

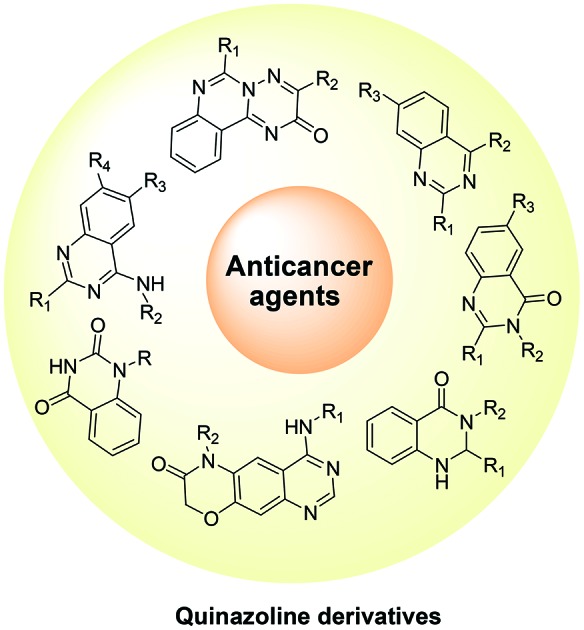
 This article reviews the recent advances in the development of quinazoline derivatives as anticancer agents.
This article reviews the recent advances in the development of quinazoline derivatives as anticancer agents.
Abstract
Cancer is one of the major causes of worldwide human mortality. A wide range of cytotoxic drugs are available on the market, and several compounds are in different phases of clinical trials. Many studies suggest that these cytotoxic molecules are also associated with different types of adverse side effects; therefore researchers around the globe are involved in the development of more efficient and safer anticancer drugs. In recent years, quinazoline and its derivatives have been considered as a novel class of cancer chemotherapeutic agents that show promising activity against different tumors. The aim of this article is to comprehensively review and highlight the recent developments concerning the anticancer activity of quinazoline derivatives as well as offer perspectives on the development of novel quinazoline derivatives as anticancer agents in the near future.
1. Introduction
Cancer has become a major cause of human mortality and is considered a major worldwide health problem. It is estimated that about 1 688 780 new cancer cases will be diagnosed in 2017, and about 600 920 Americans are expected to die of cancer in 2017, which is about 1650 people per day.1 A cancer consists of a group of cells that originated from a single cell with uncontrolled growth and rapid proliferation properties.2 Presently, a wide range of cytotoxic drugs, either alone or in combination, are used to treat cancer, and several of these drugs are in different phases of clinical trials. These cytotoxic drugs are associated with several drawbacks and are not able to discriminate between cancerous and normal cell types; therefore, they can cause serious side effects that are often cumulative and dose-limiting. The anticancer drugs in current clinical trials are mostly associated with excessive organ toxicity, lack of cell specificity, short circulating half-life, angiogenesis, metastasis, and a noticeable tendency to induce resistance in the target cells.3 Hence, to save the lives of millions of people globally, continuous efforts are being made to develop effective anti-cancer drug candidates with minimal side effects.
Different approaches are being used for the treatment of cancer. Some involve the targeting of specific molecular alterations that occur in tumor cells. This approach has yielded several molecules with significant clinical activity and minimum toxicity.4,5 Most of the chemotherapeutic agents used in cancer therapy inhibit DNA replication and transcription or inhibit various protein kinase enzymes involved in tumor growth. However, several drawbacks have been documented, including participation of a number of enzymes such as ribonucleotide reductase [RNR] and topoisomerases I and II [Topo I and II] etc. at different cancer development stages, cancer cell survival even under anaerobic conditions, and the problem of multidrug resistance that develops in cancer cells toward chemotherapeutic agents.6–9
The heterocycles are widely investigated bioactive molecules and are considered important synthetic targets for the development of novel therapeutic agents.10–12 Quinazoline is one of the heterocycles for which considerable research has been done in order to examine its biomedical applications.13 In a number of biologically active compounds and drug molecules, the quinazoline nucleus is used as a basic framework. Due to their broad range of pharmacological activities, which include antimicrobial,14,15 antimalarial,16 anti-inflammatory,17–19 anticonvulsant,20,21 antihypertensive,22 antioxidant,23 antiviral,24 anti-HIV25 and anticancer,26–29 quinazoline and its derivatives have attracted the attention of biologists and medicinal chemists. Quinazoline and its derivatives have been identified as a new class of cancer chemotherapeutic agents with significant therapeutic efficacy against solid tumors.30–42
The Food and Drug Administration (FDA) has approved several quinazoline derivatives for clinical use as anticancer drugs. These include gefitinib, erlotinib, lapatinib, afatinib, and vandetanib (Fig. 1).43 Gefitinib (Iressa®) was approved by the FDA in 2003 for the treatment of locally advanced or metastatic non-small-cell lung cancer (NSCLC) in patients after failure of both platinum-based and/or docetaxel chemotherapies. In 2004, erlotinib (Tarceva®) was approved by the FDA for treating NSCLC. Furthermore, in 2005, the FDA approved erlotinib in combination with gemcitabine for the treatment of locally advanced, unrespectable, or metastatic pancreatic cancer. Erlotinib acts as a reversible tyrosine kinase inhibitor. Lapatinib (Tykreb®) was approved by the FDA in 2012 for breast cancer treatment. It inhibits the activity of both human epidermal growth factor receptor-2 (HER2/neu) and epidermal growth factor receptor (EGFR) pathways. Vandetanib (Caprelsa®) was approved by the FDA in 2011 for the treatment of metastatic medullary thyroid cancer. It acts as a kinase inhibitor of a number of cell receptors, mainly the vascular endothelial growth factor receptor (VEGFR), EGFR, and rearranged during transfection (RET)-tyrosine kinase (TK). Afatinib (Gilotrif®) was approved by the FDA in 2013 for NSCLC treatment. It acts as an irreversible covalent inhibitor of the receptor tyrosine kinases (RTK) for EGFR and erbB-2 (HER2).
