a set of dominant T cells, which is remarkable when one considers number of potential CDR3 sequences (1010) that can be generated by genetic recombination. However, an alternative possibility is that our T cell depletion strategy was incomplete. Thus, we sought to confirm these findings using a more definitive depletion strategy.
Fig 4. The immune system is able to regenerate characteristic T cells after T cell depletion.
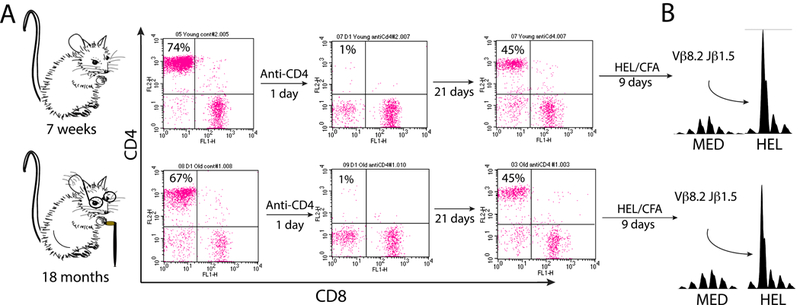
Young (7 weeks) and old (18 month) BALB/c mice were injected with 300 μg of a T cell-depleting CD4-specific monoclonal antibody. (A) One day following depletion flow cytometry was done to confirmed the selective loss of CD4+ T cells. Repeat flow cytometry 21 days later confirmed repletion of the CD4+ T cell compartment. (B) Following return of the CD4+ T cells mice were immunized subcutaneously with HEL emulsified in CFA. Lymph nodes were harvested 9 days later and primary cultures were incubated for 72h with HEL or serum-free medium alone. TCR CDR3 spectrotyping analysis revealed expansion of the characteristic HEL-specific Vβ8.2Jβ1.5 T cell. Results are representative of four independent experiments.
3.8. Dominant T cells are regenerated following hematopoietic stem cell transplantation.
Autologous hematopoietic stem cell transplantation is a promising therapy for autoimmunity and has been successfully used to treat refractory autoimmune diseases 20,21. However, after experiencing initial improvement, relapses are common 37. One possible explanation is that the transplantation protocol successfully eliminates all autoreactive clones but then, with time, the posttransplant immune system regenerates a new set of pathogenic T cells.
Thus, as a more stringent test of the ability of the immune system to regenerate dominant T cells, BALB/c mice underwent the same anti-CD4 treatment, but in addition received a lethal dose of irradiation (850cGy) followed by autologous bone marrow transplantation. After lymphocyte recovery (i.e. six weeks status post BMT) BALB/c mice were immunized with HEL/CFA and lymph nodes were harvested 9 days afterwards. Although the BMT mice responded relatively well to HEL/CFA, as measured by IFN-γ production (Fig. 5B) and clonal T cell expansions (Fig. 5C), the characteristic Vβ8.2Jβ1.5 HEL-specific T cell clone was not detected (Fig. 5A). Thus, at six weeks post BMT the T cell repertoire lacked the TCR CDR3 diversity observed in the pre-transplant immune system. One possibility was that 6 weeks was an insufficient amount of time to regenerate a full repertoire of available TCRs. We therefore repeated the experiment, this time waiting 100 days (Fig. 5D) to immunize the post-transplanted animals with HEL/CFA. Fig. 5E and5F reveals that at 100 days post BMT, BALB/c mice responded to HEL/CFA similarly to mice that had not undergone BMT. Thus, although lymphocyte recovery was present at six weeks, T cell repertoire diversity was not fully restored until 100 days post-BMT.
Fig 5. Following hematopoietic stem cell transplantation (HSCT) dominant characteristic T cells are regenerated.
