

Abstract
In this work, we developed a computational protocol that employs multiple molecular docking experiments, followed by pose clustering, molecular dynamic simulations (10 ns), and energy rescoring to produce reliable 3D models of antibody–carbohydrate complexes. The protocol was applied to 10 antibody–carbohydrate co-complexes and three unliganded (apo) antibodies. Pose clustering significantly reduced the number of potential poses. For each system, 15 or fewer clusters out of 100 initial poses were generated and chosen for further analysis. Molecular dynamics (MD) simulations allowed the docked poses to either converge or disperse, and rescoring increased the likelihood that the best-ranked pose was an acceptable pose. This approach is amenable to automation and can be a valuable aid in determining the structure of antibody–carbohydrate complexes provided there is no major side chain rearrangement or backbone conformational change in the H3 loop of the CDR regions. Further, the basic protocol of docking a small ligand to a known binding site, clustering the results, and performing MD with a suitable force field is applicable to any protein ligand system.
INTRODUCTION
Oligosaccharides (glycans) comprise a repertoire of structurally diverse biomolecules that, unlike proteins or nucleic acids, are often branched. In eukaryotes, glycans are often components of cell surfaces, where they are typically covalently bound to either proteins (glycoproteins) or lipids (glycolipids). Glycans present in these glycoconjugates play a central role in a variety of biological recognition processes, including signal transduction and protein folding.1 Because of their exposure on cell surfaces, bacterial and viral pathogens often target host glycans to initiate adhesion and infection.2 Conversely, glycans and polysaccharides present on pathogen surfaces may be targeted by the host immune system.3 Additionally, changes in glycan composition or distribution are considered hallmarks of many diseases such as rheumatoid arthritis4 and a range of cancers.5–8 The significance of glycans in disease progression, together with their cell surface accessibility, makes them attractive targets for developing pharmaceutical agents,9,10 such as carbohydrate-based vaccines,11,12 anticarbohydrate antibodies,13,14 and endogenous human lectins.15
Both lectins and antibodies can be employed to detect glycans. However, the design and development of lectin-based pharmaceuticals is challenged by multiple issues; most lectins are of plant origin and therefore suffer from unreliable availability, inconsistent activity, and high immunotoxicity. Furthermore, lectins often display a broad or complex specificity16 but have on occasion been engineered to have improved properties.17,18 Antibodies generally display high affinity and specificity toward antigens, and compared to lectins, they have the benefit of low toxicity when used as therapeutics. Although monoclonal antibody production has become commonplace19 since the advent of hybridoma technology in 1975,20,21 the generation of anticarbohydrate antibodies remains challenging due to the T-cell independent nature of carbohydrate antigens. Selection of carbohydrate-binding antibodies via biopanning of antibody combinatorial libraries has been employed to overcome this challenge,22 but it can be difficult to obtain a high affinity antibody.23 Additionally, antibodies against carbohydrate antigens can also demonstrate cross-reactivity,24 in part due to the inherent structural similarity of many glycans. Structure-based analyses of antibody–carbohydrate or lectin–carbohydrate interactions offer an alternative means to guide affinity or specificity optimization.17,25,26
In order to be effective, an anticarbohydrate antibody should be capable of differentiating between closely related glycan structures that vary in both the monosaccharide composition and glycosidic linkages that connect residues. Anticarbohydrate antibodies frequently evolve to maximize hydrophobic interactions, while forming specific hydrogen bonds to the glycan.27 The structural and energetic characterization of antibody–carbohydrate interactions is therefore essential for the rational design of diagnostic and therapeutic antibodies that target carbohydrates.28–30
X-ray crystallography and NMR spectroscopy have been used to characterize the 3D structure of antibody–carbohydrate complexes; however, there are several difficulties associated with employing these techniques. Generally, the antigen-binding fragment (Fab) must be cleaved from the fragment crystallizable (Fc) domain and purified prior to crystallization, which is a procedure that is laborious and necessitates an ample supply of the antibody. Fab fragments are typically too large to be amenable to full structural characterization by NMR, although they can be employed in STD-NMR experiments to provide insight into the region of the antigen in contact with the antibody.31–34 Both techniques are further limited by additional complexities that arise from the flexible nature of glycans35 and the difficulties involved in synthesizing or isolating complex biological glycans in sufficient quantities and purity.36
Due to the challenges associated with the experimental techniques, computational docking is often employed to generate models of the immune complex, given a structure for the antibody fragment.37,38 Multiple theoretical orientations of the carbohydrate in the binding site may be possible, each forming the same number of hydrogen bonds with the antibody.26,39 Thus, the energy scoring function must be capable of discriminating between topologically similar ligand poses. Typical scoring functions40–42 attempt to take into account the contributions from van der Waals contacts, electrostatic interactions, desolvation effects, and entropy changes. However, features specific to the ligand, such as conformational preferences, are generally ignored, as is receptor flexibility and the role played by explicit waters in the binding site. These severe approximations increase the speed of the process and permit high throughput screening; nevertheless, in many cases, there is no alternative to docking to generate an initial structure of a receptor–ligand complex. These aspects are areas of active research in the development of carbohydrate-specific docking protocols.43–45 Despite these improvements, the top-scoring pose in carbohydrate docking cannot necessarily be trusted,46,47 and further analysis of all docked poses becomes necessary. Subsequently, there are numerous established refinement methods, reviewed recently, that take into account flexibility, explicit water, and entropy.48
In this study, we examine the performance of combining two common strategies for culling unlikely poses from the ensemble of structures generated by docking, namely, clustering data from multiple docking runs and postdocking molecular dynamic (MD) simulations. Pose clustering provides an alternative to energy ranking for the identification of probable poses41 and can serve to reduce the number of unlikely poses. While MD simulations permit the inclusion of explicit solvent and molecular motions, they are costly to perform. Here, we show that by eliminating unlikely poses through a clustering analysis MD simulations may be performed on the remaining subset of potential poses, with the goal of optimizing the ligand orientation and further eliminating incorrect poses. Assessment of the MD data was based on changes in the position of the ligand, as well as on the rank of the complex based on post-MD rescoring. Docking was performed using Vina-Carb (VC), a recently modified version of AutoDock Vina40 that incorporates the conformational preferences of oligosaccharides.39 Vina-Carb was selected because it biases the docking in favor of probable oligosaccharide shapes. The Dock/Cluster/MD approach was applied to 10 antibody carboydrate co-complexes and to three unliganded (apo) anticarbohydrate antibodies. The co-crystals provide a positive control for the ability of the algorithm to place the ligand correctly when the protein is in a conformation predisposed to recognize the ligand. Docking to the apo antibodies represents the more common situation, where a co-complex does not exist.
METHODS
Selection of Test Set.
The cognate docking test set consisted of 10 crystal structures of antibody–carbohydrate complexes from the Protein Data Bank (PDB)49 (www.rcsb.org). The apo docking test set included five systems from the cognate docking test set for which an apo protein was available. Systems were chosen based on ligand size and diversity of binding site topography50 (Table 1). For systems in which multiple chains were present, the chain containing the ligand with the lowest B-factor was chosen.
Table 1.
