

Abstract
Cyclic peptides are a promising class of molecules for unique applications. Unfortunately, cyclic peptide design is severely limited by the difficulty in predicting the conformations they will adopt in solution. In this work, we use explicit-solvent molecular dynamics simulations to design well-structured cyclic peptides by studying their sequence–structure relationships. Critical to our approach is an enhanced sampling method that exploits the essential transitional motions of cyclic peptides to efficiently sample their conformational space. We simulated a range of cyclic pentapeptides from all-glycine to a library of cyclo-(X1X2AAA) peptides to map their conformational space and determine cooperative effects of neighboring residues. By combining the results from all cyclo-(X1X2AAA) peptides, we developed a scoring function to predict the structural preferences for X1-X2 residues within cyclic pentapeptides. Using this scoring function, we designed a cyclic pentapeptide, cyclo-(GNSRV), predicted to be well structured in aqueous solution. Subsequent CD and NMR spectroscopy revealed that this cyclic pentapeptide is indeed well structured in water, with NOE and J-coupling values consistent with the predicted structure.
Graphical Abstract
INTRODUCTION
Cyclic peptides (CPs) have promising applications in nanotechnology,1–3 as well as potential therapeutics, targeting a variety of protein–protein interactions (PPIs).4–12 PPIs play critical roles in important and disease-relevant biological processes.13,14 Modulating PPIs thus provides a means to control diverse cellular functions for both fundamental research and disease intervention.15,16 Despite the promise of CPs for these applications, de novo design of well-structured CPs in aqueous solution remains challenging. CPs tend to form multiple conformations in solution,17–28 making them difficult to design or even characterize. Furthermore, the limited availability of solution structural information makes it difficult to develop sequence–structure relationships for CPs. Currently, there is no tractable experimental method to synthesize and characterize the structural ensembles of hundreds of CPs to determine which CPs are well structured, and ultimately to understand how CP sequences control structures.29–32
While experimental determination of structural ensembles is difficult, there have been many promising contributions from computational simulations.33–51 Recently, we developed a novel, highly efficient enhanced sampling method custom-tailored to CPs by identifying their essential transitional motions (coupled two-dihedral angle movements).49 This enhanced sampling method has made it possible for us to quickly and efficiently sample a CP’s structural ensemble, with the inclusion of explicit water.49–51 This platform produces more rapidly converged results, allowing us to simulate more systems than was previously possible. This advance has made it much more feasible to gather the broad simulation data needed to study sequence–structure relationships for CPs. In this work, we used this method to systematically simulate over 70 head-to-tail cyclized pentapeptides, uncovered their sequence–structure relationships and ultimately designed a well-structured CP.
MATERIALS AND METHODS
Model peptides
The following model peptides were used in this study: cyclo-(GGGGG); cyclo-(X1AAAA), where X1 was any of the 20 natural amino acids; cyclo-(X1X2AAA), where X1/X2 was G, A, V, F, R, D, N or S; cyclo-(GFSEV); cyclo-(GNSRV) and cyclo-(GFNDV). Two different initial structures of each CP, S1 and S2 (structure 1 and structure 2, respectively), were prepared using the Chimera molecular modeling package.52 To construct each CP, the linear peptide was first built, followed by linkage of the N- and C- terminal residues and subsequent energy minimization.
Bias-exchange metadynamics simulations
Following preparation in Chimera, each initial structure was solvated using a pre-equilibrated box of water molecules. Minimal number of ions were added to neutralize the charge of the whole system. Each structure was then energy minimized using the steepest descent algorithm, followed by a four-step equilibration process. First, to allow the solvent molecules to equilibrate, a 50 ps isochoric–isothermal (NVT) simulation followed by a 50 ps isobaric–isothermal (NPT) simulation were performed. In both simulations, a harmonic constraint was placed on the peptide heavy atoms, with a force constant of 1,000 kJ/mol·nm2. Subsequently, a 100 ps NVT simulation followed by a 100 ps NPT simulation were performed without any restraints, to equilibrate the entire system. A thermostat of 300 K and a barostat of 1 bar were used for all equilibrations.
The NPT ensemble was used for all BE-META production simulations. The temperature was maintained at 300 K using the V-rescale thermostat,53 with a time coupling constant of 0.1 ps. The CP and the solvent were coupled to two separate thermostats, in order to mitigate the “hot solvent–cold solute” problem.54,55 The pressure was maintained at 1 bar using the Berendsen barostat56 was used to maintain the pressure at 1 bar, with a time coupling constant of 2.0 ps and an isothermal compressibility of 4.5×10−5 bar−1. All bonds involving hydrogen were constrained using the LINCS algorithm.57 Dynamics of the system were evolved using the leapfrog algorithm,58 with a time step of 2 fs. Both short-range Lennard-Jones and electrostatic nonbonded interactions were truncated at 1.0 nm. Beyond the cutoff distance, Particle Mesh Ewald (PME)59 was used for the electrostatic interactions, with a Fourier spacing of 0.12 nm and an interpolation order of 4. A long-range dispersion correction60 for the energy and pressure was used for the Lennard-Jones interaction beyond the cutoff. All trajectories were saved every 1 ps for subsequent analysis.
All simulations were performed using the RSFF2 force field61 with TIP3P water62 in Gromacs 4.6.763 with the PLUMED 2 plugin.64 The RSFF2 force field was found to accurately recapitulate several CP crystal structures48 and therefore will be used throughout this work. Conformational sampling of all CPs was enhanced using BE-META simulations. The collective variables (CVs) in the BE-META simulations consist of two types of 2D biases. The first 2D bias is along ϕi×ψi, where ϕ/ψ are the backbone dihedral angles of the same residue. The second type of 2D bias is along ψ of one residue and ϕ of the next residue, ψi×ϕi+1. These 2D biases were previously found to enhance the conformational sampling of CPs efficiently.49 Both types of the 2D biases are performed on each residue, giving a total of 10 biased replicas for a cyclic pentapeptide. Gaussian hills were deposited every 4 ps, with a height of 0.1 kJ/mol and a width of 0.314 rad. Exchanges were attempted every 5 ps between different replicas. For analysis of an unbiased structural ensemble, five neutral replicas were added, giving a total of 15 replicas per CP.
Principal component and cluster analysis
To characterize the structural ensemble of each CP, the last 50 ns of the unbiased replicas were analyzed using dihedral principal component analysis (dPCA) with the ϕ and ψ angles of all residues.65,66 However, if the peptide sequence is homogeneous (i.e. cyclo-(AAAAA)) an additional step needs to be performed prior to dPCA. To take degeneracy into account, the previously developed root-mean-square deviation (RMSD) scheme was applied.49 In brief, first the ϕ/ψ dihedral angles of the neutral replicas were calculated. For a cyclic pentapeptide with a homogeneous sequence there are five ways of reordering each frame. The RMSD to a reference structure was calculated for each of the five permutations, and the structure with the lowest RMSD to the reference structure was used to reorder the CP.
Following dPCA, the population of each cluster was calculated using a density peak-based cluster analysis.67 For cluster analysis, the principal subspace along the first three principal components (PC1, PC2 and PC3) was divided into 50×50×50 grids. Only grids with a population greater than 0.1 were used in cluster analysis. The population of each cluster was determined by summing the population of every grid the cluster contained. dPCA and cluster analysis was used to analyze the structural ensembles of cyclo-(X1AAAA), cyclo-(X1X2AAA), cyclo-(GFSEV), cyclo-(GNSRV) and cyclo-(GFDNV).
Simulation convergence was monitored using the normalized integrated product (NIP)38 of the density profiles along PC1, PC2 and PC3 of the S1 and S2 simulations. Convergence was achieved when the density profiles were similar. Final simulation lengths ranged from 100 ns to 300 ns. All further analysis was performed on the S1 simulations.
Cut-off turn analysis
Due to their highly constrained nature, cyclic pentapeptides typically form structures containing a β-turn plus a tight turn on the opposite side of the CP. The four commonly observed β-turns and tight turns give a total of 16 turn combinations (Figure 1A). However, for cyclo-(GGGGG), dPCA and cluster analysis did not identify all 16 turn combinations. To determine whether all turn combinations were present in cyclo-(GGGGG), turns were identified if the ϕ/ψ dihedral angles were within 35° of the ideal values for a specific type of turn (Figure 1A). Similarly, this cut-off based turn type analysis was used to compare all 16 types of turn combinations in the cyclo-(X1AAAA) and cyclo-(X1X2AAA) pentapeptides (Figures 3A and S1), as the dPCA–cluster analysis was unable to pick up combinations with very low populations.
