

 A biphenyl amide–tryptamine hybrid as a selective 5-HT2B antagonist.
A biphenyl amide–tryptamine hybrid as a selective 5-HT2B antagonist.
Abstract
We designed and synthesized a new biphenyl amide–tryptamine hybrid molecule 7 utilizing a pharmacophore-based approach as a 5-HT2B antagonist. The hybrid compound 7 was evaluated for its affinity to a panel of seven 5-HT receptors, demonstrating high selectivity for the 5-HT2B receptor. Functional assays revealed potent antagonism of 5-HT2B by 7 with an IC50 value of 14.1 nM. Moreover, compound 7 possessed a desirable in vitro pharmacokinetic profile and maintained its antagonistic potency in the presence of physiological concentrations of serum proteins. The design approach implemented in this investigation would facilitate the development of a second generation of highly selective and potent 5-HT2B antagonists.
Introduction
G protein-coupled receptors (GPCRs) represent the most prominent class of drug discovery targets owing to their key role in cell signalling events, and approximately one third of approved drugs rely on interactions with GPCRs.1 The 5-hydroxytryptamine 2B (5-HT2B) receptor, a member of the GPCR family, participates significantly in the regulation of cardiac function, gastrointestinal motility and CNS activity.2 Consequently, selective 5-HT2B antagonists have been explored as promising therapeutics for migraines, pulmonary arterial hypertension (PAH) and cardiac failure.3–5 Additionally, recent studies showed that the 5-HT2B receptor mediates the excitatory effects of serotonin in the human colon indicating that antagonists of this receptor might be valuable for the treatment of irritable bowel syndrome (IBS).5
Continuous research efforts have advanced several 5-HT2B antagonists into clinical trials, however, there have been no 5-HT2B antagonists clinically approved up to now.6 This is mainly because of the insufficient selectivity displayed by reported 5-HT2B antagonists against 5-HT2 receptor subtypes.7 In addition, the poor pharmacokinetic profiles of lead 5-HT2B antagonists have hindered their clinical translational studies.3a We report herein a biphenyl amide–tryptamine hybrid scaffold as a selective 5-HT2B antagonist utilizing a pharmacophore map of the recently identified structure of the 5-HT2B receptor. Moreover, the likelihood of success of the hybrid scaffold is further investigated through in vitro pharmacokinetic profiling.
Rational design
GPCR structures display a common topology characterized by seven transmembrane (TM) helices interspersed with three intra-(ILs) and extracellular loops (ELs). The high degree of homology between the 5-HT2B receptor and other relevant GPCR structures in the transmembrane (TM) segments has significantly limited the development of selective 5-HT2B antagonists.5a Recent identification of the crystal structure of the 5-HT2B receptor (PDB ID: ; 4IB4) has enabled structure-based design strategies seeking selective antagonists against this protein.8 Taking into account the pronounced conformational changes of agonist-bound and antagonist-bound GPCR structures, a doxepin induced-fit model was recently introduced for the 5-HT2B receptor which contributed to the discovery of triazine-based ligands as selective 5-HT2B antagonists.9 The triazine-based ligands occupy an orthosteric binding pocket in the 5-HT2B–ergotamine complex without completely overlapping with the ergotamine binding site. The orthosteric binding site identified for 5-HT2B antagonists based on the doxepin induced-fit model of the 5-HT2B receptor (PDB ID: ; 4IB4) and the receptor-based pharmacophore map are shown in Fig. 1A and B, respectively. The required pharmacophoric features for binding include: (a) formation of a salt bridge interaction between a protonated amine group in the ligand and an Asp3.32 residue; (b) a hydrophobic group in the ligand that occupies the region of unfavourable water in the receptor; (c) additional hydrogen-bond formation with an Asn6.55 residue of the binding pocket. The selectivity of this pharmacophore model arises from its ability to form additional hydrophobic interactions with binding region 1 near ECL II (Fig. 1A). The 5-HT2B receptor is characterized by uniquely possessing three additional residues located in the ECL II region which enables the formation of the adjacent binding region 1 consisting of Trp3.28, Leu3.29 and Val7.39 residues that other 5-HTRs lack. In addition, the ability to form an additional hydrogen bond with an Asn6.55 residue of the receptor further contributes to the selectivity in comparison with other 5-HTRs lacking this residue (for example 5-HT1B). Finally, the hydrophobic binding region 2 (Fig. 1A) consisting of Phe6.51, Phe6.52 and Trp6.48 residues is identified in the generated pharmacophore map. Incorporation of sterically expansive groups into 5-HT2B antagonists that are capable of exerting hydrophobic interactions with binding region 2 was found to be crucial for the antagonist activity.9
Fig. 1. (A) Orthosteric binding site for 5-HT2B antagonists based on the doxepin induced-fit model of the 5-HT2B receptor (PDB ID: ; 4IB4). (B) Receptor-based pharmacophore map generated for 5HT2B antagonist interaction. The pharmacophore color coding is yellow for hydrophobic regions, blue for ionisable groups and green for hydrogen donors. TM: transmembrane; ECL: extracellular loop.