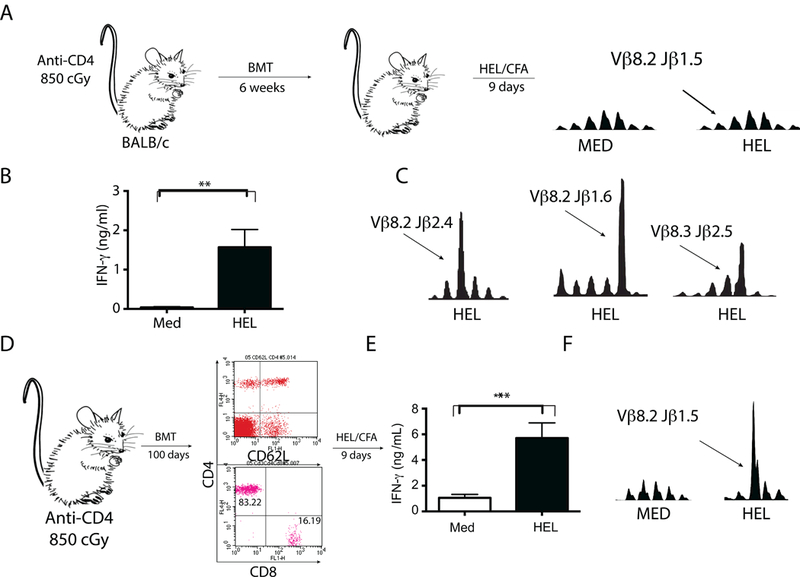
(A) BALB/c mice were lethally irradiated and injected with 300 μg of a T cell- depleting CD4-specific monoclonal antibody. They were then rescued via intravenous transplantation of T cell-depleted syngeneic bone marrow cells (106 cells). Following lymphocyte recovery (6 weeks) mice were immunized subcutaneously with HEL emulsified in CFA. Lymph nodes were harvested 9 days later and primary cultures were incubated for 72h with HEL or serum-free medium alone. TCR CDR3 spectrotyping analysis reveald that the characteristic HEL-specific Vβ8.2Jβ1.5 T cell clonotype was not significantly exanded. Results are representative of four independent experiments. (B) ELISA was performed for IFN-γ detection on the 72-hour supernatants. Although the characterisitc expansion was not observed, mice were able to secrete significant amounts of IFN-γ (p = 0.007, Student’s t-test) and (C) were able to expand other HEL-specific T cell clones. (D) A second group of mice were treated exactly as described in “A” with exception that their immune system was allowed to recover for a longer period of time (100 days). Flow cytometry was done to confirmed the repletion of the CD4+ T cell compartment. Mice were immunized subcutaneously with HEL emulsified in CFA. Lymph nodes were harvested 9 days later and primary cultures were incubated for 72h with HEL or serum-free medium alone. (E) ELISA was performed for IFN-γ detection on the 72-hour supernatants. Results are representative of four independent experiments. Significant increase in IFN-γ secretion (p = 0.0014, Student’s t-test) was detected in culture supernantants when lymphocytes were incubated with HEL and (F) TCR CDR3 spectrotyping analysis demonstrated that the characteristic HEL-specific Vβ8.2Jβ1.5 T cell clonotype was strongly exanded in respone to culture with HEL. Results are representative of four independent experiments.
3.9. Autoreactive T cells are regenerated following hematopoietic stem cell transplantation.
In the cases just described the regenerated T cell was specific to a foreign antigen, HEL. We next sought to repeat these findings in a model with more relevance to autoimmunity, the MBP:Ac1–9 B10.PL experimental autoimmune encephalomyelitis model of multiple sclerosis. It is well-known that B10.PL mice respond to MBP:Ac1– 9 by expanding an encephalitogenic VP8.2JP2.7 IFN-γ-secreting T cell clone (CDR3 sequence XXXXDAGGGYXXXX ) 14,34. This clone is known to migrate into the central nervous system and cause experimental autoimmune encephalomyelitis (EAE) 14,32,36. Corroborating its dominant role in this disease is the finding that its elimination protects animals from EAE induction and its adoptive transfer induces severe EAE 14, 36. The MBP:Ac1–9-specific Vβ8.2Jβ2.7 T cell clone is also known to express CD4 and has a high affinity for MBP:Ac1–9 when presented by the I-Au MHC class II molecule 38,39. Similar to the results obtained with the BALB/c mouse, Fig. 6A demonstrates that at 100 days post BMT, transplanted B10.PL mice respond to MBP:Ac1–9 priming by expanding the characteristic MBP:Ac1–9-specific VP8.2JP2.7 clone (Fig. 6C). Correlating well with the observed VP8.2JP2.7 T cell expansion, the post BMT MBP:Ac1–9-primed mice developed severe EAE and produced large amounts of IFN-γ (Fig. 6D and6B).
Fig. 6. Following hematopoietic stem cell transplantation (HSCT) dominant autoreactive T cells are regenerated.
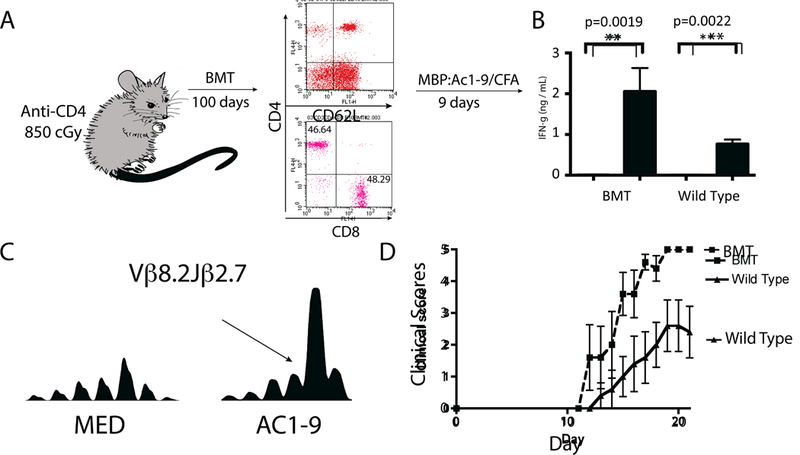
(A) B10.PL/J mice were lethally irradiated and injected with a CD4-depleting monoclonal antibody, GK1.5. They were then rescued via intravenous transplantation of T cell-depleted syngeneic bone marrow cells (106 cells). Flow cytometry was done to confirmed the repletion of the CD4+ T cell compartment. 100 days status-post transplantation mice were immunized with the self antigen P:Ac1–9 and lymph nodes were harvested 9 days afterwards. Non-transplanted mice were also immunized with MBP as control. Following MBP priming draining lymph nodes were harvested and incubated with either MBP or medium alone. (B) ELISA was performed for IFN-γ detection on the 72-hour supernatants. Results are representative of four independent experiments. Significant increase in IFN-γ secretion was detected in lymphocyte cultures incubated with autoantigen MBP:Ac1–9. (C) TCR CDR3 spectrotyping analysis demonstrated that the characteristic dominant Vβ8.2Jβ2.7 T cell clonotype was strongly expanded when lymphocytes were cultured with 20 mcg/ml MBP:Ac1–9. Results are representative of four independent experiments. (D) Experimental autoimmune encephalomyelitis (EAE) was induced in five mice that had undergone syngeneic HSCT and five non- transplanted control mice. Following EAE induction, mice that had undergone syngeneic HSCT developed worse EAE (p=0.01, Student’s t-test] than their none transplanted counterparts.
