

 Simplified aminothienopyridazine analogues were synthesised and their inhibition of tau protein aggregation assessed.
Simplified aminothienopyridazine analogues were synthesised and their inhibition of tau protein aggregation assessed.
Abstract
Aminothienopyridazines (ATPZs) have demonstrated efficacy, in vitro, as tau protein aggregation inhibitors. Modifications were made to the ATPZ scaffold to determine the importance of certain structural features for activity. More specifically, ring-opened analogues detached at the nitrogen–nitrogen bond of the pyridazine, were synthesized and their inhibitory activity evaluated. Preliminary data suggests that the ring-opened structures retain inhibitory activity, independent of tau oxidation. The structures detailed represent the beginnings of a deconstruction–reconstruction–elaboration study, with the aim of identifying simpler scaffolds, which retain activity and can be optimized in terms of physiochemical properties.
Introduction
Alzheimer's disease is the most common neurodegenerative disease worldwide.1 With an increasingly ageing population, it can be expected that the number of cases will continue to rise. Drugs that can prevent or cure the disease are currently not available, with only symptom-alleviating treatments available. There is an urgent need for new and effective treatments for Alzheimer's disease.
Alzheimer's disease is characterized by the presence of plaques composed of amyloid-β protein and tangles of tau protein.2,3 The majority of academic focus has centered around the amyloid cascade hypothesis as the cause of neurodegeneration, and hence most strategies employed thus far to treat the disease aim to target amyloid-β.4 It is thought that tau protein plays an important role in the neurodegenerative processes underlying Alzheimer's disease progression.5–7 The distribution and abundance of tau pathology correlates well with nerve cell degeneration and clinical symptoms.5 If misfolded, the usually very soluble tau protein can form insoluble aggregates that have been shown to contribute to a number of neurodegenerative conditions, including Alzheimer's disease.8 Research and clinical development of treatments that target tau have been seen as a viable alternative to the amyloid-β consensus. It is thought that if the aggregation of tau could be prevented, or even reversed, then neurodegeneration associated with tau pathology could potentially be avoided. Small molecule tau aggregation inhibitors are attracting interest as potential therapeutic agents for the treatment of Alzheimer's disease. Targeting the aggregation of tau is an attractive option, as the aggregation process is exclusively associated with disease, that is, healthy brain tissue only contains soluble monomeric tau. However, finding small drug-like molecules that disrupt protein–protein interactions over large areas of contact is challenging. Precise atomic scale knowledge of fibril composition and the aggregation mechanism has yet to be achieved. Tau is a natively unfolded protein, and hence structure elucidation by X-ray crystallography is not possible. NMR analysis, however has provided some structural information.9 Solution NMR studies of full length tau, revealed a mainly disordered structure with transient secondary structure.10 The lack of structural information means that identifying high-affinity ligand binding sites is inherently difficult. The most straightforward strategy to overcome the challenges associated with designing a small-molecule inhibitor of protein aggregation is to screen a large library of compounds for inhibitory activity. Identification of hits allows for the use of subsequent medicinal chemistry hit-to-lead optimization strategies which aim to improve the pharmacokinetic and physicochemical properties of the drug, while increasing inhibitory activity. Several small molecule compound types have been shown to be effective at inhibiting the aggregation of tau in vitro.11
Recently Ballatore and coworkers identified aminothioenopyridazines (ATPZs) as potent inhibitors of tau aggregation after screening a library of 292 000 compounds.12 We envisage that ring-opened analogues of ATPZs, detached at the nitrogen–nitrogen bond of the pyridazine ring (Fig. 1), would be expected to have similar arrangement of groups in space, and hence would have similar or improved binding to tau monomers or aggregates. Further, there is opportunity to improve drug like properties such as solubility, by such modifications.
Fig. 1. A series of ring-opened analogues of the lead ATPZ structure, 1, where the pyridazine is disconnected at the nitrogen–nitrogen bond.
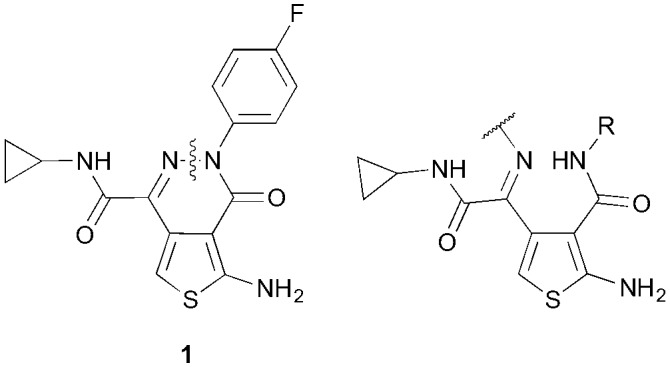
ATPZ 1, has been identified as a suitable candidate for further in vivo testing based on its favorable physicochemical properties.13 The structure is thought to be an ideal lead compound, for further modifications to explore the importance of certain structural features for activity, chiefly, the pyridazine heterocycle. As a point of comparison, the lead compound 1 was synthesized according to the protocol reported by Ballatore and coworkers.13
Results and discussion
Disconnection of the nitrogen–nitrogen bond of the pyridazine heterocycle of the ATPZ structure would result in an imine, in the simplest case. Therefore, a more stable alternative would be an oxime, or oxime ether. Based on this a series of oxime based 2-aminothiophenes were therefore synthesized, via a Gewald reaction as the key step in the synthetic protocol (Scheme 1). The oxime 2 was formed by nitrosation of tert-butyl acetoacetate with sodium nitrite in glacial acetic acid. Only one isomer was observed by NMR, with most literature reporting the Z-isomer as the major product of the oximation of acetoacetates.14 Protection of the oxime with allyl bromide afforded oxime allyl ether 3. To achieve the Gewald reaction we found that a two-step protocol was required, first with a Knoevenagel condensation to form intermediate 4, utilizing trimethylsilyl acetate as an organic dessicant.15 The crudely isolated intermediate could then be converted to thiophene 5 using aqueous sodium bicarbonate as a base for the Gewald cyclization.
Scheme 1. Reagents and conditions (a) NaNO2, AcOH, 0 °C-rt, 1 h; (b) allyl bromide, KOH, DMF, rt, 2 h; (c) HMDS, AcOH, ethyl 2-cyanoacetate, AcOH, 65 °C, 16 h; (d) S8, NaHCO3 (aq), THF, 35 °C, 5 h.