PDB IDs of Systems Employed in This Study
| PDB IDa | Binding site topography | Ligand | Number of carbohydrate residues |
|---|---|---|---|
| 1OP3 (1OM3) | Canyon | DManpa1–2DManpa1-ROH | 2 |
| 1M7D (1M71) | Canyon | LRhapa1–3(2-deoxy)LRhapa1–3GlcpNAcpb-OMe | 3 |
| 1MFA | Crater | DAbepa-13[Dgalpa1–2]DManpa-OMe | 3 |
| 1CLY | Valley | LFucpa1–2DGalpb1–4[LFucpa1−3]DGlcpNAca-OMe | 4 |
| 1S3K | Valley | LFucpa1–2DGalpb1–4[LFucpa1–3]DGlcNAcb1-ROH | 4 |
| 1UZ8 (1UZ6) | Canyon | DGalpb1–4[LFucpa1–3]DGlcpNAcb1-OME | 3 |
| 1M7I (1M71) | Canyon | LRhaap1–2LRhapa1–3LRhapa1−3DGlcpNAcb1–2LRhapa-OMe | 5 |
| 1MFB | Crater | DAbepa1–3[DGalpa1–2DManpa1–4LRhapa1–3DGalpa1–2]DManpa1–4LRhapa1-ROH | 7 |
| 3TV3 | Canyon | DManpa1–2DManpa1–6[DMAnpa1–3]DManpa1–6DManpa1–3DManpa1–2DManpa1–2DManpa-OMe | 8 |
| 3C6S (3C5S) | Crater | DGlcpa1–4[DGlcpa1–4[LRhapa1–2LRhapa1–3]LRhapa1–3DGlcpNAcb1–2LRhapa1–2LRhapa1–3] LRhapa1–3DGlcpNAcb1– 2LRhapx | 11 |
PDB IDs in parentheses represent the apo structure.
Automated Docking Protocol.
Molecular docking was performed using Vina-CARB51 with 20 docked models generated for each experiment. Coordinates for the variable domains of the antibodies were obtained for each system from the PDB and aligned based on a previously published protocol45 in order to ensure consistent placement of the grid box relative to the CDR regions across all the systems. The 3-D structures of the ligands for each system were built using the GLYCAM-Web server (www.glycam.org). The protein and ligand files for docking were prepared using Autodock tools (ADT)42 with Gassteiger52 partial atomic charges assigned to both the protein and ligand residues. Crystallographic water molecules were removed prior to docking, and hydrogen atoms were added to the protein using ADT.42 The hydrogen atoms in the ligand were assigned based on the GLYCAM residue templates. The protein was maintained rigid for both cognate and apo rigid receptor docking. The glycosidic ϕ, φ angles, and hydroxyls in the ligand were allowed to be flexible in all of the docking experiments. The exhaustiveness and energy range parameters were set to 12 and 10, respectively. The chi coefficient and cutoff value were set to 1 and 2, respectively.
MD Simulations.
MD simulations were performed with the GPU implementation of the pmemd code, pmemd.cuda53 from AMBER12,54 using the ff99SB55 parameters for the protein and GLYCAM06h56 parameters for the carbohydrate. The systems were solvated in a cubic water box using a TIP3P57 water model (12.0 Å per side), and counterions were added to neutralize the system. The following protocol was performed for each system. First, the water molecules were subjected to energy minimization (10,000 steps steepest decent followed by 10,000 steps conjugate gradient). During this minimization, the protein and the ligand were restrained with a force constant of 100 kcal/mol Å2. Full systems were then subjected to further energy minimization (10,000 steps steepest decent, 10,000 steps conjugate gradient) during which the backbone atoms of the protein and all the atoms of the ligand were restrained with a force constant of 5 kcal/mol Å2 allowing only the side chains to relax. This was followed by heating from 5 to 300 K over the course of 50 ps at constant volume. A 1 ns constant pressure (NPT ensemble) MD was used to ensure proper water and ligand equilibration. Then, 10 ns production MD simulations were performed at constant pressure (NPT ensemble) with the temperature being held constant at 300 K using the Langevin thermostat. The backbone atoms of the protein were restrained with a force constant of 5 kcal/mol Å2 during heating, equilibration, and production simulations, while the protein side chains and ligand atoms were allowed to be flexible. The backbone atoms were restrained to ensure that the protein fold remained stable during the course of the simulation. The SHAKE58 algorithm was used to constrain all covalent bonds involving hydrogen atoms, thereby allowing a time step of 2 fs. A nonbonded cutoff of 8 Å was used, and long-range electrostatics were accounted for by the particle mesh Ewald59 method.
Analysis.
Docked models were compared to their respective experimental structures by calculating RMSD values of the ring atom positions of the ligand. For the pose clustering analysis, the protocol implemented in Autodock 4.041 was employed with a cutoff value of 2.0 Å. In this method, the lowest energy docked model forms the seed for the first cluster, and the second lowest energy model is then compared to it by calculating the RMSD between them. If the RMSD is less than 2.0 Å, the second model is added to the first cluster else it becomes the seed for a second cluster. The next best docked structure is then compared to the lowest energy structure. If the RMSD value is less than 2.0 Å, this pose is added to the first cluster, else it is compared to the seed of the second cluster. If the RMSD value is less than 2.0 Å, it is added to the second cluster, else it forms the seed for the third cluster. This continues until each docked model becomes part of a cluster. To assess the stability of the complexes during the course of the simulation, the RMSD values for the ring atoms of the ligand position relative to both the first time step of the simulation and the crystal ligand were calculated. For the post-MD score-in-place calculations, snapshots of the protein and the ligand were obtained at 10 ps intervals from the final 1 ns of the simulation. Both RMSD values and snapshots were generated using the ptraj module of AMBERTOOLS 12.60 The extracted snapshots were prepared for score-in-place calculations using ADT42 with the same parameters that were employed in the initial docking protocol. Structural images were created using the Visual Molecular Dynamics program61 and 3D-SNFG plugin.62
RESULTS
Docking to Co-Crystals (Cognate Docking).
There is a potential benefit in performing multiple docking experiments when using stochastic algorithms like the genetic algorithm used in Vina-Carb (VC) since employing different random seeds can lead to different distributions of docked poses in each experiment. To assess whether this approach could be beneficial when docking carbohydrates to antibodies, five independent docking experiments were performed using VC on each of 10 antibody systems; 20 models were obtained from each experiment, resulting in a total of 100 docked models for each system. The root-mean-squared deviation (RMSD) between the positions of the ring atoms of the docked ligands and the corresponding positions in the crystallographic co-complexes were computed for all of the systems. An RMSD of 2 Å or less was considered an acceptable pose51 and an indication of a successful docking result. In 8 of the 10 systems, an acceptable pose was found consistently in each independent docking experiment, and it was also the top-ranked pose in six of the systems (Table 2). In systems 1MFB (heptasaccharide) and 1S3K (tetrasaccharide), an acceptable pose was found in the top five ranked poses but was never the top-ranked pose. Notably, multiple docked models within the top five ranked poses of the 1CLY ligand (tetrasaccharide) contained conformations similar to that of the crystal co-complex; however, the orientations within the binding pocket were incorrect (Figure 1). The docking scores of these models differed by less than 0.5 kcal/mol from that of the top-ranked acceptable pose. VC was designed to improve the accuracy of carbohydrate conformations by penalizing docked poses with energetically unfavorable glycosidic linkages; however, the results for 1CLY demonstrate that even a correct ligand shape may not guarantee that the orientation of the ligand in the top-ranked poses is acceptable.
Table 2.
