

Abstract
Genome duplication is essential for cell proliferation, and the mechanisms regulating its execution are highly conserved. These processes give rise to a spatiotemporal organization of replication initiation across the genome, referred to as the replication program. Despite the identification of such programs in diverse eukaryotic organisms, their biological importance for cellular physiology remains largely unexplored. We address this fundamental question in the context of genome maintenance, taking advantage of the inappropriate origin firing that occurs when fission yeast cells lacking the Rad3/ATR checkpoint kinase are subjected to replication stress. Using this model, we demonstrate that the replication program quantitatively dictates the extent of origin de-regulation and the clustered localization of these events. Furthermore, our results uncover an accumulation of abnormal levels of single-stranded DNA (ssDNA) and the Rad52 repair protein at de-regulated origins. We show that these loci constitute a defining source of the overall ssDNA and Rad52 hotspots in the genome, generating a signature pattern of instability along the chromosomes. We then induce a genome-wide reprogramming of origin usage and evaluate its consequences in our experimental system. This leads to a complete redistribution of the sites of both inappropriate initiation and associated Rad52 recruitment. We therefore conclude that the organization of genome duplication governs the checkpoint control of origin-associated hotspots of instability and plays an integral role in shaping the landscape of genome maintenance.
Genome duplication is highly regulated to promote the faithful transmission of the genetic material during cell proliferation. In eukaryotes, DNA synthesis requires the coordinated assembly of conserved factors at the sites of replication initiation, or origins, distributed throughout the genome. Origins are licensed through the formation of the pre-replicative complex (pre-RC), which is followed by the assembly of the pre-initiation complex (pre-IC) that activates them for replication (Fragkos et al. 2015). This multistep process ensures that the genetic material is entirely copied once and only once during each cell cycle. Different initiation sites in a genome possess reproducibly distinct characteristics of usage: At the population level, each origin is activated with a particular frequency, or efficiency, and individual origins fire at given times during S phase (Heichinger et al. 2006). Moreover, a large body of work has characterized the presence of spatial and temporal patterns of replication, in which distinct genomic regions duplicate with particular timing during S phase, in diverse organisms (Hiratani et al. 2008; McCune et al. 2008; Muller and Nieduszynski 2012; Rhind and Gilbert 2013; Rivera-Mulia and Gilbert 2016). Collectively, the efficiencies, timing of firing, and positions of all of the origins in a genome define the program, or organization, of DNA replication.
The replication program has been shown to be highly flexible, and signature profiles have been established for diverse cell types and physiological states. For example, conserved replication timing domains have been identified in different human embryonic stem cell (ESC) lines, suggesting that they are a shared feature of pluripotency (Ryba et al. 2010). The differentiation of both mouse and human ESCs is then accompanied by alterations in the timing of duplication of large portions of the genome (Hiratani et al. 2008, 2010). The profile of origin usage is also sensitive to external stimuli, as seen in fission yeast cells that show changes in replication initiation after undergoing temporary nitrogen depletion (Wu and Nurse 2014). However, despite these observations, the biological importance of the replication program remains unclear. In particular, although the genome is particularly fragile when it is being copied (Aguilera and García-Muse 2013; Zeman and Cimprich 2014), little is known about whether and how the organization of DNA replication along the chromosomes may contribute to genome integrity. Potential evidence for such an interplay derives from genetic analyses that have correlated mutation frequency with replication timing on Chromosome VI in budding yeast (Lang and Murray 2011). In addition, whole-genome sequencing of normal and cancerous human cells has shown that late-replicating chromosomal regions often harbor point mutations and copy number losses, while copy number increases and other rearrangements occur more frequently in early-replicating domains (Koren et al. 2012; Liu et al. 2013). Finally, similar approaches have suggested a relationship between replication timing and differences in mutation rates across the genome in human cells (Stamatoyannopoulos et al. 2009; Chen et al. 2010). Altogether, these findings indicate that the replication program may be intimately coupled to genome maintenance.
The processes that preserve genome integrity are particularly essential when cells are under replication stress, as challenges to DNA synthesis can lead to the generation of lesions and errors. The S phase/ATR checkpoint response to such stress conditions comprises distinct functions, including transcriptional regulation, cell cycle arrest, stabilization of stalled replication forks, and inhibition of replication initiation at a subset of origins (Santocanale and Diffley 1998; Shirahige et al. 1998; Labib and De Piccoli 2011). While the importance of many of these mechanisms is well-described, this is not the case for the regulation of origin firing, despite a large body of work that has characterized the molecular players involved and initiation sites that are sensitive to checkpoint control (Feng et al. 2006; Heichinger et al. 2006; Hayashi et al. 2007; Mickle et al. 2007; Lopez-Mosqueda et al. 2010; Zegerman and Diffley 2010; Karnani and Dutta 2011; Poli et al. 2012). Indeed, it remains unknown how the origins that are targeted for inhibition in this context are selected, the extent to which inappropriate initiation occurs when the checkpoint is defective, and the consequences of de-regulated replication initiation. In fact, the impact of this mechanism is still contested: For instance, using SLD3 and DBF4 budding yeast mutants that allow the firing of checkpoint-inhibited origins in replication stress conditions, Zegerman et al. found no effect on viability following exposure to the alkylating agent methyl methanesulfonate (MMS) (Zegerman and Diffley 2010); however, Lopez-Mosqueda and colleagues showed, via the same approach, that there is a 50% loss of viability upon nucleotide depletion by hydroxyurea (HU) treatment (Lopez-Mosqueda et al. 2010). These observations thus gave rise to contrasting views on the importance of the control of origin firing for the cellular response to replication stress. Nevertheless, the conservation of this regulation from yeast to humans strongly suggests that it is an integral feature of the processes that promote genome stability (Dimitrova and Gilbert 2000; Syljuåsen et al. 2005; Ge and Blow 2010; Karnani and Dutta 2011).
In this study, we have investigated the fundamental interplay between the program of DNA replication and genome maintenance, exploiting the checkpoint regulation of origin firing in fission yeast cells exposed to replication stress as a model. Our findings lead us to propose that the organization of genome duplication governs the checkpoint control of origin-associated hotspots of genome instability and shapes the landscape of genome integrity.
Results
Regional de-regulation of origin firing in checkpoint-defective cells exposed to replication stress
As a model for our studies, we used the checkpoint control of origin firing in replication stress conditions, which provided us with a unique entry into the potential links between the organization of DNA replication and genome maintenance. To establish our system, we first addressed previous contrasting findings regarding this regulation in the fission yeast (Feng et al. 2006; Heichinger et al. 2006; Hayashi et al. 2007; Mickle et al. 2007). Indeed, various methods have identified significantly different subsets of checkpoint-regulated initiation sites, with 2%–28% of the origins in the genome proposed to be sensitive to inhibition. One common feature of these studies is the use of high concentrations of the ribonucleotide reductase inhibitor HU (10–15 mM) to deplete nucleotide pools, and we hypothesized that this may have hindered the accurate detection of checkpoint-inhibited sites and contributed to the differences between these earlier reports. Furthermore, a quantitative assessment of the alterations in origin firing that are induced by replication stress in the absence of the checkpoint was lacking. We therefore began by characterizing the programs of origin usage in wild-type and checkpoint-defective rad3/ATRΔ fission yeast cells that replicate their DNA in the presence of moderate levels of HU (6 mM). Cells were synchronized in G2 using the cdc25-22 temperature-sensitive mutation and allowed to undergo S phase in HU (Supplemental Fig. S1A; see Methods). Genomic DNA samples were collected 5 min after the release from G2 (unreplicated DNA) and after a time when cells would normally have completed S phase in the absence of HU (replicated DNA) (Supplemental Fig. S1B). Replication origins were then mapped using microarrays as previously described (Wu and Nurse 2014), and peaks of increased copy number in the replicating DNA samples were identified as initiation sites. Copy number was then converted to efficiency, which represents the frequency of firing of a given origin in a population of cells (see Methods; details and validation of our copy number analyses are provided in the Supplemental Materials). In the context of our experiments, the major contribution to “efficiency” is replication initiation; however, our measurements do not exclude a minor contribution from passive replication. Nevertheless, the comparable widths of origin peaks between wild-type and rad3Δ cells indicate that origins are similarly affected by this potential input in the two genetic backgrounds. For simplicity, we will continue to refer to “% efficiency” to describe origin behavior.
Our data demonstrate substantial differences in replication initiation between wild-type and rad3Δ cells in replication stress conditions (we will refer to the cdc25-22 background without any other mutations as wild type) (Fig. 1A; Supplemental Fig. S1C). While the usage of a majority of origins was unaltered, a number of sites were markedly increased in their efficiencies in checkpoint-defective cells. Applying a cutoff of 8% absolute difference in efficiency between wild type and rad3Δ (see Supplemental Methods for threshold selection), we identified 176 de-regulated sites out of 876 total origins, with increases of up to 25% efficiency (Fig. 1B; Supplemental Tables S1, S2). These inappropriate initiation sites are clearly distinguishable peaks and do not arise as a result of passive replication from neighboring origins (Supplemental Fig. S1C). Given that the average origin efficiency across the genome in wild-type cells is 29.3% and reaches a maximum of 66%, these alterations represent major changes in origin usage. We will refer to these de-regulated origins as Checkpoint-Inhibited Origins, or CIOs.