Once the amine 5 had been protected as the trifluoroacetamide 6, the subsequent acid mediated hydrolysis of the tert-butyl ester to acid 7 proceeded smoothly using trifluoroacetic acid (Scheme 2). The ethyl ester was stable to the reaction conditions, allowing for chemoselective amide coupling to give amide 8. Following this, saponification of the ethyl ester afforded the carboxylic acid 9, with the trifluoroacetamide protecting group proving to be stable to the basic conditions. The acid 9 was then used as a common intermediate for the synthesis of a range of amides at the C-3 position of the thiophene. The 4-fluoroaniline amide 10 was synthesized to replicate the lead compound 1 at this amide position via a PyBop mediated coupling. In difference to the methods employed in the original synthesis of 1, the current synthetic protocol employed imparts no limitations on the identity of the amide at the C-3 position of the thiophene. The synthesis of 1, and related ATPZs, involves the diazotization of an aniline as the first step, which cannot be easily applied to aliphatic amines, meaning the amides present in the final target must be aromatic. Taking advantage of the newly developed protocol the 2-aminothiophene 11 with an aliphatic adamantyl amide at the C-3 position was synthesized (Scheme 2).
Scheme 2. Reagents and conditions (a) TFAA, DIPEA, CH2Cl2, 0 °C, 1.5 h; (b) TFA, CH2Cl2, 0 °C-rt, 16 h; (c) cyclopropylamine, PyBOP, DIPEA, DMF, 0 °C-rt, 16 h; (d) LiOH·H2O, THF, MeOH, water, rt, 16 h; (e) 4-fluoroaniline or adamantylamine, PyBOP, DIPEA, DMF, 0 °C-rt, 16 h; (f) Et3N–HCOOH, MeOH, water, reflux, 24 h; (g) Pd(OAc)2, PPh3, Et3N–HCOOH, MeOH, water, reflux, 24 h.

It was found that the removal of the trifluoroacetamide was not possible using conventional deprotection strategies. Instead, a global deprotection of the trifluoroacetamide and allyl ether group was achieved using triethylammonium formate and tetrakis(triphenylphosphine)palladium(0) to obtain 13 and 14.16 In the absence of the palladium catalyst, it was found that selective deprotection of the trifluoroacetamide group could be achieved, in order to afford 12.
Compounds 12, 13, 14 and the lead compound 1 were assessed for their ability to inhibit the aggregation of tau, in vitro, via both, a fluorescence assay and an aggregate trap assay (Fig. 2). As thioflavin-T binds to paired helical filaments of tau protein but not monomeric tau, the resulting changes in the fluorescence spectrum allows for the monitoring of tau fibrillization.17 High-order fibrillary tau aggregates can be vacuum-trapped on membranes followed by detection with tau-specific antibodies, allowing assessment of inhibition of tau fibril formation.18 This method allows for very high sensitivity and also compatibility with optically active additives.18 Tau protein (4R) underwent heparin-induced polymerization in the absence or presence of an aggregation inhibitor for 48 hours, after which, thioflavin-T fluorescence was measured, or it was subjected to vacuum-trapping and immune-detection. The thioflavin-T assay showed that, in addition to the lead compound 1, the ring-opened analogues 12 and 14 also appear to inhibit the aggregation of tau (Fig. 2a). Analysis of the fluorescence intensity at the wavelength of maximum fluorescence emission of the aggregated tau protein suggests that the lead compound 1 results in a 51% inhibition of aggregation, while 12 and 14 achieved 33% and 30% inhibition, respectively. The aggregate trap assay revealed that compound 12 and the lead compound 1 had similar efficacy in decreasing the amount of higher-order tau fibrils (Fig. 2b). However, the propensity of compounds 13 and 14 to reduce higher-order tau aggregates was significantly higher compared to compounds 1 and 12. While all compounds showed effects on inhibiting tau aggregation, differences between the thioflavin-T and the aggregate trap assays, in particular for compound 13, may indicate differential effects on higher- and lower-order tau aggregates. Alternatively, compound 13 may interfere with the thioflavin-T assay, despite absence of differences in auto-fluorescence between compounds (not shown). Taken together, the aggregation assays suggest that the pyridazine ring of the lead structure is not imperative for activity. Furthermore, it was shown that an aliphatic, rather than aromatic amides at the 3-position of the thiophene was also tolerated. This finding therefore justifies future work, based on the simplified scaffold, to further explore the structural features required to improve inhibitory activity and other properties.
Fig. 2. Tau aggregation assay using fluorescence emission of thioflavin T and trapping of aggregated tau. (a) 4R refers to tau protein, in the absence of an aggregation inhibitor. A lower intensity than the standard 4R correlates to a lower concentration of ordered β-sheet structures, indicative of inhibition of tau aggregation. (b) Aggregated tau trapped and detected on membranes. Lower spot intensity is consistent with decreased amounts of fibrillar tau aggregates. Quantification of 7 independent experiments (*p < 0.05, ****p < 0.0001).

It has been proposed that compound 1 mediates its anti-aggregation effects by oxidation of cysteine residues in tau.19 Therefore, we tested for the level of oxidation of tau induced by compounds 1, 12, 13 and 14, using Oregon green binding to non-oxidized cysteine (Fig. 3). We confirmed the previously reported oxidizing properties of compound 1. Both, compounds 13 and 14 showed oxidizing activity, while compound 12 did not induce oxidation of tau. Since compound 12, and more so 13 and 14 inhibited tau aggregation, while compounds 13 and 14, but not 12 oxidized tau, our data suggests that, in our hands, the tau aggregation inhibiting activity of compounds 12, 13 and 14 (and possibly 1) is independent of the oxidizing effects.
Fig. 3. Oxidation of tau measured by completion for the binging of Oregon green. 4R refers to tau protein, in the absence of compounds. Lower band intensities of IAA-OG Oregon green indicate increased oxidation. Band intensities were quantified from 5 independent experiments and normalized to total recombinant tau protein, stained by Coomassie blue (**p < 0.01, ***p < 0.001).

