

SUMMARY
HIV-1 expresses several accessory proteins to counteract host anti-viral restriction factors to facilitate viral replication and disease progression. One such protein, Vpr, has been implicated in affecting multiple cellular processes, but its mechanism remains elusive. Here we report that Vpr targets TET2 for polyubiquitylation by the VprBP-DDB1-CUL4-ROC1 E3 ligase and subsequent degradation. Genetic inactivation or Vpr-mediated degradation of TET2 enhances HIV-1 replication and substantially sustained expression of the proinflammatory cytokine interleukin-6 (IL-6), correlated with reduced recruitment of histone deacetylase 1 and 2 to and enhanced histone H3 acetylation of the IL-6 promoter during resolution phase. Blocking IL-6 signaling reduced the ability of Vpr to enhance HIV-1 replication. We conclude that HIV-1 Vpr degrades TET2 to sustain IL-6 expression to enhance viral replication and disease progression. These results suggest disrupting the Vpr-TET2-IL6 axis may prove clinically beneficial to reduce both viral replication and inflammation during HIV-1 infection.
eTOC
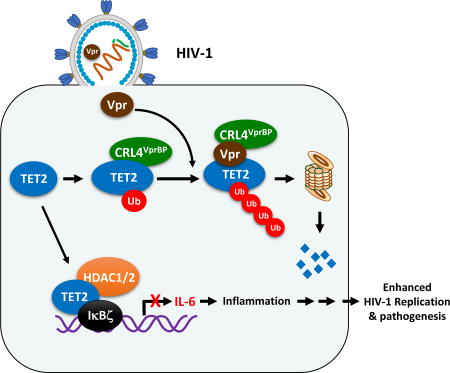
HIV-1 Vpr protein counteracts host anti-viral restriction factors to facilitate viral replication and disease progression. Lv et al. demonstrate that Vpr promotes degradation of TET DNA dioxygenases. TET2 deletion or depletion enhances HIV-1 replication and sustains proinflammatory cytokine IL-6, while blocking IL-6 reduces Vpr’s ability to enhance HIV-1 replication.
INTRODUCTION
Human immunodeficiency virus 1 (HIV-1) expresses multiple accessory proteins, including viral protein R (Vpr), to counteract host restriction inhibitors (Simon et al., 2015; Strebel, 2013). The functional importance of Vpr to viral propagation was initially shown in rhesus macaques infection using Vpr-deficient SIV, which exhibited a decrease in viral replication and delay in AIDS disease progression (Lang et al., 1993). In the humanized mouse model, Vpr enhances acute HIV-1 infection (Sato et al., 2013). Vpr has been shown to affect several cellular processes of host cells, including cell cycle, DNA damage response and apoptosis (Romani and Cohen, 2012). However, it remains unclear how a change in these host cellular processes by Vpr facilitates HIV-1 replication remains unclear.
A number of reports support that Vpr facilitates HIV-1 replication through its interaction with VprBP (Simon et al., 2015; Strebel, 2013). Mutations in Vpr disrupting its binding with VprBP, such as Q65R, reduced cell-to-cell spread of HIV-1 from macrophages to CD4+ T lymphocytes (Collins et al., 2015) or the ability of HIV-1 to escape from innate immune sensing (Laguette et al., 2014). VprBP binds near stoichiometrically with DNA damage-binding protein (DDB1) and, via DDB1, with cullin 4 and small RING finger protein ROC1/RBX1 to assemble a VprBP-DDB1-CUL4-RING E3 ubiquitin ligase (CRL4VprBP) complex (Jackson and Xiong, 2009). The finding that VprBP is a subunit of an E3 ligase complex has led to the identification of several host proteins targeted by Vpr for the ubiquitylation by the CRL4VprBP E3 ligase. These include base excision repair enzyme uracil DNA glycosylase UNG2 (Ahn et al., 2010), telomerase catalytic subunit TERT (Wang et al., 2013), DNA endonuclease and repair factor MUS81 (Laguette et al., 2014), DNA replication factor MCM10 (Romani et al., 2015), transcription factor HLTF (Lahouassa et al., 2016), and histone deacetylase HDACs (Romani et al., 2016). Whether or how these factors inhibit HIV-1 replication is unclear, in particularly whether loss of function of any of these host genes rescues the replication of Vpr-deficient HIV-1 are yet to be determined.
In a search for the substrate of VprBP, we previously discovered that it binds to the TET family of DNA dioxygenases and targets them for monoubiquitylation by the CRL4VprBP E3 ligase to promote TET binding to chromatin (Nakagawa et al., 2015). This finding led us to determine whether Vpr affects CRL4VprBP-mediated TET2 ubiquitylation.
RESULTS
HIV-1 Vpr promotes TET2 degradation in a VprBP dependent manner
To determine the effect of Vpr on TET2 protein, we first transfected HIV-1 Vpr (Clade B) into human THP1 monocytic cells. We found, surprisingly, that the level of TET2 protein, but not mRNA, was significantly decreased (Figure 1A). Moreover, Vpr from HIV-1 clade A Q23, HIV-2 ROD9 and SIV-mac239 were all capable of decreasing TET2 proteins (Figure 1B), suggesting that targeting TET2 for degradation represents a conserved function of Vpr. Vpx, a homologue of Vpr that is encoded by HIV-2 and most simian lentiviruses, also binds to VprBP and targets a host restriction factor, SAMHD1, for ubiquitylation by CRL4VprBP and subsequent degradation by the proteasome (Hrecka et al., 2011; Laguette et al., 2011). We found that in contrast to Vpr, expression of Vpx had no effect on TET2 protein (Figure 1B and Figure S1A), indicating a different specificity of Vpr from Vpx in promoting host protein degradation. We also found that Vpr can promote the degradation of both TET1 and TET3 (Figure S1B), indicating that Vpr promotes the degradation of all three TET proteins. Since TET2 is the predominant TET gene expressed in THP1 cells (Figure S1C), we therefore focused on TET2 in this study.
Figure 1. Vpr promotes TET proteins degradation in a VprBP-dependent manner.

(A) Vpr decreases levels of TET2 protein, not mRNA. THP1 cells were transfected with plasmid expressing HA-Vpr. Western blots were performed with the indicated antibodies, and mRNA level was analyzed by quantitative RT-PCR; error bars represent ±SD for triplicate experiments, n=2.
(B) Vpr from different subtypes of HIV-1, HIV-2 and SIV, but not Vpx, decrease TET2 protein levels. HEK293T cells were transfected with the indicated plasmids and whole-cell extracts were prepared 24hrs after transfection, followed by immunoblotting analysis, n=2.
(C) Wild-type, but not Vpr-deficient, HIV-1 infection leads to TET2 degradation. THP1 and PBMCs were infected with Vpr+ HIV-1 and Vpr- HIV-1 and TET2 levels were measured.
(D) THP1 cells were infected with HIV-1 in the presence of various HIV inhibitors. TET2 proteins were measured at 48h post-infection, n=2.
(E) Virion-associated Vpr in Virus-Like-Particles (VLP) degrades TET2. THP1 cells were treated with the indicated amount of VLP (as quantified by p24). TET2 proteins were analyzed 12 h after treatment.
(F) Vpr promotes TET2 degradation requires Vpr-VprBP binding. Cells were transfected with indicated plasmids, followed by Flag-TET2 immunoblotting, n=2.
(G) Knocking down VprBP blocks Vpr induced TET2 degradation. Cells were transfected with siRNA targeting VprBP for 48h and then treated with VLP-Vpr for another 24h, followed by immunoblotting with indicated antibodies, n=2.
