

 Bioactivities of quinoides 1–5 and VEGFR2 TKIs 6–10 in hepatocellular cancer (HCC) and cancer stem cells (HCSCs) were studied. Quinoide 3 was able to eradicate cancer stem cells.
Bioactivities of quinoides 1–5 and VEGFR2 TKIs 6–10 in hepatocellular cancer (HCC) and cancer stem cells (HCSCs) were studied. Quinoide 3 was able to eradicate cancer stem cells.
Abstract
Bioactivities of quinoides 1–5 and VEGFR2 TKIs 6–10 in hepatocellular cancer (HCC) and cancer stem cells (HCSCs) were studied. The compounds exhibited IC50 values in μM concentrations in HCC cells. Quinoide 3 was able to eradicate cancer stem cells, similar to the action of the stem cell inhibitor DAPT. However, the more cytotoxic VEFGR TKIs (IC50: 0.4–3.0 μM) including sorafenib, which is the only FDA approved drug for the treatment of HCC, enriched the hepatocellular cancer stem cell population by 2–3 fold after treatment. An aggressiveness factor (AF) was proposed to quantify the characteristics of drug candidates for their ability to eradicate the CSC subpopulation. Considering the tumour heterogeneity and marker positive cancer stem cell like subpopulation enrichment upon treatments in patients, this study emphasises the importance of the chemotherapeutic agent choice acting differentially on all the subpopulations including marker-positive CSCs.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most frequent and second most deadly cancer worldwide.1 HCC patients are resistant to chemotherapy and radiotherapy, because conventional therapies can only reduce the bulk of the tumour mass but are unable to restrain tumour regrowth and relapse. HCC is a highly heterogeneous tumour in terms of morphology and clinical outcome.2 The enrichment of cancer stem cells is one of the main reasons behind failure in treatment of HCC patients. Sorafenib (6), a multi-kinase inhibitor acting on VEGFR2-TK and also the only FDA approved drug for the treatment of HCC patients, inhibits proliferation and migration of tumour cells and angiogenesis. Recently, it has been reported that many patients develop resistance to sorafenib. This was explained by the enrichment of cancer stem cells that have the capacity to self-renew, differentiate into cancer cells and acquire resistance to chemotherapy in most of the cancer types.3,4 Hence, it is crucial to develop novel drugs against the differentiated cancer cells as well as liver cancer stem cells in order to successfully eradicate liver cancer. Liver cancer stem cells could be identified and isolated by several surface markers including CD133, CD90, CD44, CD13, EpCAM, OV-6, CD24, DLK1 and ICAM-1.5 Cells that carry one or two of these markers were shown to possess CSC features.
The epithelial–mesenchymal transition (EMT) is a critical step for stemness. In this study, we have selected liver cancer cell lines with different phenotypic properties based on their genotypes. Huh7 and Hep3B cells are known to have epithelial features (well-differentiated), whereas Mahlavu and SNU-475 cells were characterized as mesenchymal-like (poorly-differentiated) cells.6 Huh7 and Hep3B cells express genes associated with epithelial like properties. These cells express the HCC marker AFP along with the epithelial marker E-cadherin, while Mahlavu and SNU-475 cells don't express AFP and E-cadherin genes while they express high levels of vimentin protein.6 Furthermore, Huh7 and Hep3B cells have normal migratory properties, but Mahlavu and SNU-475 cells have high migratory properties which may be due to their differential PI3K/Akt pathway activities. It was shown that Mahlavu and SNU-475 cells have constitutively active Akt protein due to loss of the PTEN tumor suppressor.7 Current studies and clinical trials focus on the anti-CSC compounds targeting extracellular mediators or cell surface molecules as well as molecules involved in EMT and metastasis.8 Thus, it is crucial to define which marker is efficient to detect and analyse cancer stem cell markers in each cell line, which we studied by flow cytometry.
Quinones and quinone-like compounds seem to be promising candidates against cancer stem cells; however, they are mainly organotoxic. It has been reported that minor changes in the side chains of the quinone structure can lead to a strong variation in the biological toxicity;9–11 thus, modelization of various quinoid derivatives with strong inhibitory activity and low toxicity is conceivable. Therefore, we prepared a series of quinoides, 1–5 (Fig. 1), and evaluated their activities in different HCC cell lines and their cancer stem cell (HCSC) population (Tables 1, 2A and B and Fig. 5). Furthermore, VEGFR2 TKIs 6–10 (Fig. 2) were also screened in HCC cell lines (Tables 1 and 2A). Compound 6 is sorafenib tosylate (Nexavar®), and compound 9 was developed by GlaxoSmithKline as a drug candidate to inhibit VEGFR2 TK. Compounds 7, 8 and 10 were designed by Biomagi, Ltd. to modulate VEGFR2 TK activity.
