

SUMMARY
Activity-dependent changes in neuronal function require coordinated regulation of the protein synthesis and protein degradation machinery to maintain protein homeostasis, critical to proper neuronal function. However, the biochemical evidence for this balance and coordination is largely lacking. Leveraging our recent discovery of a neuronal-specific 20S membrane proteasome complex (NMP), we began exploring how neuronal activity regulates its function. Here, we found that the NMP degrades exclusively a large fraction of ribosome-associated nascent polypeptides that are being newly synthesized during neuronal stimulation. Using deep-coverage and global mass spectrometry, we identified the nascent protein substrates of the NMP, which included products encoding immediate-early genes, such as c-Fos and Npas4. Intriguingly, we found that turnover of nascent polypeptides and not full-length proteins through the NMP occurred independent of canonical ubiquitylation pathways. We propose that these findings generally define a neuronal activity-induced protein homeostasis program of coordinated protein synthesis and degradation through the NMP.
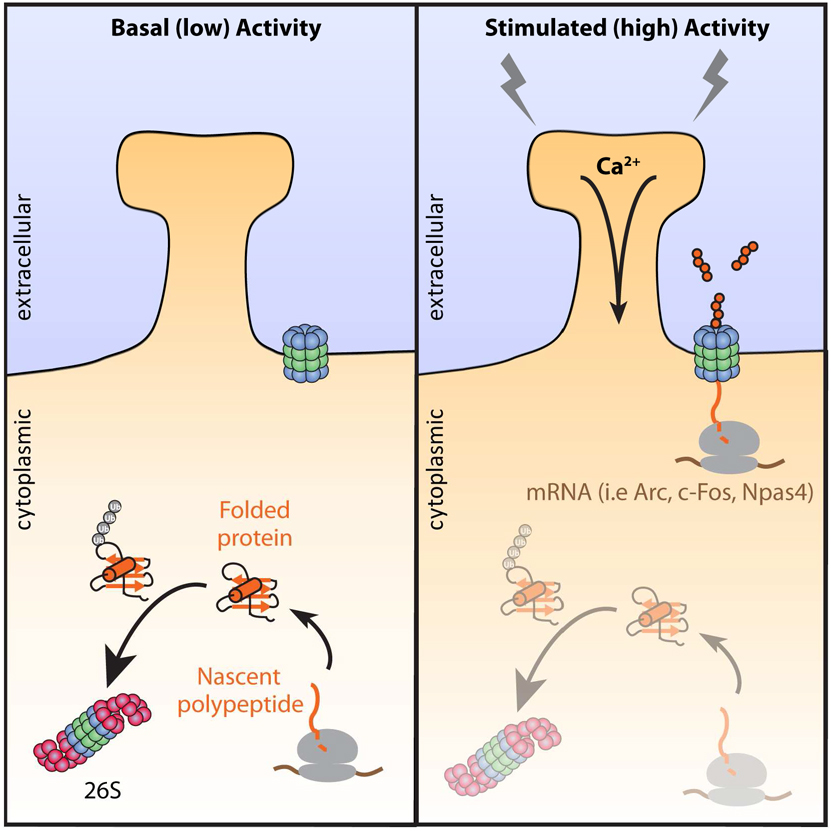
INTRODUCTION
Neuronal activity-dependent processes have been shown by many laboratories to be dependent upon new protein synthesis and proteasome-dependent protein degradation (Djakovic et al., 2009; Fonseca et al., 2006a; Kelleher et al., 2004). Moreover, the ribosome and proteasome independently localize to sites of synaptic activity and are important for activity-mediated synaptic remodeling (Bingol and Schuman, 2006; Ehlers, 2003; Ostroff et al., 2017; Tcherkezian et al., 2010). These two complexes are hypothesized to coordinate their functions to modulate neuronal signaling (Deglincerti et al., 2015; Fonseca et al., 2006b; Klein et al., 2015). However, the biochemical evidence for the existence and mechanism of this coordination remains to be elucidated.
We considered that activity dependent coordination of protein synthesis and degradation might simultaneously engage the actions of both the ribosome and proteasome in neurons. This could manifest as co-translational degradation, which is the direct proteasome-mediated degradation of a nascent polypeptide while still associated with the ribosome. Indeed, classic experiments in monitoring protein fate reported a significant portion of proteins underwent rapid degradation either during or immediately following their synthesis (Robertson and Wheatley, 1979; Wheatley et al., 1982; Wheatley and Inglis, 1980). Further investigation of these observations in yeast and in vitro revealed that some proteins undergoing synthesis are degraded co-translationally (Anton and Yewdell, 2014; Schubert et al., 2000; Turner and Varshavsky, 2000; Vabulas and Hartl, 2005). However, how these events are regulated remains unknown, the extent to which these events occur is debated, and these events have never been monitored in neurons.
Co-translational degradation mechanisms have been shown to rely on the concerted actions of the ubiquitin-proteasome system (Ben-Nissan and Sharon, 2014; Ciechanover, 1998; Coux et al., 1996). In the canonical paradigm, ubiquitylated proteins are delivered to and degraded by the 26S proteasome, a large multisubunit protease with two major functional holoenzymes (20S and 19S). The 20S core complex, is a chamber with α7β7β7α7 stacked subunit configuration that contains the catalytic domains required to cleave protein substrates (Ciechanover, 1998). The 19S cap complex, contains multiple subunits which recognize ubiquitylated substrates and ATPases which unfold proteins (de Poot et al., 2017; Finley et al., 2004; Schmidt and Finley, 2014). These 19S ATPases are generally required since the 20S core proteasome can only degrade already unfolded proteins, or those with large unstructured domains such as intrinsically disordered proteins (Ben-Nissan and Sharon, 2014; Coux et al., 1996; Tsvetkov et al., 2008; Tsvetkov et al., 2009). While the majority of studies on proteasome-dependent degradation have been attributed to the actions of the 26S, roles for the 20S proteasome are emerging.
Recently, we discovered an uncapped 20S proteasome complex tightly associated with the neuronal plasma membrane, where it degrades intracellular proteins into extracellular peptides (Ramachandran and Margolis, 2017). The mechanisms for substrate delivery to this neuronal membrane proteasome (NMP) remained unknown, but we presumed they must be unfolded to some extent since the NMP is a 20S proteasome that does not contain capping structures important for recognition and unfolding of ubiquitylated full-length substrates (de Poot et al., 2017; Finley et al., 2004; Ramachandran and Margolis, 2017; Schmidt and Finley, 2014). Protein synthesis produces a significant source of unfolded proteins, initially as nascent polypeptides, making this process ideal for 20S-mediated degradation (Balchin et al., 2016; Duttler et al., 2013; Pechmann et al., 2013; Sontag et al., 2017). Therefore, we hypothesized that neuronal activity may coordinate protein synthesis and degradation in the nervous system through the NMP. Here, we present biochemical and proteomic data in support of this hypothesis.
RESULTS
Neuronal stimulation induces NMP-dependent degradation of newly synthesized proteins into extracellular peptides
To determine whether neuronal activity modulates NMP function, we measured NMP dependent peptide release during KCl-induced membrane depolarization to induce elevated activity of the majority of neurons in culture (Lin et al., 2008; West et al., 2001). Mouse primary cortical neuron cultures at days in vitro (DIV) 10–14 were treated with either a KCl stimulation (KCl) or NaCl control buffer (Con) and were concomitantly radiolabelled with 35S-methionine/cysteine for 10 minutes. We observed a KCl-induced increase in release of radiolabelled extracellular peptides compared to release from unstimulated neurons. Peptide release was occluded in all conditions by addition of pan-proteasome inhibitors (MG132 or epoxomicin) or the NMP-specific inhibitor biotin-epoxomicin (Bio-Epox)(Figure 1A, S1A, S1B, S1C). Peptide release was occluded in unstimulated neurons by addition of neuronal activity inhibitors, consistent with neuronal activity being essential for NMP function (Figure S1D).
Figure 1. Neuronal stimulation induces NMP-dependent degradation of newly synthesized proteins into extracellular peptides.

(A) Concomitant radiolabelling during neuronal stimulation induces NMP-mediated peptide release. Scintillation data at the indicated time points are shown normalized to control at the 5-minute time point. Data are presented as mean ± SEM (n = 3). *p < 0.01 (KCl), ‡p < 0.01(MG132), §p < 0.01 (Bio-Epox)(two-way ANOVA).
(B) Neuronal stimulation induces NMP-mediated degradation of intracellular proteins made during stimulation. Left, representative autoradiograph of lysates from neurons radiolabelled during either control (C) or KCl (K) stimulation and treated with MG132 or biotin-epoxomicin (Bio-Epox). Right, quantification of densitometry signal normalized to control. Data are presented as mean ± SEM (n = 3). *p < 0.01 (Control), ‡p < 0.01 (MG132), §p < 0.01 (Bio-Epox)(two-way ANOVA).