To demonstrate TET2 degradation in HIV-1 infected cells, we infected THP1 cells or human peripheral blood mononuclear cells (PBMC) with wildtype and Vpr-deficient mutant HIV-1 and examined the level of TET2 protein at different time points after infection. TET2 protein levels decreased significantly after infection by the wildtype, but not Vpr-deficient HIV-1, in both cells (Figure 1C). To determine at which stage of the HIV-1 life cycle Vpr promotes TET2 degradation, THP1 cells were infected with HIV-1 in the presence of inhibitors of viral fusion and entry (enfuvirtide, T20), reverse transcriptase (Nevirapine, NVP), integrase (Raltegravir, RAL) or protease (Amprenavir, AMP). Only the fusion inhibitor inhibited TET2 degradation (Figure 1D), indicating that the virion-associated Vpr is sufficient to degrade TET2 protein soon after viral entry, and de novo Vpr protein production is not required for this function. Supporting this notion, we found TET2 is rapidly degraded by Vpr as early as 6 hours after HIV-1 infection (Figure S1D). In addition, we generated Virus-Like-Particles (VLP) carrying HIV-1 Vpr and found, consistently, that virion-associated Vpr can induce TET2 degradation after addition to THP1 cells (Figure 1E).
To determine if VprBP is involved in the Vpr mediated TET2 degradation, we generated Vpr Q65R mutant, which cannot bind with VprBP (Figure S1E), and tested its effect on TET2 degradation. Result showed that Q65R mutation abolished the ability of Vpr to promote TET2 degradation (Figure 1F). Consistently, knocking down VprBP blocked the Vpr-induced TET2 degradation (Figure 1G). These results demonstrate that HIV-1 Vpr promotes TET2 degradation in a VprBP dependent manner.
Vpr reprograms CRL4VprBP E3 ligase to catalyze polyubiquitylation of TET2
Two proteolytic cleavages, one by caspase (Ko et al., 2013) and one by calpain family of calcium-dependent proteases (Wang and Zhang, 2014), have previously been shown to cleave TET proteins. We treated Vpr-expressing THP1 cells with inhibitors of caspase, calpeptin and lysosome, and found that none of them affected Vpr-promoted TET2 degradation (Figure 2A). Instead, treatment with MG132, an inhibitor of the 26S proteasome, effectively blocked TET2 reduction, indicating that Vpr promotes TET2 degradation via the proteasomal pathway. Consistent with this notion, MG132 treatment accumulated polyubiquitylated TET2 in cells expressing Vpr, but not Vpr Q65R mutant (Figure 2B) or Vpx (Figure S2A).
Figure 2. Vpr reprograms CRL4VprBP E3 Ligase to catalyze polyubiquitylation of TET2.

(A) Vpr-mediated TET2 degradation is blocked by the proteasome inhibitor, MG132. THP1 cells were transfected with HA-Vpr, and then treated with inhibitors of lysosome (NH4Cl, 20mM), calpain (Calpeptin, 50µM), caspase (Z-VAD-FMK, 100µM), or 26S proteasome (MG132, 10µM) for 24hrs, followed by immunoblotting analyses, n=2.
(B) Wildtype, but not Q65R mutant Vpr, promotes TET2 polyubiquitylation in vivo. Cells were transfected with indicated plasmids. Whole cell lysates were prepared under denaturing conditions and TET2 ubiquitylation was examined by coupled IP-western, n=2.
(C) Knockdown of individual component of the CRL4VprBP E3 ligase complex inhibits Vpr-promoted TET2 ubiquitylation in vivo. Cells were transfected with indicated plasmids and siRNA targeting the indicated genes for 72h. In vivo TET2 ubiquitylation was determined by IP-western blot analysis under denaturing conditions, n=2.
(D) Wildtype, but not VprBP-binding deficient Q65R mutant Vpr, promotes TET2 polyubiquitylation in vitro. Immunopurified TET2 protein was incubated with VprBP immune-complex and purified wild-type or Q65R mutant Vpr. Reactions were terminated by adding SDS loading buffer, followed by immunoblotting with indicated antibodies, n=2.
(E) Vpr-promoted TET2 degradation by the CRL4VprBP E3 ligase is not dependent on either the monoubiquitylation site in TET2 or the VprBP-TET2 binding. HEK293T cells were transfected with the plasmids indicated and TET2 levels were determined by western blot.
(F) Vpr alters TET2-VprBP interaction. HEK293T cells were transfected with indicated plasmids, followed by IP-western analysis with the indicated antibodies. Chromo, chromo-like domain; LisH, homology to Lis1, n=2.
(G) Q65R mutant Vpr cannot bind VprBP to bridge TET2 binding to the C-terminal portion of VprBP. HEK293T cells were transfected with indicated plasmids and protein-protein interactions were determined by IP-western analysis, n=2.
To test if Vpr-promoted TET2 ubiquitylation via VprBP based DDB1-CUL4-ROC1 (CRL4VprBP) E3 complex, we knocked down individual component of CRL4VprBP E3 ligase by siRNA in Vpr-expressing cells and determined TET2 polyubiquitylation. Knocking down either VprBP or DDB1 substantially reduced the ubiquitylation of TET2 in cells (Figure 2C). Note that VprBP was down regulated when DDB1 was targeted by the siRNA, as DDB1 is the major binding protein of VprBP and the binding stabilizes VprBP as we observed previously (Nakagawa et al., 2015). Knockdown either CUL4A or CUL4B partially reduced TET2 polyubiquitylation while simultaneous knockdown both CUL4A and CUL4B almost completely inhibited TET2 polyubiquitylation (Figure 2C). In vitro, incubation of immunopurified TET2 with a CRL4VprBP immune complex resulted in robust TET2 polyubiquitylation in the presence of Vpr (Figure 2D), which is dependent on the addition of E1, E2 and ubiquitin (Figure S2B). These results demonstrate that Vpr-promoted TET2 degradation is mediated by CRL4VprBP E3 ligase via the 26S proteasome.
CRL4VprBP, in the absence of Vpr, catalyzes TET2 monoubiquitylation on K1299 (Nakagawa et al., 2015). We found that mutations in TET2 disrupting the monoubiquitylation site (K1212N or K1212E of mouse Tet2, corresponding to human K1299N or K1299E) or abolishing monoubiquitylation (C1211Y in mouse Tet2, corresponding to C1298Y in human TET2) did not affect Vpr-promoted TET2 degradation (Figure2E), suggesting that Vpr is unlikely acting as a processivity or chain elongation factor to extend the monoubiquitylation at K1299 to a polyubiquitin chain. We also found that F1213S mutation (corresponding to F1300S in human TET2) that disrupts its binding with VprBP had little effect on Vpr-promoted TET2 degradation (Figure 2E). On the other hand, the Q65R mutation abolished the ability of Vpr to promote TET2 degradation (Figure 1F) and to promote TET2 polyubiquitylation either in cells (Figure 2B) or in vitro (Figure 2D). These results indicate that Vpr-promoted polyubiquitylation by CRL4VprBP E3 ligase may involve a change of the VprBP-TET2 binding. In the absence of Vpr, TET2 binds to a sequence within the N-terminal 909 residues of VprBP (1 – 909, N909), but not the C-terminal region of VprBP (918 – 1507, C590). The expression of wild-type Vpr (but not Q65R mutant), however, enabled TET2 to bind the VprBP-C590 (Figures 2F and 2G), indicating that Vpr changes TET2-VprBP binding and tethers TET2 to a different binding site in the C-terminal region of VprBP. This is consistent with the structural analyses showing that Vpr and Vpx bridges the binding of substrate UNG2 or SAMHD1 to a sequence (residues 1050 – 1396) within the C-terminal region of VprBP (Schwefel et al., 2014; Wu et al., 2016). Structural analysis of SAMHD1-Vpx-VprBP/DCAF1 complex suggests that Vpx places SAMHD1 in the proximity of the ROC1 RING domain, ideally located for ubiquitin transfer. Our study provides a clear example where the switch between monoubiquitylation and polyubiquitylation, which lead to different paths of protein function, can be achieved by a single factor. Further structural studies would not only gain insight into the mechanism of Vpr, but also monoubiquitylation vs. polyubiquitylation.