4. Discussion
Antigen-naïve CD4+ T cells differentiate into memory and effector T cells after they encounter their cognate antigen. Following antigen recognition and T cell activation, changes to the T cell’s surface phenotype occur, marking the transition from a naive T cell to either an effector or memory cell. The goal of this manuscript was to challenge the currently accepted notion that naive and memory CD4+ T cells can be easily distinguished from one another by their differential expression patterns of cell surface markers. Based on their expression of cell-surface markers CD4+ T cells are often classified into different “compartments”: naïve (CD4+ CD25low CD62L+ CD44low), effector (CD4+ CD25low/medium CD62L- CD44high), and effector memory (CD4+ CD25low/medium CD62L+ CD44high) T cells. Although it is clear that the recent thymic emigrants are housed within the naive T cell compartment (CD4+ CD25low CD62L+ CD44low), this does not exclude the possibility that antigen-inexperienced CD4+ T cells may possess alternative cell surface phenotypes. The impetus for our research was the finding that regulatory and memory T cell repertoires actually have a surprisingly diverse set of TCR CDR3 sequences. If T cell activation was the sole mechanism by which a T cell can acquire a memory phenotype, then the naive T cell compartment should be markedly more diverse than the memory compartment, and this is not the case. Also, as an animal ages its naive T cells occupy a progressively smaller T cell population, yet for the majority of life adults retain the remarkable ability to mount robust T cell responses to new antigen encounters. Thus, to resolve these discrepancies, we sought to answer a simple but fundamental question: where do antigen inexperienced CD4+ T cells reside? This question also has direct implications to autoimmunity because theoretically memory T cells require less stimulation to mount robust proliferative responses, making the possibility of expanding a clone to a poorly stimulating self antigen more likely. As our first step we investigated how an aged animal responds to a novel foreign antigen.
In both humans and mice it has been established that T cell repertoire diversity is greatly diminished with extreme age [17, 18]. Specifically, large T cell expansions dominate the T cell repertoire in extreme age, resulting in a loss of TCR diversity. These findings have been best replicated for CD8+ T cells, and the loss of diversity has been attributed to thymic atrophy as well as the expansion of virus- specific T cells [18]. In some cases, the expanded memory population has been described as “senescent,” which is a state of cellular inactivity thought to predispose the elderly to infection and cancer [19, 20]. However, an alternative view is that age- related loss of T cell diversity is the result of a small number of T cell clones gaining a “fitness advantage” through genetic mutation [21]. The experiments presented herein do not focus on T cell repertoire perturbations associated with extreme age. Instead we chose to study 18-month-old mice because they have a dramatically decreased naive T cell reservoir but are otherwise healthy. Our 18-month-old BALB/c animals appeared normal with respect to the distribution of their CDR3 sequences (Fig. 1B), which is consistent with the belief that loss of T cell repertoire diversity is a fairly sudden event [21].
When immunized with HEL-CFA, 18-month-old BALB/c mice mounted T cell responses that were similar to their younger, 7-week-old counterparts; they expanded the exact same HEL-specific T cell clone (Fig. 1B), which produced comparable amounts of IFN-γ (Fig. 1C). Given that HEL was a novel antigen to these animals, we hypothesized that prior to immunization some antigen-inexperienced T cells may have resided outside of the “naïve” T cell compartment, as the aged animals had very few naive T cells. To directly demonstrate the existence of antigen- inexperienced T cells within the memory T cell compartments, we selected CD25, CD44, and CD62L for our sorting strategy as these are the most common cell surface markers used to identify memory T cells in mice. By conducting deep sequencing, we were able to detect the HEL-specific CD4+ T cells within naive, regulatory, central memory, and effector memory T cell compartments at relatively similar frequencies in mice that had never-before-seen HEL. Given that our results were reproduced in three separate biological replicates, we can confidently state that antigen- inexperienced T cells reside in the naive, regulatory, central memory, and effector memory T cell compartments.