Fig. 1. The structures of tested quinoides 1–5.

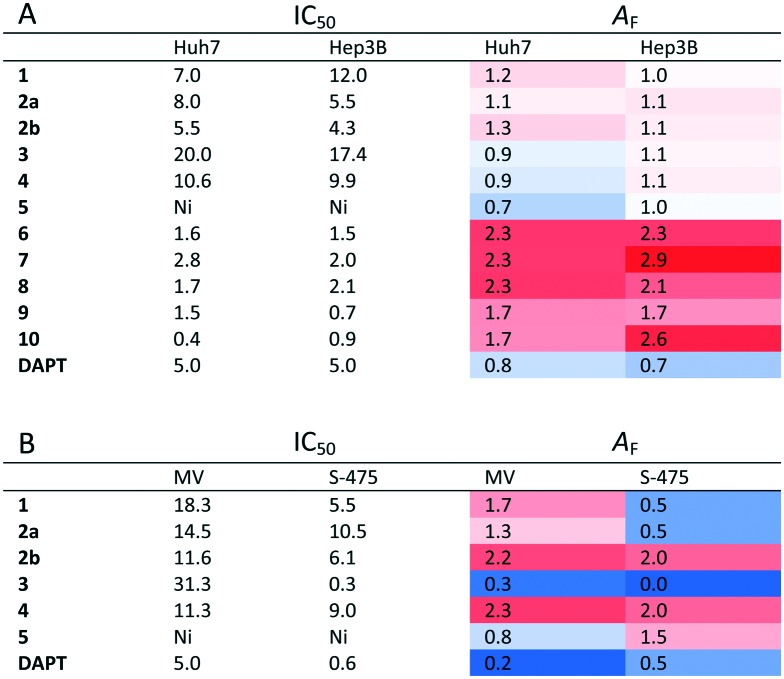
Table 1. Screening of quinoides 1–5 and VEGFR2-TKIs 6–10 in HCC cell lines.
| Compound | IC50 (μM)
a
|
||
| Huh7 | Hep3B | Mahlavu | |
| 1 | 7.0 ± 0.8 | 12.0 ± 1.7 | 18.3 ± 0.8 |
| 2a | 8.0 ± 0.9 | 5.5 ± 1.8 | 14.5 ± 1.3 |
| 2b | 5.5 ± 0.4 | 4.3 ± 0.7 | 11.6 ± 0.2 |
| 3 | 20.0 ± 1.7 | 17.4 ± 2.1 | 31.3 ± 0.3 |
| 4 | 10.6 ± 2.0 | 9.9 ± 2.3 | 11.3 ± 0.6 |
| 5 | Ni | Ni | Ni |
| 6 | 1.6 ± 0.8 | 1.5 ± 0.3 | 0.7 ± 0.4 |
| 7 | 2.8 ± 1.3 | 2.0 ± 0.9 | 3.0 ± 0.5 |
| 8 | 1.7 ± 0.7 | 2.1 ± 0.3 | 0.8 ± 0.5 |
| 9 | 1.5 ± 0.7 | 0.7 ± 0.1 | 1.2 ± 0.4 |
| 10 | 0.4 ± 0.3 | 0.9 ± 0.3 | 1.1 ± 0.7 |
| DAPT | 5.0 ± 1.2 | 5.0 ± 2.0 | 5.0 ± 1.5 |
aExperiments were performed in triplicate; Ni: no growth inhibition.
Table 2. IC50 values and AF factors of quinoides 1–5 and VEGFR2-TKIs 6–10 in (A) Huh7 and Hep3B epithelial cells and of quinoides 1–5 in (B) Mahlavu (MV) and SNU-475 (S-475) mesenchymal cells.
|
Fig. 5. Flow cytometry analysis of HCC cell lines after 72 hours of treatment with quinoides 1–5. (A) Huh7, (B) Hep3B, (C) Mahlavu, and (D) SNU-475. Cancer stem cells were defined by their positivity for CSC markers, CD133 together with EpCAM, or CD90. Each treatment was compared to its corresponding DMSO control to define the changes in percentage of double positive population. DAPT is used as positive control for CSC inhibition. The bar graphs indicate the fold change in positivity for markers between DMSO control and experimental groups.