Genetic inactivation of TET2 enhances HIV-1 replication
Vpr is known to increase HIV-1 replication in myeloid cells (Connor et al., 1995). We found that ectopic expression of TET2 significantly inhibited HIV-1 replication in THP1 cells (Figure 3A). Next, we generated TET2 knockout THP1 cells using the CRISPR-Cas9 system (Figures 3B and S3A). Deletion of TET2 did not noticeably affect THP1 cell proliferation (Figure S3B), but significantly increased HIV-1 infection at different multiplicity of infections (MOI), as measured by FACS analysis of intracellular p24 expression (Figures 3C and S3C). TET2 deletion also increased HIV-1 viremia in the cellular supernatant as measured by p24 enzyme-linked immunosorbent assay (ELISA) over multiple rounds of infection (Figures 3D and S3D).
Figure 3. TET2 inhibits HIV-1 replication.

(A) Over expression of TET2 inhibits HIV-1 replication. THP1 cells were transfected with TET2 for 24h and then infected with HIV-1 (Vpr-)/HSA reporter virus at MOI of 0.1. Viral replication was analyzed by HSA reporter expression, n=2.
(B) CRISPR-Cas9 mediated knockout of TET2 in THP1 cells. CTRL cells were transduced by non-targeting gRNA, n=2.
(C) TET2 deletion enhances HIV-1 replication. Control and TET2-KO THP1 cells were infected with HIV-1 (Vpr-, MOI = 0.05). Percentage of infected cells were analyzed at 2 days post infection (dpi) by intracellular p24 FACS (left panel). Different HIV-1 MOIs were used to infect control and TET2-KO THP1 cells and analyzed by intracellular p24 FACS (right panel).
(D) HIV-1 infection and replication kinetics in HIV-1 (Vpr-) infected control and TET2 knockout THP1 cells were measured over 9 days by p24 ELISA. This experiment was repeated twice with same clone and once with another TET2 KO clone, n=2.
(E) Re-expression of TET2 in TET2-KO THP1 cells restored HIV-1 inhibition. Empty vector or vector expressing wildtype or mutant TET2 (1 µg) were transfected into parental or TET2-KO cells and protein expression verified by Western blot 24 hr after transfection. The transfected cells were infected with HIV-1 (Vpr-), and virus replication was analyzed by p24 ELISA at 6 dpi, n=2.
(F) TET2 inhibits HIV-1 replication in human primary macrophages. shRNA-mediated knockdown of TET2 in MDMs and confirmed by RT-qPCR. MDMs were then infected with a macrophagetropic HIV-1 (Vpr-) and viral replication was analyzed by p24 FACS or p24 ELISA (donor n=3). Statistical analysis was performed using Student’s t-test. *, ** and *** indicated a p-value of less than 0.05, 0.01 or 0.001, respectively. n.s= not significant. Error bars were calculated from technical replicates.
To confirm the inhibitory effect of TET2 toward HIV and exclude the off-target effects associated with the CRISPR-Cas9 system, we performed a rescue experiment by ectopically expressing either full length or the cysteine-rich, dioxygenase (CD) domain of TET2 in TET2-KO THP1 cells. By expressing different amounts of TET2, we found that ectopically expressed TET2 inhibited HIV-1 infection in a dose-dependent manner and that this inhibition was counteracted by Vpr (Figure S3E). Expression of either the full length or CD domain of TET2 resulted in near complete restoration of inhibition on HIV-1 (Figure 3E), while the catalytic inactivating mutant in the context of either full length or CD domain of TET2 partially restored the inhibition of HIV-1 infection. These results suggest that TET2-mediated inhibition of HIV-1 involves both catalytic-dependent and –independent mechanisms. Notably, the function of TET2 in inhibiting HIV-1 was abolished by the infection of Vpr+ HIV-1 (Figure S3F).
To confirm the inhibitory activity of TET2 toward HIV-1 in primary human cells, we knocked down TET2 in primary human monocyte-derived-macrophages (MDM) by two different shRNAs. HIV-1 replication was significantly enhanced after TET2 knockdown in MDMs (Figures 3F and S3G). We also tested the effect of TET2 degradation on Vpr-enhanced HIV-1 replication in macrophages. HIV-1 or HIV-1ΔVpr were used for infection in shCTRL and shTET2-treated MDMs and levels of p24 in the supernatant were assessed. Vpr enhanced HIV-1 replication by 12 – 15 folds in control cells but only by 4 – 5 folds in TET2 knock-down cells (Figure S3H), indicating that TET2 is an important, but not the only, target of Vpr to enhance HIV-1 replication.
Vpr degrades TET2 to enhance HIV-1 replication independently of G2 cell cycle arrest
One notable alteration of cellular function by Vpr is the induction of G2 cell cycle arrest, but its cause and functional relevance to HIV-1 replication remains unclear (Romani and Cohen, 2012). Deletion of TET2 did not affect the cell cycle phase distribution and Vpr caused similar G2 cell cycle arrest in both control and TET2-KO cells (Figure S4A), indicating that TET2 does not play a significant role in cell cycle control and is not required for Vpr-induced G2 arrest. We then established a THP1 cell stable cell lines with tetracycline-inducible expression of Vpr (THP1/Tet-Vpr) and induced Vpx expression at different stages of the cell cycle. We found that Vpr degrades TET2 with an indistinguishable efficiency in cells at different phases of the cell cycle (Figure S4B), suggesting that Vpr-promoted TET2 degradation is not linked to a specific cell cycle phase such as G2/M population.
To determine functionally whether Vpr-enhanced HIV-1 replication is genetically linked to TET2 degradation and/or G2/M cell cycle arrest, we characterized a single amino acid substitution mutant of Vpr, R80A. Unlike the Q65R mutation which disrupts Vpr’s ability to bind with VprBP, to cause G2/M cell cycle arrest and to enhance HIV-1 replication, the VprR80A mutant is reported to be defective in causing G2-arrest (DeHart et al., 2007), but retains Vpr’s activity of binding with VprBP and enhancing viral replication (Rajan et al., 2006) (our confirmatory results in Figures S4C and S4D). We found that VprR80A is active to promote TET2 degradation (Figure 4A) and importantly, retains the ability to enhance HIV-1 replication similarly as wildtype Vpr, when delivered into target cells via VLP (Figures 4B and S4E). These results indicate that the two functions of Vpr in promoting TET2 degradation and causing G2/M cell cycle arrest are genetically separable and that TET2 degradation, but not the G2/M cell cycle arrest, is linked to the enhancement of HIV-1 replication.
Figure 4. Vpr degrades TET2 to sustain IL-6 expression and enhance HIV-1 replication.

(A) Vpr R80A mutant promotes TET2 degradation. Flag-TET2 protein levels were analyzed 24h after transfection with indicated plasmids by western blot, n=2.
(B) Separation of Vpr-mediated TET2 degradation and HIV-1 replication enhancement from G2 arrest. VLP incorporated with wild type or Vpr mutants Q65R and R80A were generated and quantified by p24 ELISA. THP1 cultures were treated with equal amount of VLPs (based on p24 capsid) for 6h and then infected with HIV-1 (Vpr-)/HSA (or luc) reporter virus, followed by analysis of HSA reporter expression, n=2.
(C) HIV-1 Vpr enhances induction of IL-6 expression in MDMs. Left panel: VSV-G pseudotyped HIV-lucΔVpr vectors complemented with different Vpr mutants were used to infect MDMs. Right panel: Relative Vpr-enhanced IL-6 expression with MDMs derived from 6 donors is summarized by setting wild type Vpr-enhanced IL-6 expression at 1.0 (donor n=6).
(D) Vpr impairs the termination of IL-6 expression during HIV-1 infection. MDMs were infected with VSV-G pseudotyped HIV-lucΔVpr complemented with different Vpr mutants (donor n=3). IL-6 mRNA levels were analyzed at indicated time points after infection.
(E) TET2 genetic inactivation sustains IL-6 expression similarly as Vpr. VSV-G pseudotyped HIV-lucΔVpr vectors complemented with WT or VprQ65R Vpr were used to infect the Ctrl, TET2-KO cells, and mRNA level of IL-6 was determined, n=3.