Compound 1 (Fig. 2) was identified as a lead 5-HT2B antagonist from a high throughput screening campaign owing to its potent antagonism of the 5-HT2B receptor3a which is comparable to the SmithKline antagonist SB215505 2 (ref. 10) and the Roche antagonist RS-127445 3 (ref. 11) (Fig. 2). Interestingly, compound 1 also displayed minimal activity against 5-HT2A and 5-HT2C receptors rendering it a valuable lead to achieve selective antagonism of 5-HT2B.
Fig. 2. Chemical structures of potent 5-HT2B antagonists.

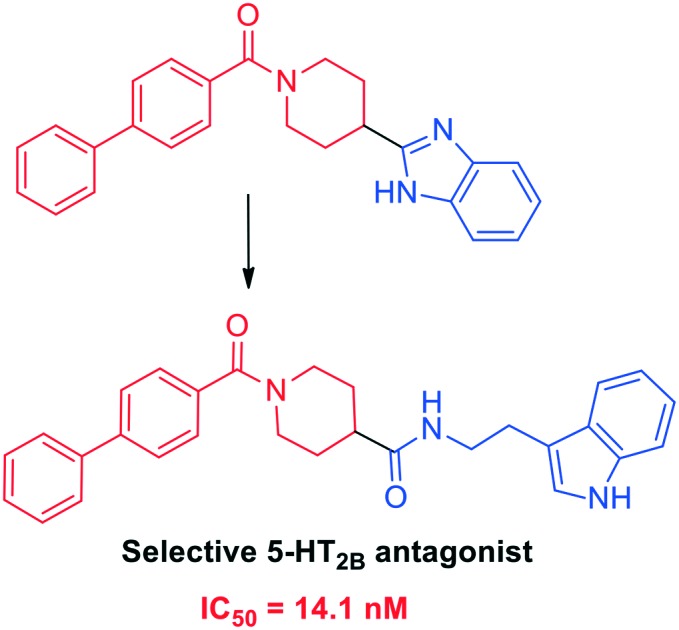
In the current study, overlaying the pharmacophore of the lead compound 1 with the receptor-based pharmacophore (Fig. 1B) directed us to maintain the biphenyl amide fragment of 1 (Fig. 3) and further modify fragment B to achieve improved alignment to the receptor-based pharmacophore. To achieve this target, tryptamine was selected to replace fragment B of 1 as it is capable of hydrogen bonding to Asp3.32 and Asn6.55 residues which is crucial for potency and selectivity to the 5-HT2B receptor. In addition, the tryptamine motif in the hybrid scaffold 7 would be an ideal fit to achieve the desired hydrophobic interactions at binding region 1 in ECL II of the target receptor (Fig. 1A). On the basis of the predicted binding mode, compound 8 was synthesized in which a tryptamine fragment is replaced with a benzyl group in order to elucidate the role of the tryptamine motif in binding the target receptor. Moreover, the effect of replacing the biphenyl group of fragment A in 7 with a phenyl ring was also studied via synthesizing the structural analogue 11 utilizing a similar synthetic approach.