Figure 1.

Regional de-regulation of origin firing in checkpoint-defective cells exposed to replication stress. (A) Origin efficiencies in a representative region of the genome for cdc25-22 (black, WT) and cdc25-22 rad3Δ (red, rad3Δ) cells in 6 mM HU (see Supplemental Fig. S1A,B for experimental design and flow cytometry analyses). Full profiles are shown in Supplemental Figure S1C. Asterisks mark CIOs. x-axis: chromosome coordinates; y-axis: origin efficiency. (B) Analysis of the absolute efficiency differences between rad3Δ and wild-type (WT) cells as a function of origin usage in WT (data from A and Supplementary Fig. S1C). x-axis: origin efficiency in WT; y-axis: differences in origin efficiency (rad3Δ − WT). Open circles: unaffected origins; red circles: CIOs (see Methods for CIO selection). Lines mark 8% differences in absolute origin efficiency. (C) Clustering of de-regulated origins in late-firing and inefficient chromosomal domains. Top: Red bars indicate positions of CIOs. Middle: Comparison of origin de-regulation in rad3Δ with the program of replication in WT. The percentages of CIOs in 1000-probe (∼250-kb) windows were determined across the genome (blue line, scale on left y-axis). The averages of origin efficiencies in WT were assessed for the same continuous windows (black line, scale on right y-axis). Bottom: The profiles of replication timing and efficiency in a wild-type background show identical regional characteristics. The average timings of origin firing were assessed for the same continuous windows as above (purple, left y-axis) (timing data from Heichinger et al. 2006). The profile of efficiency domains is as in the middle panel (black, right y-axis). x-axis: chromosome coordinates. Note that regions containing CIO clusters are also enriched in sites of inappropriate origin firing that were identified in previous studies (Supplemental Fig. S1E). (D) Time course of Cdc45 binding in cdc25-22 (WT, black) and cdc25-22 rad3Δ (rad3Δ, red) cells at an unaffected origin (ori2004, closed circles) and a CIO (ori2060, open circles). Cells were released from G2 arrest and sampled at the indicated time points. In replication stress conditions, ori2004 is used with an efficiency of ∼50% in wild type and rad3Δ, while the efficiency of ori2060 increases from 14% in wild type to 26% in rad3Δ (Supplemental Table S1). Top: −HU; bottom: 6 mM HU. x-axis: time after release; y-axis: Cdc45 occupancy (%IP).
Next, we evaluated the characteristics and distribution of CIOs. CIOs span a broad spectrum of origin activities (Fig. 1B), contrasting with previous reports that only late-firing and inefficient origins in the genome are subject to checkpoint inhibition (Santocanale and Diffley 1998; Shirahige et al. 1998). Indeed, many inefficient sites showed no changes in usage, while origins with efficiencies up to 40% in wild-type cells were found to be de-regulated (Fig. 1B). In fact, 30% of CIOs are among the first half of the origins that fire during S phase (Supplemental Fig. S1D), consistent with the known relationship between the timing and efficiency of origin firing (Heichinger et al. 2006; Wu and Nurse 2009). Notably, we observed that CIOs are clustered in distinct chromosomal regions (Fig. 1C, top panel), and we assessed what might underlie this distribution. To this end, we first ascertained the average efficiencies and timings of origin firing in a wild-type background in continuous 1000-probe (∼250-kb) windows (Fig. 1C, bottom panel), which showed that these two parameters are organized in virtually identical domains along the chromosomes. We then determined the percentages of origins that are de-regulated in the same windows (Fig. 1C, middle panel). Our findings uncover a strong inverse relationship between the program of origin usage and inappropriate initiation at a regional level. Thus, our results demonstrate that (1) regardless of their individual efficiencies, CIOs are enriched in late-firing and inefficient regions of the genome, (2) a major fraction of the origins in these regions (up to ∼70%) are de-regulated in HU-treated rad3Δ cells, and (3) there are only a few CIOs outside of these regions, even in areas that contain some inefficient and late-firing origins. This distribution of CIOs is not biased by our specific experimental conditions, as we obtained similar results using a completely different method of synchronization (Supplemental Fig. S1A,F,G). Our results therefore provide a quantitative characterization of de-regulated initiation and reveal a distinctive profile of checkpoint-inhibition of origin firing along the chromosomes that is linked to the replication program.
Finally, given the close link between the timing and efficiency of origin usage mentioned above (Heichinger et al. 2006; Wu and Nurse 2009), we asked if the increased initiation at CIOs is accompanied by earlier firing in S phase. For this analysis, we performed time courses of chromatin immunoprecipitation (ChIP) of the pre-IC component Cdc45 in cells undergoing a synchronous S phase in the absence or presence of HU after release from G2 arrest. We compared an early-firing and efficient origin whose activity is unaffected in HU-treated rad3Δ cells (ori2004; oriBGE2105 in our study) with a late-replicating and inefficient CIO (ori2060; oriBGE2122 in our study) (see Fig. 1D legend for efficiencies). In the absence of HU, rad3Δ did not affect the timing of firing of these origins (Fig. 1D). Moreover, while we observed a higher level of Cdc45 recruitment at ori2060 exclusively in HU-treated rad3Δ cells, the timing of Cdc45 binding at ori2060 was comparable between wild-type and rad3Δ cells. This indicates that the increased firing of CIOs is not accompanied by an early activation, consistent with previous work in budding yeast suggesting that the checkpoint does not regulate the temporal pattern of replication (Alvino et al. 2007).
Collectively, our findings demonstrate that, in replication stress conditions, the Rad3/ATR checkpoint inhibits origin firing based on the timing and efficiency of distinct genomic regions rather than the activity of individual origins. The unique distribution of de-regulated replication initiation in checkpoint-defective cells subjected to nucleotide depletion, together with our extensive and quantitative assessment of these events, makes this an ideal system for investigating whether the organization of genome duplication impacts genome maintenance.
De-regulation of origin firing locally induces the aberrant accumulation of single-stranded DNA
During DNA synthesis, single-stranded DNA (ssDNA) is transiently generated upon origin unwinding. In replication stress conditions, higher levels of ssDNA are produced, in particular, when the checkpoint is compromised (Shechter et al. 2004; Feng et al. 2006). This represents a precursor to DNA damage (Feng et al. 2011), and we hypothesized that de-regulated replication initiation may strongly contribute to generating such vulnerable sites. We therefore assessed the accumulation of ssDNA in wild-type and rad3Δ cells exposed to replication stress, taking advantage of the ssDNA binding protein Ssb1, the largest subunit of heterotrimeric RPA, as a marker (Wold 1997). For these experiments, cells were synchronized in G2 and released to undergo S phase in the presence or absence of 6 mM HU. RPA binding was monitored by ChIP, comparing the unaffected efficient origin ori2004 with ori2060, an inefficient CIO. At ori2004, we observed similar levels of RPA binding in both genetic backgrounds, either with or without HU exposure (Supplemental Fig. S2A). In contrast, we found a substantial increase in ssDNA at ori2060 only in HU-treated rad3Δ cells: This did not occur either in wild-type cells or when HU was not applied. Furthermore, this effect was not simply due to the increase in initiation at ori2060: While ori2004 has an efficiency of 50% and ori2060 is at 26% in rad3Δ cells in HU, the level of RPA at ori2060 was more than threefold higher than at ori2004. These data thus suggest that not all origins accumulate higher levels of ssDNA in rad3Δ cells in stress conditions and point to a specific dysfunction at de-regulated origins.
We then determined the contribution of inappropriate origin firing to ssDNA formation genome-wide. We performed ChIP of RPA followed by microarray analysis (ChIP-chip) for wild-type and rad3Δ cells synchronized in G2 and released into 6 mM HU. Samples were collected early in S phase (60 min) and at a time in late S phase when we observed high RPA binding at ori2060 (80 min) (Supplemental Figs. S1B, S2A). We note that replication has already initiated at a number of origins at 60 min (for simplicity and by comparison with the 80-min sample, we will refer to this time point as “early S”), as indicated by RPA binding (Fig. 2A–C; Supplemental Fig. S2B) and flow cytometry (Supplemental Fig. S1B). Our results showed that in early S, wild-type and rad3Δ cells possess very similar profiles of RPA binding along the chromosomes (Fig. 2A, Early S; Supplemental Fig. S2B). Consistent with the origin unwinding that is a part of the normal initiation process, we found a comparable enrichment of RPA at most origins that directly correlates with efficiencies in both backgrounds (Fig. 2B; Supplemental Fig. S2C,D, Early S). In late S, ssDNA levels at early-firing and efficient origins were reduced, as expected from the duplication of these sites (Fig. 2B, Late S; Supplemental Fig. S2B). Importantly, in HU-treated rad3Δ cells, RPA was recruited at high levels to a subset of origins with a wide range of efficiencies (Fig. 2B, Late S), revealing a problem in replication initiation at these loci.