(F) Vpr degrades TET2 and prevents recruitment of HDAC1 and HDAC2 to and enhances acetylation of the IL-6 promoter during resolution phase. THP1 cells were infected with viruses followed by ChIP assays. Each ChIP DNA fraction’s Ct value was normalized to the IgG DNA fraction’s Ct value (ΔCt) at the same time point. IgG is defined as 1. Error bars stem from three technical replicates, n=2.
(G) IL-6 contributes to Vpr-enhanced HIV-1 replication in macrophages. Left 3 panels: HIV-1 or HIV-1ΔVpr were used to infect MDMs from different 3 donors treated with isotype control or anti-IL-6 neutralizing antibody. p24 levels in the supernatant were measured at day 6 after infection. Right panel: Relative ability of Vpr to enhance HIV-1 replication in cells treated with isotype control and IL-6 blocking antibody at Day 6 was calculated (donor n=3). Data is summarized by setting Vpr-enhanced replication in isotype control MDMs at 1.0. Error bars represent the mean ± SD for triplicate experiments. * and ** indicate p-values of <0.05 and <0.01, respectively.
Vpr degrades TET2 to sustain IL-6 expression and enhance HIV-1 replication
Persistent inflammation is a hallmark of HIV-1 pathogenesis and is linked to enhanced viral replication, but the mechanism that leads to persistent inflammation remains elusive (Deeks et al., 2013). It was recently reported that during inflammation resolution phase Tet2 was recruited by IκBζ to IL-6 promoter and Tet2 in turn recruits HDAC1/2 to actively repress the transcription of IL-6 (Zhang et al., 2015), a pleiotropic cytokine that has pro- and anti-inflammatory properties and its increased levels have been associated with HIV-1 disease progression risk (Connolly et al., 2005). We hypothesized that Vpr, via degrading TET2, may block the HDAC recruitment to IL-6 gene, thereby maintaining elevated levels of IL-6 and contributing to persistent inflammation. To test this idea, we produced VSV-G pseudotyped HIV-1 expressing luciferase (HIV-luc) complemented with either empty vector (HIV-lucΔVpr), Flag-tagged wildtype (HIV-luc+Vpr) or Q65R and R80A mutants Vpr, and infected primary human MDMs from different donors. We detected significantly higher production of IL-6 in MDMs infected with HIV-luc +Vpr or VprR80A when compared with MDMs infected with HIV-lucΔVpr or +VprQ65R (Figures 4C, S4F and S4G). The ability of Vpr to increase IL-6 levels is disrupted by the Q65R, but not R80A mutation, indicating that Vpr can elevate IL-6 expression and that this function of Vpr is dependent on its binding with VprBP, but not G2/M cell cycle arrest. Kinetic studies indicate that Vpr-induced IL-6 mRNA in MDMs occurs during the resolution but not induction phase (Figures 4D and S4H). We performed similar experiments in THP1 cells and found that infection of HIV-luc+Vpr virus caused similar IL-6 increase as HIVluc+ VprQ65R virus during early infection with a somewhat faster kinetics than that seen in MDMs (Figure 4E), but sustained a higher IL-6 expression during late resolution phase (16–36 h.p.i.) when compared to the cells infected with HIV-luc+VprQ65R. In contrast, in TET2-KO THP1 cells, IL-6 expression stayed high during resolution phase regardless whether cells were infected with HIV-luc+Vpr or HIV-luc+VprQ65R virus-like particles. These data indicate that virion-associated Vpr sustains IL-6 expression during resolution phase in a TET2-dependent fashion.
To explore the mechanism of Vpr-mediated persistent expression of IL-6, we performed ChIP-qPCR experiments to measure the binding of IκBζ, HDAC1, HDAC2 to and histone acetylation of the IL-6 promoter in the parental and TET2-KO THP1 cells at different time points after infection with HIV-luc+Vpr or HIV-luc+VprQ65R viruses. Consistent with a previous report (Zhang et al., 2015), IκBζ bound to the IL-6 promoter at both early and late phases during HIV-1 infection in both parental and TET2-KO THP1 cells (Figure S4I). TET2 was not detected at IL-6 promoter during early time of viral infection (4 h.p.i.), but bound to IL-6 later after viral infection (8 and 24 h.p.i.) corresponding to the resolution phase in cells expressing Q65R mutant Vpr (Figure 4F). Binding of TET2 to IL-6 promoter, however, was not detected in THP1 cells infected with HIV-Luc expressing wildtype Vpr or in TET2-KO THP1 cells, supporting the specific binding of TET2 to IL-6 promoter. HIV-1 infection did not change the protein level of HDAC1 or HDAC2 (Figure S4J). Two HDAC proteins bound to IL-6 promoter with distinct kinetics in THP1 cells infected with HIV-Luc expressing Q65R mutant Vpr. HDAC1 binding to IL-6 promoter was nearly undetectable at early time of viral infection, increased during the infection of HIV-Luc expressing Vpr Q65R mutant and reached the peak at the end of the experimentation (24 h.p.i.). Binding of HDAC2 to IL-6 promoter, on the other hands, was readily detected prior to viral infection, decreased to a background low level during early infection (4 h.p.i.) and rebound later during the infection of HIV-Luc expressing Vpr Q65R mutant (Figure 4F). Notably, binding of both HDAC1 and HDAC2 to IL-6 promoter was substantially reduced in THP1 cells expressing wildtype Vpr or in TET2-KO THP1 cells (Figure 4F), indicating that TET2 is required for the binding of both HDAC enzymes to IL-6 promoter. The level of H3 acetylation (H3Ac) in IL-6 promoter was increased initially in parental THP1 cells regardless the genotype of Vpr at 4 h.p.i., remained relatively high in THP1 cells expressing wildtype Vpr, but was significantly reduced in cells expressing Q65R mutant Vpr later during the infection. In TET2-KO THP1 cells, H3Ac levels in IL-6 promoter were similarly increased following infection and stayed relatively high later during the infection regardless the genotype of Vpr (Figure 4F). These results support that transcription factor IκBζ recruits TET2 to IL-6 promoter later during the resolution phase and TET2 in turn recruits HDACs to promote the deacetylation and down regulation of IL-6 expression. Loss of TET2 function, resulting from either genetic deletion or Vpr-promoted degradation, blocks HDAC recruitment to IL-6 promoter and leads to persistent IL-6 expression.
Finally, to test whether the elevated IL-6 is linked to Vpr-enhanced HIV-1 replication, we infected MDMs with HIV-1 or HIV-1ΔVpr viruses and then treated infected cells with either isotype control or IL-6 neutralizing antibody, followed by the measurement of p24 in supernatant six days after infection. Vpr enhanced p24 production by average 17.2 folds in MDM cells from three donors treated with isotype control antibody, but only 8.3 folds in cells treated with IL-6 neutralizing antibody (Figures 4G and S4K), indicating that elevated IL-6 contributes to ~50% of the enhancement of viral replication by Vpr. This result suggests that Vpr degrades TET2 to enhance HIV-1 replication in part through IL-6, and Vpr has an additional, IL-6-independent activity in facilitating HIV-1 replication.
DISCUSSION
The results presented here demonstrate that TET2 is an inhibitor of HIV-1 replication and substrate of Vpr-hijacked CRL4VprBP ligase. This conclusion is supported by two lines of evidences. First, Vpr directly targets TET2 for polyubiquitylation by the CRL4VprBP E3 ligase and subsequent degradation by the proteasome. We demonstrated that Vpr-targeted degradation of TET2 proteins requires Vpr-VprBP interaction, occurs as early as 6 hours after infection, in human cell lines as well as primary cells, represents a conserved function of Vpr and can be sufficiently carried out by the Vpr protein incorporated in the HIV virion particle. Second, we showed that overexpression of TET2 inhibited HIV-1 replication and, conversely, loss of function of TET2 enhances HIV-1 replication in THP1 cells and primary macrophages. Mutations in Vpr disrupting its binding with VprBP abolished its ability to promote HIV-1 replication and this defect can be rescued by the deletion of TET2, linking directly the function of Vpr in promoting HIV-1 replication to TET2 degradation. Collectively, these results demonstrate that TET2 is a restriction factor of HIV-1 and a major functional target of Vpr.