Conclusions
With limited knowledge of the mechanism of action of tau aggregation inhibitors, screening of libraries of compounds and subsequent structure–activity relationship studies are the best methods for identifying active compounds. From the identification of lead compounds, exploitation of bioisoteric groups, and modification of key structural features provides an insight into the relationship between these motifs and inhibitory activity. A new synthetic methodology has been developed to access ring-opened analogues of the lead ATPZ 1. Furthermore this allowed for the incorporation of aliphatic amides at the C3 position of the thiophene. The tau aggregation assays suggest that the ring-opened analogues retain inhibitory activity, yet independent of oxidizing activity. Further, the presence of aliphatic, rather than aromatic amides at the 3-position of the thiophene does not have an effect on the inhibition of tau aggregation.
Further work will be devoted to determining the minimal structural features required for activity. Starting with the active ring-opened structures, motifs like the amides could be interconverted for simpler functionalities such as esters, or removed completely to determine their importance for activity. In an ideal case, a simple, easily accessible structure could be identified that retains inhibitory activity. From here, typical medicinal chemistry strategies could be applied in an effort to further improve the drug-like characteristics of these compounds. This work has identified a novel scaffold for the future development of tau aggregation inhibitors.
Experimental
Chemistry
General
All reactions were performed under an atmosphere of nitrogen unless otherwise specified. Diethylamine, cyclopropylamine and 4-fluoroaniline were distilled from calcium hydride. Anhydrous N,N-dimethylformamide, dichloromethane, tetrahydrofuran and toluene were obtained from a PureSolv MD 7 solvent purification system (Innovative Technology, Inc.). All other solvents and reagents were used as received from commercial sources. Microwave-assisted reactions were performed in sealed-tube systems using a Discover® S-Class (Ai Scientific CEM Corporation) instrument. Analytical thin-layer chromatography (TLC) was performed using Merck aluminium backed silica gel 60 F254 (0.2 mm) plates which were visualised with shortwave (254 nm) ultraviolet light. Products were also visualised with potassium permanganate, vanillin, cerium moylbdate, bromocresol green or ninhydrin stains. Flash column chromatography was performed using Grace Davisil 60 (230–400 mesh) silica gel, with the eluent mixture reported as the volume : volume ratio. Melting points were measured in open capillaries using a Stanford Research System Optimelt Automated melting point apparatus and are uncorrected. Infrared absorption spectra were recorded on a Bruker ALPHA FT-IR spectrometer as a solid or a thin film from ethanol, and the data are reported as vibrational frequencies (cm–1). Nuclear magnetic resonance spectra were recorded at 300 K using a Bruker AVANCE DRX300 (300 MHz) spectrometer. 1H chemical shifts are expressed as parts per million (ppm) with residual chloroform (δ 7.26) and dimethyl sulfoxide (δ 2.50) as reference and are reported as chemical shift (δ); relative integral; multiplicity (s = singlet, bs = broad singlet, d = doublet, dd = doublet of doublets, dt = doublet of triplets, t= triplet, q = quartet, m = multiplet); coupling constants (J) reported in Hz. 13C chemical shifts are expressed as parts per million (ppm) with residual chloroform (δ 77.16) and dimethyl sulfoxide (δ 39.52) as reference and reported as chemical shift (δ); multiplicity, coupling constants (J) reported in Hz. Proton decoupled 19F chemical shifts are reported as parts per million (ppm). Low-resolution mass spectra (LRMS) were recorded using electrospray ionisation (ESI) recorded on a Bruker AmaZon SL ion trap spectrometer. High resolution mass spectrometry was performed on a Bruker Apex Qe 7T Fourier transform ion cyclotron resonance mass spectrometer equipped with an Apollo II ESI/MALDI dual source. Samples were run with syringe infusion at 150 μL h–1 on a Cole Palmer syringe pump into the electrospray ionisation (ESI) source. High performance liquid chromatography (HPLC) analysis of organic purity was conducted on a Waters Alliance 2695 instrument using a SunFire™ C18 column (5 μm, 2.1 × 150 mm) and detected using a Waters 2996 photodiode array (PDA) detector set at 254 nm. Separation was achieved using water (solvent A) and acetonitrile (solvent B) at a flow rate of 0.2 mL min–1 and a gradient of 0% B to 100% B over 30 minutes. HPLC data is reported as percentage purity and retention time (RT) in minutes.
tert-Butyl 2-(hydroxyimino)-3-oxobutanoate 2
tert-Butyl acetoacetate (30.6 mL, 0.187 mol) was dissolved in glacial acetic acid (200 mL) and cooled to 0 °C. The cooled solution was treated, dropwise, with a solution of sodium nitrite (14.8 g, 0.215 mmol) in water (50 mL), such that the temperature did not exceed 10 °C. The mixture was then stirred at room temperature for 1 h, before it was quenched with an 18% NaCl (aq) solution (250 mL) and stirred for a further 20 minutes. The mixture was extracted with chloroform (3 × 150 mL) washed with saturated sodium bicarbonate solution (2 × 150 mL), water (2 × 150 mL) and brine (150 mL) and the organic phase dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure to afford the title compound 6 (35.0 g, 100%) as a yellow oil, Rf: 0.53 (3 : 7, ethyl acetate, hexane); IR (νmax (film)): 3220, 3002, 2981, 2936, 1730, 1682, 1397, 1369, 1339, 1271, 1149, 1075, 997, 957, 831 cm–1; 1H NMR (300 MHz, CDCl3): δ 1.54 (9H, s), 2.36 (3H, s) ppm, (OH peak not observed); 13C NMR (75 MHz, CDCl3): δ 25.4, 28.2, 85.2, 151.6, 161.4, 194.4 ppm; LRMS (+ESI) m/z: 397 ([2 M + Na]+, 100%), 210 ([M + Na]+, 89%).
