

Abstract
The SK-BR-3 cell line is one of the most important models for HER2+ breast cancers, which affect one in five breast cancer patients. SK-BR-3 is known to be highly rearranged, although much of the variation is in complex and repetitive regions that may be underreported. Addressing this, we sequenced SK-BR-3 using long-read single molecule sequencing from Pacific Biosciences and develop one of the most detailed maps of structural variations (SVs) in a cancer genome available, with nearly 20,000 variants present, most of which were missed by short-read sequencing. Surrounding the important ERBB2 oncogene (also known as HER2), we discover a complex sequence of nested duplications and translocations, suggesting a punctuated progression. Full-length transcriptome sequencing further revealed several novel gene fusions within the nested genomic variants. Combining long-read genome and transcriptome sequencing enables an in-depth analysis of how SVs disrupt the genome and sheds new light on the complex mechanisms involved in cancer genome evolution.
Genomic instability is one of the hallmarks of cancer, leading to widespread copy number variations, chromosomal fusions, and other sequence variations (Hanahan and Weinberg 2011). Structural variations, including insertions, deletions, duplications, inversions, or translocations at least 50 bp in size, are especially important to cancer development, as they can create gene fusions, amplify oncogenes, delete tumor suppressor genes, or cause other critical changes to contribute to the evolution of a cancer genome (Mitelman et al. 2007). Detecting and interpreting these structural variations is therefore a crucial challenge as we try to gain a more complete understanding of cancer genomes (Nik-Zainal et al. 2012).
Cancer genomics has been greatly aided by the advances in DNA sequencing technologies over the last 10 yr (Watson et al. 2013). The first whole-genome analysis of a cancer genome was reported in 2008 (Ley et al. 2008), and today large-scale efforts such as The Cancer Genome Atlas (Kandoth et al. 2013) or the International Cancer Genome Consortium (The International Cancer Genome Consortium 2010) have sequenced thousands of samples using short-read sequencing to detect and analyze commonly occurring mutations, especially single nucleotide and other small variations. However, these projects have performed somewhat limited analysis of structural variations, as both the false positive rate and the false negative rate for detecting structural variants from short reads are reported to be 50% or more (Sudmant et al. 2015; Huddleston et al. 2017). While short-read sequencing has no doubt revolutionized cancer genomics, this latter observation is troubling. Furthermore, the variations that are detected are rarely close enough to determine whether they occur in phase on the same molecule, limiting the analysis of how the overall chromosome structure has been altered.
Addressing this critical void, we sequenced the HER2 (ERBB2)-amplified breast cancer cell line SK-BR-3 using long-read sequencing from Pacific Biosciences. SK-BR-3 is one of the most widely studied breast cancer cell lines, with applications ranging from basic to preclinical research (Lewis Phillips et al. 2008; Navin et al. 2011; Ichikawa et al. 2012). SK-BR-3 was chosen for this study due to its importance as a basic research model for cancer and because the SK-BR-3 genome contains many of the common features of cancer alterations including a number of gene fusions, oncogene amplifications, and extensive rearrangements. Critically, the amplifications and genome complexity observed in SK-BR-3 have been demonstrated to be representative of patient tissues as well (Neve et al. 2006).
Taking full advantage of the benefits of the new long-read sequencing technology, we applied a split-read and within-read mapping approach to detect variants of different types and sizes. This allows us to develop a comprehensive map of structural variations in the cancer and study for the first time how the rearrangements have occurred with base-pair level accuracy. Furthermore, combining genomic variant discovery with Iso-Seq full-length transcriptome sequencing, we discover new isoforms and characterize several novel gene fusions, including some that required the fusion of three separate chromosome regions. Finally, using the reliable mapping and coverage information from long-read sequencing, we show that we can reconstruct the progression of rearrangements resulting in the amplification of the ERBB2 oncogene, including a previously unrecognized inverted duplication spanning a large portion of the region. Using long-read sequencing, we document a great variety of mutations including complex variants and gene fusions far beyond what is possible with alternative approaches.
Results
We sequenced the genome of SK-BR-3 using Pacific Biosciences (PacBio) SMRT long-read sequencing (Eid et al. 2009) to 71.9× coverage (based on the reference genome size) with an average read-length of 9.8 kb (Supplemental Fig. S1). For comparison, we also sequenced the genome using short-read Illumina paired-end and mate-pair sequencing to similar amounts of coverage. To investigate the relevant performance of long and short reads for cancer genome analysis, we perform an array of comparisons in parallel using both technologies.
Read mapping and copy number analysis
Long reads have more information to uniquely align to the genome than short reads do, resulting in overall better mapping qualities for long reads (Supplemental Fig. S2; Lee and Schatz 2012). Using BWA-MEM (Li 2013) to align both data sets, 69% of Illumina short paired-end reads (101-bp reads, 550-bp fragment length) align with a mapping quality of 60 compared to 91.61% of reads from the PacBio long-read sequencing library (Supplemental Fig. S2; Supplemental Table S1). We also observed a smaller GC bias in the PacBio sequencing compared to the Illumina sequence data which enables more robust copy number analysis and generally better variant detection overall (Supplemental Fig. S3).
The average aligned read depth of the PacBio data set across the genome is 54×, although there is a broad variance in coverage attributed to the highly aneuploid nature of the cell line (Supplemental Fig. S4). The short reads showed a few regions of extreme amplification (>100-fold) that were not detected by the long reads, although subsequent analysis showed these regions were highly enriched for low mappability regions (Dolgalev et al. 2017) in the genome and therefore most likely to be mapping artifacts (Supplemental Fig. S5). Using the long-read alignments, we segmented the genome into 4083 segments of different copy number states with an average segment length of 747.0 kbp. The unamplified chromosomal regions show an average coverage of 28×, which we consider the diploid baseline for this analysis. Thus, the average copy number is approximately twice the diploid level, which is consistent with previous results characterizing SK-BR-3 as tetraploid on average (Navin et al. 2011), and with any given locus being heterogeneous in copy number across the cell population.