Fig. 1. FDA approved quinazoline derivatives as anticancer drugs.

The development of novel quinazoline derivatives as anticancer drugs is considered a promising area, and researchers around the world are continuously investigating this area in order to develop novel drug candidates. The present review article was written as an effort to compile and discuss recently published studies concerning the therapeutic potential of quinazoline derivatives as anticancer agents.
2. Therapeutic potential of quinazoline derivatives as anticancer agents
2.1. Quinazoline derivatives as protein kinase inhibitors
Protein kinases constitute the most important human enzyme class that controls the sequence of events such as cell cycle progression, cell division, and cell proliferation. The protein kinases, when expressed in mutated, unregulated forms or when produced in abnormally high levels, are capable of transforming normal cells into cancer cells and thus play important roles in tumorigenesis. Protein kinases not only control cell division but also support the angiogenesis process that is required for tumor growth and metastasis. In tumor cells, therefore, the development of non-toxic and selective protein kinases inhibitors is considered as a promising target for cancer treatment. The most important protein kinases involved in a cancer state, overexpressed in neoplastic cells, and commonly targeted include several kinases: a) receptor tyrosine kinases, including EGFRs, insulin-like GFRs, platelet-derived GFRs, fibroblast GFRs, and vascular endothelial GFRs and b) serine/threonine kinases, including GC, CAMK, CK1, CMGC, STE and TKL kinase. Extracellular signal-regulated kinases (ERK 1 and 2) are related protein-serine/threonine kinases that participate in the Ras-Raf-MEK-ERK signal transduction cascade. c) Histidine kinases.44–49
In the early 2000s, discovery of erlotinib and gefitinib as anticancer drugs encouraged researchers to investigate 4-anilinoquinazoline compounds, which led to the development of new and promising compounds such as lapatinib, vandetanib, and afatinib. In previous studies, several patents and articles have been published that discuss the feasibility of the anilinoquinazoline scaffold for the development of tyrosine kinase inhibitors (TKIs).43 The main biomolecular target of this class of compounds remains the epidermal growth factor receptor (EGFR). Some compounds, however, do not show high selectivity for EGRF such as lapatinib, which is a dual EGFR/Her-2 inhibitor, whereas vandetanib inhibits the kinase activities of both EGFR and VEGFR-2. Therefore, continuous and international efforts are being undertaken in order to develop more selective and efficient TKIs.
EGFR is a very promising molecular target for cancer therapy; it has been observed, however, that most of the patients developed resistance to the EGFR inhibitors.50–53 Therefore, continuous efforts are being undertaken to design and develop new and more potent EGFR inhibitors with improved anti-tumor activities. In this regard, several novel compounds were synthesized by introducing substituents on the benzene ring of the EGFR inhibitor gefitinib. However, replacement of the benzene ring with another aromatic ring has rarely been reported in the literature. Therefore, in 2010, X. Wu et al. designed and synthesized two series of 4-benzothienyl amino quinazoline derivatives as new analogues of gefitinib.54 The anti-tumor activity of these novel gefitinib analogues in six human cancer cell lines was examined. Most of the compounds exhibited increased cytotoxicity to cancer cells when compared with the parental compound. The compounds containing ethyl or methyl groups as side chains at position 7 exhibited good pan-RTK inhibitor activity with enhanced apoptosis-inducing capabilities. In comparison with parental gefitinib, analogues 1 and 2 (Fig. 2) exhibited promising and selective apoptosis-inducing capabilities and enhanced anti-tumor activities in cancer cells with HER-overexpression and were considered as promising lead compounds for further development.