Results from Cognate Docking
| 1OP3 | 1M7D | 1MFA | 1CLY | 1S3K | 1UZ8 | 1M7I | 1MFB | ||
|---|---|---|---|---|---|---|---|---|---|
| Run 1 | N.A.Pa | 5 | 4 | 3 | 1 | 1 | 2 | 3 | 4 |
| (1,2,9,14,15)c | (1,2,4,17) | (1,2,3) | (1) | (2) | (1,2) | (1,2,3) | (3,4,8,13) | ||
| L.R.A. PRMSDb | 0.9 (1) | 0.4 (1) | 0.8 (1) | 0.6 (1) | 0.7 (2) | 0.7 (1) | 0.6 (1) | 0.7 (3) | |
| Run 2 | N.A.P | 4 | 4 | 3 | 1 | 1 | 2 | 2 | 3 |
| (1,2,3,19) | (1,2,3,4) | (1,2,4) | (1) | (2) | (1,9) | (1,2) | (2,7,10) | ||
| L.R.A PRMSD | 0.9 (1) | 0.4(1) | 0.8 (1) | 0.9(1) | 0.7 (2) | 0.4(1) | 0.6 (1) | 0.7 (2) | |
| Run 3 | N.A.P | 6 | 2 | 3 | 1 | 3 | 1 | 5 | 2 |
| (1,2,3,6,12,15) | (1,2) | (1,2,3) | (1) | (2,7,19) | (1) | (1,2,3,4,9) | (3,4) | ||
| L.R.A PRMSD | 0.9 (1) | 0.4 (1) | 0.8 (1) | 0.9(1) | 0.7 (2) | 0.9(1) | 0.6 (1) | 0.7 (3) | |
| Run 4 | N.A.P | 5 | 5 | 1 | 1 | 1 | 1 | 3 | 5 |
| (1,2,8,11,14) | (1,2,3,4,6) | (1) | (1) | (1) | (1) | (1,2,3) | (3,4,5,10,13) | ||
| L.R.A PRMSD | 0.9 (1) | 0.3 (1) | 0.8 (1) | 0.9(1) | 0.7 (1) | 0.9(1) | 0.6 (1) | 0.7 (3) | |
| Run 5 | N.A.P | 5 | 3 | 2 | 1 | 2 | 1 | 3 | 4 |
| (1,2,7,13,15) | (1,2,3) | (1,2) | (1) | (2,15) | (1) | (1,2,4) | (2,5,9,12) | ||
| L.R.A PRMSD | 0.9 (1) | 0.4 (1) | 0.8 (1) | 0.6(1) | 0.7 (2) | 0.6(1) | 0.6 (1) | 0.7 (2) |
N.A.P represents the total number of acceptable poses in each individual docking experiment.
L.R.A. PRMSD represents the lowest ranked acceptable pose RMSD (Å) in each individual docking experiment.
Numbers in parentheses represent the ranks of the poses in each individual docking experiment.
Figure 1.
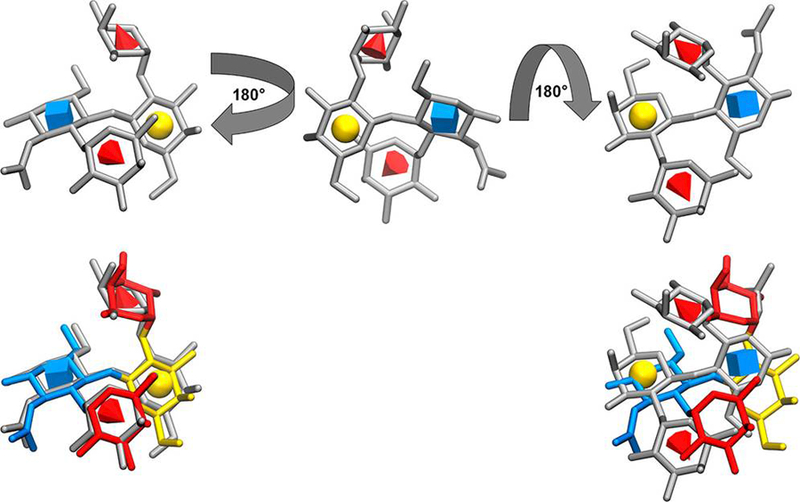
Upper left: Docked model of the 1CLY ligand in the correct orientation in the binding pocket. Residues are depicted in gray with symbols according to the Symbol Nomenclature for Glycans (GlcNAc, Fuc, and Gal are blue cube, red cone, and yellow sphere, respectively).63 Upper middle: Ligand in an intermediate flipped orientation. Upper right: Final inverted ligand pose. Lower: Two orientations of the docked ligand are superimposed on the co-crystal structure, which is colored according to SNFG.
For the two largest oligosaccharide systems (3C6S, dodecasaccharide; 3TV3, octasaccharide), VC failed to find an acceptable pose; the top-ranked poses for 3TV3 and 3C6S had RMSD values of 16 and 22 Å, respectively. Difficulties in predicting the position of these large ligands suggests that the search algorithm is less effective when evaluating a large number of rotatable bonds, which has been recognized previously.64,65 To overcome this problem, docking experiments were repeated for 3C6S and 3TV3, in which only the oligosaccharide components that directly interact with the protein were docked. Such a fragment of an oligosaccharide is often referred to as the minimal binding determinant (MBD).26 In the case of 3TV3 and 3C6S, the MBDs are a disaccharide66 and pentasaccharide,67 respectively (Figure 2). Docking of the MBD generally improved the results (Table 3); however, an acceptable pose was only found in two of the five docking experiments for 3TV3, demonstrating the importance of multiple docking experiments. There was also considerable variability in ranking of the acceptable poses between docking experiments, suggesting a lack of convergence in the sampling, and indicating a need to examine all docked poses.
Figure 2.
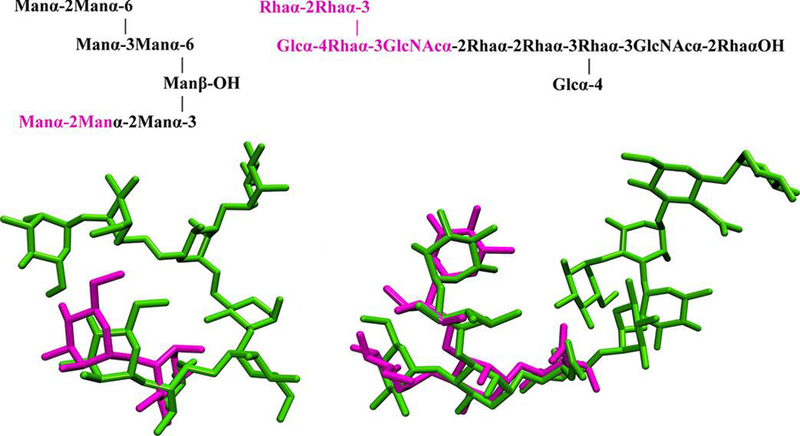
Top: Oligosaccharide sequences for the ligands in the 3TV3 (left) and 3C6S (right) co-crystals; the fragments (MBDs) chosen for docking are shown in magenta. Bottom: Acceptable docked poses from MBD docking to 3TV3 and 3C6S relative to the intact ligands in the co-crystals (green).
Table 3.
Results from Five Docking Experiments of Fragment (MBD) Docking
RMSD (Å) value of the lowest ranked acceptable pose.
Inability to generate an acceptable pose indicated by “−”.
The present results indicate that when applied to a positive control (cognate), docking a flexible oligosaccharide can lead to the identification of a top-ranked acceptable pose in approximately 6/10 cases, and this increases to 8/10 if the top five ranked poses are considered. However, in approximately 2/10 of the systems, the acceptable pose was not ranked in the top five, and there can be significant variability in ranking between independent docking experiments. Although an acceptable pose was found in the top five for 8/10 of the systems, additional acceptable poses were poorly ranked (Table 2). The docking scores of these poses were within 0.5 kcal/mol of incorrect (high RMSD) docked models, detracting from confidence in the ranking system. In order to assess the ability of the scoring function to discriminate between correct and incorrect models, the differences in docking scores between the best model (lowest RMSD relative to the crystal structure) and the worst model (highest RMSD relative to the crystal structure) were calculated (Table 4). The docking scores differed by less than 1.2 kcal/mol across all 10 systems. In four systems, the difference in docking scores was less than 0.5 kcal/mol between the best and worst models, while the RMSD differed by more than 4.5 Å. Although the success rate could be as high as 6/10, the existence of diverse poses that are energetically similar to the top-ranked pose suggests that the relative ranking is not a reliable metric for identification of the optimal pose. To avoid the issue of pose ranking all together, a clustering analysis of the poses was undertaken. The clusters were then examined to test the hypothesis that an acceptable pose should be present in the most populated cluster.
Table 4.
Differences between Docking Scores and RMSD Values for Best and Worst Poses Ranked from Smallest RMSD Difference to Largest
| PDB ID | Difference in interaction energya | RMSD differenceb |
|---|---|---|
| 1M7D | 0.51 | 1.47 |
| 1MFB | 0.31 | 1.91 |
| 1OP3 | 0.19 | 2.75 |
| 1MFA | 0.25 | 4.52 |
| 1S3K | 0.05 | 4.54 |
| 1UZ8 | 1.13 | 4.74 |
| 1CLY | 0.39 | 4.75 |
| 1M7I | 1.09 | 10.52 |
| 3C6S | 0.40 | 11.59 |
| 3TV3 | 0.20 | 21.52 |
In kcal/mol.