IL-6 has long been reported to stimulate HIV-1 replication in HIV-infected primary macrophages and is linked to enhanced HIV-1 gene expression (Poli et al., 1990). Macrophages are a main source of IL-6 during HIV infection and also a well-documented target cell for Vpr to enhance HIV-1 replication. Our findings show that Vpr degrades TET2, thereby preventing it from binding to and repressing IL-6 expression, which leads to elevated IL-6 expression during resolution phase of inflammation and is linked to the ability of Vpr to enhance viral replication. These results reveal a pathway for specific enhancement of viral replication induced by Vpr in MDMs and provide a molecular explanation for the persistent IL-6 and inflammation that has long been recognized as a hallmark of HIV-1 pathogenesis. These results also identify a target—Vpr-TET2-IL-6 axis—for therapeutic intervention of HIV-1 infection and inflammation.
STAR★METHODS
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
EXPERIMENTAL MODEL AND SUBJECT DETAILS
- METHOD DETAILS
- Plasmids
- Cell cultures and cell transfection
- In vivo and in vitro ubiquitylation assay
- RNAi, shRNA and CRISPR-Cas9
- HIV-1 proviral constructs and virion stock production
- HIV-1 infection
- Flow cytometry analysis
- ChIP assay
QUANTIFICATION AND STATISTICAL ANALYSIS
DATA AND SOFTWARE AVAILABILITY
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yue Xiong (yxiong@email.unc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Source of cell lines used in the study is reported in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HA HRP | Roche | Clone 3F10 |
| Anti-Myc HRP | Roche | Clone 9E10 |
| Anti-FLAG M2 HRP | Sigma | A8592 |
| Anti-IκBζ | Cell Signaling | Cat# 9244 |
| Anti-HDAC1 | Abcam | Cat# ab7028 |
| Anti-HDAC2 | Abcam | Cat# ab7029 |
| Anti-acetyl-Histone H3 | Millipore | Cat# 06–599 |
| Anti-TET2 | Abcam | Cat# ab94580 |
| Anti-TET2 | Millipore | Cat# MABE462 |
| Anti-Tubulin | Santa Cruz | Cat# sc-23948 |
| Anti-Actin | Santa Cruz | Cat# sc-47778 |
| Anti-HIV-1 p24 | NIH AIDS Reagent Program | Cat# 6458 |
| Anti-Vpr | NIH AIDS Reagent Program | Cat# 11836 |
| Anti-SAMHD1 | Abcam | Cat# 67820 |
| Anti-p21 | Santa Cruz | Cat# sc-397 |
| Anti-p27 | Santa Cruz | Cat# sc-71813 |
| Anti-CUL4A | Abcam | Cat# ab72548 |
| Anti-CUL4B | Sigma | Cat# HPA011880 |
| Anti-DDB1 | Home made | N/A |
| Anti-ROC1 | Home made | N/A |
| Anti-VprBP | ProteinTech | Cat# 11612-1-AP |
| anti-mCD24-PE | Biolegend | Cat# 138503 |
| Bacterial and Virus Strains | ||
| pNL4-3-HSA | NIH AIDS Reagent Program | Cat# 3419 |
| pNL4-3-HSA-R- | NIH AIDS Reagent Program | Cat# 3421 |
| pNL4-LUC-E-R- | NIH AIDS Reagent Program | Cat# 3418 |
| X4/R5 dual tropic HIV-1 | Meissner et al., 2004 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Calpeptin | Sigma | Cat# C8999 |
| Z-VAD-FMK | MBL International Corporation | Cat# 50-446-69 |
| MG132 | Peptides Intl. | Cat# 3175-v |
| CD3/CD28 activation beads | Thermo dynabeads | Cat# 11205D |
| Recombinant IL-2 | NIH AIDS Reagent Program | Cat# 136 |
| M-CSF | Peprotech | Cat# 300-25 |
| GM-CSF | Peprotech | Cat# 300-03 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| Doxycycline | Sigma | Cat# D9891 |
| Critical Commercial Assays | ||
| Enzo Ubiquitinylation (Ubiquitin Conjugation) Kit | Enzo Life Sciences | BML-UW9920-0001 |
| Chromatin Immunoprecipitation (ChIP) Assay Kit | Millipore | 17–295 |
| Human Monocyte Isolation Kit | STEMCELL TECHNOLOGIES | Cat#19359 |
| HIV-1 p24 antigen capture assay kit | Leidos Biomedical Research | AIDS and Cancer Virus Program |
| Luciferase Assay System | Promega | Cat# E1500 |
| Amaxa Cell Line Nucleofector Kit | LONZA | VVCA-1003 |
| Deposited Data | ||
| Raw imaging data | This paper | http://dx.doi.org/10.17632/s77chxfn8t.1 |
| Experimental Models: Cell Lines | ||
| 293T | ATCC | CRL 11268 |
| THP-1 | ATCC | TIB 202 |
| U2OS | ATCC | HTB 96 |
| Jurkat T cells | ATCC | TIB-152 |
| Oligonucleotides | ||
| siCUL4A: 5'-GAACUUCCGAGACAGACCU-3' | Nakagawa et al., 2015 | N/A |
| siCUL4B: 5'-AAGCCUAAAUUACCAGAAA-3' | Nakagawa et al., 2015 | N/A |
| siDDBI: 5'-CCUGUUGAUUGCCAAAAAC-3' | Nakagawa et al., 2015 | N/A |
| siVprBP: 5'-UCACAGAGUAUCUUAGAGA-3' | Nakagawa et al., 2015 | N/A |
| TET2 shRNA: TAAGTAATACAATGTTCTT | Dharmacon, GE Healthcare | clone ID: V3LHS_363201 |
| TET2 sgRNA: GATTCCGCTTGGTGAAAACG | This paper | N/A |
| ChIP-qPCR primer: IL-6 Forward: 5'-ACTTCGTGCATGACTTCAGC-3' | This paper | N/A |
| ChIP-qPCR primer: IL-6 Reverse: 5'-AGTGCAGCTTAGGTCGTCAT-3' | This paper | N/A |
| qPCR IL-6 primer, Forward:5'-ACTCACCTCTTCAGAACGAATTG-3' | This paper | N/A |
| qPCR IL-6 primer, Reverse:5'-CCATCTTTGGAAGGTTCAGGTTG | This paper | N/A |
| qPCR TET2 primer, Forward:5'-GATAGAACCAACCATGTTGAGGG-3' | This paper | N/A |
| qPCR TET2 primer, Reverse:5'-TGGAGCTTTGTAGCCAGAGGT-3' | This paper | N/A |
| Recombinant DNA | ||
| pcDNA3-Flag-TET2 WT | Nakagawa et al., 2015 | N/A |
| pcDNA3-Myc-VprBP WT | Nakagawa et al., 2015 | N/A |
| pcDNA3-HA-HIV-1 Vpr WT | This paper | N/A |
| pcDNA3-Flag-TET2 H1881Q | This paper | N/A |
| pcDNA3-Flag-TET2 R1896S | This paper | N/A |
| p3Flag-HIV-1 Vpr WT | This paper | N/A |
| p3Flag-HIV-1 Vpr Q65R | This paper | N/A |
| p3Flag-HIV-1 Vpr R80A | This paper | N/A |
| Flag-HIV-1 Q23 Vpr | A gift from Michael Emerman of University of Washington, Seattle | N/A |
| Flag-HIV-2 ROD9 Vpr | A gift from Michael Emerman of University of Washington, Seattle | N/A |
| Flag-SIV-mac 239 Vpr | A gift from Michael Emerman of University of Washington, Seattle | N/A |
| Flag-HIV-2 ROD9 Vpx | A gift from Michael Emerman of University of Washington, Seattle | N/A |
| Flag-SIV-mac 239 Vpx | A gift from Michael Emerman of University of Washington, Seattle | N/A |
| pCMV-VSV-G | Addgene | #8454 |
| ANRF | Addgene | #12263 |
| pMDLg/pRRE | Addgene | #12251 |
| pRSV-Rev | Addgene | #12253 |
| Software and Algorithms | ||
| GraphPad Prism 5 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | ImageJ Software | https://imagej.net/Downloads |
| FlowJo | Software | Version 10 |
METHODS DETAILS
Plasmids
Expression constructs for TET proteins and VprBP were previously described (Nakagawa et al., 2015; Wang et al., 2015). Full length human TET2 cDNA (2,002 residues, NCBI reference number NP_001120680.1) was subcloned to the p3XFLAG-CMV destination expression vector (Sigma-Aldrich). HIV-1 Vpr was also cloned into p3XFLAG-CMV destination expression vector. Point mutations in TET2 (H1881Q) and Vpr (Q65R and R80A) were generated by site-directed mutagenesis and verified by DNA sequencing. The C-terminal cysteine-rich, dioxygenase (CD) domain (residues 1128 – 2002) of TET2 (TET2-CD) was also cloned into p3XFLAG-CMV and it consists of amino acid 1128–2002 of the full-length TET2. Vectors expressing HIV-1 Q23 Vpr, HIV-2 ROD9 Vpr, SIV-mac 239 Vpr, HIV-2 ROD9 Vpx and SIV-mac 239 Vpx was a gift from Michael Emerman of University of Washington, Seattle. HIV-2 Vpx vector was a gift from Ronald Swanstrom of University of North Carolina at Chapel Hill and was cloned to a pcDNA3 vector.