Fig. 2. Quinazoline derivatives 1–10 as protein kinase inhibitors.

In order to develop novel RTK inhibitors with improved anticancer activity, X. Wu et al. designed and synthesized two series of 4-pyrrylamino quinazolines.55 Gefitinib was used as a parent compound in which the benzene ring was replaced by a pyrrole ring. All of the prepared compounds were evaluated against pancreatic (Miapaca2) and prostate (DU145) cancer cell lines for kinase inhibitory and antitumor activities. In vitro results suggested that most of the compounds exhibited increased antitumor activity in comparison with the parental gefitinib. The most promising compounds were 3–7 (Fig. 2). The structure–activity results suggested that the replacement of the benzene ring with a pyrrole ring increased the anticancer activity. In addition, the presence of a basic side chain at position 6 or 7 of the quinazoline nucleus plays a significant role in determining these compounds' cytotoxicity.
Y. Zhang et al. initially prepared a series of 5,6,7-trimethoxy-N-phenyl(ethyl)-4-aminoquinazoline that exhibited anticancer activities. Several compounds displayed strong inhibitory activities against ERK1/2 phosphorylation.56 Next, a series of 4-(4-substituted piperazine)-5,6,7-trialkoxy quinazoline derivatives were synthesized by replacing the aniline moiety with a piperazine moiety and then examined for inhibitory activity against ERK1/2 phosphorylation.57 In addition, to study the effects of alkoxy substitution, a few trimethoxy and triethoxy substitution patterns were prepared. Most of the compounds showed inhibitory activities against tumor cell proliferation, and some of these compounds exhibited broad spectrum inhibition activities. Against tumor cells, the ethoxy series of compounds exhibited higher inhibitory activity than did the methoxy series of compounds. The most active compound of the series was 8 (Fig. 2), which showed IC50 values in the micromolar range against most of the cell lines. Compound 8 exhibited a weaker effect on normal cells and demonstrated that the inhibitory activity was lower against NH3T3 cells. Therefore, compound 8 was selected in order to study the mechanism of antitumor activity, and cell configuration and cell cycle results showed that the compound might have caused cells to remain at the G0/G1 phase after inhibition of cell proliferation for 24 h. Compound 8 also inhibited ERK1/2 and P38 phosphorylation in a concentration-dependent manner.
In previous studies, EGFR and its family members have been described as attractive targets for anticancer therapy, mainly for the treatment of non-small-cell lung cancer.58 In the last decade, the discovery of novel EGFR inhibitors has been the main focus of researchers, and consequently, in 2016, X. Qin et al. designed and synthesized a novel series of morpholin-3-one fused quinazoline derivatives by intramolecular cyclization and evaluated their biological activities with regard to EGFR inhibition.59 The most active compound of the series was 9 (Fig. 2), which showed promising inhibitory activity in the nanomolar range against EGFRwt kinase. In addition, compound 9 exhibited antiproliferative activity against H358 and A549 cell lines and good inhibitory activity against mutant EGFRT790M/L858R as compared to gefitinib and erlotinib. In addition, to understand the possible binding mode of the target compounds, molecular docking studies using the interaction of compound 9 with the ATP binding site of EGFR kinase were performed. Compound 9 overlaid well with the binding conformation of gefitinib in the EGFR kinase domain and demonstrated hydrogen bonding with the active site through the quinazoline ring's nitrogen and oxygen.
In 2016, by following a structure-based molecular hybridization approach, J. N. Chen et al. designed and synthesized a series of novel quinazolinyl-diaryl urea derivatives using sorafenib and 4-aminoquinazolinyl moieties.60 The synthesized compounds were screened for their antiproliferative activities against three human cancer cell lines, namely, HepG2 (liver), MGC-803 (stomach), and A549 (lung). In comparison with the positive reference drugs (sorafenib and gefitinib), some of the compounds showed significant antiproliferative activities against certain cell lines. The most active compound (10) (Fig. 2), consisting of a 6,7-dimethoxyquinazolinyl moiety, a 3-benzylamino linker and a tert-butyl substituent group on the distal phenyl ring, showed the maximum activity. Compound 10 induced A549 apoptosis, arrested the cell cycle at the G0/G1 phase, elevated the intracellular reactive oxygen species level, decreased the mitochondrial membrane potential, and effectively intervened with the Raf/MEK/ERK pathway. Additionally, molecular docking studies revealed that compound 10 could bind efficiently to the active site of c-Raf.