In angstrom.
Clustering Analysis.
Pose clustering potentially provides an alternative way to identify acceptable poses without relying on the ranking. Additionally, it reduces the number of poses that need to be examined since further analysis need only consider a representative pose from each cluster. Pose clustering may be employed to identify acceptable poses that occur repeatedly across multiple docking experiments. The clustering method implemented here was based on protocols from earlier Autodock versions,42 specifically, poses within 2 Å of each other were considered to be members of the same cluster. The average docking score of each cluster was calculated, and the clusters were ranked from most to least energetically favorable (Figure S1). In 8/10 systems, an acceptable pose was either in the most populated or the lowest energy cluster, and overall in 4/10 systems, the lowest energy cluster was also found to be the most populated cluster. In two systems, additional acceptable poses were identified in clusters that were neither the most populated nor the lowest energy. The dispersion of acceptable poses among the clusters suggested that additional analysis would be required in order to increase the likelihood of identifying an acceptable pose. To address this, we selected representative poses from various clusters for further analysis via MD.
Pose Filtering Using MD Simulations.
MD simulations account for solvent effects, as well as protein and ligand dynamics, potentially correcting some of the deficiencies associated with the docking protocol or scoring function. Representative poses were chosen from clusters that contained more than two members for analysis by fully solvated MD simulation (10 ns). The pose that was most similar to the geometric average of the clustered poses was chosen for MD rather than the lowest energy in the cluster, given the poor correlation between the VC scores and pose-correctness.
Average RMSD values were calculated for the ligand ring–atom positions relative to those in the reference crystal structure at 10 ps intervals from the final 1 ns of the 10 ns trajectories. In order to determine the extent to which MD simulations altered the position of the ligand, the difference between the RMSD value from MD and the RMSD values from docking were computed (DRMSD). Low values of DRMSD were observed for acceptable poses, indicating that these poses remained stable throughout the simulation (Table 5). Simulations of unacceptable poses could, in principle, lead to either an improvement (DRMSD < 0) or a worsening (DRMSD > 0) in the ligand position. In practice, the majority of unacceptable poses did not transition into acceptable poses; however, the RMSD improved from 3.7 to 0.7 Å for 1UZ8, from 2.2 to1.1 Å for 1M7D, 2.4 to 2.0 Å for 1OP3, and from 3.9 to1.1 Å for 1CLY (Table S2). Notably, MD simulation was able to identify very poor poses (docked RMSD > 5 Å), as they generally drifted significantly from the initial binding pose during the simulation (Table S1). Additionally, in all but one case, those poses that drifted by more than 2 Å from the known binding site also ranked worse after rescoring (Table S1). It thus appears that pose drift combined with worsening rank after rescoring can be used to discriminate likely from unlikely poses. These results support the hypothesis that even a relatively short MD simulation can enhance the discrimination between acceptable and unacceptable poses, a feature that is particularly helpful for identifying unacceptable poses that nevertheless received reasonable docking scores.
Table 5.
DRMSD Values for 10 Cognate Systems
| System | Total clusters subjected to MD refinement | DRMSD for single best-ranked, acceptable pose | Number of unacceptable poses modestlya improved/worsened | Number of unacceptable poses substantiallyb improved/worsened |
|---|---|---|---|---|
| 1CLY | 12 | 0.44 | 3/9 | −/−c |
| 1M7D | 11 | 0.74 | 4/6 | −/1 |
| 1M7I | 9 | 1.10 | 2/5 | −/2 |
| 1S3K | 11 | 1.80 | 1/7 | −/3 |
| 1UZ8 | 13 | 0.03 | 1/7 | 1/4 |
| 1MFA | 14 | 0.32 | 3/6 | 1/4 |
| 1MFB | 9 | 1.90 | 3/5 | −/1 |
| 1OP3 | 12 | -0.09 | 3/5 | −/4 |
| 3C6S | 9 | 0.56 | 2/7 | −/− |
| 3TV3 | 10 | -0.53 | 4/6 | −/− |
|DRMSD| < 2 Å.
|DRMSD| > 2 Å.
“−”indicates that the simulation neither significantly improved nor worsened the pose.
In addition to RMSD values, average docking scores for the MD-generated poses were computed using the score-in-place option in VC. This method for computing the energies was selected because it allowed the poses from MD data to be ranked analogously to the data from docking. After MD, an acceptable pose was found as the top-ranked pose for 8/10 systems, in contrast to the pre-MD ranking for which the top-ranked pose was acceptable in only 6/10 systems. In 3C6S, although the RMSD of the acceptable pose worsens slightly (1.9–2.5 Å) during the course of the simulation, the top two ranked models post-MD rescoring are populated with models closer to the crystal structure when compared to molecular docking alone where an acceptable pose is not found within the top five models. Examples of the effect of MD simulation on pose ranking and RMSD is shown for 1S3K and 3TV3 in Table 6 and the remaining systems in Table S2. The rescored docking scores show a marked improvement in the correlation between docking score and pose position (Figure 3).
Table 6.
Ligand RMSD (Å) Values of Top 10 Poses after Pose Clustering, MD Simulation, and Post-MD Rescoring for 1S3K and 3TV3a
| 1S3K | 3TV3 | ||||
|---|---|---|---|---|---|
| RMSD before MD (sorted by rank) | RMSD after MD (rescored rank) | RMSD after MD (sorted by rank) | Before MD (sorted by rank) | RMSD after MD (rescored rank) | After MD (sorted by rank) |
| 6.2 (1) | 5.9 (4) | 1.1 (1) | 17.4 (1) | 18.2 (S) | 0.8 (1) |
| 0.7 (2) | 1.1 (1) | 6.7 (2) | 6.5 (2) | 6.0 (7) | 23.4 (2) |
| 5.4 (3) | 7.0 (S) | 7.5 (3) | 1.4 (3) | 0.8 (1) | 23.0 (3) |
| 6.4 (4) | 6.7 (2) | 5.9 (4) | 22.8 (4) | 23.4 (2) | 17.7 (4) |
| 5.6 (5) | 7.6 (6) | 7.0 (5) | 22.2 (5) | 23.0 (3) | 18.2 (5) |
| 6.9 (6) | 8.0 (9) | 7.6 (6) | 17.3 (6) | 17.7 (4) | 18.3 (6) |
| 7.0 (7) | 7.5 (3) | 4.9 (7) | 22.1 (7) | 24.1 (8) | 6.0 (7) |
| 4.2 (8) | 4.9 (7) | 7.6 (8) | 24.3 (8) | 18.3 (6) | 24.1 (8) |
| 6.3 (9) | 7.6 (8) | 8.0 (9) | 23.6 (9) | 24.9 (9) | 24.9 (9) |
| 5.0 (10) | 9.1 (10) | 9.1 (10) | 6.7 (10) | 43.0 (10) | 43.0 (10) |
Acceptable poses and poses that worsened by 2 Å from their initial position during MD are bolded.
Figure 3.