(C) Neuronal stimulation does not induce NMP-mediated degradation of proteins made prior to stimulation. Left, Representative autoradiograph of lysates from neurons previously radiolabelled and then chased into either control (C) or KCl (K) stimulation buffers for indicated times. Input shows sample collected immediately following labeling. Right, quantification of densitometry signal normalized to control. Data are presented as mean ± SEM (n = 3). Statistically significant differences between samples was not observed (two-way ANOVA).
(D) Neuronal stimulation does not induce NMP-mediated degradation of proteins made prior to stimulation. Experiments done as described in (A), note neurons were radiolabelled prior to instead of during stimulation as in (A). Data are presented as mean ± SEM (n = 3). Statistically significant differences between samples was not observed (two-way ANOVA).
(E) Peptide release following 30 second (dotted lines) or 10 minutes (solid lines) of radiolabeling as in (A). Data are presented as mean ± SEM (n = 3). Line graph, *p < 0.01 (two-way ANOVA) for Control compared to KCl for either 30 second or 10 minute labeling. No significance was detected between similar treatments at 30 seconds versus 10 minutes.
(F) Concomitant radiolabelling during neuronal stimulation induces NMP-mediated radiolabeled peptide release that is sensitive to cycloheximide and puromycin treatment. (Left) Translating ribosomes (grey) on mRNA. AUG start site shown just prior to tRNA (small structure with codon recognition loops, in ribosome P site) and growing radioactive polypeptide (growing red line out of translating ribosomes). Puromycin (hexagon) modifies and releases the nascent polypeptide (red) from actively translating ribosomes. Experiments done as described in (A), note, washout with fresh media containing Cycloheximide (CHX) or puromycin (Puro). Data are presented as mean ± SEM (n = 3). *p < 0.01 (Control), #p < 0.01 (puromycin) (two-way ANOVA).
See also Figure S1.
We next measured the intracellular pool of proteins made during KCl stimulation using SDS-PAGE and autoradiography. Equal loading was confirmed by coomassie (Figure S1E). In lysates from neurons treated as in Figure 1A, we noticed a decrease in radioactive intracellular protein signal from neurons that had been concomitantly radiolabelled during stimulation (Figure 1B). This effect was induced by various stimulation methods that depolarize neurons, but not by serum (Figures S1F-H)(Fortin et al., 2010; Lin et al., 2008; Marin et al., 1997; Scheetz et al., 2000). Treating these neurons with Bio-Epox or MG132 during radiolabelling blocked the stimulation-induced loss of radiolabelled protein signal (Figure 1B). This enhanced degradation of intracellular substrates was not due to increased catalytic activity of the NMP (Figure S1I).
These experiments monitored activity-dependent turnover of proteins made during stimulation. In contrast, proteins from neurons that had been radiolabelled prior to or following the onset of stimulation were not degraded, even after sustained KCl stimulation (Figure 1C and S1J). Consequentially, we did not detect an increase in peptide release when neurons were radiolabeled prior to, instead of during KCl-stimulation (Figure 1D). These data illustrate that neuronal stimulation does not simply promote the turnover of all proteins, but specifically enhances NMP-mediated turnover of proteins being newly synthesized during neuronal stimulation. These observations raised a fundamental question – why were proteins made during stimulation, as compared to all other proteins, being turned over? We hypothesized that this answer was linked to the properties of NMP substrates.
Incorporating radiolabel for 10 minutes allows us to monitor actively translating nascent polypeptides, polypeptides just completing synthesis, and full-length proteins. To determine which of these protein populations were being targeted for degradation we shortened our radiolabeling time to only 30 seconds during stimulation to preferentially label nascent polypeptides before they finish synthesis (Duttler et al., 2013; Ito et al., 2011). We noted the kinetics of peptide release observed under these conditions were highly similar to those observed in Figure 1A (Figure 1E). These data were consistent with a model where these peptides were derived from nascent polypeptides during synthesis. Indeed, the protein synthesis inhibitor, cycloheximide, abrogated this peptide release (Figure 1F). To test this further, we used puromycin which is an aminoacyl-tRNA structural analog that covalently modifies and dissociates the growing polypeptide from the ribosome (Nathans and Neidle, 1963). Addition of puromycin to neuronal cultures in the release assay resulted in loss of peptide release from both KCl-stimulated and unstimulated neurons (Figure 1F).
Neuronal stimulation induces NMP-mediated co-translational degradation of ribosome-associated nascent polypeptides
Nascent polypeptides are bound to a tRNA within the ribosome during translation (ribosome-nascent chain complex, or RNC)(Duttler et al., 2013). To determine whether the NMP was targeting nascent polypeptides while still associated with ribosomes (during translation), we performed ribosome-pelleting assays to isolate RNCs (Brandman et al., 2012). Following 30 seconds of radiolabeling with KCl-stimulation, neurons were lysed in the presence of proteasome inhibitors to block degradation and either cycloheximide (CHX) to stall RNCs or puromycin to dissociate nascent chains from RNCs (Figure 2A - model). Radiolabelled RNCs were pelleted, and quantified by scintillation (Figure 2A, S2A). Radioactivity in RNCs was highly sensitive to puromycin treatment, indicating that the radiolabel signal represents nascent polypeptides (Figure 2A). We noticed a decrease in radioactive signal in RNCs from neurons that had been radiolabelled during KCl-stimulation (Figure 2A). Treating neurons with Bio-Epox or MG132 during radiolabelling blocked this decrease in radioactive signal and showed elevation in labeling of RNCs from both stimulated and unstimulated neurons (Figure 1A). We interpret these data to reflect an increase in nascent chain length upon proteasome inhibition, potentially because the nascent chain is no longer being proteolyzed. We do not observe a proteasome-sensitive increase in radiolabel incorporation (Figure S2B) from RNCs isolated from HEK293 cells (which do not express NMPs (Ramachandran and Margolis, 2017)).
Figure 2. Neuronal stimulation induces NMP-mediated co-translational degradation of ribosome-associated nascent polypeptides.

(A) Neuronal stimulation induces NMP-mediated degradation of ribosome-associated polypeptides. (Left) Experimental setup. Translating ribosomes (grey) on mRNA, tRNA (small structure with codon recognition loops, in ribosome P site), and growing radioactive polypeptide (growing red line out of translating ribosomes). Puromycin (hexagon) modifies and releases the nascent polypeptide from actively translating ribosomes. (Right) Graph shows quantification of radiolabelled RNC complexes by liquid scintillation. Normalized against control alone. Data are presented as mean ± SEM (n = 3). *p < 0.01 (control), #p < 0.01 (KCl) (two-way ANOVA). See also Figures S2. All puromycin treatments were statistically significantly lower than controls, but not significant amongst each other.
(B) Experimental setup. Growing radiolabeled polypeptide (red) either bound to tRNA or free. Ribosome (gray)
(C) Elongating tRNA-bound nascent polypeptides are degraded by the NMP during stimulation. Pelleted RNCs from (A) were processed by 2D SDS-PAGE as described in (B).
(D) Immunoblots of the 2-D samples in (C).
(E) Experimental setup.
(F) NMP associates with ribosomal subunits from bicuculline-stimulated neurons. Samples prepared as described in (E). Immunoblots of neuronal membrane inputs (left) and proteasome IPs (right).
See also Figure S2.
To extend these analyses, we leveraged previously described 2D-gel electrophoresis approaches to visualize labeled nascent polypeptides (Ito et al., 2011). Pelleted RNCs from neurons radiolabeled for 30 seconds were separated in the first dimension by SDS-PAGE (Figure 1B). Next, individual gel lanes were treated with base to hydrolyze tRNAs from their bound nascent polypeptides, and subsequently separated by SDS-PAGE in the second dimension (Figure 1B). The fast-migrating band consisted of nascent polypeptides separated from their tRNAs in the second dimension. The slow-migrating band consisted of polypeptides that were not bound to tRNA in the first dimension (e.g. full-length proteins and nascent polypeptides separated from their tRNAs during processing in the first dimension). Importantly, all RNC-associated radioactivity came from nascent polypeptides, based on their sensitivity to puromycin (Figure 2C). We observed approximately a 40% reduction in radiolabel signal intensity of both the fast- and slow-migrating bands from KCl-stimulated versus unstimulated samples (Figure 2C). This stimulation-induced loss of radiolabel signal was entirely recovered by treating neurons with Bio-Epox or MG132 (Figure 2C, S2C). Immunoblotting these samples using an antibody against ubiquitin revealed detectable signal in the slower migrating band of the 2D-gel which was undetectable in the fast migrating nascent polypeptide band (Figure 2D). Ubiquitin immunoblot signal was unaffected by puromycin treatment (Figure 2D). Based on these data, we concluded that the nascent chain was most likely not ubiquitylated, at least at detectable levels, yet still targeted for NMP-mediated degradation. These data were consistent with the NMP operating as a 20S proteasome, which degrades unfolded polypeptides in an ubiquitin-independent manner (Ben-Nissan and Sharon, 2014; Coux et al., 1996). These data supported the hypothesis that neuronal stimulation induces NMP-mediated co-translational degradation of ribosome-associated nascent polypeptides.