Fig. 3. Design strategy of biphenyl amide–tryptamine hybrid 7 based on the lead compound 1.

Results and discussion
Pharmacophore analysis
3D and 2D pharmacophore maps were generated for compound 7 using LigandScout (Fig. 4). The investigated pharmacophoric features included hydrogen bond donors and acceptors as directed vectors, positive and negative ionizable regions and lipophilic areas that are represented by spheres. The pharmacophore scores listed in Table 1 were calculated for the alignment of the 3D pharmacophore of 7 and the receptor-based pharmacophore generated for the 5-HT2B antagonists. Compound 7 displayed impressive alignment with the receptor-based pharmacophore with a pharmacophore score value of 92.1. The overlay of compound 7 with the receptor-based pharmacophore reveals the comparable orientation of two hydrogen bond donors which are crucial for key interactions with the target receptor (ESI† Fig. S1). Moreover, the tryptamine fragment of compound 7 was overlaid with binding region 1 of the receptor pharmacophore map. The biphenyl moiety exhibited a satisfactory overlay with binding region 2. The 3D and 2D pharmacophore maps of the lead compound 1 (ESI† Fig. S2) displayed only one hydrogen bond donor and showed a pharmacophore score of 65.9 based on similarity to the receptor-based pharmacophore. It is worth mentioning that compounds 8 and 11 displayed pharmacophore scores of 14.5 and 76.4, respectively (Table 1).
Fig. 4. The 3D and 2D pharmacophoric maps of compound 7; (A) the 3D pharmacophoric map of compound 7. The pharmacophore color coding is yellow for hydrophobic regions and green for hydrogen donors; (B) the 2D pharmacophoric map of compound 7. H, hydrophobic center; HBD, hydrogen bond donor; AR, aryl.

Table 1. Pharmacophore scores of compounds 7, 8 and 11.
| Comp. no. | Pharmacophore score |
| 1 | 65.9 |
| 7 | 92.1 |
| 8 | 14.5 |
| 11 | 76.4 |
Chemistry
The initial targets of this study were synthesized according to the approach outlined in Scheme 1. As shown in Scheme 1, the starting isonipecotic acid 4 reacted with biphenyl-4-carbonyl chloride 5 to afford compound 6 in serviceable yield. Further coupling of 6 with tryptamine and benzyl amine proceeded smoothly under microwave irradiation to furnish target compounds 7 and 8. Similarly, compound 11 was synthesized via coupling of 9 (ref. 12) and tryptamine (Scheme 2).
Scheme 1. Synthesis of compounds 7 and 8.

Scheme 2. Synthesis of compound 11.

Biological screening
Cellular functional assays were performed to validate the antagonist activity of the synthesized compounds against the 5-HT2B receptor. To our delight, compound 7 displayed >95% inhibition of 5-HT2B activity at 10 μM and 1 μM (Fig. 5A). Replacement of the tryptamine motif with a benzyl group in 8 completely eliminated the antagonist activity which validates our hypothesis of the pharmacophore-based design of tryptamine-based compounds. Replacing the biphenyl subunit with a phenyl group resulted in an ∼10-fold decrease in potency (compound 11). These results indicate that hydrophobic interactions exhibited by the biphenyl unit of 7 in binding region 2 of the receptor contribute significantly to the potency displayed by compound 7. Further determination of the dose–response inhibition activity of compound 7 revealed that it exhibits an IC50 value of 14.1 nM (Fig. 5B).
Fig. 5. (A) Inhibitory activities of 7, 8 and 11 against the 5-HT2B receptor at 10 μM and 1 μM concentrations. (B) The dose-dependent inhibitory response curve of compound 7 with respect to the 5-HT2B receptor.