Assuming a diploid baseline of 28×, the locus spanning the important ERBB2 oncogene (17q12) is one of the most amplified regions of the genome with an average of 33.6 copies (average read coverage of 470×). A few other regions show even greater copy number amplification, including the region surrounding MYC, with 38 copies. Other oncogenes are also amplified, with EGFR at seven copies and BCAS1 at 16.8 copies, while TPD52 lies in the middle of an amplification hotspot on Chromosome (Chr) 8 and is spread across eight segments with an average copy number of 24.8. The locus 8q24.12 containing the SNTB1 gene is the most amplified region of the genome with 69.2 copies (969× read coverage). In addition to being the most amplified protein-coding gene in this cell line, SNTB1 is also involved in a complex gene fusion with the KLHDC2 gene on Chr 14 (see below). Copy number amplifications are distributed throughout the genome across all chromosomes (Supplemental Fig. S5). Every chromosome has at least one segment that is tetraploid or higher, and these amplified regions account for about one third (1.07 Gbp) of the genome. Extreme copy number amplifications, above 10-ploid (>140× coverage), appear on 15 different chromosomes for a total of 61.1 Mbp, with half on Chr 8 (30.1 Mbp). There is a total of 21.3 Mbp of 20-ploid sequences across five chromosomes, with 20.0 Mbp on Chr 8 and 1.3 Mbp distributed across Chromosomes 17, 7, 21, and 1. In addition to containing the greatest number of base pairs of 20-ploid sequence, Chr 8 also has 101 segments of 20-ploid sequence compared to only four total segments from Chromosomes 7, 16, 17, and 21. Chr 8 thus has far higher levels of extreme copy number amplification than all other chromosomes combined.
Structural variant analysis
We aligned the long reads to the reference using NGMLR (Sedlazeck et al. 2018), a read mapping algorithm optimized for long single-molecule sequencing reads, and analyzed the alignments for structural variations using Sniffles (Sedlazeck et al. 2018). Sniffles was specifically designed for long-read SV analysis and identifies them from both split-read and within-read alignments requiring at least 10 split reads to call a structural variant. Sniffles found a total of 76,776 variants that were 10 bp or larger, and of these, 17,313 variants were structural variants (50 bp or larger), composed of 8909 (51%) insertions, 6947 (40%) deletions, 1018 (6%) duplications, 279 (2%) inversions, and <1% total of translocations and special combined variant types (Fig. 1). Our work with several other genomes shows that the vast majority of variants called using this combination of algorithms are correct (also see below) (Sedlazeck et al. 2018). Within all of the variants detected using the long reads, 1725 variants intersect transcribed regions of 361 of the 616 genes in the COSMIC Cancer Gene Census (Futreal et al. 2004), and 172 of these genes are hit by structural variants with a minimum size of 50 bp (Supplemental Tables S4, S8). Counting only sequences identified in GENCODE as exons, a total of 58 variants intersect the exons of 46 different Cancer Gene Census genes, including breast cancer genes such as APOBEC3B and CDH1. The deletion in APOBEC3B is consistent with a germline variant previously observed to fuse APOBEC3A and APOBEC3B, which is suggested to confer increased cancer susceptibility (Nik-Zainal et al. 2014).
Figure 1.

Variants found in SK-BR-3 with PacBio long-read sequencing. (A) Circos (Krzywinski et al. 2009) plot showing long-range (larger than 10 kbp or inter-chromosomal) variants found by Sniffles from split-read alignments, with read coverage shown in the outer track. (B) Variant size histogram of deletions and insertions from size 50 bp up to 1 kbp found by long-read (Sniffles) and short-read (SURVIVOR 2-caller consensus) variant calling, showing similar size distributions for insertions and deletions from long reads but not for short reads, where insertions are greatly underrepresented. (C) Sniffles variant counts by type for variants above 1 kbp in size, including translocations and inverted duplications.
For comparison, we also called structural variants from a standard paired-end short-read sequencing library, using our SURVIVOR algorithm (Jeffares et al. 2017) to form a high-quality consensus call set from three different short-read variant callers (Manta [Chen et al. 2016], DELLY [Rausch et al. 2012], and LUMPY [Layer et al. 2014]), requiring that at least two of these variant callers identified the same variant. We have found this approach reduces the false positive rate without substantially reducing sensitivity (Jeffares et al. 2017). The total number of short-read structural variants in the consensus set was 4174, composed of 2481 (59%) deletions, 603 (14%) translocations, 580 (14%) inversions, 448 (10%) duplications, and 62 insertions (1.4%). Comparing the counts, the short-read consensus has a much smaller number (only 24%) of total variants than the long-read set (see Supplemental Figs. S9–S16 for examples of variants not detected by the short-read analysis). This difference is largely driven by the lack of insertions in the short-read call sets: DELLY and Manta report a small number of insertions, but LUMPY does not attempt to report any (Rausch et al. 2012; Layer et al. 2014; Chen et al. 2016). To further address the limited number of insertions detected, we also ran a new insertion-finding algorithm called PopIns (Kehr et al. 2016). Using the recommended settings, PopIns finds 579 insertions that it could anchor to the genome, of which six were also found by DELLY and 11 were also found by Manta (two were found by both). Overall, this raised the total number of insertions detected by two or more short-read mapping algorithms to 77 (Figs. 1B, 2B). Supplemental Table S5 shows the count of variant calls of each type, and Figure 1C shows the counts for variants that are at least 1 kbp in size. For noninsertions, we also note that the short-read SV callers are highly enriched for false positive calls, especially false translocations (see below). The disagreement between the short- and long-read variant calls does not appear to be related to coverage. Using SAMtools (Li et al. 2009) bedcov, we found the mean short-read coverage for variants found by both short and long reads was 25.6× coverage at the breakpoint, while the short-read coverage for variants only detected by long reads was 51×.