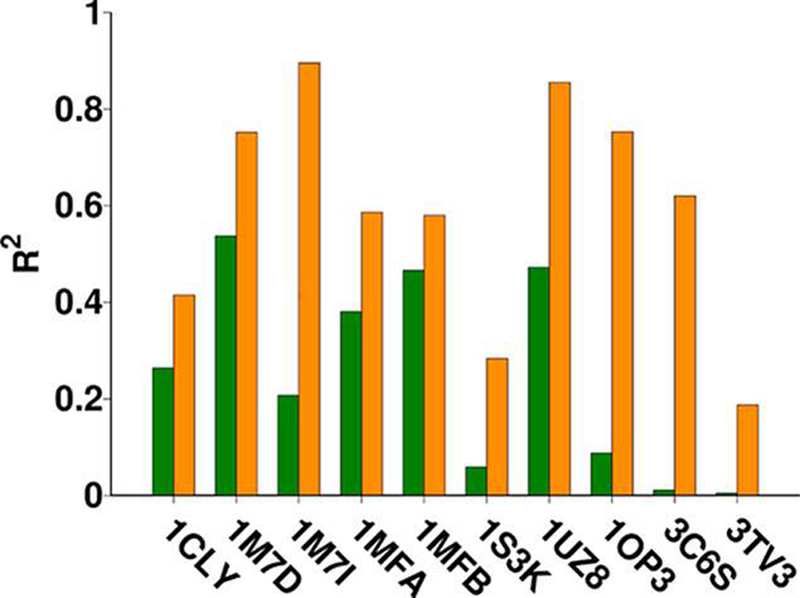
Comparison of correlation coefficient values for each of the 10 systems before (green) and after (orange) employing MD simulations. The column height corresponds to the R2 value of each system.
MD simulation in the presence of explicit water provides a sufficiently improved model for the interaction between the ligand and the protein that poor poses resulting from docking may be identified as false positives by their instability during MD. Although on the relatively short 10 ns time scale of the present simulations false positives are rarely able to access favorable binding modes, they are able to drift further away from the protein, resulting after rescoring in weaker docking scores. Thus, MD offers a complementary method to identify and eliminate false positives in the docking output. Notably, post-MD rescoring increases the likelihood that the top-ranked pose will be an acceptable pose, assuming one is present among all of the docked-poses.
Application to Apo Structures.
While cognate docking, using the receptor from a co-crystal structure of the complex with the ligand is a common method to validate docking results, it is biased toward a favorable outcome since the binding site is conformationally adapted to the ligand. Hence, cognate docking is not representative of the general application of docking to apo receptors. In order to test the ability of the (Dock-Cluster-MD-Rescore) protocol to produce accurate results in such scenarios, the protocol was applied to the apo antibody crystal structures that were available for 5 of the 10 antibodies (Table 1). During cognate docking, the protein was maintained rigid, while the glycosidic and exocyclic bonds in the ligand were permitted to be flexible. However, when docking to apo proteins, the possibility exists that ligand binding might induce conformational changes in the binding site, which would necessitate that a subset of the amino acid side chains be treated as flexible. In order to estimate the degree of induced fit in the co-complexes, the differences in positions of residues in the CDR loop regions in the apo and co-complexes were calculated (Table S3). For 3C5S, while an apo structure was available, there were several missing residues in CDR loops H1 and H3, eliminating this system from further study. In all remaining systems except 1OM3, the all-atom RMSD values for the residues in the CDRs for the apo versus cognate systems were less than 1.5 Å, with the exception of one residue (ARG H99) from loop H3 in 1UZ8/6 that had an RMSD value of 2.4 Å. The low RMSD values in 1M7D/1, 1M7I/1, and 1UZ8/6 indicate that any conformational changes in these systems resulting from binding are very subtle. In contrast, RMSD values for several residues in the H3 loop of the 1OP3/OM3 system ranged from 2.5 to 10 Å, and further analysis showed that the differences are a result of significant conformational change in the backbone conformation of these residues suggesting a large change in the loop upon ligand binding (Figure S2). Since Vina-Carb does not permit backbone flexibility, 1OM3 was also eliminated from further study. Thus, rigid protein docking was employed on three apo antibody structures (1M7D/1, 1M7I/1, 1UZ8/6). Given that the apo structures for these systems were similar to those in the co-complexes, it is not unexpected that docking to these systems resulted in essentially the same outcome as for cognate docking. For each of these systems, while the top-ranked pose was acceptable in each docking experiment, additional acceptable poses were also present that were poorly ranked (Table 7).
Table 7.
Results from Five Independent Apo Docking Experimentsa
| 1M7D/1 | 1M7I/1 | 1UZ6/8 | ||
|---|---|---|---|---|
| Run 1 | N.A.Pb | 4 (1,2,3,8)d | 3 (1,2,3) | 4 (1,4,14,15) |
| L.R.A. PRMSDc | 2.0 (1) | 1.5 (1) | 1.0 (1) | |
| Run 2 | N.A.P | 3 (1,2,3) | 4 (1,3,5,19) | 3 (1,2,11) |
| L.R.A. PRMSD | 1.9 (1) | 1.5 (1) | 1.0 (1) | |
| Run 3 | N.A.P | 4 (1,2,3,4) | 3 (1,8,14) | 3 (1,4,12) |
| L.R.A. PRMSD | 1.9 (1) | 1.5 (1) | 1.0 (1) | |
| Run 4 | N.A.P | 4 (1,2,3,18) | 1(1) | 5 (1,4,9,14,15) |
| L.R.A. PRMSD | 2.0 (1) | 1.5 (1) | 1.0 (1) | |
| Run 5 | N.A.P | 4 (1,2,3,4) | 4 (1,2,3,17) | 3 (1,4,10) |
| L.R.A. PRMSD | 1.9 (1) | 1.5 (1) | 1.0 (1) |
RMSD values are in Å.
N.A.P represents the total number of acceptable poses in each individual docking experiment.
L.R.A. PRMSD represents lowest ranked acceptable pose RMSD (Å) in each individual docking experiment.
Numbers in parentheses represent the ranks of the poses in each individual docking experiment.
A clustering analysis was employed as in cognate docking (Figure S3/Table 8), and representative poses from clusters that contain more than two members were subjected to solvated MD simulations. The average RMSD values calculated relative to the crystal structure indicate that the acceptable poses in all the three systems improved slightly (ΔRMSD ≤ 1 Å) during the course of the simulation (Table 8). These acceptable poses include poses from 1UZ6/8 and 1M71/D, which were ranked 5 and 14, respectively, in an individual docking experiment and were not present either in the lowest energy or most populated cluster. Generally, across the three systems, the majority of the unacceptable poses worsened only modestly (ΔRMSD < 2 Å), while a few poses drifted significantly further away (ΔRMSD > 5 Å) from the binding site. However, in 1M71/I, an unacceptable pose with an RMSD of 3.5 Å transitioned into an acceptable pose (1.5 Å). Post-MD rescoring places the poorly ranked acceptable poses from 1UZ6/8, 1M71/D, and the improved docked model from 1M71/I among the top three models. These results indicate that the Dock-Cluster-MD-Rescore protocol can be used reliably to predict apo complexes provided there is no significant conformational change upon binding.
Table 8.