Figure 1.

Cyclic pentapeptides typically form a β-turn plus a tight γ/γ′ or αR/αL turn opposite the β-turn. (A) Ideal dihedral angles, representative structures and hydrogen bond patterns for the four types of β-turns and four types of tight turns. (B) Populations and representative structures for the 16 turn types from our BE-META simulations of cyclo-(GGGGG). Type I, I′, II and II′ β-turns are shown in red, orange, green and blue, respectively. Tight turns γ, γ′, αR and αL are shown in cyan, magenta, purple and brown, respectively. Populations and standard deviation were calculated from the five neutral replicas of the S1 simulations.
Figure 3.
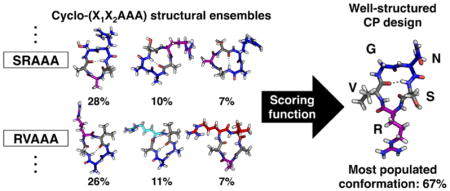
(A) Populations of 16 turn combinations from simulations of cyclo-(X1AAAA). Results use the cut-off turn analysis (see Materials and Methods for analysis details). (B) Populations and representative structures of the three most populated clusters of cyclo-(VAAAA), cyclo-(NAAAA) and cyclo-(SAAAA). Type II′ β-turn and αR turn are shown and their locations in the sequences highlighted in blue and purple, respectively. The location of distorted type I β-turn in the sequence is highlighted in red and underlined. Populations and standard deviation were calculated from the five neutral replicas of the S1 simulations.
Logo plot for individual amino acids from cyclo-(X1AAAA) simulations
To determine the most probable amino acid for each position of a given turn combination, we weighted X1 in cyclo-(X1AAAA) using the following scheme. The preference of the amino acid X1, when X1 ≠ A, for each of the five locations within a given turn combination was determined by its population from cut-off turn analysis. In the case of cyclo-(AAAAA), there are five degenerate amino acids, and the population was divided by five for the associated position in the logo plot. The logo plot for cyclo-(X1AAAA) for the βII′+αRturn combination is shown in Figure 4A.
Figure 4.

(A) Logo plot for cyclo-(X1AAAA) with the βII′+αR turn combination. Results use cut- off turn analysis of the S1 simulations (see Materials and Methods for analysis details). Type II′ β-turn and αR turn are shown and their locations in the sequences highlighted in blue and purple, respectively. (B) Population, representative structure and Ramachandran plot for the most populated cluster of cyclo-(GFSEV). Type II′ β-turn and αR turn are shown in blue and purple boxes, respectively. Population and standard deviation was calculated from the five neutral replicas of the S1 simulation.
Neighbor analysis for X1X2 from cyclo-(X1X2AAA) simulations
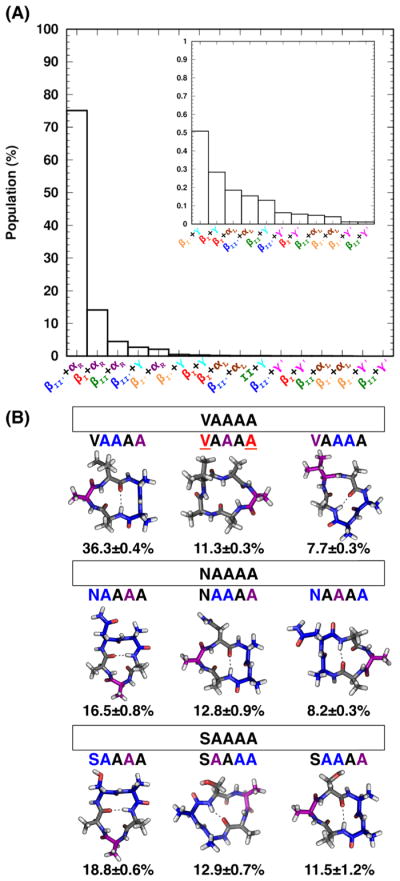
To help design well-structured cyclic peptides, a scoring function based on neighboring residues was developed using the simulation results of cyclo-(X1X2AAA), where X1/X2 is G, V, F, R, D, N and S. To evaluate the preference score for a sequence cyclo-(X1X2X3X4X5) to adopt a specific β-turn at X1X2 and a specific tight turn at X4, the sequence was broken down into five sets of nearest neighbor pairs – X1X2, X2X3, X3X4, X4X5 and X5X1, and the total preference score was the sum of the five populations for each pair to adopt the desired structure in the simulations of cyclo-(X1X2AAA), cyclo-(X2X3AAA), etc. For example, the score for sequence cyclo-(X1X2X3X4X5) adopting a type II′ β-turn at X1X2 and an αR turn at X4 was calculated as follows. We analyzed the structural ensemble of cyclo-(X1X2AAA) using the cut-off based turn analysis to evaluate the population of cyclo-(X1X2AAA) that adopts a type II′ β-turn at X1X2 and an αR turn at A4, the population of cyclo-(AX2X3AA) that adopts a type II′ β-turn at 1AX2 and an αR turn at A4, the population of cyclo-(AAX3X4A) that adopts a type II′ β-turn at 1AA2 and an αR turn at X4, etc. The score was then calculated as the sum of these five populations, when each pair is located at the desired location of the target turn combination (example for cyclo-(GNSRV) shown in Figure 5A).
Figure 5.
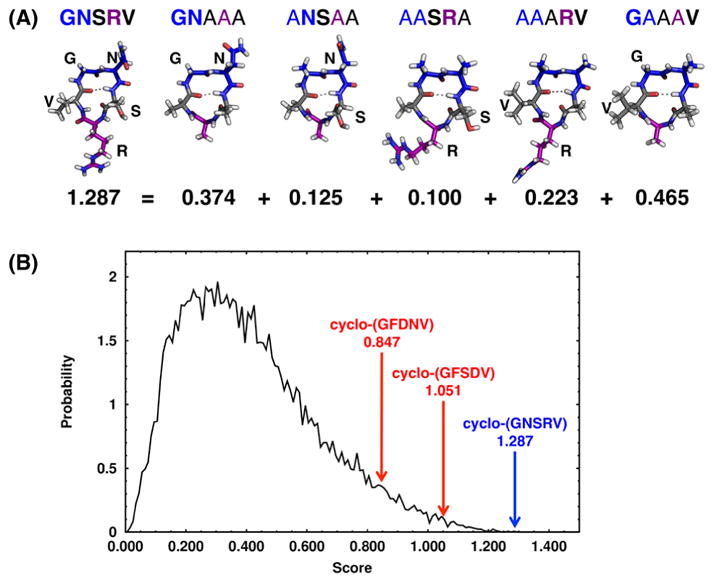
Cyclo-(GNSRV) was predicted to have high preference to adopt a βII′+αR turn combination based on neighbor pair scoring. (A) (Top) βII′+αR turn combination for cyclo-(GNSRV) and its five parent sequences: cyclo-(GNAAA), cyclo-(ANSAA), cyclo-(AASRA), cyclo-(AAARV) and cyclo-(GAAAV). Neighbor pairs are bolded, and type II′ β-turn and tight turn αR are shown in blue and purple, respectively. (Bottom) Neighbor scores, which were calculated from using cut-off analysis for the corresponding βII′+αR turn combination. (B) Distribution of scores for the βII′+αR turn combination for the 16,807 sequences of cyclo-(X1X2X3X4X5), where X1/X2/X3/X4/X5 were G, V, F, R, D, N or S. Well-structured CP cyclo-(GNSRV) and its score is highlighted in blue. Cyclo-(GFSDV), the proxy of cyclo-(GFSEV), from simulations of cyclo-(X1AAAA), and cyclo-(GFDNV) and their associated scores are highlighted in red.
Thermodynamics decomposition
To further understand the structural ensemble of a CP, we performed thermodynamics decomposition of the S1 simulation. ΔG between clusters was calculated using the Boltzmann equation, using the ratios of cluster populations and the most populated cluster as a reference. ΔG was then further separated into ΔH and ΔS, where ΔH was estimated from the difference in potential energy between clusters. ΔH was then further decomposed into peptide in vacuum ( ) and the rest (ΔHrest). To perform this decomposition, the potential energy of two groups was calculated: peptide and solvent/ions (note that ions may not be present in all systems). The peptide enthalpy ( ) consisted of peptide Lennard-Jones ( ), short-range and 1,4 electrostatics ( ), bonds ( ), angles ( ), proper dihedrals ( ) and improper dihedrals ( ). ΔS was further decomposed into configurational entropy of the peptide ( ) and solvation entropy (ΔSw). was calculated using the maximum information spanning tree (MIST)68–70 method and ΔSw was calculated using .