Figure 2.

Abnormal accumulation of ssDNA at de-regulated origins. (A) Profiles of RPA binding and origin usage in a representative region of the genome. Full RPA and origin profiles are in Supplemental Figure S2B. Top and middle: RPA recruitment in early (60 min, top) and late (80 min, middle) S phase in wild type (WT, gray) and rad3Δ (orange). y-axis: RPA binding (IP/input). Bottom: origin efficiencies for WT (black) and rad3Δ (red). Asterisks mark CIOs. y-axis: origin efficiency. x-axis for all panels: chromosome coordinates. (B) Heat maps of RPA recruitment at origins in HU-treated wild-type and rad3Δ cells in early and late S phase. Data are from ChIP-chip experiments in A and Supplemental Figure S2B. All origins are shown in order of increasing efficiency; the most efficient origins are at the bottom of the plot. Position 0: central probe for each origin; values in a window of 100 probes on either side of each origin are displayed. A more intense red represents a higher level of RPA binding. (C) Heat maps of RPA recruitment, with origins divided into two categories: unaffected origins and CIOs (data are the same as in B). Origins in each category are displayed according to their efficiencies. Probe positions and color codes are as in B. (D) Average signal plots of RPA binding at origins in HU. Each line represents average RPA binding at either unaffected origins (solid lines) or CIOs (dotted lines) in wild-type (black) and rad3Δ (red) cells. Position 0: central probe for each origin; values at each probe position in a 100-probe window on either side of each origin were averaged for all origins in a category. (E) Correlation between the differences in RPA binding and in origin usage between rad3Δ and wild type. Origins were divided into 12 equal subsets (73 origins/group) based on the changes in their efficiencies (rad3Δ – WT). The average differences in efficiency (x-axis) and RPA occupancy (y-axis) for each group are shown. The nonaveraged data are shown in Supplemental Figure S2E.
To evaluate the potential link between ssDNA accumulation and inappropriate initiation, we clustered origins into unaffected sites vs. CIOs and compared RPA recruitment. In early S, RPA was bound at efficient origins, but only CIOs displayed an increase in RPA binding in rad3Δ compared to wild-type cells (Fig. 2C; Supplemental Fig. S2D, Early S). This suggests that there is excess ssDNA formation at early-firing CIOs even during early S phase. Next, in late S, RPA binding to unaffected origins in either wild-type or rad3Δ cells was low, while it aberrantly accumulated at CIOs exclusively in the absence of the checkpoint (Fig. 2C; Supplemental Fig. S2D, Late S). This marked increase in RPA recruitment was centered on CIOs in HU-treated rad3Δ cells (Fig. 2D). Finally, we ascertained the relationship between the level of ssDNA formation and the extent of origin de-regulation. To this end, the full complement of 876 origins was divided into 12 equal groups based on their differences in efficiencies between rad3Δ vs. wild type in HU, and the average changes in efficiency and RPA occupancy were assessed for each group. Our analyses revealed a strong correlation between the alterations in origin usage and in the level of RPA binding (Fig. 2E). Taken together, these results implicate inappropriate initiation as a cause of abnormal ssDNA formation at these sites.
Although our studies indicate that origin de-regulation leads to increased RPA binding, ssDNA may also accumulate at other genomic sites in these conditions. We identified all loci of significant RPA recruitment in late S phase in both wild-type and rad3Δ cells treated with HU, applying a stringent cutoff that excludes the low levels of ssDNA that are produced as part of the normal replication process (see Methods). While we identified only 18 RPA sites in wild type, we found 153 peaks in rad3Δ cells, with CIOs representing 67% of these hotspots (Supplemental Fig. S2G). In addition, we noticed that changes in origin usage even below our 8% cutoff for CIOs were associated with abnormal ssDNA formation (Supplemental Fig. S2E,F); lowering our threshold to 5% resulted in the association of 78% of all RPA hotspots with this expanded set of de-regulated origins. Thus, inappropriate initiation in these conditions is responsible for most of the sites of ssDNA accumulation in the genome.
Our results demonstrate that abnormal ssDNA accumulation is not a common feature of all replication origins in checkpoint-defective cells in stress conditions but rather occurs at a subset of sites that undergoes de-regulation. This generates the large majority of the hotspots of ssDNA, providing evidence that unscheduled initiation is a key source of genome instability.
De-regulated origins are hotspots of Rad52 recruitment
The link between origin de-regulation and ssDNA accumulation led us to investigate the local consequences of these events. The Rad52 protein is important for the repair of DNA double-strand breaks and the restart of stalled replication forks (Benson et al. 1998; Symington 2002; Lambert et al. 2010; Ait Saada et al. 2017), and its recruitment is used as a marker for genome instability (Van Dyck et al. 1999; Lisby et al. 2001). We monitored Rad52 binding at representative origins by ChIP in wild-type and rad3Δ cells synchronized in G2 and allowed to undergo S phase in 6 mM HU. At the unaffected origin ori2004, we did not detect Rad52 in either genetic background, with or without replication stress (Supplemental Fig. S3A), throughout the time course. However, at the CIO ori2060, there was a clear increase in Rad52 recruitment specifically in rad3Δ cells treated with HU, demonstrating that de-regulation at this site is associated with an aberrant Rad52 accumulation. We then extended this analysis genome-wide, mapping Rad52 binding by ChIP-chip in HU-treated wild-type and rad3Δ cells (Fig. 3A; Supplemental Fig. S3B). Our results revealed a strong recruitment of Rad52 in late S (90 min) only at CIOs in rad3Δ cells exposed to HU (Fig. 3B,C, +HU; Supplemental Fig. S3C,D). This binding was also detected at low levels earlier in S phase (60 min) at a subset of CIOs but not at unaffected origins (Supplemental Fig. S3C,D). Importantly, we demonstrated that the alterations in Rad52 binding are quantitatively correlated with the extent of origin de-regulation between rad3Δ and wild type (Fig. 3D; Supplemental Fig. S3E). This is consistent with the direct correspondence between RPA and Rad52 levels at origins in late S phase (Supplemental Fig. S3F). In addition, an increase in Rad52 recruitment is observed at a small number of de-regulated origins that did not meet our CIO cutoff (Supplemental Fig. S3E), suggesting that even low levels of unscheduled origin activation may have consequences for genome maintenance. Complementary to these analyses, we also ascertained the contribution of de-regulated origin firing to overall Rad52 accumulation in HU-treated rad3Δ cells. To this end, we selected all the sites of significant Rad52 binding (see Methods), identifying 64 and 435 loci in wild type and rad3Δ, respectively. In rad3Δ cells, we found that 70% of CIOs are associated with Rad52 recruitment (Supplemental Fig. S3H) and that CIOs represent 31% of all Rad52 binding sites, constituting a key source of genome instability hotspots in these conditions. Moreover, we noted that the Rad52 levels associated with CIOs are higher than those colocalizing with unaffected origins or nonorigin sites, indicating that CIOs are more likely to be problematic (Fig. 3E). Altogether, our results establish inappropriate initiation as a direct and major contributor to DNA damage.
Figure 3.

Rad52 is recruited to de-regulated origins. (A) Rad52 binding and origin usage in replication stress conditions. A representative region of the genome is shown. Full Rad52 profiles are in Supplemental Figure S3B; full origin maps are in Supplemental Figure S1C. Top: Rad52 recruitment in wild type (WT, gray) and rad3Δ (blue). y-axis: Rad52 binding (IP/input). Bottom: origin efficiencies for wild type (black) and rad3Δ (red). Asterisks mark CIOs. y-axis: origin efficiency. x-axis for top and bottom: chromosome coordinates. (B) Heat maps of Rad52 binding at origins in HU-treated cells (left) or during an unperturbed S phase (right). Data are from ChIP-chip experiments as in A and Supplemental Figure S3G. Data analysis and presentation are as in Figure 2C. Note that there are a few red horizontal lines in rad3Δ in –HU; these represent high levels of Rad52 recruitment to the end of the right arm of Chromosome III (Supplemental Fig. S3B), which we have not further investigated in this study. (C) Average signal plots of Rad52 binding at origins in HU-treated cells (top) or during an unperturbed S phase (bottom). Data analysis and presentation are as in Figure 2D. Black: wild-type; red: rad3Δ. Solid lines: unaffected origins; dotted lines: CIOs. (D) Correlation between the differences in Rad52 binding and in origin usage between rad3Δ and wild type. Data analysis and presentation are as in Figure 2E. x-axis: difference in origin efficiency; y-axis: difference in Rad52 binding. The nonaveraged data are shown in Supplemental Figure S3E. (E) Levels of Rad52 binding at peaks associated with unaffected origins, CIOs, and nonorigin sites. Each point represents a single origin. Red lines: median values for each category of Rad52 peaks. x-axis: level of Rad52 binding (IP/input).