One prediction from this hypothesis is that ribosomes must be present at neuronal membranes, in close proximity to the NMP to provide nascent polypeptides as substrates. Indeed, ribosomes have been previously shown to be proximal to neuronal postsynaptic plasma membranes (Gardiol et al., 1999; Roberts and Flexner, 1966; Tcherkezian et al., 2010). Using previously described approaches (Figure 1E)(Ramachandran and Margolis, 2017), we determined that ribosomal S6 preferentially co-immunoprecipitated with the NMP in the presence of neuronal stimulation (Figure 1F). Bicuculline was used to stimulate neurons, as KCl membrane depolarization interfered with our membrane fractionation protocol (data not shown). The addition of puromycin and high salt, which knocks off nascent chain-associated proteins, to the immunoprecipitates eliminated the interaction between the ribosome and the NMP (Figure 1F). These data support an activation mechanism for NMP-mediated degradation in which translating ribosomes feed nascent polypeptides to the NMP in response to neuronal activity.
Identification of activity-dependent nascent NMP substrates
To identify substrates of co-translational degradation through the NMP in an unbiased manner, we turned to mass spectrometry-based quantitative global proteomic analysis using tandem mass tag (TMT) technology (Figure 3A). Similar to Figure 2F, neuronal cultures were incubated with bicuculline for one hour and treated with vehicle (DMSO), Bio-Epox, or Bio-Epox-CHX in the last 10 minutes of the one hour stimulation. Following these treatments in biological triplicates, proteins were extracted from the samples and derivatized using TMT tags following an enzymatic digestion. The labeled peptides were then fractionated followed by mass spectrometry analysis, database search, and statistical analysis (Figure 3A). Overall, the combined analysis of the replicates across treatment groups yielded 141,295 peptides that were mapped to 8,223 proteins (Figure 3B, Table S1). The reproducibility across biological replicates was robust, with coefficients of variation of <10% observed for >99% of the proteins. We defined a co-translationally degraded substrate of the NMP as one with higher protein levels in bicuculline/Bio-Epox-treated neurons as compared to both bicuculline and bicuculline/Bio-Epox/CHX. The CHX group serves as an essential control to demonstrate that these are newly synthesized proteins, and not preexisting proteins which are simply stabilized due to inhibition of the NMP. Statistically significant differences between Bio-Epox treated samples compared to the other groups were observed for 1,339 proteins at p < 0.05, and 408 for p < 0.01 (Table S1). Taking multiple comparisons testing into account yielded a list of 191 differentially expressed proteins, of which 122 were up-regulated, and therefore considered co-translationally degraded NMP substrates (Figure 3B,C, Table S2). Of significant note, we found IEG products c-Fos, Fosb, Npas4, and Egr1 were significantly upregulated in response to bicuculline/Bio-Epox treatment (Figure 3D).
Figure 3. Quantitative 10-plex mass spectrometry experiment to identify newly synthesized NMP substrates.

(A) Experimental setup.
(B) Scatterplot of normalized log2 bicuculline/Bio-Epox treated compared to both bicuculline alone and bicuculline/Bio-Epox/cycloheximide, versus q-values (p-values after multiple comparisons testing). NMP-targets (orange). Non-NMP targets (blue).
(C) Heat map of proteins differentially expressed in bicuculline/Bio-Epox treated compared to bicuculline and bicuculline/Bio-Epox/cycloheximide. Coloring indicated percentage of maximum fold change. Top 60 statistically significant targets are shown.
(D)Individual targets are shown, with replicates in scatterplot format. Data are presented as mean ± SEM (n = 3). ***p < 0.001, q < 0.1 (two-way ANOVA (p), adjusted for multiple corrections (q) for biotin-epoxomicin (BEp) treatment compared to other samples). NMP targets previously shown to be UPS targets in top row, orange. NMP-targets (orange). Non-NMP targets (blue).
We initially attributed the upregulation observed in our MS data to canonical activity-induced IEG induction. However, by immunoblot analysis, bicuculline stimulation for one hour does not lead to significant increases in IEG protein expression (Figure S3). In contrast, following two hours of bicuculline stimulation, we observed the canonical induction of IEG protein expression that was dependent on neuronal activity, transcription, and translation (Figure S3).
Nascent IEG products, and not full-length proteins, are substrates of the NMP
To validate our MS/MS data, cortical neurons were stimulated with bicuculline for one hour, and treated with either Bio-Epox or MG132 for the final 10 minutes. This led to an accumulation of IEG proteins, c-Fos and Npas4 but no change in the protein levels of UPS targets such as PSD95 (Figure 4A). This increase in IEG protein levels was blocked by co-incubation with cycloheximide, but transcriptional inhibitor actinomycin D had no effect (Figure 4A). While we did not detect a change in Arc levels in the MS analysis, we did observe significant changes by immunoblot. This likely reflects the differences in detection sensitivity between the two methods. Notably, in the absence of stimulation, Bio-Epox or MG132 treatment also led to a small, but reproducible increase in IEG products (Figure 4A and S4A). Addition of CHX or neuronal activity inhibitors blocked this proteasome inhibitor-mediated increase, suggesting that the effect depends on translation and baseline activity present in neuronal cultures (Figure S4A). In all of these experiments, the effects of Bio-Epox or MG132 on IEG protein expression were nearly identical (Figures 4A and S4A), which reflects that the majority of the observed degradation is through the NMP, and not the cytosolic proteasome.
Figure 4. Nascent, not full length, immediate-early gene products are activity-dependent NMP substrates.

(A) IEG products are degraded by the NMP during bicuculline stimulation in a translation-dependent and transcription-independent manner. Experimental timeline shown above. Cycloheximide (CHX) added during proteasome inhibition. Actinomycin D (ActD) added for entire hour. Neuronal lysates were immunoblotted. Representative immunoblots shown. Data are presented as mean ± SEM (n = 3).
(B) Turnover of full-length IEG protein is inhibited by MG132 and not Bio-Epox. Experimental timeline shown above. Bicuculline (Bic) treated neurons were chased into DMSO (Veh), cycloheximide (CHX) or CHX + Bic for one hour in presence of indicated drugs. Neuronal lysates were immunoblotted. Representative immunoblots shown. Data are presented as mean ± SEM (n = 3).
(C) IEG products are degraded by the NMP during bicuculline stimulation without the need for ubiquitylation. Experimental timeline shown above. MLN-7243, or DMSO was applied during the final 30 minutes of bicuculline treatment prior to indicated drug addition. Neuronal lysates were immunoblotted.
These data did not distinguish between co-translational degradation and full-length protein degradation of IEG products. To monitor turnover only of the full-length protein, we took advantage of IEG protein expression following two hours of bicuculline stimulation (Figure S3). Following stimulation, we washed out the bicuculline and added cycloheximide to prevent any further protein expression. This allows us to monitor the fate of these IEG protein products that had completed synthesis. As expected, we observed robust induction of IEG proteins following two hours of bicuculline stimulation that was largely turned over in one hour in the absence of sustained translation (Figure 3B). This turnover was inhibited by the addition of MG132, consistent with the canonical turnover of IEG proteins by the ubiquitin-proteasome pathway (Figure 4B) (Carle et al., 2007; Ito et al., 2005; Mabb et al., 2014; Peebles et al., 2010). In contrast, Bio-Epox does not prevent the turnover of these full-length IEG products (Figure 4B). These data were the clearest demonstration that the NMP co-translationally degrades nascent polypeptides during elevated activity, but does not degrade a fully synthesized substrate (Figure 4B).
We next directly tested whether NMP-mediated degradation of these putative NMP substrates occurred independent of ubiquitylation. To do this, we utilized a recently described inhibitor of E1 ubiquitin activating enzymes, MLN-7243 (Hyer et al., 2018). We find that 30 minutes of MLN-7243 treatment in primary neurons significantly reduced detectable ubiquitin immunoblot signal (Figure 3C). This duration of treatment is sufficient to discharge a large fraction of free ubiquitin in the ubiquitin-conjugating cascade, while not allowing further charging on E1 (An and Harper, 2018; Hyer et al., 2018). The addition of MLN-7243 did not have any effect on NMP-dependent degradation of IEG proteins (Figure 4C). In contrast, turnover of full-length IEG proteins was inhibited by the addition of epoxomicin or MLN-7243, but not by Bio-Epox (Figure S4B). Taken together, these data are consistent with the role for the canonical ubiquitin proteasome system, and not the NMP, in degrading full-length proteins. This provides further support that the NMP may exclusively target non-ubiquitylated nascent chains.