tert-Butyl 2-((allyloxy)imino)-3-oxobutanoate 3
To a solution of potassium hydroxide powder (14.7 g, 0.263 mol) and oxime 2 (37.8 0.202 mol) in dimethylformamide (400 mL) was added allyl bromide (26.2 mL, 0.303 mol), dropwise at 0 °C and the mixture stirred for 2 h. The reaction mixture was poured into cold water (600 mL) and extracted with ethyl acetate (3 × 400 mL). The combined organic fractions were washed with water (4 × 300 mL) and brine (300 mL), dried over anhydrous magnesium sulfate and the solvent removed to obtain a deep orange oil, which was purified by flash column chromatography using ethyl acetate, hexane 1 : 9 as an eluent to afford the title compound 3 (33.0 g, 72%) as a light yellow oil, Rf: 0.71 (1 : 4 ethyl acetate, hexane); IR (νmax (film)): 3258, 2981, 2936, 1734, 1687, 1368, 1322, 1252, 1157, 1074, 997, 956, 875, 836 cm–1; 1H NMR (300 MHz, CDCl3): δ 1.53 (9H, s), 2.36 (3H, s), 4.76 (2H, d, J = 5.6 Hz), 5.27 (1H, d, J = 10.5 Hz), 5.33 (1H, d, J = 17.2 Hz), 5.90–6.00 (1H, m) ppm; 13C NMR (75 MHz, CDCl3): δ 25.3, 28.3, 77.2, 84.4, 118.8, 132.6, 150.9, 160.5, 193.2 ppm; LRMS (+ESI) m/z: 477 ([2 M + Na]+, 100%), 250 ([M + Na]+, 40%).
Ethyl 4-(1-((allyloxy)imino)-2(tert-butoxy)-2-oxoethyl)-2-aminothiophene-3-carboxylate 5
Hexadimethyldisilazane (44.3 mL, 211 mmol), was added to glacial acetic acid (126 mL, 2.20 mol) at a rate, such that, the internal temperature did not exceed 75 °C. The mixture was then added, dropwise, to solution of the ketone 3 (32.0 g, 141 mmol) and ethyl cyanoacetate (34.2 mL, 280 mmol) in glacial acetic acid (50 mL) and stirred at 65 °C for 16 h. The mixture was cooled to room temperature and poured into water (200 mL), extracted with ethyl acetate (3 × 100 mL) and washed with saturated sodium bicarbonate solution (aq) (3 × 100 mL), water (3 × 100 mL) and brine (150 mL). The organic phase was dried over anhydrous magnesium sulfate and the solvent removed to afford the alkene 4 as a light yellow oil (33.0 g, 73%), which was used without further purification. The oil was dissolved in tetrahydrofuran (300 mL) and to this was added sulfur (3.94 g, 123 mmol) and the mixture warmed to 35 °C. A solution of sodium bicarbonate (8.6 g, 102 mmol) in water (45 mL) was added dropwise and the mixture stirred for 5 h. The reaction mixture was quenched with 12.5% NaCl solution (aq) (150 mL) and the separated organic phase washed with 25% NaCl solution (aq) (200 mL), dried over anhydrous magnesium sulfate and the solvent removed under reduced pressure. The crude oil was purified by flash column chromatography using ethyl acetate, hexane 0 : 1 to 1 : 4 as an eluent to obtain a yellow solid which was dissolved in dichloromethane and triturated with hexane to obtain the title compound 5 (27.2 g, 75%) as an off-white solid, mp: 82.9–83.6 °C; Rf: 0.37 (1 : 4 ethyl acetate, hexane); IR (νmax (neat)): 3454, 3424, 3342, 3319, 3130, 2981, 2913, 1724, 1659, 1588, 1492, 1417, 1257, 1150, 1122, 1090, 1027, 1001, 988, 878, 842, 741 cm–1; 1H NMR (300 MHz, CDCl3): δ 1.30 (3H, t, J = 7.1 Hz), 1.45 (9H, s), 4.23 (2H, q, J = 7.1 Hz), 4.68 (2H, d, J = 5.6 Hz), 5.21 (1H, d, 10.5 Hz), 5.34 (1H, d, J = 17.1 Hz), 5.92–6.06 (1H, m), 6.04 (2H, s), 6.39 (1H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 14.4, 28.1, 60.1, 76.0, 82.8, 105.3, 110.1, 117.6, 131.5, 133.9, 147.9, 159.7, 163.4, 164.9 ppm; LRMS (+ESI) m/z: 731 ([2M + Na]+ 100%), 377 ([M + Na]+ 95%), HRMS (+ESI) calcd for C16H22N2O5S [M + Na]+: 377.1142, found: 377.1143.
Ethyl 4-(1-((allyloxy)imino-2(tert-butoxy)-2-oxoethyl)-2-oxoethyl)-2-(2,2,2-trifluoroacetamido)thiophene-3-carboxylate 6
To a stirred solution of 5 (20.0 g, 56.4 mmol) and trifluoroacetic anhydride (11.8 mL, 84.6 mmol) in dichloromethane (500 mL) was added, dropwise, N,N-diisopropylethylamine (19.6 mL, 110 mmol) at 0 °C. The reaction mixture was stirred for 1.5 h, then cold water was added (400 mL) and the separated organic phase washed with 1 M hydrochloric acid (aq) (200 mL), water (3 × 250 mL) and brine (250 mL) and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure to afford the product 6 (24.1 g, 95%) as a deep orange oil, Rf: 0.60 (3 : 17 ethyl acetate, hexane); IR (νmax (film)): 3203, 2982, 2935, 1724, 1677, 1560, 1316, 1231, 1194, 1148, 1119, 1005, 989, 948, 843, 745 cm–1; 1H NMR (300 MHz, CDCl3): δ 1.37 (3H, t, J = 7.2 Hz), 1.56 (9H, s), 4.36 (2H, q, J = 7.2 Hz), 4.72 (2H, d, J = 5.4 Hz), 5.24 (1H, d, J = 10.5 Hz), 5.36 (1H, d, J = 17.3 Hz), 5.94–6.08 (1H, m), 7.10 (1H, s), 12.08 (1H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 14.1, 28.2, 62.1, 74.1, 83.3, 114.5, 115.6 (q, JCF = 287.4 Hz), 118.0, 120.8, 130.8, 133.7, 146.3, 146.5, 154.3 (q, JCF = 41.6 Hz), 159.2, 165.0 (C1) ppm; 19F NMR (282 MHz, CDCl3): δ –75.5 ppm; LRMS (+ESI) m/z: 473 ([M + Na]+ 100%), 923 ([2 M + Na]+ 18%), HRMS (+ESI) calcd for C18H21F3N2O6S [M + Na]+: 437.0965, found: 473.0967.