Our finding that de-regulated origins are hotspots of genome instability in HU-treated rad3Δ cells led us to ask whether they may already recruit Rad52 during an unperturbed S phase. To address this question, we performed ChIP-chip of Rad52 in wild-type and rad3Δ cells as above, synchronizing cells in G2 and allowing them to duplicate their DNA in the absence of HU. Rad52 recruitment was assessed 90 min after the release from G2, at the same time point as the late S experiments above. In these conditions, we observed similar genome-wide profiles of Rad52 binding in wild-type and rad3Δ cells (Supplemental Fig. S3G, bottom panels). However, they differed significantly from the pattern in checkpoint-defective cells exposed to replication stress (Supplemental Fig. S3G, top panels). Heat map and quantitative analyses demonstrated that the distinctive accumulation of Rad52 at CIOs in HU-treated rad3Δ cells is not observed in an unperturbed S phase (Fig. 3B,C, −HU). For instance, in rad3Δ cells, only 5% of CIOs are associated with Rad52 peaks in the absence of HU, while 70% of CIOs recruit Rad52 upon HU exposure (Supplemental Fig. S3H). This suggests that CIOs are not particularly problematic in a normal S phase and uncovers a pivotal role for the checkpoint in regulating origin firing to preserve genome stability when cells are subjected to replication stress.
Origin selection in replication stress conditions delineates the profile of genome instability
We have shown that CIO de-regulation in nucleotide-depleted rad3Δ cells gives rise to problematic sites that recruit the DNA repair machinery. However, genome instability in replication stress conditions arises from a variety of sources (Aguilera and García-Muse 2013; Zhou et al. 2013; Hoffman et al. 2015), and a significant portion (38%) of the Rad52 peaks in rad3Δ cells is not associated with origins (Supplemental Fig. S3H). We therefore evaluated the relationship between the genome-wide landscape of instability and inappropriate replication initiation, taking into account all 876 origins as well as all RPA and Rad52 binding sites in HU-treated cells. Regional analyses over continuous 1000-probe (∼250-kb) windows revealed a clear correspondence between origin de-regulation vs. the densities of RPA and Rad52 loci in a rad3Δ mutant (Fig. 4A). Furthermore, the signature Rad52 profile of HU-treated rad3Δ cells was not observed in situations where origins are not de-regulated, either in (1) HU-treated wild-type cells, or (2) during a normal S phase in both wild-type and rad3Δ backgrounds (Supplemental Fig. S4). These results establish unscheduled origin firing as a critical determinant of the overall pattern of genome instability. Finally, as suggested by these data and by the clustering of CIOs in late-firing and inefficient domains (Fig. 1C), we found that origin de-regulation and Rad52 binding in rad3Δ cells under replication stress show strong negative correlations with the wild-type program of origin efficiencies (Fig. 4B). Collectively, our results allow us to propose a model in which the organization of DNA replication delimits the checkpoint regulation of origin firing in cells exposed to replication stress, with key consequences for the landscape of DNA damage in checkpoint-compromised cells.
Figure 4.

The profile of origin de-regulation delineates the profiles of RPA and Rad52 binding in replication stress. (A) Genome-wide comparisons of origin de-regulation and genome instability. This analysis takes into account all 876 identified origins as well as all RPA and Rad52 binding sites. The sum of the changes in origin efficiencies upon HU treatment (rad3Δ − WT, red) was determined in continuous 1000-probe windows. The densities of RPA (orange) and Rad52 (blue) sites were calculated for HU-treated rad3Δ cells over the same windows. x-axis: chromosome coordinates; y-axes: sum of the differences in origin efficiencies (top), density of RPA sites (middle), density of Rad52 sites (bottom). The Spearman's correlation coefficients (ρ) for the different comparisons are indicated and show strong positive correlations. (***) P-value < 0.001. (B) Relationship between the wild-type replication program, the checkpoint regulation of origin firing, and genome instability during replication stress. The Spearman's correlation coefficients are indicated. (***) P-value < 0.001. Top: The profile of average origin efficiencies in wild-type cells as in Figure 1C (black, left y-axis) is displayed together with the sum of the changes in origin usage as in A (rad3Δ − WT, red, right y-axis). x-axis: chromosome coordinates. These two parameters show a strong negative correlation. Bottom: A strong negative correlation is also observed between the replication program (black, left y-axis) and the density of Rad52 (blue, right y-axis, as in A along the chromosomes. x-axis: chromosome coordinates.
Impact of altering the program of genome duplication on origin de-regulation
While our studies suggest that the replication program regulates the inhibition of origin usage by Rad3/ATR, they do not exclude the possibility that the checkpoint may simply be more readily recruited to particular chromosomal regions that may be fragile or susceptible to insults (Aguilera and García-Muse 2013). To distinguish between these possibilities, we used a rif1Δ genetic background, in which replication initiation is altered genome-wide. Rif1 is a determinant of replication timing, and the differences in the timing of firing of early vs. late origins are reduced in rif1Δ mutants (Cornacchia et al. 2012; Hayano et al. 2012; Yamazaki et al. 2012; Foti et al. 2016). Relevant to our studies, loss of Rif1 function in the fission yeast does not alter sensitivity to replication stress (Hayano et al. 2012). First, we evaluated the effect of rif1Δ in our experimental setup and determined origin usage in cdc25-22 rif1Δ cells synchronized in G2 and released in 6 mM HU (Supplemental Fig. S5A). Consistent with previous reports, we observed major changes in the replication program compared to that in wild type (Supplemental Fig. S5B). We then assessed the regional profile of DNA replication by ascertaining average origin efficiencies in continuous 1000-probe windows, as for Figure 1C, and found that the characteristic differences between efficiency domains in wild-type cells are strongly attenuated in rif1Δ (Fig. 5A). This background thus represents an ideal system for testing our model: If the replication program delimits the regions where the checkpoint inhibits origin firing in replication stress conditions, we would predict that inappropriate replication initiation in a rif1Δ rad3Δ mutant would no longer be enriched in the same genomic domains as in rad3Δ.
Figure 5.
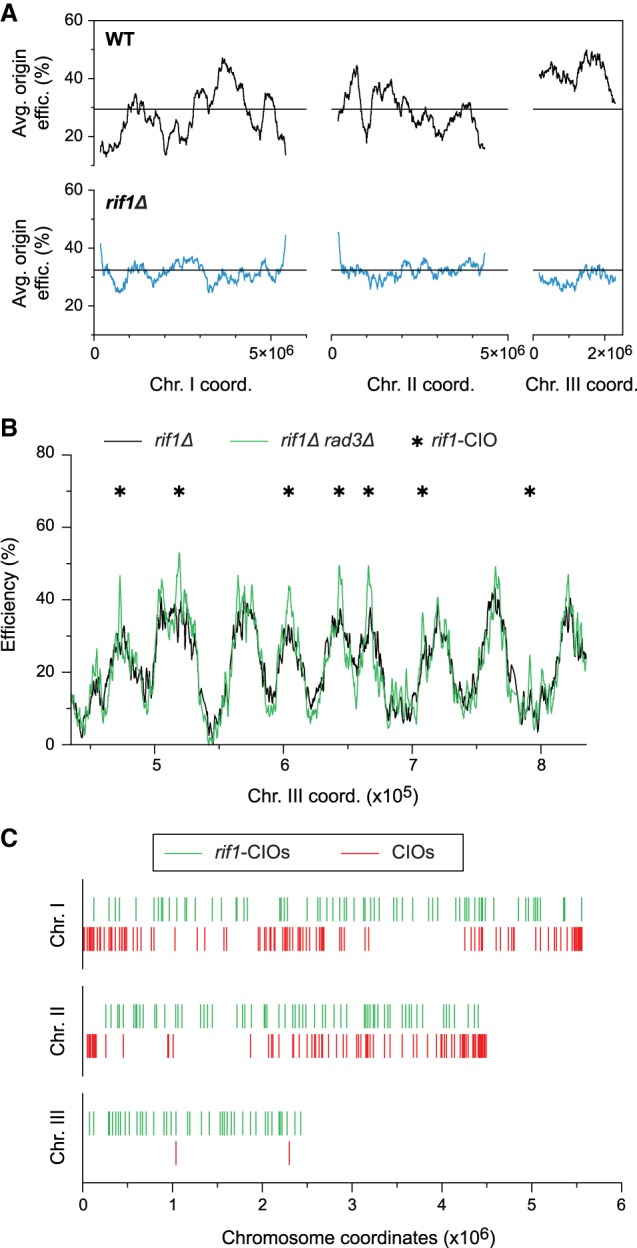
Modulation of the replication program results in redistribution of de-regulated origins. (A) Profiles of replication efficiency domains in cdc25-22 (WT, black, as in Fig. 1C) and cdc25-22 rif1Δ (rif1Δ, blue) cells. Cells were arrested in G2 and released to undergo S phase in 6 mM HU. The averages of origin efficiencies in each background were assessed for continuous 1000-probe windows. Horizontal lines: average origin efficiency genome-wide. x-axis: chromosome coordinates; y-axes: average origin efficiencies. The full maps of origin usage are shown in Supplemental Figure S5B. (B) Origin efficiencies in a representative region of the genome for rif1Δ (black) and rif1Δ rad3Δ (green) cells synchronized in G2 using the cdc25-22 mutation and released into 6 mM HU. Asterisks: rif1-CIOs. Full efficiency maps are in Supplemental Figure S5C. x-axis: chromosome coordinates; y-axis: origin efficiencies. (C) Comparison of the distribution of de-regulated origins: rif1-CIOs (rif1Δ rad3Δ, green bars) vs. CIOs (rad3Δ, red bars). x-axis: chromosome coordinates.