Linear peptide synthesis
The linear peptide was synthesized at 0.04 mmol scale on HMPB-ChemMatrix resin (PCAS BioMatrix Inc., loading = 0.5 mmol/g). The linear peptides was prepared with C-terminal glycine to facilitate head-to-tail cyclization. The HMPB resin was firstly functionalized with glycine: 5 mmol Fmoc-glycine-OH and 2.5 mmol N,N′-diisopropylcarbodiimide (DIC) were dissolved in 15 mL dimethylformamide (DMF). After 10 minutes at room temperature, the mixture was added to 0.75 g HMPB resin in a 20-mL Torviq fritted syringe. After another 1 minute, 0.05 mmol 4-dimethylaminopyridine (DMAP) was added and reacted at room temperature for 16 hours, followed by 3 times wash with DMF, twice 5 min deprotection with 20% (v/v) piperidine in DMF and 4 times wash with DMF. The resin was washed thoroughly with dichloromethane (DCM) and dried under vacuum. Following solid-phase peptide synthesis (SPPS) was carried out on a synthesizer for automated flow peptide synthesis (AFPS).71 After completion of the SPPS, the resin was washed thoroughly with DCM and dried under vacuum. The resins were transferred to a 50-mL plastic tube and the peptide was simultaneously cleaved from the resin and side-chain deprotected by treatment with 2.5% (v/v) water, 2.5% (v/v) 1,2-ethanedithiol (EDT), and 1% (v/v) triisoproprylsilane in neat trifluoroacetic acid (TFA) for 2 hours at room temperature. The resulting solution containing peptide was triturated and washed with cold diethyl ether (pre-chilled in –80° C freezer) two times. The obtained gummy-like solid was dissolved in 50% H2O: 50% acetonitrile containing 0.1% TFA and lyophilized.
Peptide cyclization and purification
The lyophilized crude peptide was directly used for cyclization. Cyclization condition: 0.5 mM peptide, 1.5 mM 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), 3 mM N,N-diisopropylethylamine (DIEA), DMF as solvent, room temperature for 1 hour. The cyclization reaction was quenched by adding TFA. After removing solvents by rotary evaporator, the reaction mixture was re-dissolved in water containing 10% dimethyl sulfoxide (DMSO) and purified on Agilent 1260 Infinity Automated LC/MS Purification System, with a semi- preparative Reverse Phase-HPLC column (Agilent Zorbax 300SB C3 column: 21.2 × 250 mm, 7 μm, linear gradient: 1–41% B over 80 min, flow rate: 4 mL/min). The purity of fractions was confirmed by LC-MS analysis. The fractions containing pure cyclized peptide were combined and lyophilized to yield cyclized peptide powder.
NMR characterization
The peptide was dissolved in H2O:D2O (90:10) at a concentration of roughly 3.5 mM. 1D and 2D 1H NMR spectra were recorded on a Bruker 600 MHz spectrometer with CryoProbe at 288 K. Complete resonance assignments were made using homonuclear 1H–1H TOCSY and ROESY experiments. Standard pulse programs available from the Bruker library were used, with mixing times of 60 ms for the TOCSY and 250 ms for the ROESY. 1H chemical shifts were referenced to DSS (δ 0.00 ppm) in water. 3JNH, CHα coupling constants were measured from 1D 1H NMR.
CD characterization
Compounds were dissolved in water to concentrations of 87.5 μM as measured by peptide weight. Equal concentrations of the peptides were verified prior to CD analysis by subjecting peptides to analytical HPLC and normalizing based on peak volumes. CD spectra were obtained on a Jasco J-815 CD Spectrometer at 20° C using the following measurement parameters: wavelength range 190–260 nm; step resolution, 0.5 nm; speed, 20 nm/min; accumulations, 3; response, 1 sec; bandwidth, 1 nm; path length, 0.1 cm.
RESULTS AND DISCUSSION
Structural ensembles of cyclo-(GGGGG) and cyclo-(AAAAA) map out the available conformational space for simple cyclic pentapeptides
Cyclic pentapeptides typically form a β-turn plus a tight turn opposite the β-turn.72–79 There are four types of β-turns (βI, βII, βI′, and βII′) and four types of tight turns (γ, γ′, αR and αL) that are commonly observed (Figure 1A), leading to 16 possible turn combinations for cyclic pentapeptides. A cyclic pentapeptide with a β-turn and a tight turn of γ or inverse-γ (γ′) tend to form two intra-peptide hydrogen bonds (Figure 1B, bottom row), while a cyclic pentapeptide having a β-turn and a tight turn with a dihedral angle of a right-handed or left-handed α-helix (αR and αL, respectively) will form only one intra-peptide hydrogen bond (at the β-turn site; Figure 1B, top row). All these 16 turn combinations were seen in our bias-exchange metadynamics (BE-META) simulations of cyclo-(GGGGG) and are shown in Figure 1B. The most populated conformation was a βII+αL, with a population of 14.7 ± 0.6%. We observed that the mirror-image turn combination, βII′+αR, has a similar population (14.3 ± 0.4%) as βII+αL. Since glycine is achiral, theoretically, conformations that are mirror images of each other should have exactly the same population. In our simulations of cyclo-(GGGGG), each conformation and its mirror image indeed had similar populations (Figure 1B), further supporting convergence of our simulation results.
Among the 16 turn combinations observed in cyclo-(GGGGG), all clusters with an αR/αL turn (top row of Figure 1B) were more populated than clusters with a γ/γ′ turn (bottom row of Figure 1B). To further understand this structural preference of cyclo-(GGGGG), we performed thermodynamics decomposition on the 16 clusters (Table S1). The thermodynamics of clusters 1 and 2 were almost identical, which is consistent with the fact that they are mirror images of each other (βII+αL vs. βII′+αR). The third and fourth most populated clusters, βI+αR and βI′+αL, had more favorable solvation enthalpy compared to the most populated clusters. However, they both had an unfavorable peptide enthalpy relative to the top clusters (most populated clusters; βII+αL and βII′+αR), arising from unfavorable electrostatics, angles and dihedrals. Clusters 5 and 6 formed the βII′+αL and βII+αR turn combinations, respectively, and were less stable than the most populated clusters due to peptide electrostatics and dihedrals. The least populated turn combinations involving αL and αR, βI′+αR and βI+αL (clusters 7 and 8) had unfavorable peptide angles and dihedrals compared to the most populated cluster.
In contrast to clusters 1–8, clusters 9–16 formed a γ/γ′ turn instead of adopting an αR/αL conformation. Since clusters with a γ/γ′ turn form two intra-peptide hydrogen bonds, their peptide electrostatics were more favorable than the most populated cluster (Table S1). However, clusters with a γ/γ′ turn all had unfavorable angles and dihedrals as well as unfavorable solvation enthalpy, relative to the top cluster. A combination of these factors makes clusters with γ/γ′ turns have lower populations than their αR/αLcounterparts.
Moving from achiral glycine to the simplest all L-amino acid peptide, cyclo-(AAAAA), we expected turn combinations involving an αR turn to be preferred over an αL turn, based on the ϕ angle preferences for an L-amino acid. The four most populated clusters of cyclo-(AAAAA) from our BE-META simulation are shown in Figure 2, and indeed all these turn combinations had a tight αR turn rather than a tight αL turn, corresponding to clusters 2, 3, 6, and 7 in Figure 1B for cyclo-(GGGGG). Similar to cyclo-(GGGGG), βII′+αR was the most favorable turn combination, with a population of 52.9 ± 0.7%. The second most populated cluster formed a distorted type βI+αR, where the distorted type I β-turn had nearly ideal dihedral angles for a type I β-turn, but deviation in the ψ angle of the i+2 residue results in a turn that does not frequently hydrogen bond.
Figure 2.