DISCUSSION
During elevated states of neural activity, protein synthesis and protein degradation are independently essential for regulating the expression level of proteins important for promoting, enhancing and maintaining neuronal activity-dependent processes. Our experiments unify these observations and elucidate an activity-dependent coordination of protein synthesis and protein degradation through the NMP. This coordination results in NMP-dependent degradation of nascent polypeptides being synthesized during neuronal stimulation. We identified immediate-early gene products as among the many activity-dependent substrates of the NMP. Taken together, our studies define a protein homeostasis program that involves the coordination of protein synthesis and NMP-mediated degradation in the nervous system.
Ubiquitin-independent co-translational degradation
Protein turnover and degradation kinetics have been studied extensively over the past few decades (Duttler et al., 2013; McShane et al., 2016; Prouty et al., 1975; Schubert et al., 2000; Schwanhausser et al., 2013; Wheatley et al., 1982). The majority of these studies have used isotope pulse labeling of proteins over at least one hour, and then monitored the fate of those synthesized proteins. Cumulatively, they have concluded that protein turnover is most well-explained by two-state degradation kinetics, which predicts that some proteins degrade at the same rate over many hours, while others have both fast (within 2 hours) and slow (>8 hour) kinetics (McShane et al., 2016). Instead of long labeling protocols like the aforementioned studies, our use of short radiolabeling protocols allowed us to capture ultrafast degradation kinetics that reflects coupled protein synthesis and degradation. Indeed, by reducing the radiolabel pulse time to within a few minutes (timeframes much more similar to our analysis), other studies have shown that immediately newly synthesized proteins are quickly turned over (Goldberg and Dice, 1974; Prouty et al., 1975; Wheatley et al., 1980; Wheatley et al., 1982). Whether these studies define co-translational degradation has not been fully elucidated and certainly, such mechanistic studies have not been carried out in the nervous system.
The NMP has previously been described to be a 20S proteasome complex and despite extensive effort, no cap to recognize a ubiquitylated substrate has yet been identified on the NMP (Ramachandran and Margolis, 2017). Given that the 20S requires an unfolded substrate, it is logical that this NMP-dependent program of co-translational degradation would be ubiquitin-independent. Consistent with NMP degradation mechanisms operating independently of ubiquitylation pathways, we do not detect in our MS analysis changes in the levels of previously well-characterized ubiquitylated substrates (Shank, PSD-95, GKAP, and AKAP79/150) of the ubiquitin proteasome system (UPS) (Colledge et al., 2003; Ehlers, 2003; Lee et al., 2008; Shin et al., 2012). The mechanisms that discriminate substrate selectivity to the UPS pathway versus NMP degradation are unknown and critical to identify. The observation that ribosomes are associated with the NMP through the nascent chain likely provides a key insight into this process. A critical question to address will be how ribosomes are targeted to the NMP and what distinguishes cytosolic ribosomes from NMP-targeted ribosomes.
Nascent polypeptides, including the immediate-early gene products, are NMP substrates
Several groups have gone on to show that proteasome inhibition using MG132 can lead to an 2–3 fold induction of IEG proteins in neurons, such as Arc, c-Fos, and Npas4 (Carle et al., 2007; Mabb et al., 2014; Peebles et al., 2010; Tsurumi et al., 1995). However, these studies have not explicitly discriminated between those IEGs being synthesized and those which have already been synthesized. In fact, the vast majority focus on those already synthesized and monitor their turnover through the UPS pathway, typically by focusing on the E3 ubiquitin ligases that target these IEGs. Our findings remain perfectly consistent with these data that cytosolic proteasomes turn full-length IEGs over, but add a layer of regulation where NMPs turn nascent IEGs over independent of ubiquitylation. This is the first such study that has discriminated between the nascent pool vs full-length pool of IEGs. Understanding the biological significance of this process is among the most important questions to address moving forward.
In the nervous system, immediate early genes are defined by their rapid response to enhanced neuronal activity at the level of new gene transcription leading to significant induction of IEG mRNAs (Flavell and Greenberg, 2008). This is known to eventually lead to translation and expression of IEG proteins, which play critical roles in the formation and function of the nervous system (Flavell and Greenberg, 2008; Lin et al., 2008; Lyford et al., 1995). In our experimental paradigm, we observed that co-translational degradation of nascent IEG proteins occurred independent of new transcription. Based on these data, we speculate that NMP-targeted nascent IEG proteins are derived from mRNA produced prior to activity induction, likely coming from spontaneous activity within our cultures. Consistent with this hypothesis, sustained inhibition of transcription prior to activity induction abolished co-translational degradation of nascent IEG proteins (KVR, unpublished data). Taken together, we believe that co-translational degradation through the NMP does not require newly transcribed mRNA being produced during the induction of neuronal activity. This raises the interesting idea that distinct pools of IEG mRNA may be specifically targeted to the NMP. The mechanisms of this targeting and relevance for neuronal function remain open and exciting questions.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Seth S. Margolis (smargol7@jhmi.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal studies were performed with protocols that are compliant and approved by the Institutional Animal Care and Use Committees of The Johns Hopkins University School of Medicine. Wild type C57BL/6 mice were purchased from Charles River Laboratories (Stock number 027; RRID: IMSR_CRL:27). These are general use multipurpose animals that are used by many laboratories in the field. These animals were used for preparing neuronal cultures.
Neuronal cultures
For primary mouse neuronal cultures, pregnant wild-type C57BL/6 mice (RRID: IMSR_CRL:27) were obtained from Charles River Laboratories, sacrificed at E17.5 and dissected in order to remove embryos. From isolated embryos brains were removed and dissected cortices were then moved into dissociation buffer (10mM MgCl2, 1mM Kynurenic Acid, 10mM HEPES, pH 7.2 in Hank’s Balanced Salt Solution) plus 16.67 U/ml Papain, which was incubated for 30 minutes at 37 °C. All neurons were mixed together and conta in both male and female neuronal tissue. It was challenging to tell the sex of a mouse embryo and there was not enough material to separate out each embryo and still be able to perform the described biochemical studies in this manuscript. Authenticity of neuronal cultures was in part determined by morphology using light microscopy and immunoblotting of lysates from neuronal cultures with well-validated antibodies to neuronal enriched markers. Proteolyzed tissue was rinsed for 5 minutes twice in 10 mg/ml Trypsin inhibitor. Tissue was then resuspended in Neurobasal and mechanically dissociated into a single-cell suspension. Neurons designated for biochemical analysis were plated in 12-well, 6-well, or 10cm dish formats at 500,000 neurons/well, 2,000,000 neurons/well, and 10,000,000 neurons/dish, respectively. Plates were previously coated with 1 mg/ml Poly-L-Lysine (PLL). Cultured cortical neurons were maintained in Neurobasal Medium supplemented with 2% B-27, penicillin/streptomycin (100 U/mL and 100 μg/mL, respectively), and 2 mM glutamine at 37 °C/5% CO2. Neurons were subsequently fed every three-five days by adding a quantity of 10% of the original culture volume of fresh medium (Ramachandran and Margolis, 2017).
HEK293 cells
HEK293 cells were maintained in DMEM supplemented with 10% FBS, 2 mM glutamine, and penicillin/streptomycin (100 U/mL and 100 μg/mL, respectively) at 37 °C/5% CO2. Sex of HEK293 cells is human female. Frozen aliquots are cultured for a limited number of passages.
METHOD DETAILS
Antibodies
The following antibodies were used according to manufacturer’s and/or published suggestions for immunoblotting: anti-β-Actin (Abcam), anti-Biotin (Cell Signaling), Streptavidin-AF647 (Invitrogen), anti-Arc (Gift from P. Worley, Johns Hopkins, verified against knockout), anti-Fos (Cell Signaling), anti-Npas4 (Gift from Y. Lin, MIT, verified against knockout), anti-PSD-95 (Pierce), anti-UBE3A (Sigma, verified against knockout), anti-Ubiquitin (FK2, Enzo), anti-S6 ribosomal subunit (Cell Signaling), anti-Transferrin receptor (Sigma), anti-β2 proteasome (Enzo), anti-α1–7 proteasome (Enzo). Standard secondary antibodies were purchased from Cell Signaling. We attempted to use antibodies that were verified by knockout controls in either our study, or by other groups. We only used antibodies that provided a signal at the appropriate molecular weight, and where minimal nonspecific bands were observed.
Immunoblot analysis
Immunoblots were performed using conventional approaches. Lysates from cultured cells were prepared directly in cold RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% Sodium Deoxycholate, 0.1% SDS, 5 mM EDTA, complete protease inhibitor cocktail tablet (Roche), 1 mM sodium orthovanadate, 1 mM β-glycerophosphate). Samples were spun down at 10000 RPM for 10 minutes, and SDS Laemmli buffer was added post lysis to the supernatants. Samples were boiled for 5 minutes before loading on to 10% or 12% Tris/Glycine SDS-PAGE gels. Proteins were transferred to nitrocellulose membranes at 100V for 2 hours in 20% methanol containing 20% MeOH tris/glycine-based transfer buffer. All antibodies were made up in 5% BSA in 0.1% TBST, except for Arc antibody which was made up in 5% Milk in 0.1% TBST. Immunoblots were incubated with appropriate secondary antibodies coupled to Horseradish Peroxidase, extensively washed, and incubated with ECL. Blots were exposed on film, and were scanned in and quantified using ImageJ by standard densitometry analysis.