We therefore analyzed the impact of the altered replication program in rif1Δ cells on origin de-regulation, comparing efficiencies in rif1Δ rad3Δ and rif1Δ cells arrested in G2 and allowed to synchronously enter S phase in 6 mM HU (Fig. 5B; Supplemental Fig. S5C). Applying the same 8% threshold as for CIOs, we identified 170 de-regulated origins in rif1Δ rad3Δ cells that we will refer to as rif1-CIOs (Fig. 5B; Supplemental Tables S4, S5). These rif1-CIOs also span a broad spectrum of activities (Supplemental Fig. S5D). However, in contrast to CIOs, rif1-CIOs are more uniformly positioned along the chromosomes and no longer clustered in distinct domains (Fig. 5C; Supplemental Fig. S5C); this reflects the “flattened” replication profile in rif1Δ. Notably, a large number of rif1-CIOs are found in regions that lack CIOs in rad3Δ. These findings demonstrate that inappropriate origin activation does not occur as a result of the chromosomal context but rather as a consequence of the program of DNA replication.
Changes in origin de-regulation redistribute hotspots of Rad52 recruitment
We next determined whether the changes in origin de-regulation in checkpoint-deficient rif1Δ cells treated with HU are accompanied by a redistribution in Rad52 recruitment. To this end, rif1Δ and rif1Δ rad3Δ cells were allowed to synchronously enter S phase in 6 mM HU, and Rad52 binding was assessed by ChIP-chip 90 min after the release, when we observed high levels of Rad52 recruitment at a representative rif1-CIO (Supplemental Fig. S6A). First, our results in rif1Δ cells showed that this mutation itself does not induce either high levels of Rad52 binding compared to wild type (Supplemental Fig. S6B) or Rad52 recruitment at origins (Supplemental Fig. S6C). Importantly, we found a genome-wide increase in Rad52 binding in HU-treated rif1Δ rad3Δ cells compared to rif1Δ (Fig. 6A; Supplemental Fig. S6D). Heat map analyses revealed a clear enrichment of Rad52 at a number of origins in rif1Δ rad3Δ that was not detected in rif1Δ (Fig. 6B, +HU). Although our 8% efficiency cutoff identified only a subset of these sites as rif1-CIOs, we established a clear positive correlation between the increases in origin efficiencies in rif1Δ rad3Δ and the changes in Rad52 binding (Fig. 6C), indicating higher levels of recruitment at rif1-CIOs. Building on these findings, we then determined the extent to which inappropriate initiation contributes to Rad52 hotspots in these cells. Using the same criteria as for our previous analyses (Supplemental Fig. S3H), we identified 618 sites of Rad52 binding in HU-treated rif1Δ rad3Δ cells compared to 94 in rif1Δ. Similar to our observations for CIOs in rad3Δ, 62% of the 170 rif1-CIOs coincide with Rad52 hotspots in rif1Δ rad3Δ, and 67% of all Rad52 loci are, in fact, associated with origins (Supplemental Fig. S6E). Together with Figure 6C, which exposes a strong correlation between origin de-regulation and Rad52 accumulation even for origins that were not initially identified as rif1-CIOs, these results indicate that inappropriate initiation is a major source of genome instability hotspots in rif1Δ rad3Δ cells exposed to replication stress.
Figure 6.

Alteration of the replication program results in genome-wide changes in Rad52 accumulation. (A–D) rif1Δ and rif1Δ rad3Δ cells were synchronized using cdc25-22 and released in 6 mM HU except where indicated. (A) Rad52 binding and origin usage in a representative region of the genome. Top: profiles of Rad52 recruitment in rif1Δ (gray) and rif1Δ rad3Δ (orange). The full Rad52 profiles are in Supplemental Figure S6D, top panel. y-axis: Rad52 binding (IP/input). Bottom: origin efficiencies for rif1Δ (black) and rif1Δ rad3Δ (green). Asterisks mark rif1-CIOs. y-axis: origin efficiency. The full origin efficiency profiles are in Supplemental Figure S5C. x-axis for top and bottom: chromosome coordinates. (B) Heat maps of Rad52 binding centered on origins in rif1Δ and rif1Δ rad3Δ cells treated with 6 mM HU (left, +HU) or in the absence of HU (right, −HU). Presentation is as in Figure 3B. Note that, as in Figure 3B, there are a few red horizontal lines in rif1Δ rad3Δ in –HU; these represent high levels of Rad52 recruitment to the end of the right arm of Chromosome III (Supplemental Fig. S6D), which we have not further investigated in this study. (C) Correlation between changes in origin usage and in Rad52 binding between rif1Δ rad3Δ and rif1Δ cells. Analysis is as in Figure 3D. x-axis: difference in origin efficiency; y-axis: difference in Rad52 binding. (D) Density of Rad52 sites in rif1Δ rad3Δ (purple) vs. rad3Δ (blue, as in Fig. 4A) calculated over continuous 1000-probe windows. Top panel: +HU; bottom panel: −HU. x-axis: chromosome coordinates; y-axes: density of Rad52 sites.
Given the large number of Rad52 binding sites in HU-treated rif1Δ rad3Δ cells, we addressed the possibility that the altered origin usage program in a rif1Δ background generates a sensitized state in which cells experience replication stress even during an unperturbed S phase. To this end, we ascertained Rad52 binding in synchronized rif1Δ and rif1Δ rad3Δ cells that proceeded through S phase in the absence of HU, taking the same time point (90 min) as for Figure 6A. Heat map analyses of data from both strains showed that in these conditions, Rad52 is not enriched at either unaffected origins or rif1-CIOs (Fig. 6B, −HU). Indeed, in contrast to HU-treated rif1Δ rad3Δ cells, where 62% of rif1-CIOs overlap with Rad52 sites, only 10% of rif1-CIOs coincide with Rad52 loci in a normal S phase (Supplemental Fig. S6E). These data therefore suggest that the altered replication program in rif1Δ is not intrinsically problematic and that absence of the checkpoint in this context does not give rise to origin-associated DNA damage.
Finally, we evaluated the overall impact of reprogramming DNA replication on the landscape of genome instability. We compared the density of Rad52 hotspots over continuous 1000-probe windows in rif1Δ rad3Δ and rad3Δ cells in the absence and presence of HU. In an unperturbed S phase, this pattern was highly similar between the two strains (Fig. 6D, bottom panel), implying that the rif1Δ mutation itself does not induce a significant alteration of the Rad52 profile in a sensitized, checkpoint-defective background. In replication stress conditions, Rad52 sites in rif1Δ rad3Δ cells were no longer clustered in the same distinct genomic domains as in rad3Δ (Fig. 6D, top panel), revealing a complete redistribution of Rad52 loci.
Taken together, these findings support our model that induced alteration of the replication program in checkpoint-defective cells subjected to replication stress leads to changes in the pattern of de-regulated replication initiation as well as associated genome instability events.
Discussion
In this study, we have identified a fundamental role for the program of genome duplication in the maintenance of genome stability. Our results demonstrate that, in checkpoint-defective cells subjected to replication stress, the significant de-regulation of Rad3/ATR-inhibited origins is clustered in late-replicating and inefficient genomic domains. We show that this selective regulation of origin usage by the checkpoint is directly and quantitatively governed by the program of genome duplication, regardless of the individual characteristics of these origins. We observe a strong correlation between the extent of origin de-regulation and the levels of RPA and Rad52 binding at these loci. These inappropriate origin firing events represent a major source of the genome-wide complement of Rad52 hotspots, and the distinctive localization of these sites delineates a signature profile of genome instability. Finally, modulation of the replication program using a rif1Δ mutant background leads to an entirely altered pattern of origin de-regulation in checkpoint-defective cells subjected to replication stress. In this context, the link between unscheduled initiation and Rad52 accumulation is nevertheless maintained, which results in a genome-wide redistribution of problematic loci. Our work therefore provides novel evidence that the organization of DNA replication establishes the landscape of checkpoint-regulated origin firing and shapes genome instability along the chromosomes.