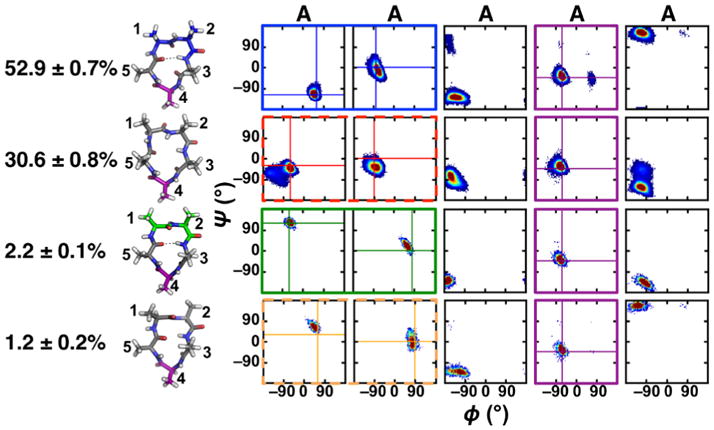
Populations, representative structures and Ramachandran plots for the top four clusters of cyclo-(AAAAA). Type II β, type II′ β and αRturns are shown and boxed in the Ramachandran plots in green, blue and purple, respectively. Distorted type I and I ′ β-turns are boxed in the Ramachandran plots by red and orange dashed lines, respectively. Populations and standard deviation were calculated from the five neutral replicas of the S1 simulation.
Breaking down the thermodynamics of cyclo-(AAAAA), the distorted βI+αR cluster had less favorable electrostatics and dihedrals compared to the most populated βII′+αR turn combination (Table S2). The third and fourth most populated clusters formed a βII+αR and a distorted type βI′+αR, and compared to cyclo-(GGGGG), the preferred order of all the αR-containing clusters remained the same. Both clusters 3 and 4 were less favorable than the most populated cluster due to entropy, specifically peptide configurational entropy. Looking at the peptide contribution to the thermodynamics, clusters 3 and 4 have poor electrostatics and cluster 4 (distorted βI′+αR) also had unfavorable angles compared to the most populated βII′+αR( Table S2).
Overall, for the thermodynamics of cyclo-(GGGGG), we found that the most populated conformation, and its mirror-image counterpart, are stabilized over the other clusters via either peptide or solvation enthalpy. The most populated conformation of cyclo-(AAAAA), on the other hand, is entropically more favorable than the other clusters due to either configurational or solvation entropy. The complex balance between peptide enthalpy, solvation enthalpy and entropy for these two simple cyclic pentapeptide systems reaffirms the necessity of using explicit water in CP simulations to accurately describe each thermodynamic factor.
Structural ensembles of cyclo-(XAAAA) show structural preferences of each amino acid
To understand how different amino acids affect structural preferences of a cyclic pentapeptide, we performed BE-META simulations of cyclo-(X1AAAA), where X1 was any of the 20 basic amino acids. The population and turn combination of the three most populated clusters for each of these 20 sequences are given in Table S3. We found that the turn combination βII′+αR was by far the most prevalent, accounting for ~75% of all the turn combinations that form for these 20 CPs (Figure 3A). The second most populated turn combination was a distorted βI+αR (~14%). Although βII′+αR predominated among the turn combinations observed for these 20 CPs, the locations of the type II′ β-turn and the αR residue varied within the X1AAAA sequence (Figure 3B and Table S3).
By combining the results from all 20 cyclo-(X1AAAA) CPs, we constructed a logo plot showing the probability of each amino acid for each location in the βII′+αR turn combination (Figure 4A). We hypothesized that combining the most probable amino acid at every position of the βII′+αR-structured cyclic pentapeptide would produce a sequence that adopts a well-structured conformation. Based on the results shown in Figure 4A, the sequence, cyclo-(GFSEV), was predicted to be the most structured for a βII′+αR turn combination, with the βII′ turn located at 1GF2 and αR turn located at E4. To verify this prediction, this designed sequence was simulated using BE-META simulations to characterize its structural ensemble. The most populated cluster (55%) of cyclo-(GFSEV) was indeed the βII′+αR turn combination, with the type II′ β-turn at 1GF2 and a tight αR turn at E4 (Figure 4B). However, this population (~55%) was not significantly higher than the most populated cluster of simple cyclo-(AAAAA) (~53%). This result suggests that the structural preferences for each of the 20 amino acids within cyclo-(X1AAAA) are not necessarily additive, implying that neighboring residues might affect each other’s structural preferences. This would explain why merely using results from cyclo-(X1AAAA) studies was unable to design a particularly well-structured cyclic pentapeptide.
Structural ensembles of cyclo-(X1X2AAA) reveal cooperative effects between neighboring amino acids
To understand the effects of neighboring amino acids on the structural ensemble of a cyclic pentapeptide, we performed BE-META simulations of cyclo-(X1X2AAA), where X1/X2 was G, A, V, F, R, D, N or S. These eight amino acids were chosen as a representative subsection of the 20 basic amino acids. The populations and turn combinations of the three most populated clusters for each of these 56 sequences is given in Table S4. Similar to cyclo- (X1AAAA), most of the top clusters formed a tight αR turn, which is consistent with the ϕ angle preferences for an L-amino acid. However, there were a few instances where a γ turn occurs in the top three clusters. Nonetheless, we observed that βII′+αR remains the most prevalent in all 56 cyclo-(X1X2AAA) CPs, similar to cyclo-(X1AAAA) (Figure S1 compared to Figure 3A). In fact, βII′+αR was the most populated cluster for 49 out of the 56 sequences. However, turn locations varied in the top clusters forming βII′+αR, depending on their sequences.
Rational design of well-structured CPs
To help design a well-structured cyclic pentapeptide, we used the structural ensemble results of cyclo-(X1X2AAA) CPs to develop a scoring function based on the observed populations for the desired conformation. The scoring function combined the structural preferences of each neighboring pair (X1X2, X2X3, etc.) in a cyclic pentapeptide cyclo-(X1X2X3X4X5) for a specific conformation (see “Neighbor analysis for X1X2 in cyclo-(X1X2AAA)” in Materials and Methods in the SI). Since alanine was a common filler amino acid in the cyclo-(X1X2AAA) data set, we first ignored A and only analyzed structural preferences and developed scoring functions for X1X2 pairs, where X1/X2 was G, V, F, R, D, N or S. The sequences and scores for the 20 highest-scoring cyclo-(X1X2X3X4X5) sequences for the βII′+αR turn combination, with a type II′ β-turn at X1X2 and an αR turn at X4, are given in Table S5. Of all 16,807 sequences, cyclo-(GNSRV) received the highest score (1.287) for the βII′+αR turn combination (Figure 5A and highlighted in blue in Figure 5B). To verify this prediction, BE-META simulations of cyclo-(GNSRV) were performed. The simulation results revealed a well-structured conformational ensemble, with the most populated cluster forming a type II′ β-turn at 1GN2 and a tight αR turn at R4, as predicted by the scoring function, with a population of 67% (Figure 6A).
Figure 6.
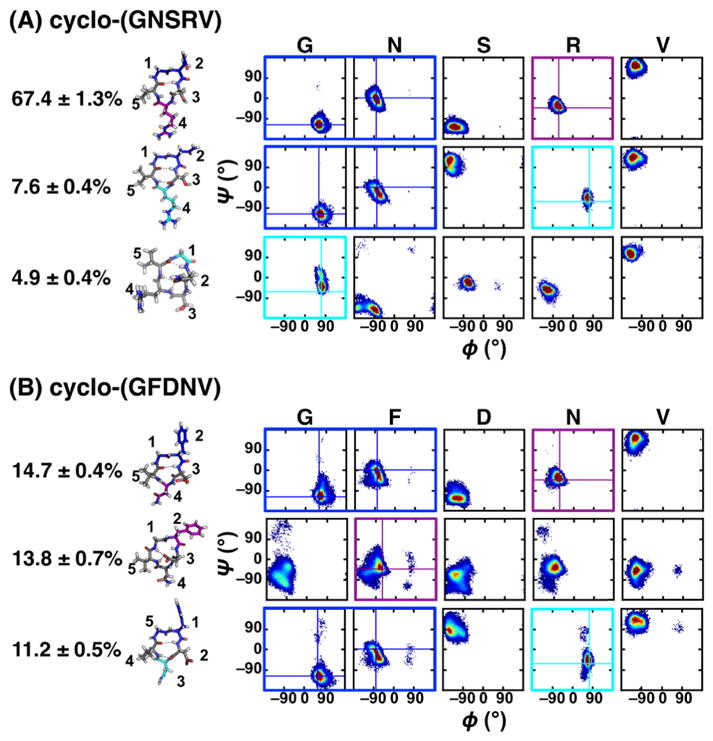
Populations, representative structures and Ramachandran plots for the top three clusters of (A) cyclo-(GNSRV) and (B) cyclo-(GFDNV). Type II′ β-turns are shown in blue boxes. Tight turns γ and αR are shown in cyan and purple boxes, respectively. Populations and standard deviation were calculated from the five neutral replicas of the S1 simulation.