2-((Allyloxy)imino)-2-(4-(ethoxycarbonyl)-5-(2,2,2-trifluoroacetamido)thiophen-3-yl)acetic acid 7
To a solution of 6 (23.4 g, 52.0 mmol) in dichloromethane at 0 °C was added trifluoroacetic acid (19.9 mL, 260 mmol), dropwise. The mixture was slowly warmed to room temperature and stirred for 16 h. The solvent was removed under a stream of nitrogen, and the residue dried under high vacuum to obtain the acid 7 (20.0 g, 98%) as a light yellow solid, mp: 115.9–118.1 °C; Rf: 0.27 (ethyl acetate); IR (νmax (neat)): 3208, 3124, 2987, 2947, 2882, 1719, 1674, 1563, 1347, 1226, 1194, 1167, 1147, 1115, 1024, 999, 985, 945 cm–1; 1H NMR (300 MHz, DMSO-d6): δ 1.26 (3H, t, J = 7.1 Hz), 4.25 (2H, q, J = 7.1 Hz), 4.65 (2H, d, J = 5.3 Hz), 5.23 (1H, d, J = 10.6 Hz), 5.34 (1H, d, J = 17.2 Hz), 5.92–6.06 (1H, m), 7.49 (1H, s), 11.90 (1H, s), 13.77 (1H, bs) ppm; 13C NMR (75 MHz, DMSO-d6): δ 13.7, 61.4, 75.4, 115.3 (q, JCF = 287.2 Hz), 117.8, 117.8, 122.8, 129.7, 133.9, 142.9, 146.5, 154.0 (q, JCF = 38.4 Hz), 162.0, 163.2 ppm; 19F NMR (282 MHz, DMSO-d6): δ –74.3 ppm; LRMS (+ESI) m/z: 417 ([M + Na]+ 100%), 439 ([M + 2Na – H]+ 67%), HRMS (+ESI) calcd for C14H13F3N2O6S [M + Na]+: 417.0339, found: 417.0340.
Ethyl 4-(1-((allyloxy)imino)-2-(cylopropylamino)-2-oxoethyl)-2-(2,2,2-trifluoroacetamido)thiophene-3-carboxylate 8
The acid 7 (20.0 g, 50.7 mmol) and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (36.9 g, 70.9 mmol) were dissolved in N,N-dimethylformamide (300 mL) and the solution cooled to 0 °C. To this was added, dropwise, cyclopropylamine (4.92 mL, 70.9 mmol), then N,N-diisopropylethylamine (13.6 mL, 75.8 mmol) and the mixture warmed to room temperature and stirred for 16 h. The reaction mixture was diluted with water (400 mL) and extracted with ethyl acetate (3 × 250 mL). The combined organic components were washed with water (4 × 200 mL) and brine (200 mL), dried over anhydrous magnesium sulfate and the solvent removed under reduced pressure. The crude residue was purified by flash column chromatography, using ethyl acetate, dichloromethane 0 : 1 to 1 : 4 as an eluent, to afford the amide 8 (20.3 g, 92%) as an off-white solid, mp: 163.8–166.1 °C; Rf: 0.46 (2 : 3 ethyl acetate, hexane); IR (νmax (neat)): 3308, 3219, 3123, 3009, 2986, 2923, 2867, 1726, 1675, 1632, 1555, 1316, 1243, 1229, 1192, 1172, 1146, 1120, 1028, 940, 919, 791 cm–1; 1H NMR (300 MHz, DMSO-d6): δ 0.70–0.45 (4H, m), 1.25 (3H, t, J = 7.0 Hz), 2.73–2.77 (1H, m), 4.22 (2H, q, J = 7.0 Hz), 4.60 (2H, d, J = 5.3 Hz), 5.23 (1H, d, J = 10.5 Hz), 5.31 (1H, d, J = 17.4 Hz), 5.92–6.06 (1H, m), 7.40 (1H, s), 8.40 (1H, d, J = 3.9 Hz), 11.88 (1H, bs) ppm; 13C NMR (75 MHz, DMSO-d6): δ 5.8, 13.7, 22.4, 61.2, 75.2, 115.2 (q, JCF = 289.3 Hz), 117.7, 119.7, 122.9, 130.3, 133.9, 141.1, 148.6, 154.2 (q, JCF = 39.4 Hz), 161.6, 163.2 ppm; 19F NMR (282 MHz, DMSO-d6): δ –74.1 ppm; LRMS (+ESI) m/z: 456 ([M + Na]+ 100%), 889 ([M + 2Na]+, 11%), HRMS (+ESI) calcd for C17H18F3N3O5S [M + Na]+: 456.0812, found: 456.0813.
4-(1-((Allyloxy)imino)-2-(cyclopropylamino)-2-oxoethyl)-2-(2,2,2-trifluoroacetamido)thiophene-3-carboxylic acid 9
To a solution of 8 (18.0 g, 41.5 mmol) in a mixture of tetrahydrofuran, methanol and water (2 : 2 : 1, 540 mL) was added lithium hydroxide monohydrate (5.23 g, 125 mmol) and the mixture stirred for 16 h. The organic solvent was removed under reduced pressure and the remaining aqueous component acidified to pH = 3 with 6 M hydrochloric acid (aq). The resulting precipitate was collected by filtration, washed with water (10 mL), ethanol (5 mL) and diethyl ether (2 × 25 mL) and dried in vacuo to obtain the acid 9 (14.9 g, 88%) as an off-white powder, mp: 161.5 °C (decomposition); Rf: 0 (2 : 3 ethyl acetate, hexane); IR (νmax (neat)): 3226, 3092, 3009, 2874, 2564, 1739, 1702, 1594, 1535, 1263, 1207, 1194, 1159, 1121, 1015, 995, 929, 743 cm–1; 1H NMR (300 MHz, DMSO-d6): δ 0.42–0.72 (4H, m), 2.69–2.81 (1H, m), 4.63 (2H, d, J = 5.3 Hz), 5.28 (1H, d, J = 10.6 Hz), 5.33 (1H, d, J = 17.1 Hz), 5.94–6.06 (1H, m), 7.46 (1H, s), 8.85 (1H, s), 12.82 (1H, bs) ppm, (COOH peak not observed); 13C NMR (75 MHz, DMSO-d6): δ 5.7, 22.7, 75.1, 115.7 (q, JCF = 286.5 Hz), 118.0, 122.3, 123.7, 127.0, 133.7, 148.2, 153.4 (q, JCF = 39.2 Hz), 163.1, 163.2, 164.5 ppm; 19F NMR (282 MHz, DMSO-d6): δ –74.0 ppm; LRMS (+ESI) m/z: 450 ([M + 2Na – H]+ 100%), 428 ([M + Na]+ 86%), HRMS (+ESI) calcd for C15H14F3N3O5S [M + Na]+: 428.0499, found: 428.0502.