Fig. 2. VEGFR2 TKIs 6–10 used in this study and their origin and VEGFR2 IC50 activities that were obtained through a commercial service in Germany (; www.proqinase.com).

Results and discussion
Chemistry
Compounds 1, 2a and 2b were prepared according to our previous work starting from commercially available 2-methylhydroquinone (S1) (Scheme 1).12
Scheme 1. Preparation of quinones 1, 2a and 2b from 2-methylhydroquinone (S1).

Compounds 3 and 5 were prepared (Scheme 2) according to Carreño's procedure with slight modifications (see the ESI‡).13–15
Scheme 2. Syntheses of quinones 3 and 5 from 1,4-dimethoxybenzene.

The new quinoide 4 was obtained from vanillin (S7) in very good overall yield using the sequence depicted in Scheme 3.
Scheme 3. Synthesis of compound 4.

VEGFR2 TKI 6 was obtained from Bayer Inc. Synthesis of 7 was described by Lintnerová et al.16 Compound 9 was prepared according the procedure described by Harris et al.17 Novel compounds 8 and 10 were synthesized using the sequence depicted in Scheme 4 (see also the ESI‡).
Scheme 4. Syntheses of 8 and 10.

Biological evaluation
Bioactivities of quinoides and VEGFR2-TKs in HCC cells
NCI-SRB assay was initially performed to define the inhibitory concentrations (IC50 values) of compounds in HCC cell lines Huh7, Hep3B, and Mahlavu. Results have shown that almost all compounds (except for 5) had IC50 values in μM concentrations for all cell lines (Table 1). DAPT (γ-secretase inhibitor) was used as a control for CSC inhibition.
Expression of cancer stem cell markers in HCC cell lines
CSC marker expression varies among cell lines of HCC, since liver CSCs exist heterogeneously and these features determine the characteristics of the cancer (tumourigenicity and metastatic tendency). It has previously been reported that the epithelial cell adhesion molecule (EpCAM) and CD133 expressing cells are more likely to execute epithelial features, whereas CD90 expressing cells are more likely to be mesenchymal.18,19 Expression of these markers in HCC cell lines was analyzed using flow cytometry. It was shown that CSCs of Huh7 cells could be identified by CD133 and EpCAM positivity (20–30%). CSCs of Mahlavu and SNU-475 could be identified by CD90 positivity (0.2–0.6%) (Fig. 3).
Fig. 3. Flow cytometry analysis of HCC cell lines indicating the positivity of HCC cell lines for cancer stem cell markers CD133, EpCAM and CD90. Top row: IgG isotype controls, middle row: cells stained for CD133 and EpCAM markers, bottom row: cells stained for CD90.
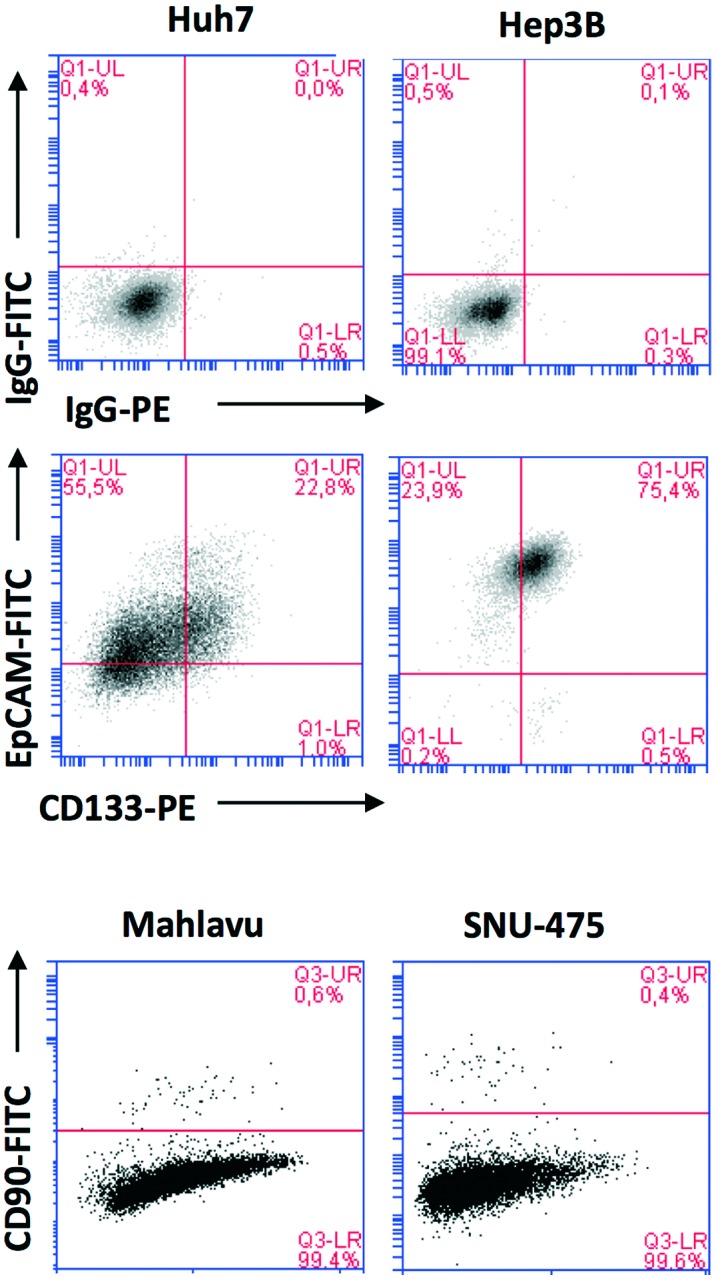