There are a number of plausible theories to account for the existence of antigen-inexperienced clones in the memory and regulatory compartments of an unprimed animal. For instance, the ability of a TCR to cross-recognize multiple antigens has been extensively studied [22]. Thus, even though the mice had no prior history of HEL exposure, the Vβ8.2Jβ1.5 HEL-specific “memory” T cells may have arisen in response to cross-recognition of another antigen that the animal happened to have encountered. In support of this possibility, it has been demonstrated in humans that memory-phenotype CD4+ T cells are abundant in antigen-unexposed adults [23]. It is believed that these T cells develop their memory phenotype through cross-recognition of other pathogens. With respect to our data (Fig. 2B and C), cross-recognition of environmental antigens cannot fully explain our findings, as the antigen-inexperienced cells were present at similar frequencies in the naive and memory pools. In addition, the “sequence rank” of the HEL-specific CDR3 sequence was relatively low in all T cell populations analyzed. Thus, we contend that since the HEL-specific T cells reside at the same low frequency within the “naive” and “memory” compartments, it is highly unlikely that their memory phenotype was a result of a clonal expansion event in response to a molecular mimic [24]. Although a low level of cross-recognition with “self” peptide-MHC complexes could not be excluded, we were unable to detect significant cross-recognition to “self” ML, the mouse analog of HEL (Fig. S1 C and D). Apart from cross-recognition, some might argue that the β-chain alone does not impart HEL-specificity. However, we were able to easily expand the same VP8.2JP1.5 HEL-specific clone by culturing lymphocytes from HEL-naive animals in vitro with HEL, which demonstrates that at least some were HEL-specific (Fig. S1C). In addition, when we cultured highly purified memory CD4+T cells with dendritic cells as APCs and soluble HEL, we clearly expanded the Vβ8.2Jβ1.5 HEL-specific clone from the memory population of antigen-naive animals (Fig. 3B).
It is known that commensal microbiota participate in the normal development of some T cells [25]. These organisms could have induced the antigen- naive HEL-specific T cells to acquire cell surface markers indicative of immunologic memory without inducing them to undergone a clonal expansion event. This possibility was not directly addressed in this manuscript. Regardless of how it occurs, it appears that as antigen-inexperienced T cells age they acquire a memory phenotype (Fig. 7). Similar observations have been reported for CD8+ T cells. These include CD8+ T cells acquiring memory surface markers as a result of homeostatic proliferation and bystander activation. Terms such as “CD8 virtual memory” and “innate CD8 memory” have been applied to CD8+ memory T cells that have acquired their memory phenotype through alternative mechanisms [26–29]. Such pathways would allow TCR diversity to be preserved in the memory compartment despite a diminishing naive T cell population, thereby ensuring that an individual maintains the ability to respond to newly encountered antigens post thymic atrophy.
Fig. 7. Pathway of conventional and unconventional naive CD4+ T cell differentiation.
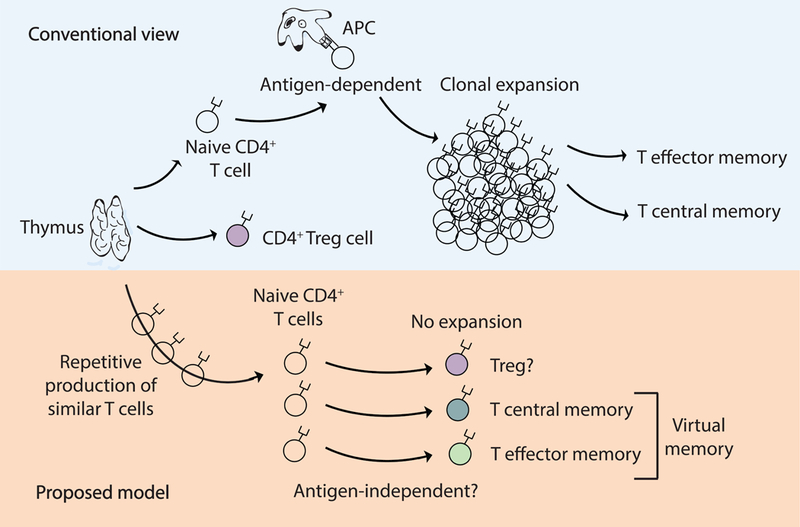
Conventional view: Upon cognate antigen recognition, naive CD4+ T cells differentiate into effector cells and form “true” antigen-experienced memory cells. Proposed model: Under physiological conditions, naive CD4+ T cells may also acquire a memory or regulatory phenotype via mechanisms that do not require strong antigen recognition and T cell expansion. Driving forces for the generation of “virtual” memory T cells from naive T cells are currently unknown but may include physiologic aging of the cell, low cross-reactivity with self, and cross-recognition with commensal microbiota. The existence of “virtual” memory CD4+ T cells has direct implication for autoimmunity as it is possible for autoreactive immune responses to arise from crossreactivity directly within the T cell memory pool.
Our results also demonstrate that the frequency of HEL-specific T cells within the regulatory compartment is similar to that of the naïve compartment (Fig. 2C and S4). Although many reports have demonstrated that the Treg compartment is biased towards self-antigen recognition, this notion is not strongly supported by T cell repertoire analysis [33, 34]. Our results also do not support the predominant belief that Treg cells are biased towards self-antigen recognition [30]. One limitation of our study is that the functionality of the sorted Treg population was not examined, although they were highly uniform with respect to their FOXP3 expression (Fig. S7).