Stimulation
For KCl stimulation experiments, neurons were depolarized with 55 mM extracellular KCl by addition of prewarmed depolarization buffer (55 mM KCl, 0.2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES pH 7.5) or a control buffer (55 mM NaCl, 0.2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES pH 7.5) in fresh neuronal growth media as previously described (Lin et al., 2008). Alternative stimuli to depolarization included chemical LTP (125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 5 mM Hepes, 33 mM Glucose, 0.2 mM Glycine, 0.02 mM Bicuculline, and 0.003 mM Strychnine), media exchange, Glutamate (100 μM), serum stimulation (5% FBS), and Bicuculline (100 μM). Media exchange was done by replacing growth media with fresh Neurobasal/B27 to account for the stress of replacing media during radiolabelling. Synaptic activity was blocked by the addition of Tetrodotoxin (1 μM, Tocris), CNQX (1 μM, Tocris), and APV (1 μM, Tocris). Tetrodotoxin (TTX) is a sodium channel antagonist, CNQX is a potent inhibitor of AMPA/kainite receptors, APV is a potent inhibitor of NMDA receptors.
For analyzing the expression of immediate-early gene products by immunoblot, special care was taken to ensure that neurons had reduced activity at baseline as measured by the expression of immediate early genes. After switching 250,000 neurons/well in 12-well format of cultured cortical neurons into 1 mL Neurobasal/B27 at DIV3, neurons were maintained in that medium, with only one 100 μl media exchange at DIV9. At DIV15, neurons were treated with pharmacological agents as indicated. Great caution was taken to minimize physical perturbation of these cultures so as not to induce any activation of IEG proteins. For example, drugs were resuspended in a small volume of growth media (media in which neurons were growing in) before addition, so cultures did not have to be shaken during treatment.
Radiolabelled peptide efflux experiments
Radiolabelled peptide collections were done as previously described (Ramachandran and Margolis, 2017). Neurons were used during days in vitro 12–14. For peptide efflux experiments during stimulation cultured cortical mouse neurons growing in Neurobasal/B27 were treated with either control or depolarization buffer for 20 minutes at 37 °C/5% CO2. Media was then removed and replaced with pre-warmed Neurobasal minus Met/Cys (Life Technologies, special order) and B27 supplement. This replacement media contained 55 mCi 35S-methionine/cysteine radiolabel with either control or depolarization buffers. Following 10 minutes or, where indicated, 30 seconds of radiolabeling during stimulation, neurons were quickly washed (<5 seconds) with pre-warmed PBS to remove free 35S-methionine/cysteine and then 10 mL of fresh Neurobasal/B27 with either control or depolarization buffer was added to washed neurons. 100 μl samples were immediately taken from the extracellular space followed by additional collections at indicated times. Samples were then quantified by liquid scintillation. For peptide efflux experiments prior to stimulation, neurons were radiolabelled for 10 minutes in control buffer and then free radiolabel was removed. Fresh Neurobasal/B27 was added with either control or depolarization buffer to washed neurons and 100 μl samples were immediately taken for quantification. Where indicated MG132 (20 μM), Bio-Epox (25 μM), Epoxomicin (20 μM), TTX/CNQX/APV (1 μM each), Puromycin (100 μg/ml), or Cycloheximide (100 μg/ml) were added during radiolabeling and collection.
Intracellular radiolabeling
Intracellular radiolabeling was done as previously described (Ramachandran and Margolis, 2017). Neurons were used during days in vitro 12–16. For intracellular radiolabelling experiments during stimulation cultured cortical mouse neurons growing in Neurobasal/B27 were treated with either control or depolarization buffer for 20 minutes at 37 °C/5% CO2. Media was then removed and replaced with pre-warmed Neurobasal minus Met/Cys (Life Technologies, special order) and B27 supplement. This replacement media contained 55 mCi 35S-methionine/cysteine radiolabel with either control or depolarization buffers. Following 10 minutes or, where indicated, 30 seconds of radiolabeling during stimulation, neuronal lysates were prepared directly in RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% Sodium Deoxycholate, 0.1% SDS, 5 mM EDTA, complete protease inhibitor cocktail tablet (Roche), 1 mM sodium orthovanadate, 1 mM β-glycerophosphate). SDS Laemmli buffer was added and samples were boiled for 5 minutes prior to loading onto SDS-PAGE gels. Autoradiographs were done by loading samples onto large SDS-PAGE gels, coomassie stained to verify equal loading, and then gels were dried down on a large gel drier onto Whatman filter paper. Dried gels were exposed to phosphorimager screens and scanned with a Typhoon FLA5500 imager.
For radiolabelling prior to stimulation, neurons were radiolabelled for 10 minutes in control buffer and then free radiolabel was removed and neurons were stimulated for indicated times before collection. For radiolabelling following stimulation, neurons were stimulated for 30 minutes followed by radiolabelling for 10 minutes in control buffer. For experiments using alternative stimuli, neurons were treated with chemical LTP buffer or Glutamate for 10 minutes during radiolabeling. For serum stimulation, 5% FBS was added for 30 minutes prior to radiolabeling and during radiolabeling. Neurons were radiolabelled for 10 minutes following media exchange. Where indicated MG132 (20 μM) or Bio-Epox (25 μM) were added during radiolabelling.
Ribosome pelleting
Ribosome-nascent chain complexes were isolated according to well-established protocols (Brandman et al., 2012; Duttler et al., 2013). Following various treatments and radiolabelling, neurons were lysed in a buffer containing either 100 μg/mL Cycloheximide or Puromycin (25 mM HEPES pH7.5, 10 mM MgCl2, 20 mM KCl, 50 mM NaCl, 2 mM ATP, 10u SuperASE-In, 20 μM MG132, 1.5% Triton X-100, protease inhibitors). Lysates were cleared by centrifugation at 10,000 RPM for 10 minutes, and the supernatant was layered onto a 1 M sucrose cushion. Ribosome-nascent chain complexes or empty ribosomes (following puromycin treatment) were pelleted via centrifugation at 70,000 RPM in a Ti 70.3 rotor. Supernatants were discarded and ribosomal pellets were washed three times with lysis buffer. 1/10 of the ribosomes were counted by liquid scintillation and the remainder was prepared in SDS loading buffer. For ribosome pelleting in HEK293 cells, cells were radiolabelled for 10 minutes prior to lysis and preparation for ribosome pelleting approaches similar to those described for neurons.
2-dimensional gels for nascent chain analysis
2-dimensional gels to analyze the ribosome-nascent chain complex were performed as previously described (Ito et al., 2011). Briefly, following 30 seconds of radiolabel incorporation at room temperature; neurons were lysed in buffers containing either Cycloheximide or Puromycin. Following lysis, RNCs were isolated as described above. Isolated RNC complexes were resuspended in SDS loading buffer, and then loaded onto neutral pH SDS-PAGE gels to minimize in-gel tRNA hydrolysis. Each samples was run with a few microliters of prestained ladder to delineate the lanes. After running in a single dimension, lanes were cut out of the gel and then incubated with 1N NaOH at 80 °C to degraded any RNA in the sample. This treatment hydrolyzes the ester bond linking the tRNA to its nascent polypeptide, generating a population of radiolabeled proteins whose mass is reduced by the weight of the tRNA (~25 kDa). Following RNA hydrolysis, samples were run in a second dimension, and then transferred onto nitrocellulose membranes. After exposure for autoradiography, membranes were blocked in BSA and immunoblotted using anti-ubiquitin antibodies.
Proteasome purification and activity assays
Cultured cortical neurons treated with Bio-Epox (25 μM) for indicated times were separated into cytosolic and membrane fractions using ultracentrifugation as previously described (Ramachandran and Margolis, 2017). Proteasomes were subsequently immunoprecipitated using resin conjugated to an antibody against the β2 proteasome subunit (Enzo). Immunoprecipitated proteasomes were then incubated with Suc-LLVY-AMC (Enzo) to test for activity. 20S activity was monitored as previously described (Ramachandran and Margolis, 2017). Samples from lysates were also prepared for SDS-PAGE and immunoblot analysis.
In addition, we used a similar protocol to determine the association of ribosomes with the membrane proteasome. Neurons stimulated with either Bicuculline (50 μM) or control were lysed into either Cycloheximide and Epoxomicin or Puromycin and Epoxomicin. These drugs were kept through the entire purification protocol. Following the membrane fractionation protocol, membranes were solubilized in 1% NP-40 and 1% Digitonin. Solubilized fractions were then adjusted to 0.4 M NaCl and then incubated with the antibody against the β2 proteasome subunit (Enzo) and washed 4x with 10x resin volume, and then prepared for SDS-PAGE and immunoblot analysis.