As stated above, the EMT is critical for cancer cell stemness. Huh7 and Hep3B cells carry epithelial features, whereas Mahlavu and SNU-475 cells carry migratory mesenchymal-like properties.6,7 Furthermore, Huh7 and Hep3B cells differ in their p53 gene status: Huh7 cells express p53(Y220S), but Hep3B cells are null for the TP53 gene. Therefore, the expression of mutant p53 causes differential activation of the β-catenin protein and its downstream mesenchymal proteins such as E-cadherin, vimentin, snail and slug. The significantly higher expression of β-catenin20 due to the lack of p53 protein in Hep3B cells correlates with the higher expression of EpCAM and CD133 markers in this cell (Fig. 3).
Effects of quinoides and VEGFR2-TKIs on hepatocellular cancer stem cells (HCSCs)
It is essential to discover novel compounds that were able to alter the enrichment of the CSC population as well as the cancer cells. For this reason, compounds 1–10 that were initially tested against HCC cell lines were further studied to determine the changes in the CSC marker positivity of Huh7 cells. Cells that were killed by the treatment were discarded, and the remaining cells were used in flow cytometry analyses. Interestingly, some of the compounds were able to reduce the percentage of CSC population, while some had the adverse effect on Huh7 cells that enriched the CSC subpopulation (Fig. 4). It was observed that unlike VEGFR2-TKIs, some of the quinoides were able to attenuate the CSC side population in Huh7 cells.
Fig. 4. Expression of stem cell markers CD133 and EpCAM in Huh7 cells treated with (A) quinoides 1–5 and (B) VEGFR2-TKIs 6–10 as shown by flow cytometry analysis.

Due to the distinct dispersion of CD90 cells, we decided to include SNU-475 cells for testing the anti-HCSC activities of quinoides. Therefore, we also determined the bioactivities of quinoides in SNU475 cells. The IC50 values were 5.5 ± 1.4, 10.5 ± 1.5, 6.1 ± 2.0, 0.3 ± 1.2, 9.0 ± 2.1, and 0.6 ± 1.1 μM for compounds 1, 2a, 2b, 3, 4, and DAPT, respectively. No growth inhibition was observed for compound 5 (Fig. 5 and Table 2B).
Although compounds 6–10 were stronger HCC inhibiting agents as quinoides 1–5, they caused enrichment of Huh7 HCSCs in the rest of the HCC after treatment. Therefore, the positive cytotoxic effect of 6–10 was discredited by their ability to leave the HCC residue fraction after treatment enriched with aggressive HCSC cells that are highly susceptible to disease relapse and that acquired drug resistance frequently observed after HCC treatment in clinics.
Further analyses were carried out on Hep3B, Mahlavu and SNU-475 cells to test whether the same effect could be observed in other HCC cell lines as well. Among all compounds tested, 3 was able to attenuate the CSCs especially in mesenchymal-like cells, surpassing the efficacy of the stem cell inhibitor, DAPT (Fig. 5). Altogether, the results have shown that compound 3 is potentially capable of impairing the cancer stem cells in HCC cells with a mechanism not yet discovered.