The results of our study have critical implications for the cellular response to genotoxic insults. While previous investigations focused on the targeting of late-firing and inefficient origins, with work in budding yeast showing that such sites are not fired when the checkpoint is functional (Poli et al. 2012), our data reveal that individual origins with a broad range of efficiencies and firing times are, in fact, subject to checkpoint inhibition. Our findings are compatible with an earlier study in which origins repressed by the checkpoint in HU eventually fire after a long period of time, hinting that the checkpoint does not specifically control a distinct set of late-firing origins (Alvino et al. 2007). In particular, we establish that, in nucleotide-depleted rad3/ATR mutants, the number of de-regulated origins in a region and the changes in their efficiencies are directly coupled to the characteristics of replication efficiency and timing domains. In addition, in the context of the stochasticity in replication initiation observed in DNA combing assays (Patel et al. 2006; Czajkowsky et al. 2008; Kaykov and Nurse 2015), we do not interpret our results to imply that every origin in late-replicating domains is de-regulated in each cell, but rather that there is a higher frequency of inappropriate initiation in these regions. Our data reveal that the control of origin usage by the Rad3/ATR pathway in replication stress conditions is a key aspect of its function in genome maintenance. Our work also presents compelling evidence for a fundamental biological importance of the replication program in genome surveillance, which may help to elucidate the rationale for the pattern of DNA replication in wild-type cells. For example, particular programs may lead to elevated local concentrations of checkpoint factors that would protect at-risk regions through more efficient signaling (Rivera-Mulia et al. 2015). This may favor the repression of neighboring sites and also locally enhance DNA repair processes. Complementary to this, there may also have been evolutionary selection for an arrangement in which potential sites of DNA damage are enriched in regions where mutations have a lower likelihood to be deleterious.
Although we have focused on the regulation of replication initiation in stress conditions, our findings may also be applicable to an S phase that is not subjected to pathological replication stress. Indeed, cells can encounter multiple challenges to DNA synthesis even in “unperturbed” conditions (Zeman and Cimprich 2014). For instance, markers of genotoxic stress are detected during replication in embryonic stem cells (Ahuja et al. 2016), and ATR function is essential in other eukaryotic systems (Eykelenboom et al. 2013). In addition, replication stress may be induced by the dependence of DNA synthesis on limiting factors that include components of the replication machinery (Cdc45, Sld2, and Sld3) (Wu and Nurse 2009; Mantiero et al. 2011; Tanaka et al. 2011) as well as those that are critical for efficient replication, such as RPA (Toledo et al. 2013). dNTPs are also limiting during genome duplication: The existing pool of dNTPs at G1/S is insufficient to complete replication, and the ribonucleotide reductase (RNR) is induced to produce the necessary nucleotides (Poli et al. 2012; Guarino et al. 2014). However, while there are multiple potential sources of challenges to replication during a normal S phase, we do not find Rad52 recruitment to origins in the absence of HU treatment in our experimental context. Nevertheless, it is possible that, in this situation, cells experience only mild intrinsic levels of stress, making inappropriate initiation events relatively rare and difficult to monitor using our short-term population-level analyses. Indeed, there are indications that the organization of DNA replication may have long-term consequences for genome architecture and maintenance. For instance, our demonstration that origins are hotspots of genome instability when inappropriately fired in stress conditions is consistent with comparative genomic analyses and laboratory evolution experiments in budding yeast that have colocalized breakpoints for gene amplifications and chromosome rearrangements with origins (Di Rienzi et al. 2009; Gordon et al. 2009). Our results also hint at a mechanism for how the replication program may delineate differences in the types and rates of mutations that have been associated with replication timing (Lang and Murray 2011; Liu et al. 2013). Altogether, these possibilities have important implications for the processes that shape the genome during its evolution.
Finally, our studies may bring insight into the mutations that accumulate when DNA synthesis is perturbed in pathological situations, such as in cancer. Early tumorigenesis is associated with replication stress and nucleotide deficiencies (Halazonetis et al. 2008; Bester et al. 2011), and a haploinsufficiency of ATR or Chk1 has been shown to contribute to cancer predisposition (Brown and Baltimore 2000; Lam et al. 2004). Furthermore, cancer cells have been found to display altered patterns of origin usage (Donley and Thayer 2013). Our experimental conditions therefore recapitulate many of these features, and our results suggest that the replication program may be responsible for the differential susceptibility of distinct genomic regions to the accumulation of DNA damage in these contexts. Our conclusions may thus inform the current understanding of how the cancer mutational landscape arises, with consequences for somatic evolution and for the heterogeneity found in individual tumors.
While our previous work coupled origin selection with programmed recombination in the context of the specialized meiotic cycle (Wu and Nurse 2014), our new findings demonstrate that the replication program has a broader and more fundamental role in cellular biology through regulating the surveillance pathways that ensure the fidelity of the genetic material. The conservation of the mechanisms for DNA replication and checkpoint regulation from yeast to human suggest that this critical function of the organization of genome duplication in genome maintenance may also extend to more complex eukaryotic systems.
Methods
Strains and cell growth
Standard media and methods for fission yeast were used (Moreno et al. 1991; Hayles and Nurse 1992). Cells were grown in minimal medium supplemented with 250 mg/L adenine, histidine, lysine, uracil, leucine, and arginine (EMM6S). Supplemental Table S6 lists the fission yeast strains used in this study. The following constructions were previously generated: Cdc45-YFP (Gregan et al. 2003), rad3Δ::ura4 (gift from the Nurse Lab), and Polδ-Flag (Taricani and Wang 2006). Deletions and tagged proteins (rad3Δ::kanMX, rif1Δ::natMX, Ssb1-HA) were constructed by homologous recombination (Bähler et al. 1998). The JW129 and JW131 strains were used for the origin mapping and ChIP-qPCR experiments in Figure 1 and Supplemental Figure S1. JW133 and JW135 were used for the Ssb1/RPA ChIP-chip experiments (Fig. 2; Supplemental Fig. S2). PN292 and JW154 were used for the Rad52 ChIP-chip experiments (Fig. 3; Supplemental Fig. S3). JW1183 and JW1184 were used for both the origin mapping and Rad52 ChIP-chip experiments in a rif1Δ background (Figs. 5, 6; Supplemental Figs. S5, S6). PN2483 and JW1135 were used for determination of origin efficiencies after synchronization with nda3-km311 (Supplemental Fig. S1). PN1 and PN1400 were used for validation of copy number experiments (Supplemental Fig. S7).
Cell cycle synchronization
Strains with the cdc25-22 mutation were grown at the permissive temperature (25°C) to a density of ∼2 × 106 cells/mL before being shifted to the restrictive temperature (36.5°C) for 4 h, leading to G2 arrest (Forsburg and Rhind 2006). Subsequent release to 25°C by rapid cooling of the culture results in synchronous entry into mitosis and S phase. For nda3-km311 strains, cells were grown in exponential phase at 32°C to a density of ∼2 × 106 cells/mL and shifted to 18°C for 6 h to induce mitotic arrest (Hiraoka et al. 1984). Release to 32°C results in synchronous re-entry into the cell cycle.
Flow cytometry
Cells were fixed in 70% cold ethanol, washed in 50 mM sodium citrate, and treated with RNase A (0.1 mg/mL) at 37°C. Samples were then stained using 2 mg/mL propidium iodide, sonicated with a Branson digital sonifier, and run on a BD Accuri C6 flow cytometer (BD Biosciences). Data were analyzed using the FlowJo software. Details for the interpretation of flow cytometry profiles in the fission yeast are provided in the Supplemental Materials.
Chromatin immunoprecipitation
ChIP experiments were performed as previously described (Wu and Nurse 2009). Cells were fixed with 1% formaldehyde, lysed in a FastPrep cell disruptor (MP Biomedicals), and sonicated with a Bioruptor Plus (Diagenode) to obtain chromatin fragments of ∼400–500 bp. Immunoprecipitations (IPs) were carried out overnight at 4°C using the following antibodies: α-GFP rabbit polyclonal antibody for Cdc45-YFP (gift from the Nurse Lab), α-Rad52/Rad22 (Ab63800; Abcam), and α-HA for Ssb1-HA (12CA5; Roche). Protein G sepharose beads (GE Healthcare) were then added to the samples and incubated for 4 h at 4°C. IPs were then washed and eluted, and crosslinking was reversed for both IP and Input samples by incubation at 65°C overnight. For quantitative PCR, IP and Input DNA were mixed with SYBR Green qPCR Master Mix (Agilent Technologies) and processed with an ABI 7900 HT. Primers used in this study are listed in Supplemental Table S7.
Origin mapping experiments and analyses
Competitive hybridizations of differentially labeled samples were performed using Agilent 4x44K S. pombe arrays (60-mer probes every ∼250 nucleotides [nt]; Agilent Technologies) as previously described (Wu and Nurse 2014). Briefly, copy number was determined by comparing genomic DNA samples from nonreplicating cells and from cells undergoing DNA replication in hydroxyurea. HU treatment limits the extension of DNA synthesis around the sites of initiation, allowing the identification of replication origins. This method has been validated in previous studies (Heichinger et al. 2006; Wu and Nurse 2014) and provides very similar origin maps to those obtained with other approaches (Hayashi et al. 2007; Daigaku et al. 2015). In cdc25-22 experiments, 6 mM HU was added 5 min after cells were released from the G2 arrest; samples were taken at this time and after a time when cells would normally have completed S phase in the absence of HU (90 min). For nda3-km311, 6 mM HU was added 5 min after cells were released from mitotic arrest; samples were taken just before the release and 80 min after release.