Biotin-epoxomicin
Biotin-epoxomicin is de-novo synthesized and purchased from Leiden University Institute of Chemistry.
Protein extraction, digestion, TMT labeling
After indicated treatments, the neurons were lysed by adding in 6 M urea and 2 M thiourea buffer with protease inhibitor cocktail. The lysates were sonicated with 35% amplitude for 1 min. Protein lysates were centrifuged at 16,000× g at 4 °C to exclude cell debris (pelleting at the bottom), and protein concentration was estimated using a SDS-PAGE method. Briefly, protein lysate was loaded with BSA standard ranging from 0.33 μg to 9 μg on a 3–12% NuPAGE gradient gel and separated for about 0.5 cm. The gel was stained with Colloidal Coomassie G-250 followed by destaining with water. The band intensities were measured by ImageJ software. A total of 200 μg of each sample was reduced with 10 mM dithiothreitol at room temperature for one hour and alkylated with 30 mM iodoacetamide at room temperature for 20 minutes in the dark. The protein samples were digested using endoproteinase LysC (Wako, 1:100 ratio of enzyme to protein) at 37 °C for 3 hours followed by sequencing-grade trypsin (Promega, 1:50 ratio of enzyme to protein) at 37 °C overnight. After the digestion, the peptide samples were subjected to desalting and labeling with 10-plex TMT reagents according to the manufacturer’s instructions (Thermo Fisher Scientific) and the 9/10 channels (126, 127N, 127C, 128N, 128C, 129N, 129C, 130N, 130C) were used for labeling. The labeling reaction was performed for one hour at room temperature, followed by quenching with 100 mM Tris-HCl (pH 8.0). The digested and labeled peptides from all 9 channels were pooled.
In order to increase protein coverage, reduce artifacts from ratio compression, and increase our signal/noise ratio, peptides were prefractionated offline before mass spectrometry (MS) analysis. Briefly, the peptides were fractionated by basic pH reversed-phase liquid chromatography (bRPLC) into 96 fractions, followed by concatenation into 24 fractions by combining every 24th fractions. Agilent 1260 offline LC system was used for bRPLC fractionation, which includes a binary pump, VWD detector, an autosampler, and an automatic fraction collector. Lyophilized samples were reconstituted in solvent A (10 mM triethylammonium bicarbonate, pH 8.5) and loaded onto XBridge C18, 5 μm 250 × 4.6 mm column (Waters, Milford, MA). Peptides were resolved using a gradient of 3 to 50% solvent B (10 mM triethylammonium bicarbonate in acetonitrile, pH 8.5) at a flow rate of 1 ml per min over 50 min collecting 96 fractions. Subsequently, the fractions were concatenated into 24 fractions followed by vacuum drying using SpeedVac. The dried peptides were suspended in 0.1% formic acid.
Mass spectrometry
The fractionated peptides were analyzed on an Orbitrap Fusion Lumos Tribrid Mass Spectrometer coupled with the UltiMate™ RSLCnano nano-flow liquid chromatography system (Thermo Fisher Scientific) (Figure 2A). The peptides from each fraction were reconstituted in 0.1% formic acid and loaded on a Acclaim PepMap100 Nano-Trap Column (100 μm × 2 cm, Thermo Fisher Scientific) packed with 5 μm C18 particles at a flow rate of 5 μl per minute. Peptides were resolved at 250-nl/min flow rate using a linear gradient of 10% to 35% solvent B (0.1% formic acid in 95% acetonitrile) over 95 minutes on an EASY-Spray column (50 cm × 75 μm ID, Thermo Fisher Scientific) packed with 2 μm C18 particles, which was fitted with an EASY-Spray ion source that was operated at a voltage of 2.0 kV.
Mass spectrometry analysis was carried out in a data-dependent manner with a full scan in the mass-to-charge ratio (m/z) range of 350 to 1550 in the “Top Speed” setting, three seconds per cycle. MS1 and MS2 were acquired for the precursor ion detection and peptide fragmentation ion detection, respectively. MS1 scans were measured at a resolution of 120,000 at an m/z of 200. MS2 scan were acquired by fragmenting precursor ions using the higher-energy collisional dissociation (HCD) method and detected at a mass resolution of 50,000, at an m/z of 200. Automatic gain control for MS1 was set to one million ions and for MS2 was set to 0.05 million ions. A maximum ion injection time was set to 50 ms for MS1 and 100 ms for MS2. MS1 was acquired in profile mode and MS2 was acquired in centroid mode. Higher-energy collisional dissociation was set to 35 for MS2. Dynamic exclusion was set to 30 seconds, and singly-charged ions were rejected. Internal calibration was carried out using the lock mass option (m/z 445.1200025) from ambient air.
Data analysis
Proteome Discoverer (v 2.1; Thermo Scientific) suite was used for quantitation and identification. During the preprocessing of MS/MS spectra, the top 10 peaks in each window of 100 m/z were selected for database search. The tandem mass spectrometry data were then searched using SEQUEST algorithms against mouse RefSeq protein database (version 84) with common contaminant proteins. The search parameters used were as follows: a) trypsin as a proteolytic enzyme (with up to two missed cleavages); b) peptide mass error tolerance of 10 ppm; c) fragment mass error tolerance of 0.02 Da; and d) carbamidomethylation of cysteine (+57.02146 Da) and TMT tags (+229.162932 Da) on lysine residues and peptide N-termini as a fixed modification and oxidation of methionine (+15.99492 Da) as a variable modification. The minimum peptide length was set to 6 amino acids. Peptides and proteins were filtered at a 1 % false-discovery rate (FDR) at the PSM level using percolator node and at the protein level using protein FDR validator node, respectively.
The protein quantification was performed with following parameters and methods. The most confident centroid option was used for the integration mode while the reporter ion tolerance was set to 20 ppm. The MS order was set to MS2 and the activation type was set to HCD. Unique and razor peptides both were used for peptide quantification while protein groups were considered for peptide uniqueness. Reporter ion abundance was computed based on signal-to-noise ratio and the missing intensity values were replaced with the minimum value. The quantification value corrections for isobaric tags and data normalization were disabled while the co-isolation threshold was set to 50%. The highest signal-to-noise ratio value from PSMs for a peptide was used to generate a peptide level abundance followed by averaging peptide level signal-to-noise ratio values for a protein to generate a protein level abundance.
Protein grouping was performed with strict parsimony principle to generate the final protein groups. All proteins sharing the same set or subset of identified peptides were grouped while protein groups with no unique peptides were filtered out. The Proteome Discoverer iterated through all spectra and selected PSM with the highest number of unambiguous and unique peptides.
Differential protein expression analysis
The list of quantified proteins exported from Proteome Discoverer 2.1 was utilized as the input for our differential expression analysis. The raw values were organized in a matrix where each column represented a sample and each row a protein. To normalize the raw expression values, we began by log2 transforming the matrix with a +1 for computation. Then we median polished the log-transformed values by subtracting the row median from each row, followed by the subtraction of the column median from each column. The resulting normalized expression values for each sample appeared normally distributed and was comparable across samples. For the detection of differential regulation, we followed the recommendation outline in (Kammers et al., 2015). An empirical Bayes method was employed on the normalized matrix to detect differences between the 3 samples of the biotin-epoxomicin treated group compared to the 6 samples of the control and cycloheximide groups. The empirical Bayes method shrinks individual protein’s sample variance towards a pooled estimate, and creates a more stable and powerful inference in differential protein abundance detection.
The output of the differential abundance analysis detected 1340 and 408 proteins to be differentially abundant at the 0.05 and 0.01 level respectively. However, due to the large number of proteins tested, we were more interested in q-values that adjust for multiple comparisons. Using a cutoff of q < 0.1, which corresponds to a false discovery rate of 10%, we detect 190 proteins to be differentially abundant in the 2 groups that we defined. Of those 190 proteins, 122 were up-regulated.
For the selection of the colors in the heatmap, we carried out feature-scaling of the normalized expression values on a gene-by-gene basis. For each gene, this process assigns the largest expression a value of 1, and the smallest expression a value of 0. The remaining values are scaled between 0 and 1 based on where they are relative to the largest and smallest expression values. For instance, a feature-scaled value of 0.5 represents an expression level that is halfway between the lowest expression and the highest expression observed for a gene. In other words, this sample’s expression is 50% of the maximum fold change away from the lowest and the highest expression values at this gene.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data shown are mean ± SEM. The n values represent biological repeats from independent neuronal cultures as specified in each figure legend. No statistical methods were used to predetermine sample size. The experiments were not randomized. All statistical analyses were performed using Origin Prism and Graphpad software, accounting for appropriate distribution and variance to ensure proper statistical parameters were applied. Experimental sample sizes were chosen according to norms within the field. The observed magnitude of differences, together with the low replicate variance, permits high power of analysis based on the sample size chosen. For remaining experiments investigators were not blinded to allocation during experiments and outcome assessment. Statistical analysis using Student’s t tests and two-way ANOVAs were performed as described in each figure legend. The p values ≤ 0.05 were considered significant. Notable exceptions to this are in the mass spectrometry data.