To gain further insight into the selectivity of 7 against 5HT2B, binding profiling tests were performed on a panel of 7 5-HT receptors (5-HT1A, 5-HT1B, 5-HT1E, 5HT2A, 5-HT2B, 5-HT2C and 5-HT3). The results revealed that compound 7 is highly selective to the 5-HT2B receptor (Table 2). Compound 7 binds the 5-HT2B receptor tightly with a Ki value of 4.5 nM. However, 7 displays Ki values of 2460 and 473 nM against 5HT2A and 5-HT2C, respectively, which are the two most highly related family members of 5-HT2B. In addition, 7 displays generally weak binding against 5-HT1B, 5-HT1E and 5-HT3. Replacement of the tryptamine moiety with the benzyl group in 8 resulted in an ∼130-fold decrease in binding affinity to the 5-HT2B receptor. Interestingly, compound 7 displayed >750-fold selectivity to 5HT2B over the closely related 5HT1B receptor in comparison to the ∼15-fold selectivity of compound 8. This significant difference in selectivity is attributed to the ability of the tryptamine moiety in 7 to form an additional hydrogen bond with the Asn6.55 residue in the 5HT2B receptor. Moreover, the presence of the biphenyl moiety in 7 which is capable of interacting with ECL II region of 5HT2B resulted in ∼11- and 8-fold improvements in selectivity over the 5HT2C and 5-HT2A receptors, respectively, in comparison with 11 possessing a phenyl group. These results are in good agreement with the proposed selectivity improvement to the 5HT2B receptor based on the pharmacophore-based design.
Table 2. 5-HTR binding profile results (Ki, nM).
| Comp. | 5-HT1A | 5-HT1B | 5-HT1E | 5-HT2A | 5-HT2B | 5-HT2C | 5-HT3 |
| 7 | 603 | 3406 | >10 000 | 2460 | 4.5 | 473 | >10 000 |
| 8 | 9389 | 8594 | >10 000 | 6794 | 569 | 986 | >10 000 |
| 11 | 5396 | 7596 | >10 000 | 5693 | 79.3 | 749 | >10 000 |
| Clozapine | Nd a | Nd | Nd | 16.1 | Nd | Nd | Nd |
| SB-206553 | Nd | Nd | Nd | Nd | 22.2 | Nd | Nd |
| Ritanserin | Nd | Nd | Nd | Nd | Nd | 1.9 | Nd |
aNd; not determined.
Pharmacokinetic profile
The ADME and toxicological parameters of the new biphenyl amide–tryptamine hybrid 7 were further examined (Table 3). The potencies of the benchmark 5-HT2B antagonists 2 and 3 drop by 50- and 27-fold, respectively, in the presence of 4% human serum albumin (HSA).3a In addition, the potency of the lead compound 1 drops by ∼500-fold under similar conditions. Thus, there is a pressing need for the development of 5-HT2B antagonists that can maintain their potency in the presence of physiological concentrations of serum proteins. Interestingly, the potency of 7 was minimally affected in the presence of 4% HSA (Table 3) which is a distinguishing positive attribute of 7. Compound 7 displayed high stability in simulated intestinal fluid of pH 6.5 and a relatively lower stability in gastric fluid of pH 1.6. Moreover, 7 demonstrated ∼97% stability in human plasma over 2 h and was found to be relatively nontoxic to HepG2 liver cells. High permeability of 7 to CACO-2 cells was observed with no significant efflux (A to B Papp = 16.3 × 10–6 cm s–1; efflux ratio = 2.01). The in vitro pharmacokinetic profile of 7 reveals its suitability for studying the role of 5-HT2B receptors.
Table 3. In vitro pharmacokinetic profile of 7.
| Test | Comp. 7 |
| Effect of HSA | |
| 5-HT2B IC50 | 14.1 |
| 5-HT2B IC50 + 4% HSA | 18.7 |
| Stability in simulated fluids (t1/2, min) | |
| Gastric (pH 1.6) | 259 |
| Intestinal (pH 6.5) | >500 |
| Stability in human plasma (% remaining at 120 min) | 97.4 |
| CACO cell permeability | |
| P app, A → B (10–6 cm s–1) | 16.3 |
| P app, B → A (10–6 cm s–1) | 32.8 |
| Stability in rat liver microsomes (t1/2, min) | 179 |
| Cytotoxicity in HepG2 cells (IC50, μM) | >50 |
Conclusions
In summary, a biphenyl amide–tryptamine hybrid scaffold 7 displayed selective and potent antagonism of the 5-HT2B receptor. The design strategy embraced structural tailoring of the potent 5-HT2B antagonist 1 to the receptor-based pharmacophore map of the doxepin induced-fit model of the 5-HT2B receptor. Additional exploration of the scaffold would furnish new tool compounds that should have great utility for mapping the binding surfaces of the 5-HT2B receptor. Initial assessment of the in vitro pharmacokinetic profile of 7 suggests that it would serve as the foundation for the development of 5-HT2B antagonists as potential therapeutics.