Ligand RMSD (Å) Values after Pose Clustering, MD Simulation and Post-MD Rescoring for apo Systemsa
| 1M71/D | 1M71/I | 1UZ6/8 | ||||||
|---|---|---|---|---|---|---|---|---|
| RMSD before MD (sorted by rank) | RMSD after MD (rank) | RMSD after MD (sorted by rank) | RMSD before MD (sorted by rank) | RMSD after MD (rank) | RMSD after MD (sorted by rank) | RMSD before MD (sorted by rank) | RMSD after MD (rank) | RMSD after M (sorted by rank) |
| 2.0 (1) | 1.4 (2) | 1.2 (1) | 1.5 (1) | 1.5 (2) | 1.5 (1) | 1.0 (1) | 0.8 (1) | 0.8 (1) |
| 7.4 (2) | 8.9 (6) | 1.4 (2) | 12.3 (2) | 12.0 (7) | 1.5 (2) | 4.6 (2) | 4.8 (4) | 4.8 (2) |
| 1.9 (3) | 1.2 (1) | 7.0 (3) | 11.6 (3) | 10.7 (5) | 10.5 (3) | 4.8 (3) | 4.7 (5) | 0.9 (3) |
| 6.5 (4) | 7.0 (3) | 6.9 (4) | 12.2 (4) | 17.3 (13) | 6.1 (4) | 2.9 (4) | 5.7 (10) | 4.7 (4) |
| 8.0 (5) | 8.9 (S) | 8.9 (S) | 11.8 (S) | 12.4 (12) | 10.7 (5) | 5.3 (5) | 4.8 (2) | 4.7 (5) |
| 6.5 (6) | 7.6 (7) | 8.9 (6) | 3.5 (6) | 1.5 (1) | 10.7 (6) | 5.5 (6) | 37.1 (12) | 5.2 (6) |
| 3.6 (7) | 28.5 (13) | 7.6 (7) | 11.4 (7) | 10.7 (6) | 12.0 (7) | 4.0 (7) | 5.2 (6) | 6.2 (7) |
| 8.2 (8) | 10.1 (11) | 9.1 (8) | 4.0 (8) | 6.1 (4) | 9.3 (8) | 4.9 (8) | 6.8 (8) | 6.8 (8) |
| 6.6 (9) | 6.9 (4) | 7.7 (9) | 7.5 (9) | 9.3 (8) | 6.8 (9) | 5.8 (9) | 6.2 (7) | 6.7 (9) |
| 7.8 (10) | 9.1 (8) | 9.0 (10) | S.5 (10) | 6.8 (9) | 9.5 (10) | 4.8 (10) | 6.7 (9) | 5.7 (10) |
| 7.2 (11) | 7.7 (9) | 10.1 (11) | 11.6 (11) | 10.5 (3) | 10.2 (11) | 1.9 (11) | 0.9 (3) | 24.2 (11) |
| 7.1 (12) | 18.6 (12) | 18.6 (12) | 7.2 (12) | 10.2 (11) | 12.4 (12) | 5.3 (12) | 68.5 (13) | 37.1 (12) |
| 7.5 (13) | 9.0 (10) | 28.5 (13) | 4.3 (13) | 9.5 (10) | 17.3 (13) | 6.3 (13) | 24.2 (11) | 68.5 (13) |
Acceptable poses are bolded.
CONCLUSION
In terms of objectively identifying the correct pose of a ligand in a binding site, molecular docking, followed by clustering and MD, enriched the number of poses that were acceptable and improved their rank compared to molecular docking alone for both cognate and the limited number of available apo receptors which show no significant conformational change upon binding. This protocol therefore is a viable approach in cases when the binding pose is unknown, where traditional docking alone is less likely to lead to the correct outcome. The success of this protocol shows that subjecting poses from docking to solvated MD simulations provides a promising method to identify correct poses by permitting unfavorable poses to dissociate from the protein surface (Table S1), while at the same time retaining acceptable poses. Further, we have shown that when docking large oligosaccharides the probability of generating an acceptable pose can be significantly increased by trimming the ligand down to the minimal binding determinant (MBD). It may be that significantly increasing the number of runs and the number of poses output by VINA might lead to the discovery of acceptable poses for large oligosaccharides. However, there will always be ever larger ligands; so we propose that the present approach offers a balance between the benefits of docking (throughput) with the accuracy of MD. The composition of the MBD can be inferred from complementary studies, such as by chemical modification of the ligand,68–70 by saturation transfer difference NMR spectroscopy,71–74 or by glycan array screening.16,26,75,76 Lastly, the observation that the docking scores correlated well with pose-correctness, after the poses were subjected to MD simulation, indicates that weaknesses in the performance of docking with Vina-Carb are not related primarily to the scoring function but to inaccuracies in the generation of the poses.
Supplementary Material
Acknowledgments
Funding
The authors thank the National Institutes of Health (U01 CA207824 and P41 GM103390) for support.
ABBREVIATIONS
- MD
Molecular Dynamics
- PDB
Protein Data Bank
- VC
Vina-Carb
- ADT
Autodock Tools
- RMSD
Root Mean Square Deviation
Associated Content
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.7b00588.
Additional tables and figures that aid in understanding the results. (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1). Varki A; Lowe JB Biological Roles of Glycans In Essentials of Glycobiology, 2nd ed.; Varki A; Cummings RD; Esko JD; Freeze HH; Stanley P; Bertozzi CR; Hart GW; Etzler ME, Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2009. [PubMed] [Google Scholar]
- (2). Karlsson KA Bacterium-Host Protein-Carbohydrate Interactions and Pathogenicity. Biochem. Soc. Trans 1999, 27, 471–474. [DOI] [PubMed] [Google Scholar]
- (3). Ceravolo IP; Sanchez B.a.M.; Sousa TN; Guerra BM; Soares IS; Braga EM; Mchenry AM; Adams JH; Brito CFA; Carvalho LH Naturally Acquired Inhibitory Antibodies to Plasmodium Vivax Duffy Binding Protein Are Short-Lived and Allele-Specific Following a Single Malaria Infection. Clin. Exp. Immunol 2009, 156, 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4). Arnold JN; Wormald MR; Sim RB; Rudd PM; Dwek RA The Impact of Glycosylation on the Biological Function and Structure of Human Immunoglobulins. Annu. Rev. Immunol 2007, 25, 21–50. [DOI] [PubMed] [Google Scholar]
- (5). Brooks SA; Carter TM; Royle L; Harvey DJ; Fry SA; Kinch C; Dwek RA; Rudd PM Altered Glycosylation of Proteins in Cancer: What Is the Potential for New Anti-Tumour Strategies. Anti-Cancer Agents Med. Chem 2008, 8, 2–21. [DOI] [PubMed] [Google Scholar]
- (6). Dube DH; Bertozzi CR Glycans in Cancer and Inflammation [Mdash] Potential for Therapeutics and Diagnostics. Nat. Rev. Drug Discovery 2005, 4, 477–488. [DOI] [PubMed] [Google Scholar]
- (7). Fuster MM; Esko JD The Sweet and Sour of Cancer: Glycans as Novel Therapeutic Targets. Nat. Rev. Cancer 2005, 5, 526–542. [DOI] [PubMed] [Google Scholar]
- (8). Hakomori S-I; Vande Woude GF; Klein G Aberrant Glycosylation in Tumors and Tumor-Associated Carbohydrate Antigens. Adv. Cancer Res. 1989, 52, 257–331. [DOI] [PubMed] [Google Scholar]
- (9). Abbott KL; Aoki K; Lim JM; Porterfield M; Johnson R; O’regan RM; Wells L; Tiemeyer M; Pierce M Targeted Glycoproteomic Identification of Biomarkers for Human Breast Carcinoma. J. Proteome Res 2008, 7, 1470–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10). Block TM; Comunale MA; Lowman M; Steel LF; Romano PR; Fimmel C; Tennant BC; London WT; Evans AA; Blumberg BS; Dwek RA; Mattu TS; Mehta AS Use of Targeted Glycoproteomics to Identify Serum Glycoproteins That Correlate with Liver Cancer in Woodchucks and Humans. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11). Slovin SF; Keding SJ; Ragupathi G Carbohydrate Vaccines as Immunotherapy for Cancer. Immunol. Cell Biol 2005, 83, 418–428. [DOI] [PubMed] [Google Scholar]
- (12). Keding SJ; Danishefsky SJ Prospects for Total Synthesis: A Vision for a Totally Synthetic Vaccine Targeting Epithelial Tumors. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 11937–11942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13). Pai LH; Wittes R; Setser A; Willingham MC; Pastan I Treatment of Advanced Solid Tumors with Immunotoxin Lmb-1: An Antibody Linked to Pseudomonas Exotoxin. Nat. Med 1996, 2, 350–353. [DOI] [PubMed] [Google Scholar]