Cell cultures and cell transfection
THP1 and HEK293T cells were purchased form UNC Lineberger Tissue Culture Facility. HEK293T cells were cultured in DMEM containing 10% FBS and 1X Antibiotic-Antimycotic (Invitrogen™). THP1 cells were maintained in RPMI 1640 medium containing 10% FBS, 1% Pen/Strep antibiotics, 2mM glutamine, 10mM HEPES and 1× non-essential amino acids (all from GIBCO). Human buffy coats were obtained from Gulf Coast Regional Blood Center. Primary human peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats using Ficoll-paque gradient. PBMCs that are used for HIV-1 infection experiments were first activated with CD3/CD28 activation beads (Thermo dynabeads) and IL-2 (300 U/mL) for 3 days, then maintained in 20 U/mL IL-2 during experiments.
Primary monocytes were isolated from PBMCs using negative magnetic selection (EasySep™ Human Monocyte Isolation Kit # 19359). Purified monocytes were seeded at a concentration of 1×106 cells/mL in complete RPMI, and differentiated into Monocytes-Derived-Macrophages (MDM) using 50ng/mL M-CSF and 50ng/mL GM-CSF (Peprotech) for 6 days, changing medium and cytokines every two days.
Transfection of 293T was performed using lipofectamine 2000 (Invitrogen™) following manufacturer’s protocol. Transfection of THP1 was performed using Amaxa Cell Line Nucleofector Kit V according to manufacturer’s protocol (Lonza VCA-1003). Briefly, 1×106 THP1 cells were incubated with 300 ng DNA in 100 µL nucleofector solution V, and immediately inserted into the nucleofector device and use nucleofector program U-001. Transfected cells are cultured at 0.5×106 cells/mL for 24h, before use for further experiments.
In vivo and in vitro ubiquitylation assay
In vitro ubiquitylation assays were performed as described previously (Nakagawa et al., 2015). Briefly, CUL4-VprBP immune complexes were purified from HEK293T cells transfected with Flag-VprBP. Flag tagged VprBP was immunoprecipitated with anti-FLAG M2 agarose beads for 3 h in a NP-40 lysis buffer (0.3% Nonidet P-40, 50 mM Tris pH 7.5, 150 mM NaCl). Immobilized Flag-VprBP complexes were washed three times in the same lysis buffer and eluted with an excess of Flag peptide (Sigma). Ubiquitylation reactions were performed in a 50 µL reaction volume, containing 100 nM E1 (Enzo Life Sci.), 1 µM E2 (Enzo Life Sci.), 1 µM human recombinant ubiquitin (Boston Biochem), 1 unit inorganic pyrophosphatase, 1 mM DTT and 5 mM Mg-ATP, 100 ng of eluted CUL4-VprBP complexes as the source of E3 and 100 ng of human Flag-TET2 as substrate. Reactions were incubated at 37 °C for 30 min, terminated by addition of an equal volume of SDS sample buffer and resolved by SDS-PAGE.
For the in vivo ubiquitylation assay, HEK293T cells were transfected with indicated plasmids and siRNA for 48hrs, and were treated with MG132 (10 µM) for 5hrs before collecting the cells. Cells were lysed under denaturing conditions in a SDS buffer (50 mM Tris-HCl, pH 7.5, 0.5 mM EDTA, 1 mM DTT, 1% SDS) by boiling for 10min. Lysate was clarified by centrifugation at 13,000 rpm for 10min and diluted 10-fold with an NP-40 buffer (50mM Tris pH 7.5, 150mM NaCl, 0.3% Nonidet P-40) and then subjected to immunoprecipitation by anti-Flag M2 agarose beads and subsequent SDS-PAGE. Ubiquitylated TET2 was detected with HA antibody.
RNAi, shRNA and CRISPR-Cas9
All siRNA oligonucleotides were synthesized with 3’ dTdT overhangs by Sigma in a purified and annealed duplex form. The sequences targeting each gene were as follows: human CUL4A, 5’-GAACUUCCGAGACAGACCU-3’; Human CUL4B, 5’-AAGCCUAAAUUACCAGAAA-3’; Human DDB1, 5’-CCUGUUGAUUGCCAAAAAC-3’; Human VprBP, 5’-UCACAGAGUAUCUUAGAGA-3’. For transfection of siRNA, OPTI-MEM medium (250 µL) was mixed with Lipofectamine 2000 reagent (Life Technologies, 10 µL) for 5 min and then incubated with another 250 µL OPTI-MEM medium containing 10 µL (20 mM) of siRNA for 20 min at room temperature. The mixtures were added to cells cultured on a 60-mm plate at 30%–40% confluence. The knocking down efficiency was determined 48–72 h after transfection.
pGIPZ-lentiviral vectors expressing control (non-targeting), TET2-targeting shRNA were purchased from Dharmacon, GE Healthcare (Lafayette, CO). shRNA targeting human TET2 has the mature antisense sequence: TAAGTAATACAATGTTCTT (clone ID: V3LHS_363201). Lentiviruses expressing GIPZ-shRNA were produced by CaCl2-BES transfection in 293T cells (10cm plate) using 15ug pGIPZ-shRNA vector, 10 µg packaging construct ΔNRF and 5 µg pCMV-VSV-G. Lentiviruses were titered on 293T cells. Transductions of THP-1 cells were performed with a multiplicity of infection (M.O.I.) of 0.5. For transduction of primary MDM, 50 ng of VLPVpx were first treated to 4×105 cells MDM for 6h, then the cells were washed once with PBS and incubated with GIPZ-shRNA lentivirus (100 ng of p24). All lentiviral transductions were performed by adding 8ug/mL polybrene, and spin inoculated with the cells for 2h, 1500g at 37 °C. Transduced THP1 and MDM cells after 3 days were selected with 1–2 ug/mL puromycin for 3–7 days. The transduction efficiency was assessed by %GFP expression. All MDM donors presented in the data has achieved >90% transduction efficiency. TET2 knockdown efficiency were subsequently measured by RT-qPCR.