On the other hand, treatments of cells with VEGFR2-TKIs 6–10 were shown to enrich the cancer stem cell population of Huh7 and Hep3B cells at about 1.7–2.9 times as opposed to the effect of DAPT (Fig. 4 and Table 2A).
Conclusions
There is only one literature report describing macrocyclic benzoquinone herbimycin A [CAS: 70563-58-5, BRN: 4834067] that inhibits (61–78%) human bone marrow mesenchymal stem cells at 1–5 μM concentration.21 We present here for the first time the screening of benzoquinone compounds in CSCs. One of the achievements of our pivotal study is identification of quinoide 3 as an exceptional compound to treat HCSCs. Although its mechanism is not yet known, we hypothesize different behaviours of screened quinoides 1–5 based on their structural differences, benzoquinone reactivity and general medicinal chemistry knowledge. All quinoides 1–5 possess a chemically reactive benzoquinone fragment that can likely be an irreversible inhibitor binding to a biological target e.g. by the Michael reaction with a cysteine amino acid residue. The most active quinoide 3 in Mahlavu and SNU-475 is the one that misses most of substitutions on its benzoquinone skeleton compared to the other tested quinoides. The steric and electronic properties of 3 could be a reason why quinoide 3 is the most active one included (Fig. 5). In the case of the SNU-475 tumour, there are three active quinoides (1, 2a and 3); therefore, other mechanism(s) can also be included. As could be seen, all VEGFR2 TKIs 6–10 are cytotoxic to HCC cell lines (IC50 = 3.0–0.4 μM, Table 1), and at the same time, they are less toxic towards HCSCs since it was observed that cancer stem cells were enriched up to 3 times after treatment (Fig. 5 and Table 2A). The most synergic case would be to identify cytotoxic compounds for both HCCs and HCSCs. The results obtained by screening of VEGFR2 TKIs can deliver two positive consequences: i) VEGFR2 TKIs can be used to enrich the % of stem cells in the final fraction before stem cell isolation for research purposes. Moreover, such stem cells can remember VEGFR2 TKI treatment and can be used for the development of the next generation of inhibitors. ii) It is obvious that experimental compounds can be differently cytotoxic towards heterogeneous tumour cells. Their different influence on CSCs should be quantified separately because of its practical meaning.
In cancer therapeutics, it is important to identify treatment regimens against cancer stem cells as well as tumor cells, because patients suffer from relapse or incomplete recovery after treatment if it fails to eliminate CSCs. The percentage of DMSO treated cells in flow cytometry analysis represents the initial CSC marker positivity of the cells before treatment (Fig. 4) Thus, we suggest that once the cells were treated with a compound, the number of cells with marker positivity can be compared with the DMSO control in order to define the fold change in the percentage of cancer stem cells. Since only the cells that stay alive after treatment are analyzed (see methods), it is important to emphasize the density of the cancer stem cells before and after treatment. Therefore, the compounds that cause a decrease or enrichment in cancer stem cell population can be quantified simply by normalizing the treated cell population by DMSO controls.
Here, we suggest the aggressiveness factor (AF), a new characteristic quantifying the risk for certain compounds to be able to develop a more aggressive disease: AF (exp. cmpd.) = [(total number of CSCs after exp. cmpd. treatment)/(total number of CSCs before treatment)].
The low AF value is an indication for the quality of a drug candidate toward cancer cells. Such molecules don't possess drug resistance or induce more aggressive tumours (Table 2). AF reflects the fold increase of CSCs that survived the compound treatment independently of the shrinkage of the tumour itself.
The compounds with AF values below 1 can be regarded as molecules which reduce the cancer stem cell population, whereas AF values above 1 signify molecules that enrich the cancer stem cell population. Thus, the usage of AF values defines the success or failure of the compounds in affecting cancer cells toward cancer stem cell population.
Cancer stem cell-like subpopulations carry behaviors such as higher tumor-forming and metastasis capacities along with resistance to antitumor drugs which allows tumors to survive and relapse.8 The analysis of the action of the chemotherapeutics that deplete the CSC-like population by the following surface markers may enumerate the qualities of the compounds and may allow one to assess their differential action. Our results represent parallel findings with sorafenib (6) and other compounds and allow comparative analysis of the compounds toward cancer stem cell marker positive cell populations. The AF concept introduced in this study clearly demonstrates the differential action of the compounds.