To further understand why cyclo-(GNSRV) is well-structured, we performed thermodynamics decomposition on this peptide and on the five parent cyclo-(X1X2AAA) peptides that contain the five relevant neighbor pairs (Table S6). Comparing the location of the turns in cyclo-(GNSRV) to the locations of the turns in the five parent sequences, cyclo-(GNAAA), cyclo-(AAARV) and cyclo-(GAAAV) all had the βII′+αR turns at the same location as cyclo-(GNSRV) in the most populated cluster. However, in cyclo-(ANSAA) and cyclo-(AASRA), the βII′+αR turn combination corresponding to cyclo-(GNSRV) occurred as the third and second most populated clusters, respectively. The comparison of the thermodynamics of cyclo-(GNSRV) and the five parent sequences shows that a variety of factors stabilized the most populated clusters, and that these factors are generally consistent among the five neighbor-pair peptides and cyclo-(GNSRV). For example, the most populated cluster of cyclo-(GNSRV), cyclo-(AAARV) and cyclo-(GAAAV) formed a βII′+αR, while the second most populated cluster formed a βII′+γ with the γ turn is at the same residues as the αR turn. In all three simulations, the top clusters were stabilized over the second clusters due to enthalpy. Specifically, even though the βII′+γ clusters have more favorable intra-peptide electrostatics, they have more unfavorable dihedrals and much more unfavorable solvation enthalpy as compared to the most populated cluster. Interestingly, the most populated conformation of cyclo-(GNSRV) is both enthalpically and entropically stabilized over clusters 2 and 3. This is unlike any of the other five parent sequences, whose most populated cluster is stabilized by either enthalpy or entropy alone.
Previously, using the simulation results of cyclo-(X1AAAA), where X1 was any of the 20 basic amino acids, cyclo-(GFSEV) was predicted to have the highest preference for the βII′+αR turn combination. However, BE-META simulations of cyclo-(GFSEV) showed that the population of the desired structure (βII′+αR) was only 55%, a modest improvement from the 53% in cyclo- (AAAAA). Indeed, when using the scoring functions derived from the cyclo-(X1X2AAA) simulations results, the score for the βII′+αR conformation for cyclo-(GFSDV), the proxy of cyclo-(GFSEV) since in the cyclo-(X1X2AAA) data set X1/X2 could only be G, V, F, R, D, N or S, ranked poorly at number 277, with a score of 1.051 (highlighted in red in Figure 5B). This observation verified the importance of incorporating the neighboring effects in scoring the cyclic peptide’s structural preference.
The predicted II′+αR structure of cyclo-(GNSRV) is supported by NMR
To corroborate our simulation predictions, we synthesized cyclo-(GNSRV) and characterized its structure in aqueous solution. NMR spectroscopy provided high-resolution data describing the structural ensemble of cyclo-(GNSRV) in water. The 1D spectrum and 2D spectra from ROESY and TOCSY experiments provided ample evidence that cyclo-(GNSRV) was well structured in water (Figures S2–S5), and these data were wholly consistent with the predicted type II′ β-turn at 1GN2 and αR turn at R4 (Table S7–S8). Overall, protons had unique, well-resolved and well-dispersed chemical shifts, consistent with a single predominant structure. We observed several NOEs that support the predicted βII′+αR structure. For example, for types I and I′ β-turns, the distance between the HN protons of the i+1 and i+2 residues and the distance between the HN protons of the i+2 and i+3 residues are both small (< 3 Å); on the other hand, in types II and II′ β-turns, only the distance between the HN protons of the i+2 and i+3 residues is small (< 3 Å) (Figure 1A). In the ROESY spectra of cyclo-(GNSRV) we observed a strong NOE between Asn2(HN) and Ser3(HN), but no NOE was observed between Gly1(HN) and Asn2(HN) (Figure 7A; Figure S6; Table S7). The presence of this specific HN–HN NOE and the lack of the other provides strong support for a type II or II′ β-turn at residues 1GN2, rather than a type I or I′ β-turn. Similarly, the distance between the HN protons of the i+1 and i+2 residues is only small in a αR or αL turn, but not in a γ or γ′ turn (Figure 1A). We observed a strong NOE between Arg4(HN) and Val5(HN) (Figure S6; Table S7), which supports a αR turn centered at Arg4. J-values allowed estimation of the ϕ angle for Arg4 at –60 ± 30° (Figure S6; Table S8), which is consistent with an αR turn rather than an αL turn. Similarly, the ϕ angles for Ser3 and Val5 were estimated at –120 ± 30° based on the J-values, which are also in agreement with the predicted structure (Figure 6A; Figure S6 and Table S8).
Figure 7.
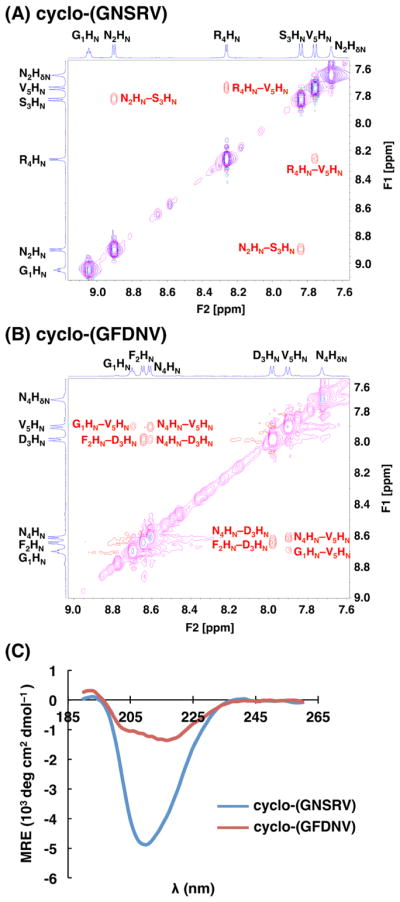
Structural characterization of cyclo-(GNSRV) and cyclo-(GFDNV). (A) HN region of ROESY spectrum (red, magenta) for cyclo-(GNSRV). TOCSY spectrum (blue, cyan) is overlaid to highlight separate spin systems. Two ROESY cross-peaks were observed in this region, between N2HN and S3HN and between R4HN and V5HN. (B) HN region of ROESY and TOCSY spectra for cyclo-(GFDNV). Four different ROESY NH-NH cross-peaks were observed. (C) Circular dichroism spectra for both cyclic peptides at 87.5 μM in aqueous solution at 20 °C, plotted as mean residue ellipticity (MRE).
Design of an unstructured control for cyclo-(GNSRV)
We observed that one of the parent sequences of the well-structured cyclo-(GNSRV), cyclo-(VGAAA), adopted a relatively structured βII′+αR configuration (58%; Table S4), suggesting that it might be possible that any cyclic pentapeptide containing the VG motif would have a rather strong preference for the βII′+αR configuration regardless of the rest of the sequence. To test this hypothesis, based on our scoring function, we designed a negative control, cyclo-(GFDNV) (highlighted in red in Figure 5B), which contained the VG motif but had a low score for the βII′+αR configuration. BE-META simulations showed that although the most populated cluster of cyclo-(GFDNV) still adopted a type II′ β-turn at residues 1GF2 and an αR turn at N4, its population was, however, a mere 14.7% (Figure 6B).
To corroborate the simulation results of our negative control, we synthesized and characterized cyclo-(GFDNV) using NMR spectroscopy (Figures S7–S10; Tables S9–S10). Overall, specific NOEs and J-values consistent with a single predominant structure, as observed for cyclo-(GNSRV), were not observed for cyclo-(GFDNV). For instance, instead of a distinct, selective pattern of HN–HN NOEs, we observed four out of the five possible HN–HN NOEs (Figure 7B; Table S9). This indicates that no one single structure predominates in solution, but that multiple different structures with various underlying turn combinations are present. Also, J-values allowed estimation of ϕ angles for Asp3 and Val5. Both of these values were estimated at –120 ± 30°, which was not consistent with an α-turn at either residue (Table S10). Finally, we observed an additional set of peaks within the NMR spectrum of cyclo-(GFDNV) that integrated to roughly 33% of the total peak volume (Figure S11). These are consistent with the presence of two or more conformations within the ensemble that are stable on the NMR timescale. This is another clear indicator that this peptide does not have a single predominant structure in aqueous solution.