CRISPR-Cas9 lentiviral vector lentiCRISPR v2 were purchased from Addgene. The gRNA sequence used for targeting TET2 is GATTCCGCTTGGTGAAAACG. Lentiviruses for CRISPR were produced by CaCl2-BES transfection of 293T cells (10 cm plate) using 10 ug vector, 15 ug pMDLg/pRRE, 4 ug pRSV-Rev, and 5 ug pCMV-VSV-G. One million THP1 cells were transduced with ~100ng p24 of lentiviruses, and after 3 days selected with 2ug/mL puromycin for 7 days. For single clone isolation of TET2-KO THP1, the pool of knockout cells was limiting diluted to 2 cells per well in 96-well plates, and the recovered KO clones were validated by DNA sequencing and western blot.
HIV-1 proviral constructs and virion stock production
NL4-3-HSA, NL4-3-HSA-R− and NL4-LUC-E−R− proviral constructs expressing heat stable antigen (HSA) reporter or firefly luciferase reporter, respectively, were obtained from the NIH AIDS Reagent program. X4/R5 dual tropic HIV-1 NL4-R3A was generated by the Su lab as previously described (Meissner et al., 2004). Vpr-mutant R3A was generated by insertion of heat stable antigen (HSA) reporter into the Vpr gene.
HIV-1 virions were produced by CaCl2-BES transfection of proviral plasmids in 293T cells. 293T cells cultured on a 10 cm plate were transfected with 30 µg DNA of pNL4-3 or pNL4-R3A for production of replication competent HIV-1 viruses, 25ug pNL4-LUC-E−R− with 5 µg pVSV-G for the production of VSV-G pseudotyped firefly luciferase reporter virus (HIV-LUC-G). For the production of virus-like-particles (VLP) carrying Vpr or Vpr-mutants, we transfected 293T with 6µg p3XFLAG-CMV-Vpr, 15ug pMDLg/pRRE, 4ug pRSV-Rev, and 5ug pCMV-VSV-G. For the production of VLP-Vpx particles, we used a Gag/Pol expressing vector pMDL-X, containing the Vpx-packaging motifs in the p6 region, as described before (Bobadilla et al., 2013).
Virion stocks (both HIV-1 and VLPs) were harvested at 48 h post transfection, and purified by ultra-centrifugation (20,000 g, 4 °C, 2 h). Concentrated particles were reconstituted with 1/10 times the amount of starting medium (i.e. resulting in 10× concentration of virus stocks). Concentration of virion stocks were quantified by p24 ELISA assay (Frederick National Laboratory for Cancer Research – AIDS and Cancer Virus Program).
HIV-1 infection
The titer of HIV-1 virion stocks was determined by infecting a known number of Jurkat T cells with titrated amount (p24) of virion particles, and evaluated by flow cytometry the percentage of infected cells (%p24 or %HSA) 2 days later. HIV-1 infection experiments were typically conducted at an M.O.I of 0.1, unless otherwise indicated in the figure legend. All HIV-1 infections were typically performed in 96-well format (triplicates), by spin inoculation for 2 hr at 1500g, 37 °C containing 8ug/mL polybrene. Virus input are removed after spin inoculation, and fresh medium are added to infected cells. When indicated, anti-viral drugs sCD4 (10 ug/mL) T20 (5 ug/mL), Nevirapine (5 uM), Raltegravir (10 uM), or Amprenavir (5 uM) were added to the cells right before infection and maintained after infection.
Infections of freshly purified PBMCs with HIV-LUC-G were performed with 10ng p24 virus per 1×105 cells in 96-well format. Firefly luciferase activity were measured at 48 hr post infection using Luciferase Assay System from Promega and normalized to total protein concentration. Infection with VSV-G pseudotyped VLP, VLP-Vpr or VLP-Vprmutant in THP-1 and PBMCs were performed using 500ng p24 amount of VLPs per 1×106 cells by spin inoculation for 2 hr at 1500g, 37 °C containing 8ug/mL polybrene.
Flow cytometry analysis
HIV-1 infection efficiency was analyzed by HSA reporter expression or intracellular p24 expression at 48 hr post infection. For HSA detection, surface antigen staining was performed in live infected cells with anti-mCD24-PE antibody (biolegend, 1:50 dilution in 2%FBS PBS buffer) for 20 minutes. For intracellular p24 detection, infected cells were first permeabilized using Cytofix/cytoperm™ from BD following manufacturer’s protocol, and then incubated with anti-p24-PE antibody (1:50 dilution, Beckman Coulter KC-57). Stained samples were subsequently fixed in 1× formalin and analyzed with CyAn ADP (DAKO) or GUAVA ExpressPlus instrument. Cell cycle analysis was performed with genomic DNA staining by fixing cells with 75% ethanol at −20 °C overnight, followed by Propidium Iodide staining of cellular DNA in 1% BSA containing RNase A. DNA levels were analyzed by FACS.
ChIP assay
For ChIP assay, cells were collected in an Eppendorf tube and resuspended in 7.5 ml cold PBS. Cellular proteins and DNA were cross-linked by adding 0.5 ml of 16% formaldehyde into the tube (final concentration: 1%) and incubated at 37 °C for 15 min. Cells were centrifuged at 1,000 rpm for 5 min and cell pellets were resuspended in 5ml PBS buffer containing 0.125 M Glycine and 1X protease inhibitor cocktail (PIC). The cells were then incubated at room temperature with shaking for 5 min to quench unreacted formaldehyde. The reaction was centrifuged and cell pellets were quickly rinsed with 10 ml of cold PBS and 1X PIC. Cells were collected after centrifugation and transferred into a new Eppendorf tube with 1ml cold PBS with 1X PIC, followed by centrifugation at 1,000 rpm for 2 min to pellet the cells. Cell pellets were resuspended in 100 µL of SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris pH8.1, 1X PIC) and incubated on ice for 15 min. DNA was sheared by sonication using a Covaris Sonicator (13 min at 4°C). Insoluble material was removed by centrifugation at 12,000 g at 4°C for 10 min. The supernatants were transferred to fresh tubes and 900 µL ChIP Dilution Buffer containing 1X PIC and 5ul antibody or 20 µL fully resuspended anti-FLAG M2 Affinity Gel (Sigma) was added to each tube. Mixtures were incubated overnight at 4°C with rotation. Flag beads were precipitated by centrifuging at 2,000 rpm for 2 min at 4°C. The bead-protein-chromatin complexes were washed in 0.5 ml each of the cold buffers in the following order: (1) low Salt Wash Buffer, (2) high Salt Wash Buffer, (3) LiCl Wash Buffer, and (4) TE Buffer (all buffers from Millipore). All washes were performed by incubating complexes for 3–5 minutes with rotation followed by centrifuging at 2,000 rpm for 2 minutes. 100 µL of ChIP Dilution Buffer and 1 µL proteinase K were then added into each tube, followed by two incubations, first at 62 °C for 2 h and then a second at 95 °C for 10 min, both with constant shaking on an Eppendorf ThermoMixer (700 rpm). Samples were cooled down to room temperature and centrifuged at 2,000 rpm for 2 min at 4°C. Supernatants were carefully transferred to a new tube containing 0.5 ml of Binding Reagent A. The sample-Binding Reagent A mixtures were transferred to the spin filter in the collection tube, centrifuged for 30 s at 12,000 g and decanted (this might be an easier way of saying pouring out supernatant). 500 µL of Wash Reagent B were added to the same spin filter in the collection tube and centrifuged for 30 s at 12,000 g. The aqueous was carefully discarded, followed by centrifugation for 30 s at 12,000 g. The spin filter was put into a new collection tube and 50 µL of Elution Buffer C (Millipore) was added to the center of the spin filter membrane. After centrifugation for 30 s at 12,000 g, purified DNA was collected in the eluate for qPCR analysis. IL-6 primers used for qPCR analysis of IL-6 promoter binding IκBζ, TET2, HDAC1 and HDAC2 were as follows: Forward: 5’-acttcgtgcatgacttcagc-3’, Reverse: 5’-agtgcagcttaggtcgtcat-3’.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis was performed with unpaired Student’s t-test, and are considered significant when the p-value is less than 0.05. *, ** and *** indicate p-values of less than 0.05, 0.01 and 0.001 respectively. n.s = not significant. N.D = not detected. The number of times each experiment is repeated is indicated in the figure legends.