- (14). Posey JA; Khazaeli MB; Bookman MA; Nowrouzi A; Grizzle WE; Thornton J; Carey DE; Lorenz JM; Sing AP; Siegall CB; Lobuglio AF; Saleh MN A Phase I Trial of the Single-Chain Immunotoxin Sgn-10 (Br96 Sfv-Pe40) in Patients with Advanced Solid Tumors. Clin. Cancer Res 2002, 8, 3092–3099. [PubMed] [Google Scholar]
- (15). Valdimarsson H Infusion of Plasma-Derived Mannan-Binding Lectin (Mbl) into Mbl-Deficient Humans. Biochem. Soc. Trans 2003, 31, 768–769. [DOI] [PubMed] [Google Scholar]
- (16). Grant OC; Tessier MB; Meche L; Mahal LK; Foley BL; Woods RJ Combining 3d Structure with Glycan Array Data Provides Insight into the Origin of Glycan Specificity. Glycobiology 2016, 26, 772–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17). Arnaud J; Audfray A; Imberty A Binding Sugars: From Natural Lectins to Synthetic Receptors and Engineered Neolectins. Chem. Soc. Rev 2013, 42, 4798–4813. [DOI] [PubMed] [Google Scholar]
- (18). Olausson J; Åström E; Jonsson B-H; Tibell L.a.E.; Påhlsson P Production and Characterization of a Monomeric Form and a Single-Site Form of Aleuria Aurantia Lectin. Glycobiology 2011, 21, 34–44. [DOI] [PubMed] [Google Scholar]
- (19). Trebak M; Chong JM; Herlyn D; Speicher DW Efficient Laboratory-Scale Production of Monoclonal Antibodies Using Membrane-Based High-Density Cell Culture Technology. J. Immunol. Methods 1999, 230, 59–70. [DOI] [PubMed] [Google Scholar]
- (20). Kohler G; Milstein C Continuous Cultures of Fused Cells Secreting Antibody of Predefined Specificity. Nature 1975, 256, 495–497. [DOI] [PubMed] [Google Scholar]
- (21). Kohler G; Milstein C Derivation of Specific Antibody-Producing Tissue Culture and Tumor Lines by Cell Fusion. Eur. J. Immunol 1976, 6, 511–519. [DOI] [PubMed] [Google Scholar]
- (22). Fukuda MN Peptide-Displaying Phage Technology in Glycobiology. Glycobiology 2012, 22, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23). Vodnik M; Zager U; Strukelj B; Lunder M Phage Display: Selecting Straws Instead of a Needle from a Haystack. Molecules 2011, 16, 790–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24). Manimala JC; Roach TA; Li Z; Gildersleeve JC High-Throughput Carbohydrate Microarray Profiling of 27 Antibodies Demonstrates Widespread Specificity Problems. Glycobiology 2007, 17, 17C–23C. [DOI] [PubMed] [Google Scholar]
- (25). Lak P; Makeneni S; Woods RJ; Lowary TL Specificity of Furanoside–Protein Recognition through Antibody Engineering and Molecular Modeling. Chem. - Eur. J 2015, 21, 1138–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26). Tessier MB; Grant OC; Heimburg-Molinaro J; Smith D; Jadey S; Gulick AM; Glushka J; Deutscher SL; Rittenhouse-Olson K; Woods RJ Computational Screening of the Human Tf-Glycome Provides a Structural Definition for the Specificity of Anti-Tumor Antibody Jaa-F11. PLoS One 2013, 8, e54874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27). Cygler M; Rose DR; Bundle DR Recognition of a Cell-Surface Oligosaccharide of Pathogenic Salmonella by an Antibody Fab Fragment. Science 1991, 253, 442–445. [DOI] [PubMed] [Google Scholar]
- (28). Arnaud J; Audfray A; Imberty A Binding Sugars: From Natural Lectins to Synthetic Receptors and Engineered Neolectins. Chem. Soc. Rev 2013, 42, 4798–4813. [DOI] [PubMed] [Google Scholar]
- (29). Romano PR; Mackay A; Vong M; Desa J; Lamontagne A; Comunale MA; Hafner J; Block T; Lec R; Mehta A Development of Recombinant Aleuria Aurantia Lectins with Altered Binding Specificities to Fucosylated Glycans. Biochem. Biophys. Res. Commun 2011, 414, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30). Brummell DA; Sharma VP; Anand NN; Bilous D; Dubuc G; Michniewicz J; Mackenzie CR; Sadowska J; Sigurskjold BW Probing the Combining Site of an Anti-Carbohydrate Antibody by Saturation-Mutagenesis: Role of the Heavy-Chain Cdr3 Residues. Biochemistry 1993, 32, 1180–1187. [DOI] [PubMed] [Google Scholar]
- (31). Lak P; Makeneni S; Woods RJ; Lowary TL Specificity of Furanoside,Äìprotein Recognition through Antibody Engineering and Molecular Modeling. Chem. - Eur. J 2015, 21, 1138–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32). Makeneni S; Ji Y; Watson DC; Young NM; Woods RJ Predicting the Origins of Anti-Blood Group Antibody Specificity: A Case Study of the Abo a- and B-Antigens. Front. Immunol 2014, 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33). Johnson MA; Pinto BM Saturation-Transfer Difference Nmr Studies for the Epitope Mapping of a Carbohydrate-Mimetic Peptide Recognized by an Anti-Carbohydrate Antibody. Bioorg. Med. Chem 2004, 12, 295–300. [DOI] [PubMed] [Google Scholar]
- (34). Herfurth L; Ernst B; Wagner B; Ricklin D; Strasser DS; Magnani JL; Benie AJ; Peters T Comparative Epitope Mapping with Saturation Transfer Difference Nmr of Sialyl Lewis(a) Compounds and Derivatives Bound to a Monoclonal Antibody. J. Med. Chem 2005, 48, 6879–6886. [DOI] [PubMed] [Google Scholar]
- (35). Demarco ML; Woods RJ Structural Glycobiology: A Game of Snakes and Ladders. Glycobiology 2008, 18, 426–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36). Boltje TJ; Buskas T; Boons G-J Opportunities and Challenges in Synthetic Oligosaccharide and Glycoconjugate Research. Nat. Chem 2009, 1, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37). Paula S; Monson N; Ball WJ Jr. Molecular Modeling of Cardiac Glycoside Binding by the Human Sequence Monoclonal Antibody 1b3. Proteins: Struct., Funct., Genet 2005, 60, 382–391. [DOI] [PubMed] [Google Scholar]
- (38). Makeneni S; Ji Y; Watson DC; Young NM; Woods RJ Predicting the Origins of Anti-Blood Group Antibody Specificity: A Case Study of the Abo a- and B-Antigens. Front. Immunol 2014, 5, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39). Nivedha AK; Thieker DF; Makeneni S; Hu H; Woods RJ Vina-Carb: Improving Glycosidic Angles During Carbohydrate Docking. J. Chem. Theory Comput 2016, 12, 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40). Trott O; Olson AJ Autodock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41). Morris GM; Goodsell DS; Halliday RS; Huey R; Hart WE; Belew RK; Olson AJ Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem 1998, 19, 1639–1662. [Google Scholar]
- (42). Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ Autodock4 and Autodocktools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43). Gauto DF; Petruk AA; Modenutti CP; Blanco JI; Di Lella S; Marti MA Solvent Structure Improves Docking Prediction in Lectin-Carbohydrate Complexes. Glycobiology 2013, 23, 241–258. [DOI] [PubMed] [Google Scholar]
- (44). Pyrkov TV; Pyrkova DV; Balitskaya ED; Efremov RG The Role of Stacking Interactions in Complexes of Proteins with Adenine and Guanine Fragments of Ligands. Acta Naturae 2009, 1, 124–127. [PMC free article] [PubMed] [Google Scholar]
- (45). Nivedha AK; Makeneni S; Foley BL; Tessier MB; Woods RJ Importance of Ligand Conformational Energies in Carbohydrate Docking: Sorting the Wheat from the Chaff. J. Comput. Chem 2014, 35, 526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46). Shoichet BK Virtual Screening of Chemical Libraries. Nature 2004, 432, 862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47). Schulz-Gasch T; Stahl M Scoring Functions for Protein–Ligand Interactions: A Critical Perspective. Drug Discovery Today: Technol. 2004, 1, 231–239. [DOI] [PubMed] [Google Scholar]