CD spectroscopy is complementary to NMR and provides a low-resolution but informative measurement of the extent of peptide structure in solution. As shown in Figure 7C, the CD spectra of cyclo-(GNSRV) and cyclo-(GFDNV) show broad minima between 208 and 218 nm, consistent with mixed α-helical and β-sheet structures. The negative ellipticity from cyclo-(GNSRV) is roughly fourfold more intense than that of cyclo-(GFDNV). Though it is difficult to calculate the degree of structure accurately from CD data for cyclic peptides this small, these data do indicate a much higher degree of structure for cyclo-(GNSRV) compared to cyclo-(GFDNV).
CONCLUSIONS
In summary, using an efficient enhanced sampling method tailored for CPs, we characterized the structural ensembles of more than 70 head-to-tail cyclized pentapeptides. We demonstrated the value of these data by using them to rationally design a CP with a high degree of structure in water. We note that although this designed CP sequence is well structured despite a lack of proline residues. Inclusion of proline is a common strategy to stabilize turn structures but limits the design space for CP development.80
While the scoring function derived from cyclo-(X1X2AAA) enabled us to design well-structured CPs that adopt the βII′+αR turn combination, the distribution of the scores shows that most sequences are likely not well-structured (Figure 5B). Furthermore, the scores for the other turn combinations (such as βI+αR) are all minimal, suggesting that it would be extremely unlikely to favor these conformations using simple sequence substitutions. For example, cyclo-(AFDAG) has the highest score to adopt βI+αR, with the type I β-turn at 1AF2 and the tight αR turn at A4. However, this score is only 0.208. BE-META simulation of cyclo-(AFDAG) shows that indeed the top cluster of this CP was βI+αR, however, with a population of < 30%. Therefore, to design well-structured cyclic pentapeptides that adopt conformations other than the βII′+αR turn combination demonstrated here, non-natural amino acids are likely needed. D-amino acids are a logical choice, though incorporating other non-natural amino acids, such as N-methylated amino acids, β-amino acids and proline analogs may also prove to be useful tools for CP design. The effects of incorporating these non-natural amino acids are currently being investigated.
Having a simulation method which exploits the essential transitional motions of CPs has allowed us to efficiently simulate more than 70 head-to-tail cyclized pentapeptides and thereby better understand their sequence–structure relationships. We demonstrated the value of these data by using them to rationally design a CP with a high degree of structure in water. This method is not limited to cyclic pentapeptides or head-to-tail cyclized peptides, and can be readily extended to larger systems or macrocycles cyclized by other linking chemistries. This work is a clear demonstration of how rigorous, explicit-solvent simulation methods are opening new opportunities to rationally design CPs with desired structures, providing a platform to rationally design novel CPs.
Supplementary Material
Acknowledgments
We thank the support of the Tufts start-up fund, the Knez Family Faculty Investment Fund, and the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM124160 for Y.-S. L, Sontag Foundation Distinguished Scientist Award to B. L. P., and the National Science Foundation under Grant No. (1507456) for J. A. K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Dr. Kamlesh Makwana for assistance with NMR data acquisition and interpretation.
Footnotes
Notes
The authors declare no competing financial interests.
The Supporting Information is available free of charge on the ACS Publications website.
Populations of 16 turn combinations from simulations of cyclo-(X1X2AAA); 1H 1D, 1H–1H 2D TOCSY and 1H–1H 2D ROESY NMR spectra of cyclo-(GNSRV) and cyclo-(GFDNV); comparison of 1H 1D NMR spectra of cyclo-(GNSRV) and cyclo-(GFDNV) after 6 week incubation; thermodynamics decompositions for cyclo-(GGGGG) and cyclo-(AAAAA); populations and turn combinations for the top three most populated clusters of cyclo-(X1AAAA) and cyclo-(X1X2AAA); highest scoring sequences from neighbor analysis; thermodynamic decomposition for cyclo-(GNSRV) and five parent sequences; NOEs and J-coupling values for cyclo-(GNSRV) and cyclo-(GFDNV) (PDF)
References
- 1.Sánchez-Quesada J, Kim HS, Ghadiri MR. A Synthetic Pore-Mediated Transmembrane Transport of Glutamic Acid. Angew Chem Int Ed. 2001;40:2503–2506. doi: 10.1002/1521-3773(20010702)40:13<2503::AID-ANIE2503>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 2.Brea RJ, Castedo L, Granja JR, Herranz MA, Sanchez L, Martin N, Seitz W, Guldi DM. Electron Transfer in Me-Blocked Heterodimeric α,γ-Peptide Nanotubular Donor-Acceptor Hybrids. Proc Natl Acad Sci US A. 2007;104:5291–5294. doi: 10.1073/pnas.0609506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hourani R, Zhang C, van der Weegen R, Ruiz L, Li C, Keten S, Helms BA, Xu T. Processable Cyclic Peptide Nanotubes with Tunable Interiors. J Am Chem Soc. 2011;133:15296–15299. doi: 10.1021/ja2063082. [DOI] [PubMed] [Google Scholar]
- 4.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Crystal Structure of the Extracellular Segment of Integrin αvβ3 in Complex with an Arg-Gly-Asp Ligand. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 5.Horswill AR, Benkovic SJ. Cyclic Peptides, A Chemical Genetics Tool for Biologists. Cell Cycle. 2005;4:552–555. doi: 10.4161/cc.4.4.1585. [DOI] [PubMed] [Google Scholar]
- 6.Demmer O, Frank AO, Hagn F, Schottelius M, Marinelli L, Cosconati S, Brack-Werner R, Kremb S, Wester HJ, Kessler H. A Conformationally Frozen Peptoid Boosts CXCR4 Affinity and Anti-HIV Activity. Angew Chem Int Ed. 2012;51:8110–8113. doi: 10.1002/anie.201202090. [DOI] [PubMed] [Google Scholar]
- 7.Joo SH. Cyclic Peptides as Therapeutic Agents and Biochemical Tools. Biomol Ther. 2012;20:19–26. doi: 10.4062/biomolther.2012.20.1.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miranda E, Nordgren IK, Male AL, Lawrence CE, Hoakwie F, Cuda F, Court W, Fox KR, Townsend PA, Packham GK, et al. A Cyclic Peptide Inhibitor of HIF-1 Heterodimerization That Inhibits Hypoxia Signaling in Cancer Cells. J Am Chem Soc. 2013;135:10418–10425. doi: 10.1021/ja402993u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kling A, Lukat P, Almeida DV, Bauer A, Fontaine E, Sordello S, Zaburannyi N, Herrmann J, Wenzel SC, Konig C, et al. Targeting DnaN for Tuberculosis Therapy using Novel Griselimycins. Science. 2015;348:1106–1112. doi: 10.1126/science.aaa4690. [DOI] [PubMed] [Google Scholar]
- 10.Morse RP, Willett JLE, Johnson PM, Zheng J, Credali A, Iniguez A, Nowick JS, Hayes CS, Goulding CW. Diversification of β-Augmentation Interactions Between CDI Toxin/Immunity Proteins. J Mol Biol. 2015;427:3766–3784. doi: 10.1016/j.jmb.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tapeinou A, Matsoukas MT, Simal C, Tselios T. Cyclic Peptides on a Merry-Go-Round: Towards Drug Design. Biopolymers. 2015;104:453–461. doi: 10.1002/bip.22669. [DOI] [PubMed] [Google Scholar]