DATA AND SOFTWARE AVAILABILITY
Original imaging data have been deposited to Mendeley Data and are available at http://dx.doi.org/10.17632/s77chxfn8t.1
Supplementary Material
HIGHLIGHTS.
HIV-1 Vpr promotes TET2 degradation
Vpr reprograms CRL4VprBP E3 ligase to catalyze polyubiquitylation of TET2
Vpr degrades TET2 to enhance HIV-1 replication
Vpr sustains IL-6 production to promote HIV-1 replication
Acknowledgments
We thank Michael Emerman and Ronald Swanstrom for providing HIV-2/SIV Vpr and Vpx expression plasmids, Ronald Swanstrom for discussion throughout this study, and Matt Smith and Chris Murphy for reading the manuscript. This study was supported by NIH grants AI095097 and AI080432 to L.S., a Samuel Waxman grant and NIH grants GM067113 and CA163834 to Y.X.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
L.L. made the discovery of Vpr mediated TET2 degradation; YP.X. found that Vpr reprograms CRL4VprBP to catalyze TET2 polyubiquitylation; Q.W. demonstrated that Vpr sustains IL-6 expression via TET2; L.T. and H.G. demonstrated that TET2 inhibits HIV-1 replication; T. N. helped initial TET degradation assay. L.S. and Y.X. conceived the project.
DECLARATION OF INTERESTS
The authors declare no conflict of interest.
References
- Ahn J, Vu T, Novince Z, Guerrero-Santoro J, Rapic-Otrin V, Gronenborn AM. HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-ring E3 ubiquitin ligase for proteasome-dependent degradation. J Biol Chem. 2010;285:37333–37341. doi: 10.1074/jbc.M110.133181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobadilla S, Sunseri N, Landau NR. Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther. 2013;20:514–520. doi: 10.1038/gt.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins DR, Lubow J, Lukic Z, Mashiba M, Collins KL. Vpr Promotes Macrophage-Dependent HIV-1 Infection of CD4+ T Lymphocytes. PLoS Pathog. 2015;11:e1005054. doi: 10.1371/journal.ppat.1005054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly NC, Riddler SA, Rinaldo CR. Proinflammatory cytokines in HIV disease-a review and rationale for new therapeutic approaches. AIDS Rev. 2005;7:168–180. [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet. 2013;382:1525–1533. doi: 10.1016/S0140-6736(13)61809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHart JL, Zimmerman ES, Ardon O, Monteiro-Filho CM, Arganaraz ER, Planelles V. HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol J. 2007;4:57. doi: 10.1186/1743-422X-4-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S, Xiong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci. 2009;34:562–570. doi: 10.1016/j.tibs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, An J, Bandukwala HS, Chavez L, Aijo T, Pastor WA, Segal MF, Li H, Koh KP, Lahdesmaki H, et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. 2013;497:122–126. doi: 10.1038/nature12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguette N, Bregnard C, Hue P, Basbous J, Yatim A, Larroque M, Kirchhoff F, Constantinou A, Sobhian B, Benkirane M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell. 2014;156:134–145. doi: 10.1016/j.cell.2013.12.011. [DOI] [PubMed] [Google Scholar]
- Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahouassa H, Blondot ML, Chauveau L, Chougui G, Morel M, Leduc M, Guillonneau F, Ramirez BC, Schwartz O, Margottin-Goguet F. HIV-1 Vpr degrades the HLTF DNA translocase in T cells and macrophages. Proc Natl Acad Sci U S A. 2016;113:5311–5316. doi: 10.1073/pnas.1600485113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang SM, Weeger M, Stahl-Hennig C, Coulibaly C, Hunsmann G, Muller J, Muller-Hermelink H, Fuchs D, Wachter H, Daniel MM, et al. Importance of vpr for infection of rhesus monkeys with simian immunodeficiency virus. J Virol. 1993;67:902–912. doi: 10.1128/jvi.67.2.902-912.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner EG, Duus KM, Gao F, Yu XF, Su L. Characterization of a thymustropic HIV-1 isolate from a rapid progressor: role of the envelope. Virology. 2004;328:74–88. doi: 10.1016/j.virol.2004.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Lv L, Nakagawa M, Yu Y, Yu C, D'Alessio AC, Nakayama K, Fan HY, Chen X, Xiong Y. CRL4(VprBP) E3 ligase promotes monoubiquitylation and chromatin binding of TET dioxygenases. Mol Cell. 2015;57:247–260. doi: 10.1016/j.molcel.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli G, Bressler P, Kinter A, Duh E, Timmer WC, Rabson A, Justement JS, Stanley S, Fauci AS. Interleukin 6 induces human immunodeficiency virus expression in infected monocytic cells alone and in synergy with tumor necrosis factor alpha by transcriptional and post-transcriptional mechanisms. J Exp Med. 1990;172:151–158. doi: 10.1084/jem.172.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani B, Baygloo NS, Hamidi-Fard M, Aghasadeghi MR, Allahbakhshi E. HIV-1 Vpr Protein Induces Proteasomal Degradation of Chromatin-associated Class I HDACs to Overcome Latent Infection of Macrophages. J Biol Chem. 2016;291:2696–2711. doi: 10.1074/jbc.M115.689018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani B, Cohen EA. Lentivirus Vpr and Vpx accessory proteins usurp the cullin4-DDB1 (DCAF1) E3 ubiquitin ligase. Curr Opin Virol. 2012;2:755–763. doi: 10.1016/j.coviro.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani B, Shaykh Baygloo N, Aghasadeghi MR, Allahbakhshi E. HIV-1 Vpr Protein Enhances Proteasomal Degradation of MCM10 DNA Replication Factor through the Cul4-DDB1[VprBP] E3 Ubiquitin Ligase to Induce G2/M Cell Cycle Arrest. J Biol Chem. 2015;290:17380–17389. doi: 10.1074/jbc.M115.641522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Misawa N, Iwami S, Satou Y, Matsuoka M, Ishizaka Y, Ito M, Aihara K, An DS, Koyanagi Y. HIV-1 Vpr accelerates viral replication during acute infection by exploitation of proliferating CD4+ T cells in vivo. PLoS Pathog. 2013;9:e1003812. doi: 10.1371/journal.ppat.1003812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwefel D, Groom HC, Boucherit VC, Christodoulou E, Walker PA, Stoye JP, Bishop KN, Taylor IA. Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature. 2014;505:234–238. doi: 10.1038/nature12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon V, Bloch N, Landau NR. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol. 2015;16:546–553. doi: 10.1038/ni.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strebel K. HIV accessory proteins versus host restriction factors. Curr Opin Virol. 2013;3:692–699. doi: 10.1016/j.coviro.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Singh S, Jung HY, Yang G, Jun S, Sastry KJ, Park JI. HIV-1 Vpr protein inhibits telomerase activity via the EDD-DDB1-VPRBP E3 ligase complex. J Biol Chem. 2013;288:15474–15480. doi: 10.1074/jbc.M112.416735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xiao M, Chen X, Chen L, Xu Y, Lv L, Wang P, Yang H, Ma S, Lin H, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell. 2015;57:662–673. doi: 10.1016/j.molcel.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang Y. Regulation of TET protein stability by calpains. Cell Rep. 2014;6:278–284. doi: 10.1016/j.celrep.2013.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zhou X, Barnes CO, DeLucia M, Cohen AE, Gronenborn AM, Ahn J, Calero G. The DDB1-DCAF1-Vpr-UNG2 crystal structure reveals how HIV-1 Vpr steers human UNG2 toward destruction. Nat Struct Mol Biol. 2016;23:933–940. doi: 10.1038/nsmb.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, Zhao D, Liu Y, Wang C, Zhang X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–393. doi: 10.1038/nature15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.