Figure 2.
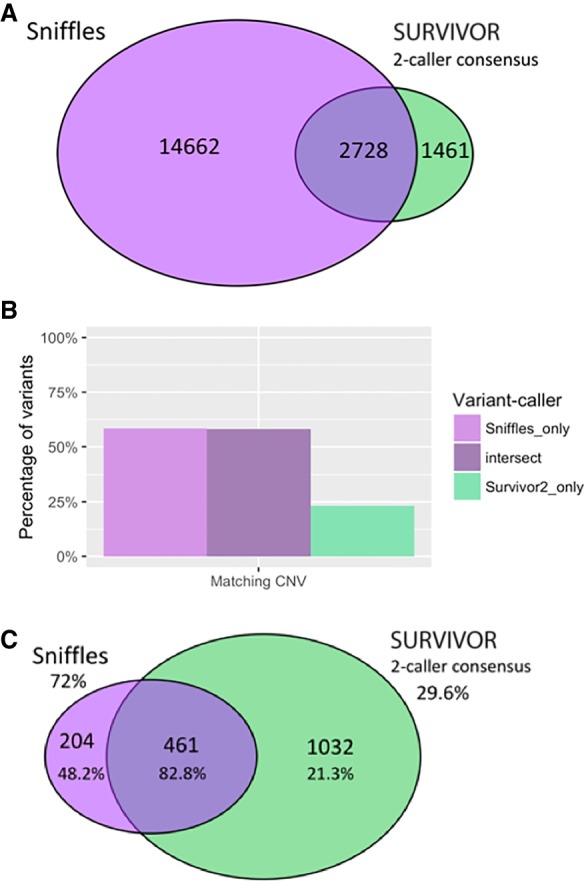
Comparing results of mapping and variant calling between PacBio and Illumina paired-end sequencing. (A) Venn diagram showing the intersection of structural variants between the Sniffles call set versus the SURVIVOR 2-caller consensus, with counts indicated. (B) Percentage of variant calls in each area of Venn diagram in A that have matching CNV calls within 50 kbp (the smallest segment allowed in segmentation), where a CNV is a difference in copy number (long-read sequencing) between segments of at least 28×, the diploid average. (C) Venn diagram showing the intersection of long-range variants between the Sniffles call set versus the SURVIVOR 2-caller consensus. Validation rates are shown as percentages below the counts for each category, and extrapolated overall validation rates are shown for Sniffles and SURVIVOR.
In parallel, we performed assembly-based variant calling with Assemblytics (Nattestad and Schatz 2016) using a de novo assembly of the long reads using FALCON (Chin et al. 2016) and the short reads using ALLPATHS-LG (Supplemental Note S1; Gnerre et al. 2011). The long-read assembly achieved a 2.4-Mbp contig N50 and showed good sensitivity for many structural variant types, especially insertions and deletions <1 kbp in size (Supplemental Figs. S6–S8) and found variations within many hundreds of ALU sequences (Supplemental Table S3). However, we also found this approach misses many long-range variants due to splitting of the assembly graph into contigs at or near the branch points caused by large variants. Therefore, the long-range variants are not always captured well within the contigs, and the evidence can be skewed to the end of the contigs where alignment and assembler errors are more common. Consequently, assembly-based variant calling is therefore not ideal for this class of variants. The short-read assembly was even more limited, as the contig N50 was only 3.2 kbp, and only a small number of SVs could be found (Supplemental Tables S2, S3).
Our initial expectation was that approximately the same number of insertions and deletions would be present due to normal human genetic variation. However, the long-read variant call set has a ratio of 1.28:1 insertions to deletions. This insertional bias has been seen previously and suggests an underestimate of the lengths of low-complexity regions in the human reference genome (Chaisson et al. 2015). In support of this analysis, using the repeat annotation tracks from the UCSC Genome Browser, we found that 52 Sniffles insertions are within annotated microsatellites, 5015 insertions are within simple repeats, and 6027 are within regions identified by RepeatMasker (Smit et al. 2013–2015), in agreement with the prior studies.
As long-range variants are of particular interest in cancer genomics, we performed several analyses specific to this subset of variants. We define long-range variants as those that are either (1) between different chromosomes, (2) connecting breakpoints at least 10 kbp apart within the same chromosome, or (3) inverted duplications. These long-range variants indicate novel adjacencies joining chromosomal regions that were originally distant in the genome. This causes novel sequences to be formed at the junction, potentially leading to gene fusions, large deletions or duplications, and other aberrant genomic features. Split reads provide a robust signal for detecting these long-range variants and chromosomal rearrangements. Within the long-read Sniffles call set, we found 665 variants in this long-range class (Fig. 1A; Supplemental Table S6), 125 of which were between different chromosomes. From the SURVIVOR short-read consensus calls, 1493 are long-range variants, with 603 of these being between different chromosomes.