4-(1-((Allyloxy)imino)-2-(cyclopropylamino)-2-oxoethyl)-N-(4-fluorophenyl)-2-(2,2,2-trilfluroracetamido)thiophene-3-carboxamide 10
The acid 9 (2.00 g, 4.93 mmol) and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (3.59 g, 6.91 mmol) were dissolved in N,N-dimethylformamide (10 mL) and the solution cooled to 0 °C. To this was added, dropwise, 4-fluoroaniline (655 μL, 6.91 mmol), then N,N-diisopropylethylamine (1.72 mL, 9.86 mmol) and the mixture warmed to room temperature and stirred for 16 h. The reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic phases were washed with water (4 × 30 mL) and brine (30 mL) and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure and the crude solid purified by flash column chromatography, using ethyl acetate, hexane 0 : 1 to 1 : 1 as an eluent to afford the product as an off-white solid which was recrystallized from dichloromethane, hexane to yield the title compound 10 (1.39 g, 55%) as a white crystalline solid, mp: 198.0–199.2 °C; Rf: 0.57 (4 : 1 ethyl acetate, hexane); IR (νmax (neat)): 3271, 3221, 3158, 3115, 3086, 3015, 2903, 1716, 1684, 1557, 1525, 1506, 1256, 1225, 1210, 1186, 1167, 1118, 1003, 991, 922, 739 cm–1; 1H NMR (300 MHz, DMSO-d6): δ 0.52–0.75 (4H, m), 2.78–2.81 (1H, m), 4.67 (2H, d, J = 5.4 Hz), 5.28 (1H, d, J = 10.5 Hz), 5.34 (1H, d, J = 17.4 Hz), 5.93–6.05 (1H, m), 7.17–7.24 (2H, m), 7.54 (1H, s), 7.65–7.71 (2H, m), 8.98 (1H, s), 10.78 (1H, s), 13.01 (1H, bs) ppm; 13C NMR (75 MHz, DMSO-d6): δ 5.7, 22.7, 75.1, 115.5 (q, JCF = 287.1 Hz), 115.6 (d, JCF = 22.8 Hz), 117.9, 121.6 (d, JCF = 7.8 Hz), 124.0, 124.1, 124.2, 127.5, 133.8, 134.2, 149.7, 154.1 (q, JCF = 38.0 Hz), 158.4 (d, JCF = 241.1 Hz), 160.4, 164.8 ppm; 19F NMR (282 MHz, DMSO-d6): δ –74.1, –117.9 ppm; LRMS (–ESI) m/z: 497 ([M – H]– 100%), 995 ([2M – H]– 3%), HRMS (+ESI) calcd for C21H18F4N4O4S [M + Na]+: 521.0877, found: 521.0879.
4-(1-((Allyloxy)imino)-2-(cyclopropylamino)-2-oxoethyl)-2-amino-N-(4-fluorophenyl)thiophene-3-carboxamide 12
A solution of 10 (500 mg, 1.00 mmol) and triethylammonium formate (442 μL, 3.00 mmol) in a 4 : 1 methanol water mixture (5 mL) was heated at reflux for 24 h. The solvent was removed under reduced pressure and the residue diluted with ethyl acetate (25 mL), washed with water (2 × 20 mL), dried over anhydrous magnesium sulfate and the solvent removed. The crude product was purified by flash column chromatography, using ethyl acetate, hexane 0 : 1 to 3 : 2 as an eluent, to afford an off-white solid, which was recrystallized from dichloromethane, hexane, to yield 12 (270 mg, 68%) as a white crystalline solid, mp: 194.8–195.9 °C; Rf: 0.52 (4 : 1 ethyl acetate, hexane); IR (νmax (neat)): 3466, 3306, 3253, 3203, 3151, 3119, 3050, 3013, 2903, 2859, 2923, 1678, 1643, 1556, 1504, 1481, 1452, 1408, 1200, 1181, 1021, 1010, 919, 831, 747 cm–1; 1H NMR (300 MHz, DMSO-d6): δ 0.34–0.79 (4H, m), 2.67–2.80 (1H, m), 4.62 (2H, d, J = 4.1 Hz), 5.22 (1H, J = 10.4 Hz), 5.31 (1H, J = 17.3 Hz), 5.88–6.08 (1H, m), 6.64 (1H, s), 7.11–7.31 (4H, m), 7.54–7.75 (2H, m), 8.38 (1H, s), 10.7 (1H, s) ppm; 13C NMR (75 MHz, DMSO-d6): δ 5.9, 22.4, 74.7, 105.9, 112.9, 115.6 (d, JCF = 22.3 Hz), 117.6, 141.5 (d, JCF = 7.7 Hz), 127.7, 133.9, 134.3, 151.1, 158.5 (d, JCF = 241.0 Hz), 161.1, 161.7, 166.4 ppm; 19F NMR (282 MHz, DMSO-d6): δ –118.0 ppm; LRMS (+ESI) m/z: 425 ([M + Na]+ 100%), 827 ([2M + Na]+ 15%), HRMS (+ESI) calcd for C19H19FN4O3S [M + Na]+: 425.1054, found: 425.1054, HPLC: 99.1%, RT: 23.8 min.