Experimental
General
All commercially available starting materials, reagents, and solvents were used as supplied, unless otherwise stated. The reported yields are isolated yields. Purification of all final products was accomplished by silica gel flash column chromatography. Chloroform : methanol or hexane : ethyl acetate was used as the elution solvent. Proton (1H) and carbon (13C) NMR were conducted on Bruker NMR spectrometers at 400 MHz for 1H and 100 MHz for 13C. Chemical shifts (δ) are reported in parts-per million (ppm) relative to the residual undeuterated solvent. Melting points were recorded using capillary melting point apparatus and are uncorrected. High resolution mass spectra were obtained using electron spray ionization (ESI†).
1-([1,1′-Biphenyl]-4-carbonyl)piperidine-4-carboxylic acid (6)
A suspension of biphenyl-4-carbonyl chloride 5 (10.83 g, 50.0 mmol) in dichloromethane (50 mL) was added to a solution of isonipecotic acid 4 (6.45 g, 50.0 mmol) in 2 M aqueous NaOH (50 mL). The reaction mixture was stirred for 8 h, then the reaction mixture was acidified using 1 M HCl and extracted with dichloromethane (3 × 50 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using 10% ethyl acetate in hexane, followed by 60% ethyl acetate in hexane as the eluent to yield 6 (68%) as a white solid. Mp 115–117 °C. 1H NMR (400 MHz, CDCl3) δ 1.83–1.86 (m, 2H), 2.07–2.11 (m, 2H), 2.66–2.73 (m, 2H), 3.18 (t, 2H, J = 10.6, 3.8 Hz), 3.84–3.97 (m, 1H), 4.55–4.61 (m, 1H), 7.43–7.54 (m, 5H), 7.64–7.70 (m, 4H), 9.89 (br s, 1H). 13C NMR (100 MHz, CDCl3) δ 28.0, 40.6, 45.2, 127.1, 127.2, 127.4, 127.8, 128.8, 134.3, 140.1, 142.7, 170.5, 179.0. HRMS (ESI): calcd for C19H18NO3 [M – H]–, 308.1286; found, 308.1291.
N-(2-(1H-Indol-3-yl)ethyl)-1-([1,1′-biphenyl]-4-carbonyl)piperidine-4-carboxamide (7)
To a solution of compound 6 (310 mg, 1.0 mmol) in MeCN (4 mL), PyBroP (466 mg, 1.0 mmol) and Et3N (0.32 mL, 2.5 mmol) were added and stirred at room temperature for 10 min. A solution of tryptamine (160 mg, 1.0 mmol) in MeCN (3 mL) was then added and the reaction mixture was stirred under microwave irradiation (80 W) at 90 °C for 20 min. The solvent was removed under reduced pressure, and the crude mixture was suspended in H2O (20 mL) and extracted with dichloromethane (2 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using 20% ethyl acetate in hexane, followed by 80% ethyl acetate in hexane as the eluent to yield 7 (89%) as a yellow-orange solid. Mp 160–162 °C. 1H NMR (400 MHz, CDCl3) δ 1.51–1.76 (m, 4H), 2.62–2.89 (m, 6H), 3.42–3.51 (m, 2H), 3.64–3.67 (m, 1H), 4.52–4.57 (m, 1H), 6.21 (br s, 1H), 6.90 (s, 1H), 6.95–7.13 (m, 2H), 7.24–7.29 (m, 3H), 7.32–7.39 (m, 3H), 7.44–7.51 (m, 5H), 9.02 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 20.8, 24.8, 31.1, 36.2, 39.6, 42.4, 111.3, 112.1, 118.3, 118.9, 121.6, 122.3, 126.8, 126.9, 127.1, 127.3, 127.6, 128.7, 134.2, 136.3, 139.6, 142.4, 170.1, 174.3. HRMS (ESI): calcd for C29H30N3O2 [M + H]+, 452.2338; found, 452.2336.