Focusing on the long-range variants, we analyzed the intersections between the Sniffles and SURVIVOR (two-caller consensus) call sets. Compared to the SURVIVOR consensus call set, Sniffles detects the same 461 and an additional 204 variants, whereas the short-read SURVIVOR consensus detects an additional 1032 (Fig. 2A). We randomly selected 100 variants from each subset for PCR plus Sanger validation, with 100 calls from the intersect, 100 Sniffles calls not shared by SURVIVOR (two-caller consensus), and 100 SURVIVOR 2-calls not shared by Sniffles. Within each randomly selected group of 100 variants, some variant calls could not be validated due to primer issues or other technical issues, so the final validation rates are calculated as successful Sanger validation counts out of the total valid attempts. As expected, the variants called by both Sniffles and SURVIVOR had the highest validation rate of 82.8% (77/93) (Supplemental Table S7). Of the 16 variants called by both short- and long-read approaches that failed validation, five calls were translocations, and the rest were paracentric. All of the reported variants had at least 10 supporting long reads (the minimum threshold we used for Sniffles), and ranged from 10 to 31 supporting reads (mean: 17.6×), except for one outlier with 113 supporting reads. Seven of the PCR attempts produced no recognizable product even after multiple attempts and two primer designs. The other nine attempts produced a weak product or a multibanded product but failed to report the expected sequence during Sanger validation, and so we consider them as failing validation. Given the strong support from both PacBio and Illumina sequencing, we attribute the failures as either inadequate primer design and/or other systematic errors in the validation protocol. Of the calls unique to one method, long-read Sniffles variants have a validation rate more than twice that of the short-read variants: 48.2% (26/54) compared to 21.3% (17/80). Furthermore, extrapolating the validation rates for these subsets, the overall validation rate of Sniffles calls is 72%, while the SURVIVOR 2-calls is only 29.6%. We emphasize that this is the validation rate for the most complicated long-range variants present in the genome, and our work with structural variant detection in other long-read data sets reached 94% to over 99% (Sedlazeck et al. 2018). We also note that several of these long-range variants were previously found through RNA-seq and confirmed with mate-pairs in previous studies (Table 1; Kim and Salzberg 2011).
Table 1.
Gene fusions with RNA evidence from Iso-Seq and DNA evidence from SMRT DNA sequencing where the genomic path is found using SplitThreader from Sniffles variant calls

Further supporting this higher validation rate of long-read variants, the Sniffles variants were also more likely to occur at the breakpoint of a copy number variant than their short-read counterparts. Specifically, 58.3% of the Sniffles unique variants show a matching copy number variant, compared to only 23.2% of the SURVIVOR unique consensus variants, where 58.1% of the variants shared by both sets show a matching CNV (Fig. 2B,C). Similar results were also found using the short reads for segmentation. The high rate of CNV matching for the shared set indicates that copy number evidence can serve as a measure of confidence in a variant call. CNV matching provides additional support for the majority of the Sniffles unique calls, though it does not exclude others that may be copy number neutral variants such as balanced translocations. The low rate of CNV support for the short-read consensus suggests that a larger proportion of these variants are either false positives or Sniffles is not sensitive enough to capture them. Reducing the threshold in Sniffles to five split reads (instead of the 10 split reads employed throughout this analysis) captures another 134 of the short-read consensus variants out of 1032, so there appears to be little long-read evidence of these additional variants.
Characterization of the ERBB2 copy number amplification
Chromosome 8 is the most aberrant chromosome in the genome of SK-BR-3, accounting for over half of the highly amplified sequence in the genome and almost half of the long-range variants. Most of the new connections between sequences originally on Chr 8 are clustered in three major hotspots. The ERBB2 oncogene, originally located on Chr 17, is amplified to, on average, 32.8 copies, while most of the remainder of Chr 17 is present in just two copies, consistent with selection against gains of tumor suppressor proteins on Chr 17, such as TP53 and BRCA1. The amplified region that includes ERBB2 contains five translocations (Fig. 3A) into the hotspot regions on Chr 8, as well as an inverted duplication. Each of the six variants mark the site of an abrupt change in copy number, meaning they mediated the overall amplification to 32.8-fold, and all six were validated by directed PCR and Sanger sequencing. It is notable that the inverted duplication was not identified by any of the short-read variant callers, although it is clearly visible in the long-read alignments and is automatically identified by Sniffles.
Figure 3.

Reconstruction of the copy number amplification of the ERBB2 oncogene. (A) Copy number and translocations for the amplified region on Chr 17 that includes ERBB2 showing the relations to Chr 8. Note Chr 8 has extensive rearrangements shown by the green intra-chromosomal arcs. (B) Sequence of events that best explains the copy number and translocations found in this region. Segment 1 (orange) first translocated into Chr 8, followed by the segment 2 (yellow) translocating to a different place on Chr 8. Then, the segment 3 (green) was duplicated from segment 2 by an inversion of the piece between variants D and E along with a 1.5-Mb piece of Chr 8 that was attached at variant E, all of which then attached at variant C. The whole green segment including the 1.5 Mb of Chr 8 then underwent an inverted duplication at variant D. The purple segment could have come from the orange, yellow, or green sequences since it only shares breakpoint A. Additionally, there is a deletion of 10,305 bp between breakpoints D and E.
The ERBB2 oncogene appears to have been amplified to such a great extent due to its association with the highly mutated hotspots in Chr 8 and suggests a remarkably complex and punctuated mutational history (Fig. 3B). The long-range variants within the amplified region containing ERBB2 were studied to determine whether the number of split and reference-spanning reads at each breakpoint are consistent with the copy number profile, which was found to be true for all five translocations and the inverted duplication. In order to determine the order in which these six events took place, we derived the most parsimonious reconstruction factoring in a couple of important assumptions established within population genetics, analogous to the widely used infinite sites model used in population genetics (Kimura 1969). First, we assume that variants we observe have taken place only once, since it is extremely unlikely that the same long-range variant at the same two breakpoints would recur down to base-pair resolution. Second, once a variant has occurred and created an observable breakpoint, the breakpoint would not be repaired in some copies of the sequence and not others. Therefore, all reference-spanning reads represent an ancestral state and not a repaired breakpoint. In this analysis, the long-range variants have more reliability to reconstruct the genomic history rather than SNPs because those two simplifying assumptions are extremely unlikely to be violated when two breakpoints are involved.