Another interesting aspect of our study was the finding that the foreign- specific and self-directed T cells behaved very similarly with respect to their ability to repopulate following T cell depletion despite having very different TCRs. The CDR3 sequence of the MBP:Ac1–9-specific I-Au-restricted VP8.2JP2.7 clone differed dramatically from the HEL-specific I-Ed restricted Vβ8.2Jβ1.5 clonotype. A few days after birth the immune system up-regulates the terminal deoxynucleotidyl transferase enzyme, which adds N-nucleotides to the V, D, and J exons during TCR gene recombination. Thus, the very first T cells produced by the immune system lack N addition. Interestingly, the Vβ8.2Jβ1.5 HEL-specific T cells lack N-additions within their CDR3 sequence [10]. In contrast, the MBP:Ac1–9-specific Vβ8.2Jβ2.7 clone contains N-additions within its CDR3 region [14]. Thus, the ability of the immune system to continuously replenish the same sets of T cells holds true for all types of T cell clones, not just those lacking N-additions.
Also of interest is our finding that aged animals maintained their ability to regenerate dominant T cell clones. Based on this finding we hypothesize that the skewed T cell repertoire known to accompany extreme age is not a result of normal aging but rather represents a pathologic process, possibly related to virus-specific aberrant T cell expansions, genetic mutations providing some T cells with a “fitness advantage”, or bone marrow failure. Our results also demonstrate that aged but otherwise healthy animals have a T cell repertoire, of similar composition to their younger counterparts. Regardless of age or haplotype, it appears that the immune system continuously produces the same set of dominant T cells throughout the life of the individual. These results have implications for clone-specific therapeutic strategies and the use of autogenetic HSCT for the management of autoimmune diseases.
4.1. Conclusions
In summary, by conducting highly focused TCR sequencing we have demonstrated that an antigen-inexperienced CD4+ T cell clone exists in all of the archetypical T cell compartments. Our results are unique in that we were able to reliably and reproducibly detect a particular unexpanded T cell clone from within a vastly diverse population of background T cells that reside within the same animal. This finding strongly argues against pathogen-derived antigen recognition as the sole means of developing a “memory” phenotype. They support the existence of an alternative pathway of generating immunologic “memory”, which based on the diversity of the memory repertoire appears to be the dominant pathway (Fig. 7). Our results also demonstrate that the immune system can continuously generate the same set of dominant T cells throughout the life of the individual. These results have direct implications for the pathophysiology of autoimmunity.
Supplementary Material
Spleen cells from unimmunized 12–14 week old BALB/c mice were harvested and stained with antibodies specific to CD4, CD25, CD44, CD127, and CD62L. Lymphocytes were then sorted to isolate pure populations of naive (CD4+, CD25med/low, CD44low, and CD62Lhigh], regulatory (CD4+, CD25high, CD127low], effector memory (CD4+, CD25med/low, CD44 high, CD62L-), and central memory (CD4+, CD25med/low, CD44 high, CD62L+) T cells. RNA was extracted from the isolated T cell populations for focused Vβ8.2Jβ1.5 TCR next generation repertoire analysis. Resulting reads were then analyzed using an in-house developed software or (B) the recently published MiTCR software (15). Although the frequencies were slightly different, both methods detected HEL-specific T cell clones within the naive, central memory, effector memory, and regulatory T cell populations. (C) 12 week old BALB/c mice were immunized with PBS emulsified in CFA. 9-days later lymph nodes were removed and parallel in vitro cultures were incubated with either ML1, ML2, Medium, HEL, or PPD. Cells were harvested on day 3 for focused Vβ8.2Jβ1.5 TCR next generation repertoire analysis. Abundance of HEL-specific T cell clone (CDR3=CASGTGNNQAPL) relative to other CDR3 sequences was calculated. Results represent 3 biological replicates. Significance calculated by Student’s t-test. (D) 12 week old BALB/c mice were immunized with HEL emulsified in CFA. 9-days later lymph nodes were harvested and analyzed as described in “C”. Again, only incubation with HEL resulted in a statistically significant expansion of HEL-specific T cells (CDR3=CASGTGNNQAPL) greater than medium alone. Results represent 3 biological replicates. Significance calculated by Student’s t-test.
Gene segment sequences for TRBV13–2*01 (VP8.2) and TRBJ1–5*01 (JP1.5) were obtained from the international ImMunoGeneTics information system (IMGT). (A) The exact sequence of the TRBV13–2*01 - TRBJ1–5*01 gene rearrangement that encodes the CDR3 loop of the HEL-specific TCR β chain. V and J sequences lying outside of the CDR3 region are also shown. (B) Primers used to amplify the TRBV13– 2*01 TRBJ1–5*01 TCR sequence. Note that the TRBJ1–5*01 primer does not capture a few gene rearrangements. (C) Depiction of the motifs within the V and J segments used to identify reads containing a complete CDR3 region. (D) Depiction of the motifs used to identify the 12nt region that was used to calculate the sequencing/amplification error rate.