1-([1,1′-Biphenyl]-4-carbonyl)-N-benzylpiperidine-4-carboxamide (8)
Using the procedure given for the preparation of 7, the coupling of 6 (310 mg, 1.0 mmol) and benzyl amine (107 mg, 1.0 mmol) gave 8 (75%) as a dark yellow solid after purification by flash column chromatography using 60% ethyl acetate in hexane as the eluent. Mp 132–134 °C. 1H NMR (400 MHz, CDCl3) δ 1.75–1.95 (m, 4H), 2.41–2.47 (m, 1H), 2.82–2.98 (m, 2H), 3.86–3.99 (m, 1H), 4.43 (d, 2H, J = 5.7 Hz), 4.62–4.75 (m, 1H), 6.91 (t, 1H, J = 5.7, 5.7 Hz), 7.25–7.30 (m, 2H), 7.32–7.38 (m, 3H), 7.41–7.46 (m, 3H), 7.49–7.53 (m, 2H), 7.61–7.66 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 28.7, 41.7, 42.6, 43.1, 126.9, 127.0, 127.2, 127.4, 127.5, 127.7, 128.4, 128.7, 134.4, 138.3, 139.8, 142.3, 170.0, 173.9. HRMS (ESI): calcd for C26H27N2O2 [M + H]+, 399.2072; found, 399.2073.
N-(2-(1H-Indol-3-yl)ethyl)-1-benzoylpiperidine-4-carboxamide (11)
Using the procedure given for the preparation of 7, the coupling of 9 (233 mg, 1.0 mmol) and tryptamine (160 mg, 1.0 mmol) gave 11 (80%) as a yellowish orange solid after purification by flash column chromatography using 60% ethyl acetate in hexane as the eluent. Mp 155–156 °C. 1H NMR (400 MHz, CDCl3) δ 1.54–1.81, 2.09–2.14 (m, 1H), 2.62–2.83 (m, 2H), 2.90–2.94 (m, 2H), 3.51 (d, 2H, J = 6.1 Hz), 3.61–3.64 (m, 1H), 4.57–4.62 (m, 1H), 6.55 (t, 1H, J = 5.6, 5.6 Hz), 6.87–6.89 (m, 1H), 7.04–7.15 (m, 2H), 7.21–7.24 (m, 2H), 7.28–7.36 (m, 4H), 7.56 (d, 1H, J = 7.6 Hz), 9.51 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 20.7, 24.8, 39.6, 42.2, 46.7, 111.2, 111.9, 118.1, 118.6, 121.3, 122.0, 126.2, 126.9, 128.2, 129.3, 135.4, 136.1, 170.1, 174.1. HRMS (ESI): calcd for C23H26N3O2 [M + H]+, 376.2025; found, 376.2031.
Pharmacophore analysis
The crystal structure of the histamine H1–doxepin complex (PDB ID: 3RZE) was utilized to generate the initial binding mode of doxepin. The doxepin induced-fit model of the 5-HT2B receptor was prepared as reported previously9 by overlaying the doxepin-bound GPCR structure with the 5-HT2B structure (PDB ID: ; 4IB4). The pharmacophore map of the binding mode was generated using LigandScout software, which was further used to generate the pharmacophore scores for the investigated compounds.