Given these conditions, we conclude that the orange segment (A–F) in Figure 3B must have translocated first, as the other breakpoints are shared on the leftmost edge (variant A). Next, the yellow segment shown in Figure 3B derived from the orange segment because otherwise, variant A must have occurred more than once. Applying the same logic, the green segment must have derived from the yellow segment because it shares variant E, and it is not yellow derived from green because that would violate assumption 2 by requiring that variants C and D were repaired. Variants C and D appear to co-occur in the same sequences because the copy number is the same between those two parts of the green segment and because the other sides of the variants are at breakpoints within only 1.5 Mb of each other. The only uncertainty in the ordering of events is that the purple segment could have derived from any of the segments sharing variant A: the orange, yellow, or green segments. There is not enough information to determine which of these segments it came out of, but we can conclude that it only came out of one of them, given assumption 1 that precludes multiple occurrences of the same variant.
Complex gene fusions captured fully by long reads
In addition to genome sequencing, we performed long-read transcriptome sequencing using PacBio Iso-Seq to capture full-length transcripts. Although traditional short-read RNA-seq approaches allow isoform quantification, in many cases these reads are too short to reconstruct all isoforms, even with paired-end analysis, exon abundance, or other indirect measurements. Instead, long reads overcome such limitations by spanning multiple exon junctions and often covering complete transcripts. This makes it possible to exactly resolve complex isoforms and identify large transcripts, without the need for statistical inference (Sharon et al. 2013; Weirather et al. 2015; Wang et al. 2016).
Iso-Seq reads were consolidated into isoforms using the SMRTAnalysis Iso-Seq pipeline (Gordon et al. 2015). In total, 1,692,379 isoforms (95.7%) mapped uniquely to the reference genome. The Iso-Seq RNA sequence reads indicated a total of 53 putative gene fusions each with at least five Iso-Seq reads of evidence (Supplemental Table S9). We further refined this candidate set using SplitThreader (Nattestad et al. 2016a) to exclude variants not supported by genomic structural variations, especially to account for any residual sequencing error or mapping errors in the data. Specifically, SplitThreader searches for a path of structural variations linking the pair of genes in the putative gene fusion, requiring that the variants bring the genes together within a 1-Mbp distance. Out of 53 candidate gene fusions, SplitThreader found genomic evidence for 39 of these: 15 are the high-quality gene fusions with a genomic path between the gene bodies of, at most, 10 kbp, shown in Table 1; 19 fusions overlap the first 15 (sharing the same variant and often one of the genes), and five fusions (three nonoverlapping) have paths longer than 10 kbp, leaving 14 candidate gene fusions with no genomic paths.
Three of the gene fusions had no single variant directly linking the genes, but SplitThreader discovered that the genes could be linked by a series of two or even three variants. One of these, CPNE1-PREX1 had been discovered previously using RNA-seq data and validated using genomic PCR as a two-variant gene fusion (Chen et al. 2013). We have now confirmed this by showing long reads that not only capture the two variants but capture them together in a single read along with robust alignments to both genes (Supplemental Fig. S19). CYTH1-EIF3H had been discovered previously with RNA-seq and been validated with RT-PCR (Edgren et al. 2011), but it was not known to be a “2-hop” gene fusion (taking place through a series of two variants) until now. This fusion was also captured in full by several individual SMRT-seq reads that contain both variants and have alignments in both genes (Supplemental Fig. S18). Interestingly, we discovered a novel 3-hop gene fusion between KLHDC2 and SNTB1, which has been misreported before as only taking place through two variants (Asmann et al. 2011). We observe both the previously reported 2-hop path (600,326 bp) and this additional 3-hop path (9837 bp), which would both result in the same gene fusion. Given the shorter distance for the 3-hop gene fusion, we were able to find direct linking evidence for the 3-hop fusion between these two genes. We observe 37 reads that stretch from one gene to the other through all three variants, bringing the genes within a distance of just 9837 bp across three different chromosomes (Fig. 4; Supplemental Fig. S17). Due to the long distance between the genes through the previously reported 2-hop fusion, we believe the 3-hop fusion is more likely to produce the observed fusion transcript.
Figure 4.

The KLHDC2-SNTB1 gene fusion in SK-BR-3 occurs through a series of three variants and is directly observed to link the two genes in several individual SMRT-seq reads (A), one of which is shown in detail in B.
Most of the gene fusions observed are contained within a few of the most rearranged chromosomes. Four gene fusions take place within Chr 20, which is rich in intra-chromosomal variants, while Chr 8 is involved in six gene fusions both intra- and inter-chromosomally. The genomic variant fusing TATDN1 and GSDMB is one of the variants contributing to the amplification of the ERBB2 oncogene. All of the gene fusions are captured fully with individual SMRT-seq reads that align to both genes, with some novel variants affecting important cancer genes (e.g., PVT1 and RAD51B). See long-read alignments spanning all 15 gene fusions in Supplemental Note S2 and Supplemental Figures S20–S32.
Discussion
Advances in long-read sequencing have produced a resurgence of reference quality genome assemblies and exposed previously hidden genomic variation in healthy human genomes (Chaisson et al. 2015; Pendleton et al. 2015; Seo et al. 2016). Now, we have applied long-read sequencing to explore the hidden variation in a cancer genome and have discovered nearly 20,000 structural variations present, most of which cannot be found using short-read sequencing and many are intersecting known cancer genes. More than twice as many of the copy number amplifications could be explained through long-range variants identified by long-read sequencing compared to short-read sequencing. We further found the ERBB2 oncogene to be amplified through a complex series of events initiated by a large translocation into the highly rearranged hotspots of Chr 8, where the sequence was then copied dozens of times more, with further translocations and inverted duplications resolved only by the long reads. Furthermore, we find 20 additional inverted duplications throughout the genome, highlighting the importance of this underreported structural variation type. Overall, using long-read sequencing, we see that far more bases in the genome are affected by structural variation compared to SNPs.