It is known that patients used to develop resistance towards sorafenib (6). This is consistent with our data with sorafenib (6) having an AF value that is 2 fold higher compared to that of the DMSO control while maintaining good cytotoxicity through the low IC50 values (Table 2A). However, the known CSC inhibitor (DAPT) has low AF values and its IC50 is much higher than that for sorafenib (6). Furthermore, while quinoide 5 does not possess cytotoxic actions (for each HCC line) and 3 has high IC50 values against Huh7, Hep3B or MV (Mahlavu) cells, they both significantly reduce the CSC marker positive subpopulations in mesenchymal Mahlavu and SNU-475 cells (Tables 2A and B).
The normalized aggressiveness factor allows selection of promising experimental compounds possessing lower probability to form aggressive tumours in comparison to a particular drug with known clinical behaviour. The methodology can serve as a simple and valuable tool for pre-clinical screening.
Experimental section
Syntheses of compounds 1–10
The syntheses and physicochemical properties of prepared organic compounds 1–10 can be found in the ESI.‡
Cell culture
Huh7 and Mahlavu, human hepatocellular carcinoma (HCC) cell lines, were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen/GIBCO), supplemented with 10% fetal bovine serum (FBS) (Invitrogen/GIBCO) and 0.1 mM nonessential amino acid, whereas SNU-475 cells were maintained in RPMI (Invitrogen/GIBCO), supplemented 10% fetal bovine serum (FBS) and 2 mM l-glutamine. Both media contained 100 units per mL penicillin and 100 units per mL streptomycin. Cells were grown at 37 °C in a humidified incubator under 5% CO2.
NCI-60 sulforhodamine B (SRB) cytotoxicity assay
Huh7 and Hep3B (2000 cells per well), SNU-475 (1000 cells per well) and Mahlavu (1000 cells per well) cells were inoculated into 96-well plates (150 μl per well). After 24 hours, molecules of interest and DMSO control were applied in concentrations of 40 μM to 2.5 μM in serial dilutions. After 72 h of treatment, cells were fixed with cold 10% (w/v) trichloroacetic acid (MERCK) for an hour. Then, the wells were washed with ddH2O and dried. 50 μl of 0.4% SRB dye (Sigma-Aldrich) was applied to each well and incubated at room temperature for 10 min. Each well was washed with 1% acetic acid and left for air-drying. SRB dye was solubilised using 100 μl per well 10 mM Tris-Base solution, and the absorbance was measured at 515 nm. The experiment was performed in triplicate, and the absorbance values were normalized to DMSO controls.
Flow cytometry
HCC cells are inoculated into 100 mm2 culture dishes (100 000–200 000 cells). 24 hours later, cells were treated with the compounds (IC100 conc.) for 72 hours. Dead cells that no longer remained attached to the surface of the culture plates were discarded through vacuum aspiration and cells that remained attached were collected to be fixed with 4% paraformaldehyde for 30 minutes. Huh7 and Hep3B cells were stained for cancer stem cell markers using anti-CD133/1 (AC133)–biotin (Miltenyi, 130-090-664), anti-biotin–PE (Miltenyi, 130-090-756), and anti-EpCAM–FITC (Miltenyi, 130-080-301), whereas Mahlavu and SNU-475 cells were stained using anti-CD90–FITC (Miltenyi, 130-095-403). For isotype controls, mouse IgG1 isotype control–FITC conjugate (Miltenyi, 130-092-213) and mouse IgG1 isotype control antibody–biotin conjugate (Miltenyi, 130-093-018) were used. Staining of cells was performed according to the manufacturer's protocol. Cells were analyzed using the BD Accuri C6 Flow Cytometer and Software (BD Biosciences). The same staining procedure was applied for the analysis of HCC cells in order to determine the CSC marker positivity.
Supplementary Material
Acknowledgments
Liver cancer stem cell studies and DCK supported by TUBITAK grant #113S540. COST Actions: CM1106, CA15135, VEGA 1/0634/13, APV SK-FR-2015-0014 and Biomagi, Ltd. are acknowledged for chemistry. Sorafenib tosylate (6) was obtained as a gift from Bayer, Inc.
Footnotes
†The authors declare no competing interests.
‡Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00392c
References
- Stewart B. W. and Wild C. P., World Cancer Report 2014, 2014.