DATA AND SOFTWARE AVAILABILITY
All reagents and protocols used in this study are available for sharing upon reasonable request to the authors. The raw mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaino et al., 2016) partner repository with the dataset identifier PXD009920. Table S1, Related to Figure 3 shows all unfiltered proteomics data and quantification. Table S2, Related to Figure 3 shows top filtered NMP substrates.
KEY RESOURCES TABLE
Supplementary Material
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-β-Actin (8226) | Abcam | Cat# ab8226; RRID AB_306371 |
| Rabbit anti-Biotin (D5A7) | Cell Signaling Technology | Cat# 5597S; RRID AB 10828011 |
| Streptavidin-Alexa Fluor 647 | Invitrogen | Cat# S21374; RRID AB_2336066 |
| Rabbit anti-Arc | P. Worley (Hopkins) | Lyford et al., 1995 |
| Rabbit anti-Fos (9F6) | Cell Signaling Technology | Cat# 2250S; RRID AB_2247211 |
| Rabbit anti-NPAS4 | Y. Lin (MIT) | Lin et al., 2008 |
| Mouse anti-PSD-95 (7E31B8) | Pierce | Cat# MA1046; RRID AB_2092361 |
| Mouse anti-UBE3A (330) | Sigma-Aldrich | Cat# E8655; RRID AB_261956 |
| Mouse anti-Ubiquitin (FK2) | Enzo Life Sciences | Cat# PW8810; RRID AB_2051891 |
| Mouse anti-S6 ribosomal subunit (54D2) |
Cell Signaling Technology | Cat# 2317S; RRID AB_2238583 |
| Mouse anti-Transferrin receptor (H68.4) |
Invitrogen | Cat# 136800; RRID AB_2533029 |
| Mouse anti-β2 proteasome subunit (MCP168) |
Enzo Life Sciences | Cat# PW8145; RRID AB_2052386 |
| Mouse anti-α1–7 proteasome subunit (MCP231) |
Enzo Life Sciences | Cat# PW8195; RRID AB_2052368 |
| Goat anti-Rabbit IgG, (H+L), HRP |
Cell Signaling Technology | Cat# 7074S; RRID AB_2099233 |
| Horse anti-Mouse IgG, (H+L), HRP |
Cell Signaling Technology | Cat# 7076S; RRID AB_330924 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Poly-L-Lysine (PLL) | Sigma-Aldrich | Cat# P4707 |
| Papain | Worthington | Cat# LS003127 |
| B27 | Thermo Fisher Scientific | Cat# 17504044 |
| Neurobasal | Thermo Fisher Scientific | Cat# 21103049 |
| Neurobasal minus met/cys | Thermo Fisher Scientific | Cat# 21103049* special order |
| Penicillin/streptomycin | Sigma-Aldrich | Cat# 15140122 |
| L-Glutamine | Thermo Fisher Scientific | Cat# 25030081 |
| Trypsin | Thermo Fisher Scientific | Cat# 25200056 |
| Trypsin Inhibitor | Sigma-Aldrich | Cat# T9253; CAS: 9035-81-8 |
| DMEM | Thermo Fisher Scientific | Cat# 11960069 |
| SuperAse | Thermo Fisher Scientific | Cat# AM2694 |
| Fetal bovine serum (FBS) | Thermo Fisher Scientific | Cat# 16000044 |
| PBS | Thermo Fisher Scientific | Cat# 10010049 |
| cOmplete Protease inhibitor | Sigma-Aldrich | Cat# 11836170001 |
| Proteasome substrate | Enzo Life Sciences | Cat# P802 |
| MLN-7243 | Chemie Tek | Cat# CT-M7243; CAS:1450833-55-2 |
| Epoxomicin | UBPBio | Cat# F1400; CAS: 134381-21-8 |
| MG-132 | Cell Signaling Technology | Cat# 2194S |
| Precision Plus Standards | Bio-Rad | Cat# 1610374 |
| Bicuculline | Tocris Bioscience | Cat# 013110; CAS: 53552-05-9 |
| Cycloheximide | Sigma-Aldrich | Cat# C1988; CAS: 66-81-9 |
| Puromycin | Sigma-Aldrich | Cat# P7255; CAS: 58-58-2 |
| Actinomycin D | Sigma-Aldrich | Cat# A1410; CAS: 50-76-0 |
| TTX | Tocris Bioscience | Cat# 1078; CAS: 4368-28-9 |
| CNQX | Tocris Bioscience | Cat# 0190; CAS: 115066-14-3 |
| APV | Tocris Bioscience | Cat# 010610; CAS: 79055-68-8 |
| Trypsin (MS/MS) | Promega | Cat# V5111 |
| Endoproteinase LysC (MS/MS) | Wako Chemical | Cat# 125-05061 |
| TMT reagent (MS/MS) | ThermoFisher Scientific | Cat# 90110 |
| National Diagnostics PROTOGEL (30%) |
ThermoFisher Scientific | Cat# 50-899-90118 |
| Ecoscint A | National Diagnostics | Cat # LS-273 |
| Critical Commercial Assays | ||
| Proteasome purification kit | VWR International | 76002-400 |
| EasyTag EXPRESS 35S Protein Labeling Kit |
Perkin-Elmer | NEG772014MC |
| Deposited Data | ||
| Proteomic datasets: TMT experiment |
This paper | ProteomeXchange: PXD009920 |
| Mouse RefSeq protein database (version 84) |
This paper | https://www.ncbi.nlm.nih.gov/refseq/ |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | CRL-1573 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 | Charles River | RRID: IMSR_CRL:27 |
| Software and Algorithms | ||
| Proteome Discoverer 2.1 | ThermoFisher Scientific | Cat# OPTON-30795 |
| SEQUEST | Yates’ Lab, Eng et al., 1994 | http://proteomicswiki.com/wiki/index.php/SEQUEST |
| Empirical Bayes method for protein differential abundance testing |
Kammers et al., 2015 | http://www.biostat.jhsph.edu/~kkammers/software/eupa/R_guide.html |
Highlights:
Neuronal membrane proteasomes (NMPs) are regulated by neuronal activity
NMPs degrade ribosome-associated nascent polypeptides during protein synthesis
Substrates of the NMP include immediate early gene products, Npas4, Fos and Arc
Degradation through the NMP does not require ubiquitylation
ACKNOWLEDGEMENTS
We thank Jeremy Nathans, Geraldine Seydoux, Michael Caterina, Carolyn Machamer, Rachel Green for reagents, technical assistance and advice on the project. Special thanks, to Fuad Mohammed for running 2D gels and discussion on nascent chain biology and ribosome pelleting. Thanks to Chirag Vasavda, Rebecca Keener, Leah Cairns, and members of the Margolis laboratory for critical reading and feedback. For antibodies, we thank Yingxi Lin (Npas4) and Paul Worley (Arc). We acknowledge support of the Center for Proteomics Discovery at Johns Hopkins and shared instrumentation grant S10OD021844. This work was funded by institutional funding and the following grant to S.S.M. (R01 MH102364). K.V.R. was supported by a training grant T32 GM007445 and NSF Graduate Research Fellowship DGE-1232825.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTION
K.V.R. and S.S.M. conceived study and designed experiments. K.V.R. did experiments. C.H.N. ran MS instrumentation with samples provided by K.V.R. J.M.F. performed statistical analyses for MS data. T.B.S. helped optimize reproducibility conditions for IEG immunoblots. K.V.R. and S.S.M. analyzed the data and wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- An H, and Harper JW (2018). Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat Cell Biol 20, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton LC, and Yewdell JW (2014). Translating DRiPs: MHC class I immunosurveillance of pathogens and tumors. J Leukoc Biol 95, 551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M, and Hartl FU (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. [DOI] [PubMed] [Google Scholar]
- Ben-Nissan G, and Sharon M (2014). Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 4, 862–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, and Schuman EM (2006). Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature 441, 1144–1148. [DOI] [PubMed] [Google Scholar]
- Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li GW, Zhou S, King D, Shen PS, Weibezahn J, et al. (2012). A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151, 1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carle TL, Ohnishi YN, Ohnishi YH, Alibhai IN, Wilkinson MB, Kumar A, and Nestler EJ (2007). Proteasome-dependent and -independent mechanisms for FosB destabilization: identification of FosB degron domains and implications for DeltaFosB stability. Eur J Neurosci 25, 3009–3019. [DOI] [PubMed] [Google Scholar]
- Ciechanover A (1998). The ubiquitin-proteasome pathway: on protein death and cell life. Embo J 17, 7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, and Scott JD (2003). Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40, 595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coux O, Tanaka K, and Goldberg AL (1996). Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem 65, 801–847. [DOI] [PubMed] [Google Scholar]
- de Poot SAH, Tian G, and Finley D (2017). Meddling with Fate: The Proteasomal Deubiquitinating Enzymes. J Mol Biol 429, 3525–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deglincerti A, Liu Y, Colak D, Hengst U, Xu G, and Jaffrey SR (2015). Coupled local translation and degradation regulate growth cone collapse. Nat Commun 6, 6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djakovic SN, Schwarz LA, Barylko B, DeMartino GN, and Patrick GN (2009). Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J Biol Chem 284, 26655–26665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttler S, Pechmann S, and Frydman J (2013). Principles of cotranslational ubiquitination and quality control at the ribosome. Mol Cell 50, 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6, 231–242. [DOI] [PubMed] [Google Scholar]