Using long-read transcriptome sequencing, we capture full gene fusion isoforms, and by combining this with our genomic variant discovery, we discover several novel gene fusions in this seemingly well-characterized cell line. Notably, we uncover for the first time a gene fusion that takes place through a series of three variants: KLHDC2-SNTB1 through the fusions of Chromosomes 8, 14, and 17, captured fully by 37 genomic SMRT-seq reads. In a single cancer genome, we discovered three gene fusions that take place through a series of two or more variants, suggesting that such multi-hop gene fusions could also be common in other cancers, although they will be exceedingly difficult to discover using short-read sequencing. Conducting a similar search for multi-hop gene fusions in other highly rearranged cancers could reveal other instances of a complex type of variation.
The differences between variants found with long reads versus short reads is likely due to an interplay of sequencing technology and algorithmic approach. The primary advantages of long reads are better mapping through repetitive elements that often flank SVs and an increase in the probability that a SV breakpoint will be spanned by individual reads. These advantages are offset by the increased error rate that makes them more difficult to map and analyze, although new mapping and SV detection tools are now available that can largely overcome these challenges. For short-read analysis, using paired-end or mate-pair sequencing can partially offset the short-read lengths, but they still have relatively poor sensitivity using current approaches. In addition to the results presented here, a previous study by Hillmer et al. (2011) analyzed SVs in SK-BR-3 using long-range mate-pairs. In this work, they generated 68.4× physical coverage of mate-pairs averaging 8.2 kbp and yet only found 1145 SVs, most of which were deletions (606), followed by inversions (191), and other complex intra-chromosomal rearrangements (158). This represents <10% of the variants that we could detect using long reads. Further algorithmic advances may be possible to improve accuracy, and we currently recommend using a consensus approach for short-read analysis to alleviate false positives. It may also be possible to improve sensitivity of certain types of variants using focused methods. For example, ARC-SV (Arthur et al. 2018) and SVelter (Zhao et al. 2016) were recently developed to focus on distal and inverted duplications from short-read sequencing, although these classes of variations are a minority in this sample. What is most needed is a robust method to detect insertions from short reads, as they are currently not well captured by either mapping-based or assembly-based approaches.
We have shown that long-read sequencing can expose complex variants with great certainty and context, suggesting that more multi-hop gene fusions, inverted duplications, and complex events may be found in other cancer genomes. Having observed complex variants such as inverted duplications with the increased informational context of long reads, the resulting variant signatures could make these events more observable even using standard short-read sequencing. However, there may be many other types of complex variations present in other cancer genomes that were not found in SK-BR-3, so it is essential to continue building a catalog of these variant types using the best available technologies. Long-read sequencing is an invaluable resource to capture the complexity of structural variations on both the genomic and transcriptomic levels, and we anticipate widespread adoption for research and clinical practice as the costs further decline.
Methods
Sequencing
Long-read sequencing was performed using the Pacific Biosciences Single-Molecule Real-Time (SMRT) sequencing technology with P6C4 SMRT cell chemistry. After selecting the longest subread from each polymerase PacBio read, our sequencing of SK-BR-3 yielded a mean read length of 9872 bp, where the longest read was 71,518 bp. Total coverage of the genome is 71.9× (79.0× if redundant sequences from the same polymerase reads are included) where × refers to the number of reads that cover the average genomic base. The coverage of reads at least 10 kbp long is 51.0×, and the coverage of reads at least 20 kbp long is 13.3×. These read depth values and those in Supplemental Figure S1B are based on a female genome size of 3,101,804,739 bp, the total lengths of Chromosomes 1–22 and Chr X in hg19.
For short-read variant calling, Illumina sequencing was performed on a 550-bp paired-end library (2 × 250 bp). This library produced a total of 795,942,102 reads and 64.2× genome coverage based on the same female genome size. For the short-read assembly, Illumina sequencing was performed on 180-bp paired-end overlapping library (2 × 100-bp reads), as well as 2–3 kbp and 5–10 kbp mate-pair libraries.
Alignment and variant calling
The hg19 reference genome (the 1000 Genomes version) was used for all analysis. Results aligning to GRCh38 are expected to be similar as no major differences were introduced in the ERBB2 locus and only a minority of the bases changed genome-wide. Reads were aligned to the reference using NGMLR (v0.2.1) (Sedlazeck et al. 2018), and Sniffles (v1.0.6) (Sedlazeck et al. 2018) was used to call variants from long-read split alignments using the recommended parameters. Variants were called on the short-read variant-calling Illumina sequencing data set using Manta (Chen et al. 2016), DELLY (Rausch et al. 2012), LUMPY (Layer et al. 2014), and PopIns (Kehr et al. 2016), and a consensus was taken using SURVIVOR (Jeffares et al. 2017) with the recommended parameters, requiring two of these variant callers to support the same variant , except where noted otherwise. Because there is often variability in the reported location of SVs, this process allows for merging of SVs that have breakpoints within 1 kbp of each other, as long as the type of variant is the same. For purposes of intersecting Sniffles and SURVIVOR long-range variant call sets, for which breakpoints must be 10 kbp apart, we used BEDTools (Quinlan and Hall 2010) pair-to-pair with a slop parameter of 1000 to test if variants shared both breakpoints with matching strands within a 1000-bp range. Copy number segmentation was computed using SplitThreader (Nattestad et al. 2016a), which internally uses the DNAcopy R package for circular binary segmentation (Olshen et al. 2004). The SplitThreader source code is also available as a Supplemental File. The Circos plot in Figure 1A was generated using Circa (http://omgenomics.com/circa). Cancer gene intersects were determined using BEDTools pairtobed to intersect Sniffles variants down to 10 bp in size with the GENCODE hg19 annotation (Harrow et al. 2012) and filtered by matches to the COSMIC Cancer Gene Census (Futreal et al. 2004).