- Yamashita T., Wang X. W. J. Clin. Invest. 2013;123(5):1911–1918. doi: 10.1172/JCI66024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H.-W., Ambe C. M., Hari D. M., Wiegand G. W., Miller T. C., Chen J. Q., Anderson A. J., Ray S., Mullinax J. E., Koizumi T., Langan R. C., Burka D., Herrmann M. A., Goldsmith P. K., Stojadinovic A., Rudloff U., Thorgeirsson S. S., Avital I. Gut. 2013;62:1777–1786. doi: 10.1136/gutjnl-2012-303261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow A. K.-M., Ng L., Lam C. S.-C., Wong S. K.-M., Wan T. M.-H., Cheng N. S.-M., Yau T. C.-C., Poon R. T.-P., Pang R. W.-C. PLoS One. 2013;8:e78675. doi: 10.1371/journal.pone.0078675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.-H., Luo Q., Liu L.-L., Song G.-B. World J. Gastroenterol. 2016;22:3547–3557. doi: 10.3748/wjg.v22.i13.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzugullu H., Benhaj K., Ozturk N., Senturk S., Celik E., Toylu A., Tasdemir N., Yilmaz M., Erdal E., Akcali K. C., Atabey N., Ozturk M. Mol. Cancer Ther. 2009;8:90. doi: 10.1186/1476-4598-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durmaz I., Guven E. B., Ersahin T., Ozturk M., Calis I., Cetin-Atalay R. Phytomedicine. 2016;23:42–51. doi: 10.1016/j.phymed.2015.11.012. [DOI] [PubMed] [Google Scholar]
- Marcucci F., Rumio C., Lefoulon F. Front. Radiat. Oncol. 2016;6:115. doi: 10.3389/fonc.2016.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks T. J., Hanzlik R. P., Cohen G. M., Ross D., Graham D. G. Toxicol. Appl. Pharmacol. 1992;112:2–16. doi: 10.1016/0041-008x(92)90273-u. [DOI] [PubMed] [Google Scholar]
- Monks T. J., Jones D. C. Curr. Drug Metab. 2002;3:425–438. doi: 10.2174/1389200023337388. [DOI] [PubMed] [Google Scholar]
- Ollinger K., Brunmark A. J. Biolumin. Chemilumin. 1991;266:21496–21503. [Google Scholar]
- Lanfranchi D. A., Hanquet G. J. Organomet. Chem. 2006;71:4854–4861. doi: 10.1021/jo060486n. [DOI] [PubMed] [Google Scholar]
- Carreño M. C., García Ruano J. L. Synthesis. 1992;1992:651–653. [Google Scholar]
- Carreño M. C., Ruano J. L. G., Toledo M. A., Urbano A. Tetrahedron Lett. 1994;35:9759–9762. [Google Scholar]
- Brimble M. A., Duncalf L. J., Reid D. C. W. Tetrahedron: Asymmetry. 1995;6:263–269. [Google Scholar]
- Lintnerová L., García-Caballero M., Gregáň F., Melicherčík M., Quesada A. R., Dobiaš J., Lác J., Sališová M., Boháč A. Eur. J. Med. Chem. 2014;72:146–159. doi: 10.1016/j.ejmech.2013.11.023. [DOI] [PubMed] [Google Scholar]
- Harris P. A., Cheung M., Hunter R. N., Brown M. L., Veal J. M., Nolte R. T., Wang L., Liu W., Crosby R. M., Johnson J. H., Epperly A. H., Kumar R., Luttrell D. K., Stafford J. A. J. Med. Chem. 2005;48:1610–1619. doi: 10.1021/jm049538w. [DOI] [PubMed] [Google Scholar]
- Yamashita T., Kaneko S. J. Gastroenterol. 2014;49:1105–1110. doi: 10.1007/s00535-014-0951-1. [DOI] [PubMed] [Google Scholar]
- Yang Z. F., Ho D. W., Ng M. N., Lau C. K., Yu W. C., Ngai P., Chu P. W. K., Lam C. T., Poon R. T. P., Fan S. T. Cancer Cell. 2008;13:153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Cagatay T., Ozturk M. Oncogene. 2002;21:7971–7980. doi: 10.1038/sj.onc.1205919. [DOI] [PubMed] [Google Scholar]
- Mruthyunjaya S., Manchanda R., Godbole R., Pujari R., Shiras A., Shastry P. Biochem. Biophys. Res. Commun. 2010;391:43–48. doi: 10.1016/j.bbrc.2009.10.158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.