Splenocytes from antigen-naive 18 month old BALB/c mice were sorted to isolate effector memory and central memory CD4+T cells using antibodies specific to CD4, CD25, CD44, and CD62L. RNA was then harvested from the isolated T cells and used to generate focused Vβ8.2Jβ1.5 TCR libraries that were then sequenced using the HiSeq 2000 platform. The sequences were then filtered to remove sequences with incomplete CDR3 regions, N’s, and frameshifts. Sequences were also removed if they did not meet a Phred quality score cut-off of 30, or if their forward and reverse sequences did not match perfectly. (A) In silico spectratyping of CDR3 lengths revealed Gaussian distributions for the central memory and effector memory Vβ8.2Jβ1.5 spectra. Results are representative of at lest three independent experiments. (B) Graphs of copy number vs. distinct CDR3 sequence revealed that the HEL-specific Vβ8.2Jβ1.5 CDR3 sequence was present within the effector memoryand central memory T cell populations and that the sequence was not expanded when compared with other CDR3 sequences. Results are representative of at lest three independent experiments.Graphs for nucleotide and amino acid CDR3 sequences are shown separately.
To characterize the types of errors and to estimate the frequency of the amplification/sequencing errors encountered when sequencing TCR CDR3 gene rearrangements, the germline Vβ8.2 region, which lies just upstream of the CDR3 region, was analyzed. Similarity scores for the different sequences, and the their copy number are represented graphically against the sequence’s rank order; reads were ranked based upon their copy number with “1” being the most abundant read. Likewise, the similarity scores and copy numbers of the individual sequences corresponding to the CDR3 region were compared. Red bars indicate either the correct germline Vβ8.2 sequence or the characteristic HEL-specific Vβ8.2Jβ1.5 TCR CDR3 sequence. In each case the similarity between the HEL-specific CDR3 sequence and the most abundant CDR3 sequence was low.
FACS was used to isolate CD4+ CD25low (Treg )T cells from OT-II TCR transgenic mice. The isolated cells were then added to a unsorted of wild-type lymphocytescontaining all T cell populations (naive, memory, and regulatory). The mixture of wild-type lymphocytes and transgenic CD4+ CD25low T cells was then subjected to a second round of FACS to isolate Treg and non-Treg populations. A TCR transgene- specific PCR was conducted on the Treg, non-Treg sorted populations and wild-type lymphocytes to confirm the accuracy of our sorting strategy. TBP housekeeping gene served as loading control. Transcripts matching the transgenic TCR were present only within the FACS isolated CD4+ CD25low population.
Effector memory T cells (CD4+, CD25med/low, CD44 high, CD62L-) were isolated by FACS and then added to a unsorted of lymphocytes that differed at their CD45 allele, detectable by an allele-specific anti-CD45 antibody. The mixture (containing regulatory, naive, and memory T cells) was then analyzed by flow cytometry. Results from this experiment revealed that the CD45-mismatched effector memory T cells were detected solely within their corresponding effector memory population (CD4+, CD25med/low, CD44high, CD62L- cells).
Tregs (CD4+, CD25high, CD127low cells) were isolated using FACS. The isolated T cells were then permeabilized and stained intracellularly with fluorescently labeled antibodies specific to the transcription factor Foxp3. The T cells were then reanalyzed by flow of purity of Tregs.
Highlights.
Antigen-inexperienced CD4+ T cells comprise a portion of the memory repertoire
CD4+ T cells can display a memory phenotype without clonal expansion to an antigen
Our study supports the existence of CD4+ virtual memory T cells
These findings posit a new paradigm for T cell maturation
Acknowledgments
Funding sources: EM is supported by career awards from the Howard Hughes Medical Institute and the Burroughs Wellcome Fund. This project was supported NIH 1DP2OD008752.
Footnotes
Author contributions: E.M., G.L., L.O., I.E.A. and A.I.M. designed research; Y.O., A.I.M., K.K., H.O., Y.M., I.B.V., J.B., M.S. and A.M.S. performed research; A.A.M. contributed new reagents/analytic tools; A.I.M, A.A.M. and Y.O. analyzed data; E.M., T.D.Y., A.I.M. and D.G.K. wrote the manuscript; Y.O., K.K., H.O., Y.M., E.A.W., T.D.Y., I.B.V., J.B., A.M.S., and I.E.A. revised the manuscript.