- (48). Hadden JA; Tessier MB; Fadda E; Woods RJ Calculating Binding Free Energies for Protein–Carbohydrate Complexes In Glycoinformatics; Lütteke T; Frank M, Eds.; Springer: New York, 2015; pp 431–465. [DOI] [PubMed] [Google Scholar]
- (49). Bernstein FC; Koetzle TF; Williams GJ; Meyer EF Jr.; Brice MD; Rodgers JR; Kennard O; Shimanouchi T; Tasumi M The Protein Data Bank: A Computer-Based Archival File for Macromolecular Structures. J. Mol. Biol 1977, 112, 535–542. [DOI] [PubMed] [Google Scholar]
- (50). Lee M; Lloyd P; Zhang X; Schallhorn JM; Sugimoto K; Leach AG; Sapiro G; Houk KN Shapes of Antibody Binding Sites: Qualitative and Quantitative Analyses Based on a Geomorphic Classification Scheme. J. Org. Chem 2006, 71, 5082–5092. [DOI] [PubMed] [Google Scholar]
- (51). Nivedha AK; Thieker DF; Hu H; Woods RJ; Makeneni S Vina-Carb: Improving Glycosidic Angles During Carbohydrate Docking. J. Chem. Theory Comput 2016, 12, 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52). Gasteiger J; Marsili M Iterative Partial Equalization of Orbital Electronegativity—a Rapid Access to Atomic Charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar]
- (53). Götz AW; Williamson MJ; Xu D; Poole D; Le Grand S; Walker RC Routine Microsecond Molecular Dynamics Simulations with Amber on Gpus. 1. Generalized Born. J. Chem. Theory Comput 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54). Case DA; Darden TA; Iii TEC; Simmerling CL; Wang J; Duke RE; Luo R; Walker RC; Zhang W; Merz KM; Roberts B; Hayik S; Roitberg A; Seabra G; Swails J; Götz AW; Kolossváry IW; Wong KF; Paesani F; Vanicek J; Wolf RM; Liu J; Wu X; Brozell SR; Steinbrecher T; Gohlke H; Cai Q; Ye X; Hsieh M-J; Cui G; Roe DR; Mathews DH; Seetin MG; Salomon-Ferrer R; Sagui CA; Babin V; Luchko T; Gusarov S; Kovalenko A; Kollman PA; Cheatham TE; Goetz AW; Kolossvai I AMBER12; University of California: San Francisco, 2012. [Google Scholar]
- (55). Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins: Struct., Funct., Genet 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56). Kirschner KN; Yongye AB; Tschampel SM; González-Outeiriño J; Daniels CR; Foley BL; Woods RJ Glycam06: A Generalizable Biomolecular Force Field. Carbohydrates. J. Comput. Chem 2008, 29, 622–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57). Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (58). Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of N-Alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- (59). Darden T; York D; Pedersen L Particle Mesh Ewald: An N Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (60). Roe DR; Cheatham TE Ptraj and Cpptraj: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- (61). Humphrey W; Dalke A; Schulten K Vmd: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (62). Thieker DF; Hadden JA; Schulten K; Woods RJ 3d Implementation of the Symbol Nomenclature for Graphical Representation of Glycans. Glycobiology 2016, 26, 786–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63). Varki A; Cummings RD; Aebi M; Packer NH; Seeberger PH; Esko JD; Stanley P; Hart G; Darvill A; Kinoshita T; Prestegard JJ; Schnaar RL; Freeze HH; Marth JD; Bertozzi CR; Etzler ME; Frank M; Vliegenthart JFG; Lütteke T; Perez S; Bolton E; Rudd P; Paulson J; Kanehisa M; Toukach P; Aoki-Kinoshita KF; Dell A; Narimatsu H; York W; Taniguchi N; Kornfeld S Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 2015, 25, 1323–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64). Hetényi C; Van Der Spoel D Efficient Docking of Peptides to Proteins without Prior Knowledge of the Binding Site. Protein Sci. 2002, 11, 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65). Erickson JA; Jalaie M; Robertson DH; Lewis RA; Vieth M Lessons in Molecular Recognition: The Effects of Ligand and Protein Flexibility on Molecular Docking Accuracy. J. Med. Chem 2004, 47, 45–55. [DOI] [PubMed] [Google Scholar]
- (66). Pejchal R; Doores KJ; Walker LM; Khayat R; Huang PS; Wang SK; Stanfield RL; Julien JP; Ramos A; Crispin M; Depetris R; Katpally U; Marozsan A; Cupo A; Maloveste S; Liu Y; Mcbride R; Ito Y; Sanders RW; Ogohara C; Paulson JC; Feizi T; Scanlan CN; Wong CH; Moore JP; Olson WC; Ward AB; Poignard P; Schief WR; Burton DR; Wilson IA A Potent and Broad Neutralizing Antibody Recognizes and Penetrates the Hiv Glycan Shield. Science 2011, 334, 1097–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67). Vulliez-Le Normand B; Saul FA; Phalipon A; Belot F; Guerreiro C; Mulard LA; Bentley GA Structures of Synthetic O-Antigen Fragments from Serotype 2a Shigella Flexneri in Complex with a Protective Monoclonal Antibody. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 9976–9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68). Daranas AH; Shimizu H; Homans SW Thermodynamics of Binding of D-Galactose and Deoxy Derivatives Thereof to the L-Arabinose-Binding Protein. J. Am. Chem. Soc 2004, 126, 11870–11876. [DOI] [PubMed] [Google Scholar]
- (69). Vyas NK; Vyas MN; Chervenak MC; Johnson MA; Pinto BM; Bundle DR; Quiocho FA Molecular Recognition of Oligosaccharide Epitopes by a Monoclonal Fab Specific for Shigella Flexneri Y Lipopolysaccharide: X-Ray Structures and Thermodynamics. Biochemistry 2002, 41, 13575–13586. [DOI] [PubMed] [Google Scholar]
- (70). Dam TK; Cavada BS; Grangeiro TB; Santos CF; Ceccatto VM; De Sousa FA; Oscarson S; Brewer CF Thermodynamic Binding Studies of Lectins from the Diocleinae Subtribe to Deoxy Analogs of the Core Trimannoside of Asparagine-Linked Oligosaccharides. J. Biol. Chem 2000, 275, 16119–16126. [DOI] [PubMed] [Google Scholar]
- (71). Wen X; Yuan Y; Kuntz DA; Rose DR; Pinto BM A Combined Std-Nmr/Molecular Modeling Protocol for Predicting the Binding Modes of the Glycosidase Inhibitors Kifunensine and Salacinol to Golgi A-Mannosidase Ii. Biochemistry 2005, 44, 6729–6737. [DOI] [PubMed] [Google Scholar]
- (72). Lycknert K; Edblad M; Imberty A; Widmalm G Nmr and Molecular Modeling Studies of the Interaction between Wheat Germ Agglutinin and the B-D-Glcpnac-(1→6)-A-D-Manp Epitope Present in Glycoproteins of Tumor Cells. Biochemistry 2004, 43, 9647–9654. [DOI] [PubMed] [Google Scholar]
- (73). Haselhorst T; Blanchard H; Frank M; Kraschnefski MJ; Kiefel MJ; Szyczew AJ; Dyason JC; Fleming F; Holloway G; Coulson BS; Von Itzstein M Std Nmr Spectroscopy and Molecular Modeling Investigation of the Binding of N-Acetylneuraminic Acid Derivatives to Rhesus Rotavirus Vp8* Core. Glycobiology 2007, 17, 68–81. [DOI] [PubMed] [Google Scholar]
- (74). Hoog C; Widmalm G Molecular Dynamics Simulation and Nuclear Magnetic Resonance Studies of the Terminal Glucotriose Unit Found in the Oligosaccharide of Glycoprotein Precursors. Arch. Biochem. Biophys 2000, 377, 163–170. [DOI] [PubMed] [Google Scholar]
- (75). Porter A; Yue T; Heeringa L; Day S; Suh E; Haab BB A Motif-Based Analysis of Glycan Array Data to Determine the Specificities of Glycan-Binding Proteins. Glycobiology 2010, 20 (3), 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76). Kletter D; Cao Z; Bern M; Haab B Determining Lectin Specificity from Glycan Array Data Using Motif Segregation and Glycosearch Software. Current Protocols in Chemical Biology 2013, 5, 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.