- 12.Cardote TAF, Ciulli A. Cyclic and Macrocyclic Peptides as Chemical Tools To Recognise Protein Surfaces and Probe Protein-Protein Interactions. ChemMedChem. 2016;11:787–794. doi: 10.1002/cmdc.201500450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan DP, Matthews JM. Protein–Protein Interactions in Human Disease. Curr Opin Struct Biol. 2005;15:441–446. doi: 10.1016/j.sbi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez MW, Kann MG. Chapter 4: Protein Interactions and Disease. PLoS Comput Biol. 2012;8:e1002819. doi: 10.1371/journal.pcbi.1002819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arkin MR, Tang YY, Wells JA. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milroy LG, Grossmann TN, Hennig S, Brunsveld L, Ottmann C. Modulators of Protein–Protein Interactions. Chem Rev. 2014;114:4695–4748. doi: 10.1021/cr400698c. [DOI] [PubMed] [Google Scholar]
- 17.Kopple KD, Go A, Logan RJJ, Savrda J. Conformations of Cyclic Peptides. VI. Factors Influencing Mono-, 1,4-Di-, and 1,2,4-Trisubstituted Cyclic Hexapeptide Backbones. J Am Chem Soc. 1972;94:973–981. doi: 10.1021/ja00758a042. [DOI] [PubMed] [Google Scholar]
- 18.Tonelli AE, Brewster AI. Conformational Characteristics in Solution of the Cyclic Hexapeptide Gly-Gly-D-Ala-D-Ala-Gly-Gly. J Am Chem Soc. 1972;94:2851–2854. doi: 10.1021/ja00763a052. [DOI] [PubMed] [Google Scholar]
- 19.Kopple KD, Go A, Schamper TJ. Conformation of Cyclic Peptides. 10. Conformational Averaging in Peptides with the Sequence Cyclo (Gly-D-Xxx-L-Yyy)2. J Am Chem Soc. 1978;100:4289–4295. [Google Scholar]
- 20.Blout ER. Cyclic Peptides: Past, Present, and Future. Biopolymers. 1981;20:1901–1912. [Google Scholar]
- 21.Varughese KI, Kartha G, Kopple KD. Crystal Structure and Conformation of Cyclo-(Glycyl-D-Leucyl-L-Leucyl)2. J Am Chem Soc. 1981;103:3310–3313. [Google Scholar]
- 22.Yang CH, Brown JN, Kopple KD. Crystal Structure and Solution Studies of the Molecular Conformation of the Cyclic Hexapeptide Cyclo-(Gly-L-His-Gly-L-Ala-L-Tyr-Gly) J Am Chem Soc. 1981;103:1715–1719. [Google Scholar]
- 23.Kopple KD, Wang YS, Cheng AG, Bhandary KK. Conformations of Cyclic Octapeptides. 5. Crystal Structure of Cyclo(Cys-Gly-Pro-Phe)2 and Rotating Frame Relaxation (T1ρ) NMR Studies of Internal Mobility in Cyclic Octapeptides. J Am Chem Soc. 1988;110:4168–4176. [Google Scholar]
- 24.Stradley SJ, Rizo J, Bruch MD, Stroup AN, Gierasch LM. Cyclic Pentapeptides as Models for Reverse Turns: Determination of the Equilibrium Distribution Between Type I and Type II Conformations of Pro-Asn and Pro-Ala β-turns. Biopolymers. 1990;29:263–287. doi: 10.1002/bip.360290130. [DOI] [PubMed] [Google Scholar]
- 25.Alberg DG, Schreiber SL. Structure-Based Design of a Cyclophilin-Calcineurin Bridging Ligand. Science. 1993;262:248–250. doi: 10.1126/science.8211144. [DOI] [PubMed] [Google Scholar]
- 26.Kopple KD, Bean JW, Bhandary KK, Briand J, D’Ambrosio CA, Peishoff CE. Conformational Mobility in Cyclic Oligopeptides. Biopolymers. 1993;33:1093–1099. doi: 10.1002/bip.360330711. [DOI] [PubMed] [Google Scholar]
- 27.Marshall GR, Beusen DD, Nikiforovich GV. In: Peptides: Synthesis, Structures, and Applications. Gutte B, editor. Vol. 27. Academic Press, Inc; San Diego, CA: 1995. pp. 193–245. [Google Scholar]
- 28.Beck JG, Chatterjee J, Laufer B, Kiran MU, Frank AO, Neubauer S, Ovadia O, Greenberg S, Gilon C, Hoffman A, et al. Intestinal Permeability of Cyclic Peptides: Common Key Backbone Motifs Identified. J Am Chem Soc. 2012;134:12125–12133. doi: 10.1021/ja303200d. [DOI] [PubMed] [Google Scholar]
- 29.Bhowmick A, Brookes DH, Yost SR, Dyson HJ, Forman–Kay JD, Gunter D, Head-Gordon M, Hura GL, Pande VS, Wemmer DE, et al. Finding Our Way in the Dark Proteome. J Am Chem Soc. 2016;138:9730–9742. doi: 10.1021/jacs.6b06543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brookes DH, Head-Gordon T. Experimental Inferential Structure Determination of Ensembles for Intrinsically Disordered Proteins. J Am Chem Soc. 2016;138:4530–4538. doi: 10.1021/jacs.6b00351. [DOI] [PubMed] [Google Scholar]
- 31.Best RB. Computational and Theoretical Advances in Studies of Intrinsically Disordered Proteins. Curr Opin Struct Biol. 2017;42:147–154. doi: 10.1016/j.sbi.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 32.Bonomi M, Heller GT, Camilloni C, Vendruscolo M. Principles of Protein Structural Ensemble Determination. Curr Opin Struct Biol. 2017;42:106–116. doi: 10.1016/j.sbi.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Riemann RN, Zacharias M. Reversible Scaling of Dihedral Angle Barriers During Molecular Dynamics to Improve Structure Prediction of Cyclic Peptides. J Pept Res. 2004;63:354–364. doi: 10.1111/j.1399-3011.2004.00110.x. [DOI] [PubMed] [Google Scholar]
- 34.Spitaleri A, Ghitti M, Mari S, Alberici L, Traversari C, Rizzardi GP, Musco G. Use of Metadynamics in the Design of isoDGR-Based αvβ3 Antagonists To Fine-Tune the Conformational Ensemble. Angew Chem Int Ed. 2011;50:1832–1836. doi: 10.1002/anie.201007091. [DOI] [PubMed] [Google Scholar]
- 35.Voelz VA, Dill KA, Chorny I. Peptoid Conformational Free Energy Landscapes From Implicit-Solvent Molecular Simulations in AMBER. Biopolymers. 2011;96:639–650. doi: 10.1002/bip.21575. [DOI] [PubMed] [Google Scholar]
- 36.Butterfoss GL, Yoo B, Jaworski JN, Chorny I, Dill KA, Zuckermann RN, Bonneau R, Kirshenbaum K, Voelz VA. De Novo Structure Prediction and Experimental Characterization of Folded Peptoid Oligomers. Proc Natl Acad Sci US A. 2012;109:14320–14325. doi: 10.1073/pnas.1209945109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y, Deng K, Qiu X, Wang C. Visualizing Cyclic Peptide Hydration at the Single-Molecule Level. Sci Rep. 2013;3:2461. doi: 10.1038/srep02461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Damas JM, Filipe LCS, Campos SRR, Lousa D, Victor BL, Baptista AM, Soares CM. Predicting the Thermodynamics and Kinetics of Helix Formation in a Cyclic Peptide Model. J Chem Theory Comput. 2013;9:5148–5157. doi: 10.1021/ct400529k. [DOI] [PubMed] [Google Scholar]
- 39.Oakley MT, Johnston RL. Exploring the Energy Landscapes of Cyclic Tetrapeptides with Discrete Path Sampling. J Chem Theory Comput. 2013;9:650–657. doi: 10.1021/ct3005084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oakley MT, Oheix E, Peacock AFA, Johnston RL. Computational and Experimental Investigations into the Conformations of Cyclic Tetra-α/β-Peptides. J Phys Chem B. 2013;117:8122–8134. doi: 10.1021/jp4043039. [DOI] [PubMed] [Google Scholar]
- 41.Merten C, Li F, Bravo-Rodriguez K, Sanchez-Garcia E, Xu Y, Sander W. Solvent-Induced Conformational Changes in Cyclic Peptides: A Vibrational Circular Dichroism Study. Phys Chem Chem Phys. 2014;16:5627–5633. doi: 10.1039/c3cp55018d. [DOI] [PubMed] [Google Scholar]
- 42.Quartararo JS, Eshelman MR, Peraro L, Yu H, Baleja JD, Lin YS, Kritzer JA. A Bicyclic Peptide Scaffold Promotes Phosphotyrosine Mimicry and Cellular Uptake. Bioorg Med Chem. 2014;22:6387–6391. doi: 10.1016/j.bmc.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Razavi AM, Wuest WM, Voelz VA. Computational Screening and Selection of Cyclic Peptide Hairpin Mimetics by Molecular Simulation and Kinetic Network Models. J Chem Inf Model. 2014;54:1425–1432. doi: 10.1021/ci500102y. [DOI] [PubMed] [Google Scholar]