- Finley D, Ciechanover A, and Varshavsky A (2004). Ubiquitin as a central cellular regulator. Cell 116, S29–32. [DOI] [PubMed] [Google Scholar]
- Flavell SW, and Greenberg ME (2008). Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci 31, 563–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca R, Nagerl UV, and Bonhoeffer T (2006a). Neuronal activity determines the protein synthesis dependence of long-term potentiation. Nat Neurosci 9, 478–480. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, and Nagerl UV (2006b). A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 52, 239–245. [DOI] [PubMed] [Google Scholar]
- Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, and Soderling TR (2010). Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AmPa receptors recruited by CaM-kinase I. J Neurosci 30, 11565–11575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiol A, Racca C, and Triller A (1999). Dendritic and postsynaptic protein synthetic machinery. J Neurosci 19, 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL, and Dice JF (1974). Intracellular protein degradation in mammalian and bacterial cells. Annu Rev Biochem 43, 835–869. [DOI] [PubMed] [Google Scholar]
- Hyer ML, Milhollen MA, Ciavarri J, Fleming P, Traore T, Sappal D, Huck J, Shi J, Gavin J, Brownell J, et al. (2018). A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med 24, 186–193. [DOI] [PubMed] [Google Scholar]
- Ito K, Chadani Y, Nakamori K, Chiba S, Akiyama Y, and Abo T (2011). Nascentome analysis uncovers futile protein synthesis in Escherichia coli. PLoS One 6, e28413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Inoue D, Kido S, and Matsumoto T (2005). c-Fos degradation by the ubiquitin- proteasome proteolytic pathway in osteoclast progenitors. Bone 37, 842–849. [DOI] [PubMed] [Google Scholar]
- Kammers K, Cole RN, Tiengwe C, and Ruczinski I (2015). Detecting Significant Changes in Protein Abundance. EuPA Open Proteom 7, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ 3rd, Govindarajan A, and Tonegawa S (2004). Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 44, 59–73. [DOI] [PubMed] [Google Scholar]
- Klein ME, Castillo PE, and Jordan BA (2015). Coordination between Translation and Degradation Regulates Inducibility of mGluR-LTD. Cell Rep 10, 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Lee SH, Kim H, and Kaang BK (2008). Synaptic protein degradation underlies destabilization of retrieved fear memory. Science 319, 1253–1256. [DOI] [PubMed] [Google Scholar]
- Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, and Greenberg ME (2008). Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG, Gilbert DJ, Jenkins NA, Lanahan AA, and Worley PF (1995). Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron 14, 433–445. [DOI] [PubMed] [Google Scholar]
- Mabb AM, Je HS, Wall MJ, Robinson CG, Larsen RS, Qiang Y, Correa SA, and Ehlers MD (2014). Triad3A regulates synaptic strength by ubiquitination of Arc. Neuron 82, 1299–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin P, Nastiuk KL, Daniel N, Girault JA, Czernik AJ, Glowinski J, Nairn AC, and Premont J (1997). Glutamate-dependent phosphorylation of elongation factor-2 and inhibition of protein synthesis in neurons. J Neurosci 17, 3445–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane E, Sin C, Zauber H, Wells JN, Donnelly N, Wang X, Hou J, Chen W, Storchova Z, Marsh JA, et al. (2016). Kinetic Analysis of Protein Stability Reveals Age-Dependent Degradation. Cell 167, 803–815 e821. [DOI] [PubMed] [Google Scholar]
- Nathans D, and Neidle A (1963). Structural requirements for puromycin inhibition of protein synthesis. Nature 197, 1076–1077. [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Botsford B, Gindina S, Cowansage KK, LeDoux JE, Klann E, and Hoeffer C (2017). Accumulation of Polyribosomes in Dendritic Spine Heads, But Not Bases and Necks, during Memory Consolidation Depends on Cap-Dependent Translation Initiation. J Neurosci 37, 1862–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S, Willmund F, and Frydman J (2013). The ribosome as a hub for protein quality control. Mol Cell 49, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peebles CL, Yoo J, Thwin MT, Palop JJ, Noebels JL, and Finkbeiner S (2010). Arc regulates spine morphology and maintains network stability in vivo. Proc Natl Acad Sci U S A 107, 18173–18178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prouty WF, Karnovsky MJ, and Goldberg AL (1975). Degradation of abnormal proteins in Escherichia coli. Formation of protein inclusions in cells exposed to amino acid analogs. J Biol Chem 250, 1112–1122. [PubMed] [Google Scholar]
- Ramachandran KV, and Margolis SS (2017). A mammalian nervous-system-specific plasma membrane proteasome complex that modulates neuronal function. Nat Struct Mol Biol 24, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RB, and Flexner LB (1966). A model for the development of retina-cortex connections. Am Sci 54, 174–183. [PubMed] [Google Scholar]
- Robertson JH, and Wheatley DN (1979). Pools and protein synthesis in mammalian cells. Biochem J 178, 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheetz AJ, Nairn AC, and Constantine-Paton M (2000). NMDA receptor-mediated control of protein synthesis at developing synapses. Nat Neurosci 3, 211–216. [DOI] [PubMed] [Google Scholar]
- Schmidt M, and Finley D (2014). Regulation of proteasome activity in health and disease. Biochim Biophys Acta 1843, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, and Bennink JR (2000). Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404, 770–774. [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Wolf J, Selbach M, and Busse D (2013). Synthesis and degradation jointly determine the responsiveness of the cellular proteome. Bioessays 35, 597–601. [DOI] [PubMed] [Google Scholar]
- Shin SM, Zhang N, Hansen J, Gerges NZ, Pak DT, Sheng M, and Lee SH (2012). GKAP orchestrates activity-dependent postsynaptic protein remodeling and homeostatic scaling. Nat Neurosci 15, 1655–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag EM, Samant RS, and Frydman J (2017). Mechanisms and Functions of Spatial Protein Quality Control. Annu Rev Biochem 86, 97–122. [DOI] [PubMed] [Google Scholar]
- Tcherkezian J, Brittis PA, Thomas F, Roux PP, and Flanagan JG (2010). Transmembrane receptor DCC associates with protein synthesis machinery and regulates translation. Cell 141, 632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurumi C, Ishida N, Tamura T, Kakizuka A, Nishida E, Okumura E, Kishimoto T, Inagaki M, Okazaki K, Sagata N, et al. (1995). Degradation of c-Fos by the 26S proteasome is accelerated by c-Jun and multiple protein kinases. Mol Cell Biol 15, 5682–5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov P, Asher G, Paz A, Reuven N, Sussman JL, Silman I, and Shaul Y (2008). Operational definition of intrinsically unstructured protein sequences based on susceptibility to the 20S proteasome. Proteins 70, 1357–1366. [DOI] [PubMed] [Google Scholar]
- Tsvetkov P, Reuven N, and Shaul Y (2009). The nanny model for IDPs. Nat Chem Biol 5, 778–781. [DOI] [PubMed] [Google Scholar]
- Turner GC, and Varshavsky A (2000). Detecting and measuring cotranslational protein degradation in vivo. Science 289, 2117–2120. [DOI] [PubMed] [Google Scholar]
- Vabulas RM, and Hartl FU (2005). Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 310, 1960–1963. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, 11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, and Greenberg ME (2001). Calcium regulation of neuronal gene expression. Proc Natl Acad Sci U S A 98, 11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley DN, Giddings MR, and Inglis MS (1980). Kinetics of degradation of "short-" and "long-lived" proteins in cultured mammalian cells. Cell Biol Int Rep 4, 1081–1090. [DOI] [PubMed] [Google Scholar]
- Wheatley DN, Grisolia S, and Hernandez-Yago J (1982). Significance of the rapid degradation of newly synthesized proteins in mammalian cells: a working hypothesis. J Theor Biol 98, 283–300. [DOI] [PubMed] [Google Scholar]
- Wheatley DN, and Inglis MS (1980). An intracellular perfusion system linking pools and protein synthesis. J Theor Biol 83, 437–445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.