Conflicts of Interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Sallusto F, Lenig D, Forster R, Lipp M, & Lanzavecchia A (1999) Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401(6754):708–712. [DOI] [PubMed] [Google Scholar]
- [2].Bielekova B, et al. (2000) Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med 6(10):1167–1175. [DOI] [PubMed] [Google Scholar]
- [3].Kimmig S, et al. (2002) Two subsets of naive T helper cells with distinct T cell receptor excision circle content in human adult peripheral blood. J Exp Med 195(6):789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Davis MM & Bjorkman PJ (1988) T-cell antigen receptor genes and T-cell recognition. Nature 334(6181):395–402. [DOI] [PubMed] [Google Scholar]
- [5].Warren RL, et al. (2011) Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res 21(5):790–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pannetier C, et al. (1993) The sizes of the CDR3 hypervariable regions of the murine T-cell receptor beta chains vary as a function of the recombined germ-line segments. Proc Natl Acad Sci U S A 90(9):4319–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Freeman JD, Warren RL, Webb JR, Nelson BH, & Holt RA (2009) Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res 19(10):1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cibotti R, et al. (1994) Public and private V beta T cell receptor repertoires against hen egg white lysozyme (HEL) in nontransgenic versus HEL transgenic mice. J Exp Med 180(3):861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Foucras G, Gapin L, Coureau C, Kanellopoulos JM, & Guery JC (2000) Interleukin 4-producing CD4 T cells arise from different precursors depending on the conditions of antigen exposure in vivo. J Exp Med 191(4):683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Maverakis E, et al. (2000) T cell receptor complementarity determining region 3 length analysis reveals the absence of a characteristic public T cell repertoire in neonatal tolerance. The response in the “tolerant” mouse within the residual repertoire is quantitatively similar but qualitatively different. J Exp Med 191(4):695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gapin L, et al. (1998) Antigen presentation by dendritic cells focuses T cell responses against immunodominant peptides: studies in the hen egg-white lysozyme (HEL) model. J Immunol 160(4):1555–1564. [PubMed] [Google Scholar]
- [12].Rudolph M, Hebel K, Miyamura Y, Maverakis E, & Brunner-Weinzierl MC (2011) Blockade of CTLA-4 decreases the generation of multifunctional memory CD4+ T cells in vivo. J Immunol 186(10):5580–5589. [DOI] [PubMed] [Google Scholar]
- [13].Klarenbeek PL, et al. (2010) Human T-cell memory consists mainly of unexpanded clones. Immunol Lett 133(1):42–48. [DOI] [PubMed] [Google Scholar]
- [14].Nguyen P, et al. (2011) Identification of errors introduced during high throughput sequencing of the T cell receptor repertoire. BMC Genomics 12:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bolotin DA, et al. (2013) MiTCR: software for T-cell receptor sequencing data analysis. Nat Methods 10(9):813–814. [DOI] [PubMed] [Google Scholar]
- [16].Rudd BD, et al. (2011) Nonrandom attrition of the naive CD8+ T-cell pool with aging governed by T-cell receptor:pMHC interactions. Proc Natl Acad Sci U S A 108(33):13694–13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Naylor K, et al. (2005) The influence of age on T cell generation and TCR diversity. J Immunol 174(11):7446–7452. [DOI] [PubMed] [Google Scholar]
- [18].Posnett DN, Sinha R, Kabak S, & Russo C (1994) Clonal populations of T cells in normal elderly humans: the T cell equivalent to “benign monoclonal gammapathy”. J Exp Med 179(2):609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Voehringer D, et al. (2001) Viral infections induce abundant numbers of senescent CD8 T cells. J Immunol 167(9):4838–4843. [DOI] [PubMed] [Google Scholar]
- [20].Smithey MJ, Li G, Venturi V, Davenport MP, & Nikolich-Zugich J (2012) Lifelong persistent viral infection alters the naive T cell pool, impairing CD8 T cell immunity in late life. J Immunol 189(11):5356–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Johnson PL, Yates AJ, Goronzy JJ, & Antia R (2012) Peripheral selection rather than thymic involution explains sudden contraction in naive CD4 T-cell diversity with age. Proc Natl Acad Sci U S A 109(52):21432–21437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wucherpfennig KW & Strominger JL (1995) Molecular mimicry in T cell- mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 80(5):695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Su LF, Kidd BA, Han A, Kotzin JJ, & Davis MM (2013) Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity 38(2):373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Maverakis E, et al. (2010) Molecular mimics can induce a nonautoaggressive repertoire that preempts induction of autoimmunity. Proc Natl Acad Sci U S A 107(6):2550–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mazmanian SK, Liu CH, Tzianabos AO, & Kasper DL (2005) An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122(1):107–118. [DOI] [PubMed] [Google Scholar]
- [26].Sprent J & Surh CD (2011) Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol 12(6):478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lee YJ, Jameson SC, & Hogquist KA (2011) Alternative memory in the CD8 T cell lineage. Trends Immunol 32(2):50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Haluszczak C, et al. (2009) The antigen-specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J Exp Med 206(2):435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Renkema KR, Li G, Wu A, Smithey MJ, & Nikolich-Zugich J (2014) Two separate defects affecting true naive or virtual memory T cell precursors combine to reduce naive T cell responses with aging. J Immunol 192(1):151– 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hsieh CS, et al. (2004) Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity 21(2):267–277. [DOI] [PubMed] [Google Scholar]
- [31].Menezes JS, et al. (2007) A public T cell clonotype within a heterogeneous autoreactive repertoire is dominant in driving EAE. J Clin Invest 117(8):2176– 2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Radinger M, et al. (2011) Local proliferation and mobilization of CCR3(+) CD34(+) eosinophil-lineage-committed cells in the lung. Immunology 132(1):144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cebula A, et al. (2013) Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 497(7448):258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pacholczyk R, et al. (2008) The T-cell receptor repertoire of regulatory T cells. Immunology 125(4): 450–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spleen cells from unimmunized 12–14 week old BALB/c mice were harvested and stained with antibodies specific to CD4, CD25, CD44, CD127, and CD62L. Lymphocytes were then sorted to isolate pure populations of naive (CD4+, CD25med/low, CD44low, and CD62Lhigh], regulatory (CD4+, CD25high, CD127low], effector memory (CD4+, CD25med/low, CD44 high, CD62L-), and central memory (CD4+, CD25med/low, CD44 high, CD62L+) T cells. RNA was extracted from the isolated T cell populations for focused Vβ8.2Jβ1.5 TCR next generation repertoire analysis. Resulting reads were then analyzed using an in-house developed software or (B) the recently published MiTCR software (15). Although the frequencies were slightly different, both methods detected HEL-specific T cell clones within the naive, central memory, effector memory, and regulatory T cell populations. (C) 12 week old BALB/c mice were immunized with PBS emulsified in CFA. 9-days later lymph nodes were removed and parallel in vitro cultures were incubated with either ML1, ML2, Medium, HEL, or PPD. Cells were harvested on day 3 for focused Vβ8.2Jβ1.5 TCR next generation repertoire analysis. Abundance of HEL-specific T cell clone (CDR3=CASGTGNNQAPL) relative to other CDR3 sequences was calculated. Results represent 3 biological replicates. Significance calculated by Student’s t-test. (D) 12 week old BALB/c mice were immunized with HEL emulsified in CFA. 9-days later lymph nodes were harvested and analyzed as described in “C”. Again, only incubation with HEL resulted in a statistically significant expansion of HEL-specific T cells (CDR3=CASGTGNNQAPL) greater than medium alone. Results represent 3 biological replicates. Significance calculated by Student’s t-test.