2-Amino-4-(2-(cyclopropylamino)-1-(hydroxyimino)-2-oxoethyl)-N-(4-fluorophenyl)thiophene-3-carboxamide 13
A solution of 10 (650 mg, 1.30 mmol), palladium acetate (2.9 mg, 12.9 μmol), triphenylphosphine (13.7 mg, 52.2 μmol) and triethylammonium formate (580 mg, 3.90 mmol) in an aqueous methanol solution (80%, 10 mL) was heated at reflux for 24 h. The solvent was removed under reduced pressure and the residue extracted with ethyl acetate, washed with water (2 × 20 mL) and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure and the crude product purified by flash column chromatography, using ethyl acetate, hexane 0 : 1 to 3 : 2 as an eluent, to afford the desired product 13 (120 mg, 26%) as a tan solid, mp: 201.2–203.2 °C; Rf: 0.36 (4 : 1 ethyl acetate, hexane); IR (νmax (neat)): 3437, 3301, 3151, 3010, 2836, 1649, 1614, 1556, 1506, 1454, 1409, 1212, 1014, 836, 778 cm–1; 1H NMR (300 MHz, DMSO-d6): δ 0.43–0.76 (4H, m), 2.69–2.85 (1H, m), 6.55 (1H, s), 7.19 (2H, t, J = 8.5 Hz), 7.44 (2H, s), 7.60–7.76 (2H, m), 8.86 (1H, s), 10.62 (1H, s), 11.68 (1H, s) ppm; 13C NMR (75 MHz, DMSO-d6): δ 5.9, 22.4, 105.5, 111.9, 115.5 (d, JCF = 22.5 Hz), 121.3 (d, JCF = 7.8 Hz), 128.5, 134.6, 150.7, 158.4 (d, JCF = 241.4 Hz), 162.1, 152.5, 166.6 ppm; 19F NMR (282 MHz, DMSO-d6): δ –118.4 ppm; LRMS (+ESI) m/z: 385 ([M + Na]+ 100%), 747 ([2 M + Na]+ 20%), HRMS (+ESI) calcd for C16H15FN4O3S [M + Na]+: 385.0741, found: 385.0743, HPLC: 98.8%, RT: 19.8 min.
N-(Adamantan-1-yl)-4-(1-((allyloxy)imino)-2-(cyclopropylamino)-2-oxoethyl)-2-(2,2,2-trifluoroacetamido)thiophene-3-carboxamide 11
The acid 9 (1.00 g, 2.47 mmol), benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (1.80 g, 3.45 mmol) and 1-adamantylamine (0.56 g, 3.70 mmol) were dissolved in N,N-dimethylformamide (10 mL) and the solution cooled to 0 °C. To this was added, dropwise, N,N-diisopropylethylamine (860 μL, 4.93 mmol) and the mixture warmed to room temperature and stirred for 16 h. The reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic phases were washed with water (4 × 30 mL) and brine (30 mL) and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure and the crude solid purified by flash column chromatography, using ethyl acetate, hexane 0 : 1 to 2 : 3 as an eluent to afford the title compound 11 (675 mg, 50%) as a white solid, mp: 209.6–209.9 °C; Rf: 0.69 (1 : 1 ethyl acetate, hexane); IR (νmax (neat)): 3298, 3219, 3109, 3016, 2904, 2855, 1722, 1667, 1615, 1526, 1257, 1205, 1186, 1156, 1127, 1019, 1005, 991, 739 cm–1; 1H NMR (300 MHz, DMSO-d6): δ 0.41–0.77 (4H, m), 1.51–1.68 (6H, m), 1.85–2.14 (9H, m), 2.78–2.85 (1H, m), 4.60 (2H, d, J = 4.0 Hz), 5.23 (1H, d, J = 10.4 Hz), 5.34 (1H, d, J = 17.3 Hz), 5.89–6.04 (1H, m), 7.42 (1H, s), 8.22 (1H, s), 9.19 (1H, s), 13.27 (1H, bs) ppm; 13C NMR (75 MHz, DMSO-d6): δ 5.8, 22.6, 28.7, 35.9, 40.9, 51.9, 74.5, 115.1 (q, JCF = 279.4 Hz), 117.2, 122.7, 127.6, 127.9, 128.0, 133.8, 151.6 (q, JCF = 39.4 Hz), 161.6, 165.2, 167.8 ppm; 19F NMR (282 MHz, DMSO-d6): δ –74.4 ppm; LRMS (–ESI) m/z: 437 ([M – H]), HRMS (+ESI) calcd for C25H29F3N4O4S [M + Na]+: 561.1754, found: 561.1754.
N-(Adamantan-1-yl)-2-amino-4-(2-(cyclopropylamino)-1-(hydroximino)-2-oxoethyl)thiophene-3-carboxamide 14
A solution of 11 (230 mg, 429 μmol), palladium acetate (1.0 mg, 4.29 μmol), triphenylphosphine (4.5 mg, 17.2 μmol) and triethylammonium formate (189 mg, 1.29 mmol) in a 4 : 1 methanol water mixture (10 mL) was heated at reflux for 24 h. The organic solvent was removed under reduced pressure and the residue extracted with ethyl acetate, washed with water (2 × 20 mL) and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure and the crude product purified by flash column chromatography, using ethyl acetate, hexane 0 : 1 to 7 : 3 as an eluent, to afford the desired product 14 (50 mg, 29%) as a white powder, mp: 216.6–218.1 °C; Rf: 0.22 (7 : 3 ethyl acetate, hexane); IR (νmax (neat)): 3410, 3276, 3213, 3040, 2904, 2853, 1648, 1606, 1556, 1309, 1015, 700 cm–1; 1H NMR (300 MHz, DMSO-d6): δ 0.36–0.93 (4H, m), 1.51–1.72 (6H, m), 1.89–2.11 (9H, m), 2.62–2.81 (1H, m), 6.46 (1H, s), 7.31 (2H, s), 8.00 (1H, s), 8.86 (1H, s), 11.24 (1H, s) ppm; 13C NMR (75 MHz, DMSO-d6): δ 6.0, 22.4, 28.8, 35.9, 40.9, 51.7, 106.1, 110.5, 129.5, 151.0, 161.9, 162.9, 166.6 ppm; LRMS (+ESI) m/z: 425 ([M + Na]+ 100%), 827 ([2 M + Na]+ 18%), HRMS (+ESI) calcd for C20H26N4O3S [M + Na]+: 425.1618, found: 425.1619, HPLC: 98.1%, RT: 23.2 min.
Thioflavin T fluorescence assay
The ThT fluorescence assay examining the inhibition of tau aggregation by compounds was slightly modified compared to previous reports.20 Briefly, 20 μM 4R tau, 5 μM heparin (average molecular weight of 15 000) in 100 mM sodium acetate pH 7.0, with or without 100 μM of the test compound was incubated for 3 days at 37 °C. 1 mM of dithiothreitol was added daily. 12.5 μM solution of ThT was added and the incubation continued for 1 h at room temperature prior to fluorescence reading. ThT fluorescence spectra was measured on a Fluoromax-4 spectrometer (Horiba Jobin Yvon) equipped with both excitation and emission monochromators. Excitation was at 440 nm and emission collected from 450–750 nm in 1 nm increments.