- 44.Paissoni C, Ghitti M, Belvisi L, Spitaleri A, Musco G. Metadynamics Simulations Rationalise the Conformational Effects Induced by N-Methylation of RGD Cyclic Hexapeptides. Chem Eur J. 2015;21:14165–14170. doi: 10.1002/chem.201501196. [DOI] [PubMed] [Google Scholar]
- 45.Wakefield AE, Wuest WM, Voelz VA. Molecular Simulation of Conformational Pre-Organization in Cyclic RGD Peptides. J Chem Inf Model. 2015;55:806–813. doi: 10.1021/ci500768u. [DOI] [PubMed] [Google Scholar]
- 46.Yedvabny E, Nerenberg PS, So C, Head-Gordon T. Disordered Structural Ensembles of Vasopressin and Oxytocin and Their Mutants. J Phys Chem B. 2015;119:896–905. doi: 10.1021/jp505902m. [DOI] [PubMed] [Google Scholar]
- 47.Yu H, Lin YS. Toward Structure Prediction of Cyclic Peptides. Phys Chem Chem Phys. 2015;17:4210–4219. doi: 10.1039/c4cp04580g. [DOI] [PubMed] [Google Scholar]
- 48.Geng H, Jiang F, Wu YD. Accurate Structure Prediction and Conformational Analysis of Cyclic Peptides with Residue-Specific Force Fields. J Phys Chem Lett. 2016;7:1805–1810. doi: 10.1021/acs.jpclett.6b00452. [DOI] [PubMed] [Google Scholar]
- 49.McHugh SM, Rogers JR, Yu H, Lin YS. Insights Into How Cyclic Peptides Switch Conformations. J Chem Theory Comput. 2016;12:2480–2488. doi: 10.1021/acs.jctc.6b00193. [DOI] [PubMed] [Google Scholar]
- 50.McHugh SM, Yu H, Slough DP, Lin YS. Mapping the Sequence–Structure Relationships of Simple Cyclic Hexapeptides. Phys Chem Chem Phys. 2017;19:3315–3324. doi: 10.1039/c6cp06192c. [DOI] [PubMed] [Google Scholar]
- 51.Slough DP, Yu H, McHugh SM, Lin YS. Towards Accurately Modeling N-Methylated Cyclic Peptides. Phys Chem Chem Phys. 2017;19:5377–5388. doi: 10.1039/c6cp07700e. [DOI] [PubMed] [Google Scholar]
- 52.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–A Visualization System for Exploratory Research and Analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 53.Bussi G, Donadio D, Parrinello M. Canonical Sampling Through Velocity Rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 54.Cheng A, Merz KM., Jr Application of the Nosé Hoover Chain Algorithm to the Study of Protein Dynamics. J Phys Chem. 1996;100:1927–1937. [Google Scholar]
- 55.Lingenheil M, Denschlag R, Reichold R, Tavan P. The “Hot-Solvent/Cold-Solute” Problem Revisited. J Chem Theory Comput. 2008;4:1293–1306. doi: 10.1021/ct8000365. [DOI] [PubMed] [Google Scholar]
- 56.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular Dynamics with Coupling to an External Bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 57.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A Linear Constraint Solver for Molecular Simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- 58.Hockney RW. The Potential Calculation and Some Applications. Methods Comput Phys. 1970;9:135–211. [Google Scholar]
- 59.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 60.Allen MP, Tildesley DJ, editors. Computer Simulations of Liquids. Oxford University Press; New York: 1987. [Google Scholar]
- 61.Zhou CY, Jiang F, Wu YD. Residue-Specific Force Field Based on Protein Coil Library. RSFF2: Modification of AMBER ff99SB. J Phys Chem B. 2015;119:1035–1047. doi: 10.1021/jp5064676. [DOI] [PubMed] [Google Scholar]
- 62.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 63.Hess B, Kutzner C, Van Der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 64.Tribello GA, Bonomi M, Branduardi D, Camilloni C, Bussi G. PLUMED 2: New Feathers for an Old Bird. Comput Phys Commun. 2014;185:604–613. [Google Scholar]
- 65.Mu Y, Nguyen PH, Stock G. Energy Landscape of a Small Peptide Revealed by Dihedral Angle Principal Component Analysis. Proteins. 2005;58:45–52. doi: 10.1002/prot.20310. [DOI] [PubMed] [Google Scholar]
- 66.Sittel F, Jain A, Stock G. Principal Component Analysis of Molecular Dynamics: On the Use of Cartesian vs. Internal Coordinates. J Chem Phys. 2014;141:014111. doi: 10.1063/1.4885338. [DOI] [PubMed] [Google Scholar]
- 67.Rodriguez A, Laio A. Clustering by Fast Search and Find of Density Peaks. Science. 2014;344:1492–1496. doi: 10.1126/science.1242072. [DOI] [PubMed] [Google Scholar]
- 68.King BM, Tidor B. MIST: Maximum Information Spanning Trees for Dimension Reduction of Biological Data Sets. Bioinformatics. 2009;25:1165–1172. doi: 10.1093/bioinformatics/btp109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.King BM, Silver NW, Tidor B. Efficient Calculation of Molecular Configurational Entropies Using an Information Theoretic Approximation. J Phys Chem B. 2012;116:2891–2904. doi: 10.1021/jp2068123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fleck M, Polyansky AA, Zagrovic B. PARENT: A Parallel Software Suite for the Calculation of Configurational Entropy in Biomolecular Systems. J Chem Theory Comput. 2016;12:2055–2065. doi: 10.1021/acs.jctc.5b01217. [DOI] [PubMed] [Google Scholar]
- 71.Mijalis AJ, Thomas DA, 3rd, Simon MD, Adamo A, Beaumont R, Jensen KF, Pentelute BL. A Fully Automated Flow-Based Approach for Accelerated Peptide Synthesis. Nat Chem Biol. 2017;13:464–466. doi: 10.1038/nchembio.2318. [DOI] [PubMed] [Google Scholar]
- 72.Karle IL. Crystal Structure and Conformation of Cyclo-(GlycylProlylGlycyl-D-AlanylProlyl) Containing 4→1 and 3→1 Intramolecular Hydrogen Bonds. J Am Chem Soc. 1978;100:1286–1289. [Google Scholar]
- 73.Gurrath M, Mullen G, Kessler H, Aumailley M, Timpl R. Conformation/Activity Studies of Rationally Designed Potent Anti-Adhesive RGD Peptides. Eur J Biochem. 1992;210:911–921. doi: 10.1111/j.1432-1033.1992.tb17495.x. [DOI] [PubMed] [Google Scholar]
- 74.Mierke DF, Kurz M, Kessler H. Peptide Flexibility and Calculations of an Ensemble of Molecules. J Am Chem Soc. 1994;116:1042–1049. [Google Scholar]
- 75.Nagarajaram HA, Ramakrishnan C. Stereochemical Studies on Cyclic Peptides: Detailed Energy Minimization Studies on Hydrogen Bonded All-Trans Cyclic Pentapeptide Backbones. J Bioscience. 1995;20:591–611. [Google Scholar]
- 76.Davies JS. In: Cyclic Polymers. Semlyen ER, editor. Kluwer Academic Publishers; Netherlands: 2000. pp. 85–124. [Google Scholar]
- 77.Nikiforovich GV, Kover KE, Zhang WJ, Marshall GR. Cyclopentapeptides as Flexible Conformational Templates. J Am Chem Soc. 2000;122:3262–3273. [Google Scholar]
- 78.Zhang X, Nikiforovich GV, Marshall GR. Conformational Templates for Rational Drug Design: Flexibility of Cyclo(D-Pro1-Ala2-Ala3-Ala4-Ala5) in DMSO Solution. J Med Chem. 2007;50:2921–2925. doi: 10.1021/jm070084n. [DOI] [PubMed] [Google Scholar]
- 79.Demmer O, Frank AO, Kessler H. In: Peptide and Protein Design for Biopharmaceutical Applications. Jensen KJ, editor. John Wiley & Sons, Ltd; Chichester, UK: 2009. pp. 133–176. [Google Scholar]
- 80.Hosseinzadeh P, Bhardwaj G, Mulligan VK, Shortridge MD, Craven TW, Pardo-Avila F, Rettie SA, Kim DE, Silva DA, Ibrahim YM, et al. Comprehensive Computational Design of Ordered Peptide Macrocycles. Science. 2017;358:1461–1466. doi: 10.1126/science.aap7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.