Biological assays
The CHO-K1/5-HT2B cell line was obtained from GenScript and used for cellular screening of the compounds. HepG2 cells were obtained from ATCC. Descriptions of the cellular function assay and binding assay are included in the ESI.† Briefly, cellular function assays were performed using the CHO-K1/5-HT2B cell line, using a calcium flux assay method. The binding assays were performed as previously reported.13 Radiolabeled reference compounds ([3H]8-OH-DPAT for 5-HT1A; [3H]GR127543 for 5-HT1B; [3H]5-HT for 5-HT1E; [3H]ketanserin for 5-HT2A; [3H]LSD for 5-HT2B and 5-HT2C; [3H]LY278584 for 5-HT3) were used in the Ki determination assays. All assays were performed in triplicate.
In vitro pharmacokinetic assays
Descriptions of the assays are provided in the ESI† which were performed according to standard procedures.14
Conflicts of interest
The authors declare no competing interests.
Supplementary Material
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00204e
References
- (a) Nambi P., Aiyar N. Assay Drug Dev. Technol. 2003;1:305–310. doi: 10.1089/15406580360545116. [DOI] [PubMed] [Google Scholar]; (b) Hauser A. S., Attwood M. M., Rask-Andersen M., Schioth H. B., Gloriam D. E. Nat. Rev. Drug Discovery. 2017;16:829–842. doi: 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jacobson K. A. Biochem. Pharmacol. 2015;98:541–555. doi: 10.1016/j.bcp.2015.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Filmore D. Mod. Drug Discovery. 2004;7:24–28. [Google Scholar]; (e) Kumari P., Ghosh E., Shukla A. K. Trends Mol. Med. 2015;21:687–701. doi: 10.1016/j.molmed.2015.09.002. [DOI] [PubMed] [Google Scholar]
- (a) Kapadia N., Ahmed S., Harding W. W. Bioorg. Med. Chem. Lett. 2016;26:3216–3219. doi: 10.1016/j.bmcl.2016.05.079. [DOI] [PubMed] [Google Scholar]; (b) Shyu G. K. Circ. Res. 2009;104:1–3. doi: 10.1161/CIRCRESAHA.108.191122. [DOI] [PubMed] [Google Scholar]; (c) Kantor S., Jakus R., Balogh B., Benko A., Bagdy G. Br. J. Pharmacol. 2004;142:1332–1342. doi: 10.1038/sj.bjp.0705887. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Doly S., Valjent E., Setola V., Callebert J., Herve D., Launay J.-M., Maroteaux L. J. Neurosci. 2008;28:2933–2940. doi: 10.1523/JNEUROSCI.5723-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Launay J.-M., Schneider B., Loric S., Da Prada M., Kellermann O. FASEB J. 2006;20:1843–1854. doi: 10.1096/fj.06-5724com. [DOI] [PubMed] [Google Scholar]; (f) Wouters M. M., Gibbons S. J., Roeder J. L., Distad M., Ou Y., Strege P. R., Szurszewski J. H., Farrugia G. Gastroenterology. 2007;133:897–906. doi: 10.1053/j.gastro.2007.06.017. [DOI] [PubMed] [Google Scholar]; (g) Jaffre F., Callebert J., Sarre A., Etienne N., Nebigil C. G., Launay J.-M., Maroteaux L., Monassier L. Circulation. 2004;110:969–974. doi: 10.1161/01.CIR.0000139856.20505.57. [DOI] [PubMed] [Google Scholar]