Filter trap assay
The filter trap assay for detection of tau aggregates was carried out as previously described.18 Briefly, tau fibrils were induced by heparin in PBS, in the presence of 200 μM of the test compound, at 37 °C for 24 hours. The fibrils were further diluted to a final concentration of 0.8 μM 4R tau in 2% SDS, then applied to the wells of the 96 well microfiltration apparatus (BioRad). The vacuum was applied, and subsequently for 2 washes with 2% SDS. The membrane was then incubated with 4R tau specific antibody (Dako) and detected with the ChemiDocMP system (BioRad). Dots were quantified and analyzed using Fiji software.
Oregon green oxidation assay
Recombinant 4R tau was labeled with Oregon green-Iodoacetamide (IAA-OG) as previously described,16 with the following modifications. Briefly, heparin induced 4R tau (10 μM) fibrillisation was carried out in sodium acetate buffer (pH 7), 40 μM heparin, with or without 200 μM of the test compounds at 37 °C for 2 hours. Controls were prepared in sodium acetate buffer (pH 7) and either reduced with 1 mM DTT or oxidised using 20% v/v DMSO at 37 °C for 2 hours. The solutions were further incubated with IAA-OG for 30 minutes at room temperature. The bound proteins were analysed by SDS-page and visualized using the Alexa 488 settings on a ChemiDocMP system (BioRad). Bands were quantified and analysed using Fiji software. Protein loading was determined using Coomassie Blue staining.
Supplementary Material
Footnotes
†The authors declare no competing interests.
‡Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00306k
References
- Ferri C., Prince M., Brayne C., Brodaty H., Fratiglioni L., Ganguli M., Hall K., Hasegawa K., Hendrie H., Huang Y., Jorm A., Mathers C., Menezes P., Rimmer E., Scazufca M. Lancet. 2005;366:2112. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten M. D., Lockwood A. H., Kirschner M. W. Proc. Natl. Acad. Sci. U. S. A. 1975;72:1858. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. A., Higgins G. A. Science. 1992;256:184. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hardy J., Selkoe D. J. Science. 2002;297:353. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E. Acta Neuropathol. 1991;82:239. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Ittner A., Chua S. W., Bertz J., Volkerling A., van der Hoven J., Gladbach A., Przybyla M., Bi M., Hummel A., Stevens C. H., Ippati S., Suh L. S., Macmillan A., Sutherland G., Krill J. J., Silva A. P. G., Mackay J. P., Poljak A., Delerue F., Ke Y. D., Ittner L. M. Science. 2016;354:904. doi: 10.1126/science.aah6205. [DOI] [PubMed] [Google Scholar]
- Ittner L. M., Ke Y. D., Delerue F., Bi M., Gladbach A., van Eersel J., Wölfing H., Chieng B. C., Christie M. J., Napier I. A., Eckert A., Staufenbiel M., Hardeman E., Götz J. Cell. 2010;142:387. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Hanger D. P., Gibb G. M., de Silva R., Boutagangout A., Drion J. P., Revesz T., Lees A. J., Anderton B. H. FEBS Lett. 2002;531:538. doi: 10.1016/s0014-5793(02)03611-6. [DOI] [PubMed] [Google Scholar]
- Mukrasch M. D., Marwick P., Biernat J., Bergen J., Bernado P., Griesinger C., Mandelkow E., Zweckstetter M., Blackledge M. J. Am. Chem. Soc. 2007;129:5235. doi: 10.1021/ja0690159. [DOI] [PubMed] [Google Scholar]
- Janin J., Chothia C., Lo Conte L. J. Mol. Biol. 1999;285:2177. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- Bulic B., Pickhardt M., Mandelkow E. M., Mandelkow E. Neuropharmacology. 2010;59:289. doi: 10.1016/j.neuropharm.2010.01.016. [DOI] [PubMed] [Google Scholar]
- Crowe A., Huang W., Ballatore C., Johnson R. L., Hogan A. L., Huang R., Wichterman J., McCoy J., Huryn D., Auld D. S., Smith A. B., Inglese J., Trojanowski J. Q., Austin C. P., Brunden K. R., Lee V. M.-Y. Biochemistry. 2009;48:7732. doi: 10.1021/bi9006435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C., Crowe A., Piscitelli F., James M. J., Lou K., Rossidivito G., Yao Y., Trojanowski J. Q., Lee V. M.-Y., Brunden K. R., Smith A. B. Bioorg. Med. Chem. 2012;20:4451. doi: 10.1016/j.bmc.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su K., Wang Q., Zheng G., Teng J., Liu C. Jinan Daxue Xuebao, Ziran Kexueban. 2010;24:61. [Google Scholar]
- Barnes D. M., Haight A. R., Maureen A., McLaughlin M. A., Mei J., Tedrow J. S., Toma J. D. R. Tetrahedron. 2006;62:11311. [Google Scholar]
- Yamada T., Goto K., Mitsuda Y., Tsuji J. Tetrahedron Lett. 1987;28:4557. [Google Scholar]
- Barghorn S., Biernat J., Mandelkow E. Methods Mol. Biol. 2005;299:35. doi: 10.1385/1-59259-874-9:035. [DOI] [PubMed] [Google Scholar]
- Nanavaty N., Lin L., Hinckley S. H. and Kuret J., in Tau Protein: Methods and Protocols, ed. C. Smet-Nocca, Springer New York, New York, NY, 2017, pp. 101–111, 10.1007/978-1-4939-6598-4_6. [DOI] [Google Scholar]
- Crowe A., James M. J., Lee V. M., Smith A. B., Trojanowski J. Q., Ballatore C., Brunden K. R. J. Biol. Chem. 2013;288:11024. doi: 10.1074/jbc.M112.436006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua S. W., Cornejo A., van Eersel J., Stevens C. H., Vaca I., Cueto M., Kassiou M., Gladbach A., Macmillan A., Lewis L., Whan R., Ittner L. M. ACS Chem. Neurosci. 2017;8:743–751. doi: 10.1021/acschemneuro.6b00433. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.