Genomic DNA was extracted (Hoffman and Winston 1987) and purified using the Qiagen Genomic DNA kit. Samples were labeled using the BioPrime Plus Array CGH Indirect Genome Labeling kit (Invitrogen) according to the manufacturer's instructions with either Alexa 555/647 (Agilent Technologies) or Cy3/Cy5 (GE Healthcare) dyes. One to two micrograms of labeled DNA from the unreplicated and S phase samples were hybridized onto the microarrays. Two independent hybridizations of the same samples were systematically performed in a dye-swap experiment, the ratios of replicating DNA to unreplicated DNA were assessed, and these data sets were averaged. To determine copy number, the geometric means over five consecutive probes were determined across the genome. Outliers were identified and removed prior to origin assignment. We averaged three biological repeats for cdc25-22 vs. cdc25-22 rad3Δ as well as for cdc25-22 rif1Δ vs. cdc25-22 rif1Δ rad3Δ; two biological repeats were used for the nda3-km311 vs. nda3-km311 rad3Δ comparison. To obtain origin efficiencies, the lowest ratios, which represent nonreplicated DNA (a small proportion of the genome in 6 mM HU), were adjusted to be centered on a value of 1. This resulted in a correction of 0.25 for all data sets. The application of this correction was further validated by visual inspection of the overall profiles. Copy number was converted to efficiency (for example, 1.5 = 50%), which represents the frequency of firing of a given origin in a population of cells. Supplemental Tables S1–S5 provide information regarding origin positions and efficiencies in our experimental conditions. Additional details of our analyses of origin efficiencies and the selection of de-regulated origins are provided in the Supplemental Methods and in Supplemental Figures S7–S9.
ChIP-chip analysis
For ChIP-chip assays, 150 mL of cells were collected after release from G2 arrest at either 60 min or 80 min for Ssb1/RPA and at 60 min or 90 min for Rad52. ChIPs were performed as described above. For amplification of the ChIP material and labeling for hybridization, the protocol from van Bakel et al. (2008) was used according to Wu and Nurse (2014). Each ChIP was then hybridized against its reciprocally labeled Input sample (IP/Input). For HU-treated cdc25-22 and cdc25-22 rad3Δ cells, three biological repeats were performed for RPA and Rad52. Two biological repeats were performed for Rad52 in HU-treated cdc25-22 rif1Δ and cdc25-22 rif1Δ rad3Δ. For all Rad52 experiments in the absence of HU, two biological repeats were performed.
For all quantitative analyses (measurement of binding at origins, heat maps, average signal plots, and selection of binding sites), probe values were used directly. For visual representation in Figures 2A, 3A, and 6A as well as Supplemental Figures S2B, S3, B and G, and S6, B and D, the moving geometric means of five consecutive probes were calculated across the genome and plotted.
To identify peaks of RPA or Rad52 recruitment, we designated a position as a binding site only when three consecutive probes showed values higher than our thresholds. For RPA, we established a cutoff of 2.0 based on the values of IP/Input for all the probes in a wild-type background after calculating the dispersion of the data from the median. Values were divided into quartiles, and as RPA only binds to specific sites, we considered that the median value of the data set represents the unbound DNA and that significant binding occurs only when the values are 1.5 times the interquartile range (IQR) above the third quartile (Q3 + 1.5[IQR] = 1.84). This cutoff excludes low levels of RPA that are part of the normal replication process. For Rad52, a threshold of 1.5 for IP/Input was established. This was based on the distribution of the signal IP/Input for all the probes in a wild-type background, after calculating the dispersion of the data from the median (Q3 + 1.5[IQR] = 1.48). We considered RPA and Rad52 sites to colocalize with an origin if they occurred within a distance of <3 kb.
Quantitative and statistical analyses
For the quantitative analyses in Figures 2, D and E, 3, C and D, and 6C as well as in Supplemental Figures S2C–F, S3D–F, S4, and S6C, the levels of RPA and Rad52 recruitment were determined as follows: For each origin, we took the two probes closest to the origin position and identified the larger value for either RPA or Rad52 binding. This was then used as the occupancy at that origin. Statistics were performed with RStudio (RStudio Team 2015). The correlations in Figure 4 and Supplemental Figure S4 were tested using Spearman's rank correlation coefficient. The correlations in Supplemental Figure S9A were tested using the Pearson correlation coefficient.
The heat maps in Figures 2, B and C, 3B, and 6B, and Supplemental Figure S3C were generated using RStudio (RStudio Team 2015). Levels of RPA and Rad52 binding were converted to a log2 scale. Origins were then aligned on a central position, and the log2 values within a distance of −100 to +100 probes from this position were plotted. For visualization, log2 values <0.5 were assigned a value of 0. For the average signal plots in Figures 2D and 3C, the means of the values were calculated for a distance from −100 to +100 probes centered on the origins.
Data access
All microarray data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE98462.
Supplementary Material
Acknowledgments
We thank members of the Genome Duplication and Maintenance team for helpful suggestions, in particular, Anthony Perrot for experimental as well as bioinformatics assistance and Diane Schausi-Tiffoche for technical aid. We thank Biogenouest Genomics and the Human Environmental Genomics core facility of Rennes (Biosit/OSUR) for technical support. We also thank Damien Coudreuse for valuable discussions and critical reading of the manuscript. This study was supported by funding from Worldwide Cancer Research (11-0685), the CNRS/INSERM (ATIP-Avenir program), the Fondation pour la Recherche Médicale (“Amorcage de jeunes equipes” program), the Fondation ARC pour la Recherche sur le Cancer (Projet ARC), the Ligue contre le cancer (Comités 22, 29, and 35), and the Institut National du Cancer (INCA, PLBIO 15-043). P.-Y.J.W. was also supported by the Région Bretagne and Rennes Métropole. The funding bodies had no role in the design and progress of this study.
Author contributions: B.G.-E. and P.-Y.J.W. conceived and designed the experiments, analyzed the data, and wrote the paper. B.G.-E. performed the experiments, and P.-Y.J.W. supervised the study.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.224527.117.
Freely available online through the Genome Research Open Access option.
References
- Aguilera A, García-Muse T. 2013. Causes of genome instability. Annu Rev Genet 47: 1–32. [DOI] [PubMed] [Google Scholar]
- Ahuja AK, Jodkowska K, Teloni F, Bizard AH, Zellweger R, Herrador R, Ortega S, Hickson ID, Altmeyer M, Méndez J, et al. 2016. A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat Commun 7: 10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait Saada A, Teixeira-Silva A, Iraqui I, Costes A, Hardy J, Paoletti G, Fréon K, Lambert SAE. 2017. Unprotected replication forks are converted into mitotic sister chromatid bridges. Mol Cell 66: 398–410.e4. [DOI] [PubMed] [Google Scholar]
- Alvino GM, Collingwood D, Murphy JM, Delrow J, Brewer BJ, Raghuraman MK. 2007. Replication in hydroxyurea: It's a matter of time. Mol Cell Biol 27: 6396–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, Steever AB, Wach A, Philippsen P, Pringle JR. 1998. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14: 943–951. [DOI] [PubMed] [Google Scholar]
- Benson FE, Baumann P, West SC. 1998. Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature 391: 401–404. [DOI] [PubMed] [Google Scholar]
- Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. 2011. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145: 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D. 2000. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 14: 397–402. [PMC free article] [PubMed] [Google Scholar]
- Chen C-L, Rappailles A, Duquenne L, Huvet M, Guilbaud G, Farinelli L, Audit B, d'Aubenton-Carafa Y, Arneodo A, Hyrien O, et al. 2010. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res 20: 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornacchia D, Dileep V, Quivy J-P, Foti R, Tili F, Santarella-Mellwig R, Antony C, Almouzni G, Gilbert DM, Buonomo SBC. 2012. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J 31: 3678–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czajkowsky DM, Liu J, Hamlin JL, Shao Z. 2008. DNA combing reveals intrinsic temporal disorder in the replication of yeast chromosome VI. J Mol Biol 375: 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Keszthelyi A, Müller CA, Miyabe I, Brooks T, Retkute R, Hubank M, Nieduszynski CA, Carr AM. 2015. A global profile of replicative polymerase usage. Nat Struct Mol Biol 22: 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzi SC, Collingwood D, Raghuraman MK, Brewer BJ. 2009. Fragile genomic sites are associated with origins of replication. Genome Biol Evol 1: 350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova DS, Gilbert DM. 2000. Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol 2: 686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donley N, Thayer MJ. 2013. DNA replication timing, genome stability and cancer: Late and/or delayed DNA replication timing is associated with increased genomic instability. Semin Cancer Biol 23: 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eykelenboom JK, Harte EC, Canavan L, Pastor-Peidro A, Calvo-Asensio I, Llorens-Agost M, Lowndes NF. 2013. ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep 5: 1095–1107. [DOI] [PubMed] [Google Scholar]
- Feng W, Collingwood D, Boeck ME, Fox LA, Alvino GM, Fangman WL, Raghuraman MK, Brewer BJ. 2006. Genomic mapping of single-stranded DNA in hydroxyurea-challenged yeasts identifies origins of replication. Nat Cell Biol 8: 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Di Rienzi SC, Raghuraman MK, Brewer BJ. 2011. Replication stress-induced chromosome breakage is correlated with replication fork progression and is preceded by single-stranded DNA formation. G3 (Bethesda) 1: 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsburg SL, Rhind N. 2006. Basic methods for fission yeast. Yeast 23: 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti R, Gnan S, Cornacchia D, Dileep V, Bulut-Karslioglu A, Diehl S, Buness A, Klein FA, Huber W, Johnstone E, et al. 2016. Nuclear architecture organized by Rif1 underpins the replication-timing program. Mol Cell 61: 260–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkos M, Ganier O, Coulombe P, Méchali M. 2015. DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 16: 360–374. [DOI] [PubMed] [Google Scholar]