- (a) Moss N., Choi Y., Cogan D., Flegg A., Kahrs A., Loke P., Meyn O., Nagaraja R., Napier S., Parker A., Thomas Peterson J., Ramsden P., Sarko C., Skow D., Tomlinson J., Tye H., Whitaker M. Bioorg. Med. Chem. Lett. 2009;19:2206–2210. doi: 10.1016/j.bmcl.2009.02.126. [DOI] [PubMed] [Google Scholar]; (b) Adegunsoye A., Levy M., Oyenuga O. BioMed Res. Int. 2015;2015:929170. doi: 10.1155/2015/929170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Jaffre F., Bonnin P., Callebert J., Debbabi H., Setola V., Doly S., Monassier L., Mettauer B., Blaxall B. C., Launay J. M., Maroteaux L. Circ. Res. 2009;104(1):113–123. doi: 10.1161/CIRCRESAHA.108.180976. [DOI] [PubMed] [Google Scholar]; (b) Schaerlinger B., Hickel P., Etienne N., Guesnier L., Maroteaux L. Br. J. Pharmacol. 2003;140:277–284. doi: 10.1038/sj.bjp.0705437. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Esteve J. M., Launay J.-M., Kellermann O., Maroteaux L. Cell Biochem. Biophys. 2007;47:33–44. doi: 10.1385/cbb:47:1:33. [DOI] [PubMed] [Google Scholar]; (d) Liang Y.-J., Lai L.-P., Wang B.-W., Juang S.-J., Chang C.-M., Leu J.-G., Shyu K.-G. Cardiovasc. Res. 2006;72:303–312. doi: 10.1016/j.cardiores.2006.08.003. [DOI] [PubMed] [Google Scholar]
- (a) Poissonnet G., Parmentier J. G., Boutin J. A., Goldstein S. Mini-Rev. Med. Chem. 2004;4(3):325–330. doi: 10.2174/1389557043487312. [DOI] [PubMed] [Google Scholar]; (b) Sanger G. J. Trends Pharmacol. Sci. 2008;29:465–471. doi: 10.1016/j.tips.2008.06.008. [DOI] [PubMed] [Google Scholar]; (c) Borman R. A., Tilford N. S., Harmer D. W., Day N., Ellis E. S., Sheldrick R. L., Carey J., Coleman R. A., Baxter G. S. Br. J. Pharmacol. 2002;135:1144–1151. doi: 10.1038/sj.bjp.0704571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnala S., Kapadia N., Harding W. W. Med. Chem. Commun. 2015;6:601–605. [Google Scholar]
- Ohashi-doi K., Himaki D., Nagao K., Kawai M., Gale J. D., Furness J. B., Kurebayashi Y. Neurogastroenterol. Motil. 2010;22:e69–e76. doi: 10.1111/j.1365-2982.2009.01395.x. [DOI] [PubMed] [Google Scholar]
- Wacker D., Wang C., Katritch V., Han G. W., Huang X. P., Vardy E., McCorvy J. D., Jiang Y., Chu M., Siu F. Y., Liu W., Xu H. E., Cherezov V., Roth B. L., Stevens R. C. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Ma J., Lin X., Huang X.-P., Wu K., Huang N. J. Med. Chem. 2016;59:707–720. doi: 10.1021/acs.jmedchem.5b01631. [DOI] [PubMed] [Google Scholar]
- Reavill C., Kettle A., Holland V., Riley G., Blackburn T. P. Br. J. Pharmacol. 1999;126:572–574. doi: 10.1038/sj.bjp.0702350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhaus D. W., Flippin L. A., Greenhouse R. J., Jaime S., Rocha C., Dawson M., Van Natta K., Chang L. K., Pulido-Rios T., Webber A., Leung E., Eglen R. M., Martin G. R. Br. J. Pharmacol. 1999;127:1075–1082. doi: 10.1038/sj.bjp.0702632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventre S., Petronijevic F. R., MacMillan D. W. C. J. Am. Chem. Soc. 2015;137:5654–5657. doi: 10.1021/jacs.5b02244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard J., Ruda G. F., Setola V., Abecassis K., Rodriguiz R. M., Huang X. P., Norval S., Sassano M. F., Shin A. I., Webster L. A., Simeons F. R., Stojanovski L., Prat A., Seidah N. G., Constam D. B., Bickerton G. R., Read K. D., Wetsel W. C., Gilbert I. H., Roth B. L., Hopkins A. L. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Tosh D. K., Finley A., Paoletta S., Moss S. M., Gao Z.-G., Gizewski E. T., Auchampach J. A., Salvemini D., Jacobson K. A. J. Med. Chem. 2014;57:9901–9914. doi: 10.1021/jm501021n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Carlin J. L., Tosh D. K., Xiao C., Pinol R. A., Chen Z., Salvemini D., Gavrilova O., Jacobson K. A., Reitman M. L. J. Pharmacol. Exp. Ther. 2016;356:475–483. doi: 10.1124/jpet.115.229872. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.