Mapping comparison
In order to compare the mappability of long and short reads, we aligned both the paired-end Illumina sequencing and the PacBio long-read sequencing data sets to the hg19 reference genome using BWA-MEM (Li 2013). The Illumina sequencing was performed using a 550-bp paired-end library with each read being ∼250 bp of sequence. We trimmed these reads to 101 bp and compared both of these against the PacBio data set. All three read sets were aligned using default parameters, except that the PacBio reads were aligned using the pacbio alignment mode in BWA-MEM (-x pacbio). The maximum mapping quality in BWA-MEM is 60, and the minimum is 0. Using the same aligner allows us to better compare mapping quality scores for the reads. We analyzed the mapping quality from each type of sequencing in two different ways, by individual reads and by binned windows in the genome. First, we selected the best alignment by mapping quality for each read and counted the number of reads in each category: mapping quality of 60, mapping quality between 1 and 59, mapping quality of 0, or unmapped. Alignment of PacBio sequence reads resulted in 91.6% of reads mapping with a mapping quality of 60, compared to only 71.2% of Illumina reads (69.0% of the 101-bp trimmed reads). A greater fraction of reads from PacBio long-read sequencing map uniquely to the genome compared to short reads from Illumina sequencing (Supplemental Fig. S2; Supplemental Table S1).
In order to determine the effect of GC content (the fraction of guanine and cytosine as opposed to adenine and thymine in a particular region), we counted the GC-fraction of each 10-kbp window in the genome, excluding those containing Ns in the reference, and calculated the read coverage from each data set. The read depth of each 10-kb bin is shown in Supplementary Figure S3 on a log scale versus the GC fraction, along with a Lowess fit for each data set. There is a higher GC-bias in the Illumina data sets compared to the PacBio data set, as seen by a lower read depth in bins with a higher GC fraction, while for SMRT sequencing there is a much lower bias.
To determine the read depth per chromosome, we used BEDTools to find the distribution of read depth for each chromosome for the PacBio, Illumina 250-bp, and Illumina 101-bp data sets. These are shown as a violin plot of Gaussian kernel distributions for each chromosome in Supplemental Figure S4. The shapes of the distributions are largely consistent between sequencing technologies.
Iso-Seq and gene fusion analysis
PacBio Iso-Seq sequencing was performed in four size batches (0.8–2, 2–3, 3–5, and 5–10 kb). The Iso-Seq data were processed using the SMRTAnalysis (version 2.3) Iso-Seq pipeline, which generated 441,932 high-quality (HQ), full-length Quivered consensus sequences, which were then aligned using GMAP (Wu and Watanabe 2005; Wu et al. 2016) to hg19. The GMAP alignments were filtered using quality scores from BWA-MEM (Li 2013) alignments by removing any reads that in BWA-MEM have alignments below a mapping quality of 60. The remaining GMAP alignments were used for gene fusion detection using ToFU (Gordon et al. 2015). Aligned fusion transcripts identified by ToFU were intersected with the GENCODE hg19 annotation (Harrow et al. 2012), and the total number of full-length reads supporting fusions between each pair of genes was counted. All putative gene fusions with at least five full-length Iso-Seq reads from ToFU were input into SplitThreader (Nattestad et al. 2016a) to identify those with any combination of long-range variants that place the genes within 100 kbp of each other. Gene fusion alignments were visualized and figures generated using Ribbon (Nattestad et al. 2016b). The Ribbon source code is also available as a Supplemental File.
Data access
Illumina and PacBio sequencing data from this study have been submitted to the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA476239. Variant calls from all callers are available as a Supplemental File. The alignments, assemblies, and variant calls are also available at: http://www.schatz-lab.org/publications/SKBR3/.
Competing interest statement
C.-S.C. and E.T. are full-time employees of Pacific Biosciences. W.R.M. has participated in Illumina-sponsored meetings over the past four years and received travel reimbursement and an honorarium for presenting at these events. Illumina had no role in decisions relating to the study/work to be published, data collection and analysis of data, or the decision to publish. W.R.M. has participated in Pacific Biosciences-sponsored meetings over the past three years and received travel reimbursement for presenting at these events. W.R.M. is a founder and share holder of Orion Genomics, which focuses on plant genomics and cancer genetics. W.R.M. and M.C.S. are SAB members for RainDance Technologies, Inc.
Supplementary Material
Acknowledgments
We thank DNAnexus for their assistance in assembling the genome. This work has been supported by the National Science Foundation (DBI-1350041), the National Institutes of Health (R01-HG006677, UM1-HG008898), the Cold Spring Harbor Laboratory (CSHL) Cancer Center (Support Grant 5P30CA045508), the Watson School of Biological Sciences at CSHL through a training grant (5T32GM065094) from the US National Institutes of Health, and by Pacific Biosciences. T. Baslan is supported by the William C. and Joyce C. O'Neil Charitable Trust, Memorial Sloan Kettering Single Cell Sequencing Initiative.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.231100.117.