- Ge XQ, Blow JJ. 2010. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol 191: 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JL, Byrne KP, Wolfe KH. 2009. Additions, losses, and rearrangements on the evolutionary route from a reconstructed ancestor to the modern Saccharomyces cerevisiae genome. PLoS Genet 5: e1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregan J, Lindner K, Brimage L, Franklin R, Namdar M, Hart EA, Aves SJ, Kearsey SE. 2003. Fission yeast Cdc23/Mcm10 functions after pre-replicative complex formation to promote Cdc45 chromatin binding. Mol Biol Cell 14: 3876–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarino E, Salguero I, Kearsey SE. 2014. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin Cell Dev Biol 30: 97–103. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. 2008. An oncogene-induced DNA damage model for cancer development. Science 319: 1352–1355. [DOI] [PubMed] [Google Scholar]
- Hayano M, Kanoh Y, Matsumoto S, Renard-Guillet C, Shirahige K, Masai H. 2012. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev 26: 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Katou Y, Itoh T, Tazumi A, Tazumi M, Yamada Y, Takahashi T, Nakagawa T, Shirahige K, Masukata H. 2007. Genome-wide localization of pre-RC sites and identification of replication origins in fission yeast. EMBO J 26: 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayles J, Nurse P. 1992. Genetics of the fission yeast Schizosaccharomyces pombe. Annu Rev Genet 26: 373–402. [DOI] [PubMed] [Google Scholar]
- Heichinger C, Penkett CJ, Bähler J, Nurse P. 2006. Genome-wide characterization of fission yeast DNA replication origins. EMBO J 25: 5171–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka Y, Toda T, Yanagida M. 1984. The NDA3 gene of fission yeast encodes β-tubulin: A cold-sensitive nda3 mutation reversibly blocks spindle formation and chromosome movement in mitosis. Cell 39: 349–358. [DOI] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang C-W, Lyou Y, Townes TM, Schübeler D, Gilbert DM. 2008. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol 6: e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Rathjen J, Kulik M, Papp B, Fussner E, Bazett-Jones DP, Plath K, Dalton S, et al. 2010. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res 20: 155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman CS, Winston F. 1987. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 57: 267–272. [DOI] [PubMed] [Google Scholar]
- Hoffman EA, McCulley A, Haarer B, Arnak R, Feng W. 2015. Break-seq reveals hydroxyurea-induced chromosome fragility as a result of unscheduled conflict between DNA replication and transcription. Genome Res 25: 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnani N, Dutta A. 2011. The effect of the intra-S-phase checkpoint on origins of replication in human cells. Genes Dev 25: 621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaykov A, Nurse P. 2015. The spatial and temporal organization of origin firing during the S-phase of fission yeast. Genome Res 25: 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, McCarroll SA. 2012. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet 91: 1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib K, De Piccoli G. 2011. Surviving chromosome replication: the many roles of the S-phase checkpoint pathway. Philos Trans R Soc B Biol Sci 366: 3554–3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MH, Liu Q, Elledge SJ, Rosen JM. 2004. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 6: 45–59. [DOI] [PubMed] [Google Scholar]
- Lambert S, Mizuno K, Blaisonneau J, Martineau S, Chanet R, Fréon K, Murray JM, Carr AM, Baldacci G. 2010. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell 39: 346–359. [DOI] [PubMed] [Google Scholar]
- Lang GI, Murray AW. 2011. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol Evol 3: 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Rothstein R, Mortensen UH. 2001. Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci 98: 8276–8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, De S, Michor F. 2013. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun 4: 1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Mosqueda J, Maas NL, Jonsson ZO, Defazio-Eli LG, Wohlschlegel J, Toczyski DP. 2010. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature 467: 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Mackenzie A, Donaldson A, Zegerman P. 2011. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J 30: 4805–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune HJ, Danielson LS, Alvino GM, Collingwood D, Delrow JJ, Fangman WL, Brewer BJ, Raghuraman MK. 2008. The temporal program of chromosome replication: genomewide replication in clb5Δ Saccharomyces cerevisiae. Genetics 180: 1833–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickle KL, Ramanathan S, Rosebrock A, Oliva A, Chaudari A, Yompakdee C, Scott D, Leatherwood J, Huberman JA. 2007. Checkpoint independence of most DNA replication origins in fission yeast. BMC Mol Biol 8: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. 1991. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol 194: 795–823. [DOI] [PubMed] [Google Scholar]
- Muller CA, Nieduszynski CA. 2012. Conservation of replication timing reveals global and local regulation of replication origin activity. Genome Res 22: 1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PK, Arcangioli B, Baker SP, Bensimon A, Rhind N. 2006. DNA replication origins fire stochastically in fission yeast. Mol Biol Cell 17: 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Tsaponina O, Crabbé L, Keszthelyi A, Pantesco V, Chabes A, Lengronne A, Pasero P. 2012. dNTP pools determine fork progression and origin usage under replication stress. EMBO J 31: 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Gilbert DM. 2013. DNA replication timing. Cold Spring Harb Perspect Biol 5: a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Mulia JC, Gilbert DM. 2016. Replicating large genomes: divide and conquer. Mol Cell 62: 756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Mulia JC, Buckley Q, Sasaki T, Zimmerman J, Didier RA, Nazor K, Loring JF, Lian Z, Weissman S, Robins AJ, et al. 2015. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res 25: 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio Team. 2015. RStudio: integrated development for R. RStudio, Inc., Boston, MA: http://www.rstudio.com. [Google Scholar]
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. 2010. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res 20: 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santocanale C, Diffley JF. 1998. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395: 615–618. [DOI] [PubMed] [Google Scholar]
- Shechter D, Costanzo V, Gautier J. 2004. ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol 6: 648–655. [DOI] [PubMed] [Google Scholar]
- Shirahige K, Hori Y, Shiraishi K, Yamashita M, Takahashi K, Obuse C, Tsurimoto T, Yoshikawa H. 1998. Regulation of DNA-replication origins during cell-cycle progression. Nature 395: 618–621. [DOI] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR. 2009. Human mutation rate associated with DNA replication timing. Nat Genet 41: 393–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syljuåsen RG, Sørensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J. 2005. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 25: 3553–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS. 2002. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev 66: 630–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Umemori T, Endo S, Muramatsu S, Kanemaki M, Kamimura Y, Obuse C, Araki H. 2011. Sld7, an Sld3-associated protein required for efficient chromosomal DNA replication in budding yeast. EMBO J 30: 2019–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taricani L, Wang TSF. 2006. Rad4TopBP1, a scaffold protein, plays separate roles in DNA damage and replication checkpoints and DNA replication. Mol Biol Cell 17: 3456–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask M-B, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. 2013. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103. [DOI] [PubMed] [Google Scholar]
- van Bakel H, van Werven FJ, Radonjic M, Brok MO, van Leenen D, Holstege FCP, Timmers HTM. 2008. Improved genome-wide localization by ChIP-chip using double-round T7 RNA polymerase-based amplification. Nucleic Acids Res 36: e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyck E, Stasiak AZ, Stasiak A, West SC. 1999. Binding of double-strand breaks in DNA by human Rad52 protein. Nature 398: 728–731. [DOI] [PubMed] [Google Scholar]
- Wold MS. 1997. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem 66: 61–92. [DOI] [PubMed] [Google Scholar]
- Wu P-YJ, Nurse P. 2009. Establishing the program of origin firing during S phase in fission yeast. Cell 136: 852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P-YJ, Nurse P. 2014. Replication origin selection regulates the distribution of meiotic recombination. Mol Cell 53: 655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Ishii A, Kanoh Y, Oda M, Nishito Y, Masai H. 2012. Rif1 regulates the replication timing domains on the human genome. EMBO J 31: 3667–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegerman P, Diffley JFX. 2010. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 467: 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. 2014. Causes and consequences of replication stress. Nat Cell Biol 16: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z-X, Zhang M-J, Peng X, Takayama Y, Xu X-Y, Huang L-Z, Du L-L. 2013. Mapping genomic hotspots of DNA damage by a single-strand-DNA-compatible and strand-specific ChIP-seq method. Genome Res 23: 705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.