Gene segment sequences for TRBV13–2*01 (VP8.2) and TRBJ1–5*01 (JP1.5) were obtained from the international ImMunoGeneTics information system (IMGT). (A) The exact sequence of the TRBV13–2*01 - TRBJ1–5*01 gene rearrangement that encodes the CDR3 loop of the HEL-specific TCR β chain. V and J sequences lying outside of the CDR3 region are also shown. (B) Primers used to amplify the TRBV13– 2*01 TRBJ1–5*01 TCR sequence. Note that the TRBJ1–5*01 primer does not capture a few gene rearrangements. (C) Depiction of the motifs within the V and J segments used to identify reads containing a complete CDR3 region. (D) Depiction of the motifs used to identify the 12nt region that was used to calculate the sequencing/amplification error rate.
Splenocytes from antigen-naive 18 month old BALB/c mice were sorted to isolate effector memory and central memory CD4+T cells using antibodies specific to CD4, CD25, CD44, and CD62L. RNA was then harvested from the isolated T cells and used to generate focused Vβ8.2Jβ1.5 TCR libraries that were then sequenced using the HiSeq 2000 platform. The sequences were then filtered to remove sequences with incomplete CDR3 regions, N’s, and frameshifts. Sequences were also removed if they did not meet a Phred quality score cut-off of 30, or if their forward and reverse sequences did not match perfectly. (A) In silico spectratyping of CDR3 lengths revealed Gaussian distributions for the central memory and effector memory Vβ8.2Jβ1.5 spectra. Results are representative of at lest three independent experiments. (B) Graphs of copy number vs. distinct CDR3 sequence revealed that the HEL-specific Vβ8.2Jβ1.5 CDR3 sequence was present within the effector memoryand central memory T cell populations and that the sequence was not expanded when compared with other CDR3 sequences. Results are representative of at lest three independent experiments.Graphs for nucleotide and amino acid CDR3 sequences are shown separately.
To characterize the types of errors and to estimate the frequency of the amplification/sequencing errors encountered when sequencing TCR CDR3 gene rearrangements, the germline Vβ8.2 region, which lies just upstream of the CDR3 region, was analyzed. Similarity scores for the different sequences, and the their copy number are represented graphically against the sequence’s rank order; reads were ranked based upon their copy number with “1” being the most abundant read. Likewise, the similarity scores and copy numbers of the individual sequences corresponding to the CDR3 region were compared. Red bars indicate either the correct germline Vβ8.2 sequence or the characteristic HEL-specific Vβ8.2Jβ1.5 TCR CDR3 sequence. In each case the similarity between the HEL-specific CDR3 sequence and the most abundant CDR3 sequence was low.
FACS was used to isolate CD4+ CD25low (Treg )T cells from OT-II TCR transgenic mice. The isolated cells were then added to a unsorted of wild-type lymphocytescontaining all T cell populations (naive, memory, and regulatory). The mixture of wild-type lymphocytes and transgenic CD4+ CD25low T cells was then subjected to a second round of FACS to isolate Treg and non-Treg populations. A TCR transgene- specific PCR was conducted on the Treg, non-Treg sorted populations and wild-type lymphocytes to confirm the accuracy of our sorting strategy. TBP housekeeping gene served as loading control. Transcripts matching the transgenic TCR were present only within the FACS isolated CD4+ CD25low population.
Effector memory T cells (CD4+, CD25med/low, CD44 high, CD62L-) were isolated by FACS and then added to a unsorted of lymphocytes that differed at their CD45 allele, detectable by an allele-specific anti-CD45 antibody. The mixture (containing regulatory, naive, and memory T cells) was then analyzed by flow cytometry. Results from this experiment revealed that the CD45-mismatched effector memory T cells were detected solely within their corresponding effector memory population (CD4+, CD25med/low, CD44high, CD62L- cells).
Tregs (CD4+, CD25high, CD127low cells) were isolated using FACS. The isolated T cells were then permeabilized and stained intracellularly with fluorescently labeled antibodies specific to the transcription factor Foxp3. The T cells were then reanalyzed by flow of purity of Tregs.