References
- Arthur JG, Chen X, Zhou B, Urban AE, Wong WH. 2018. Detection of complex structural variation from paired-end sequencing data. bioRxiv 10.1101/200170. [DOI]
- Asmann YW, Hossain A, Necela BM, Middha S, Kalari KR, Sun Z, Chai HS, Williamson DW, Radisky D, Schroth GP, et al. 2011. A novel bioinformatics pipeline for identification and characterization of fusion transcripts in breast cancer and normal cell lines. Nucleic Acids Res 39: e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, et al. 2015. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517: 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Navin NE, Wang Y, Schmidt HK, Wallis JW, Niu B, Fan X, Zhao H, McLellan MD, Hoadley KA, et al. 2013. BreakTrans: uncovering the genomic architecture of gene fusions. Genome Biol 14: R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, Saunders CT. 2016. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32: 1220–1222. [DOI] [PubMed] [Google Scholar]
- Chin C-S, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O'Malley R, Figueroa-Balderas R, Morales-Cruz A, et al. 2016. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13: 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgalev I, Sedlazeck F, Busby B. 2017. DangerTrack: a scoring system to detect difficult-to-assess regions. F1000Res 6: 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgren H, Murumagi A, Kangaspeska S, Nicorici D, Hongisto V, Kleivi K, Rye IH, Nyberg S, Wolf M, Borresen-Dale A-L, et al. 2011. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol 12: R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323: 133–138. [DOI] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. 2004. A census of human cancer genes. Nat Rev Cancer 4: 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci 108: 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SP, Tseng E, Salamov A, Zhang J, Meng X, Zhao Z, Kang D, Underwood J, Grigoriev IV, Figueroa M, et al. 2015. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS One 10: e0132628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22: 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillmer AM, Yao F, Inaki K, Lee WH, Ariyaratne PN, Teo ASM, Woo XY, Zhang Z, Zhao H, Ukil L, et al. 2011. Comprehensive long-span paired-end-tag mapping reveals characteristic patterns of structural variations in epithelial cancer genomes. Genome Res 21: 665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston J, Chaisson MJP, Steinberg KM, Warren W, Hoekzema K, Gordon D, Graves-Lindsay TA, Munson KM, Kronenberg ZN, Vives L, et al. 2017. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res 27: 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa T, Sato F, Terasawa K, Tsuchiya S, Toi M, Tsujimoto G, Shimizu K. 2012. Trastuzumab produces therapeutic actions by upregulating miR-26a and miR-30b in breast cancer cells. PLoS One 7: e31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Cancer Genome Consortium. 2010. International network of cancer genome projects. Nature 465: 966–966.20559394 [Google Scholar]
- Jeffares DC, Jolly C, Hoti M, Speed D, Shaw L, Rallis C, Balloux F, Dessimoz C, Bahler J, Sedlazeck FJ. 2017. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun 8: 14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. 2013. Mutational landscape and significance across 12 major cancer types. Nature 502: 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehr B, Melsted P, Halldórsson BV. 2016. PopIns: population-scale detection of novel sequence insertions. Bioinformatics 32: 961–967. [DOI] [PubMed] [Google Scholar]
- Kim D, Salzberg SL. 2011. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol 12: R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1969. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61: 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer RM, Chiang C, Quinlan AR, Hall IM. 2014. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15: R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Schatz MC. 2012. Genomic dark matter: the reliability of short read mapping illustrated by the genome mappability score. Bioinformatics 28: 2097–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blättler WA, Lambert JM, Chari RVJ, Lutz RJ, et al. 2008. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody–cytotoxic drug conjugate. Cancer Res 68: 9280–9290. [DOI] [PubMed] [Google Scholar]
- Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. 2008. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456: 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303.3997 [q-bio.GN].
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F. 2007. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 7: 233–245. [DOI] [PubMed] [Google Scholar]
- Nattestad M, Schatz MC. 2016. Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32: 3021–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattestad M, Alford MC, Sedlazeck FJ, Schatz MC. 2016a. SplitThreader: exploration and analysis of rearrangements in cancer genomes. bioRxiv 10.1101/087981. [DOI]
- Nattestad M, Chin C-S, Schatz MC. 2016b. Ribbon: visualizing complex genome alignments and structural variation. bioRxiv 10.1101/082123. [DOI]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. 2011. Tumour evolution inferred by single-cell sequencing. Nature 472: 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe J-P, Tong F, et al. 2006. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10: 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, et al. 2012. The life history of 21 breast cancers. Cell 149: 994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Wedge DC, Alexandrov LB, Petljak M, Butler AP, Bolli N, Davies HR, Knappskog S, Martin S, Papaemmanuil E, et al. 2014. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat Genet 46: 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. 2004. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5: 557–572. [DOI] [PubMed] [Google Scholar]
- Pendleton M, Sebra R, Pang AWC, Ummat A, Franzen O, Rausch T, Stütz AM, Stedman W, Anantharaman T, Hastie A, et al. 2015. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat Methods 12: 780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. 2012. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28: i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, Haeseler von A, Schatz MC. 2018. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 14: 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J-S, Rhie A, Kim J, Lee S, Sohn M-H, Kim C-U, Hastie A, Cao H, Yun J-Y, Kim J, et al. 2016. De novo assembly and phasing of a Korean human genome. Nature 538: 243–247. [DOI] [PubMed] [Google Scholar]
- Sharon D, Tilgner H, Grubert F, Snyder M. 2013. A single-molecule long-read survey of the human transcriptome. Nat Biotechnol 31: 1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2013–2015. RepeatMasker Open-4.0. http://www.repeatmasker.org. [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, et al. 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Tseng E, Regulski M, Clark TA, Hon T, Jiao Y, Lu Z, Olson A, Stein JC, Ware D. 2016. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat Commun 7: 11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson IR, Takahashi K, Futreal PA, Chin L. 2013. Emerging patterns of somatic mutations in cancer. Nat Rev Genet 14: 703–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirather JL, Afshar PT, Clark TA, Tseng E, Powers LS, Underwood JG, Zabner J, Korlach J, Wong WH, Au KF. 2015. Characterization of fusion genes and the significantly expressed fusion isoforms in breast cancer by hybrid sequencing. Nucleic Acids Res 43: e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. 2005. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21: 1859–1875. [DOI] [PubMed] [Google Scholar]
- Wu TD, Reeder J, Lawrence M, Becker G, Brauer MJ. 2016. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. Methods Mol Biol 1418: 283–334. [DOI] [PubMed] [Google Scholar]
- Zhao X, Emery SB, Myers B, Kidd JM, Mills RE. 2016. Resolving complex structural genomic rearrangements using a randomized approach. Genome Biol 17: 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.