2.2. Quinazoline derivatives as tubulin polymerization inhibitors
Tubulin has multiple drug binding sites and is considered as an important target for anticancer drugs. The most comprehensively examined binding sites were for taxoid, vinca, and colchicine.61 In previous studies, colchicine and its analogues have been shown to have promising activity by inhibiting tubulin polymerization into microtubules. However, the clinical uses of these compounds are limited due to their high toxicity. Recently, several small molecules that act at the colchicine site on tubulin have been discovered. These molecules not only inhibit the growth of a wide variety of human cancer cell lines but they also show vascular-disrupting effects on tumor endothelial cells required for cancer growth, and are therefore considered as vascular-disrupting agents (VDAs).62
In 2013, A. Chilin and his coworkers reported biphenylaminoquinazoline (11) (Fig. 3) as an inhibitor of EGFR, FGFR-1, PDGFRβ, Abl1, and Src kinase activities at submicromolar concentrations; it also exhibited EC50s in the nanomolar range against several cancer cell lines.63 After investigations of the biphenylaminoquinazolines' mechanism of action, it was observed that these molecules not only showed anti-TK activity but also inhibited tubulin polymerization. To identify the structural features required for dual TK/tubulin inhibition or to be selective anti-tubulin compounds, several heterobiaryl analogues of compound 11 were designed, synthesized, and evaluated as TKIs and as tubulin polymerization inhibitors.64 The compounds were synthesized by replacing one of the two benzene rings of the biphenylamino moiety with a 5- or 6-atom heterocycle. The computational analysis and biological data of these compounds revealed some important structural features required for dual activity: 1) the dioxygenated function is required on the benzene of the quinazoline ring, however, the carbon atom linker between the oxygen atoms can be varied; 2) the aniline benzene is important for the dual activity, and its replacement with heteroaryl is not feasible; and 3) the terminal ring attached to the aniline benzene was considered as a switch between the two activities. Selective anti-tubulin activity was observed with the heterocycle whereas dual anti-tubulin/anti-TK activity was observed with phenyl rings.
Fig. 3. Quinazoline derivatives 11–16 as tubulin polymerization inhibitors.

In 2013, X.-F. Wang et al. selected compounds 12 and 13 (Fig. 3), which exhibited low nanomolar GI50 values against a human tumor cell line panel, inhibited tubulin assembly, and inhibited colchicine binding to tubulin.65 For further modifications and structure–activity studies, a series of 4-(N-cycloamino)phenylquinazolines were designed, synthesized, and evaluated in cellular and tubulin inhibition assays.66 The compound showed significant activity and thus resulted in the discovery of new tubulin-polymerization inhibitors. The most promising compound of the series was 14 (7-methoxy-4-(2-methylquinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one [Fig. 3]), which showed both low nanomolar antiproliferative activity in cellular assays and significant inhibition of tubulin assembly with an IC50 in the micromolar range. The compound also showed exceptionally potent inhibition of colchicine binding to tubulin. The mechanistic study suggested that compound 14 arrested most cells in the G2/M phase of the cell cycle, disrupted cellular microtubules, and competed mostly at the colchicine site on tubulin.
Through random screening for anticancer agents, K. Kuroiwa et al. discovered a quinazoline derivative, PVHD121 (15) (Fig. 3), with promising antiproliferative activity against various tumor-derived cell lines, including the A549 (lung), NCI-H460 (lung), HCT116 (colon), MCF7 (breast), PC3 (prostate), and HeLa (cervical) cells with IC50 values in the micromolar range. In addition to developing a novel anticancer drug, a series of compounds with various substituents at positions 2 and 4 of the quinazoline core were synthesized and evaluated for anticancer activities.67 Structure–activity relationship studies suggested that the 2-chloroquinazoline derivative compound 16 was the most potent and most active compound of the series (Fig. 3) in the low micromolar range. By using in vitro tubulin polymerization and fluorescence-based colchicine site competition assays with purified tubulin, it was proven that compound 15 inhibits tubulin polymerization by binding to the colchicine site.
2.3. Quinazoline derivatives as protein lysine methyltransferase inhibitors
Protein lysine methyltransferase G9a is over-expressed in cancer cells and acts as a catalyst during methylation of lysine 9 of histone H3 (H3K9) and lysine 373 (K373) of p53. Genetic knockdown of G9a inhibits cancer cell growth via the di-methylation of p53 K373 that leads to the inactivation of p53.68–70 Initially, the 2,4-diamino-6,7-dimethoxyquinazoline template, represented by 17 (BIX01294) (Fig. 4), was discovered as a selective small molecule inhibitor of G9a and GLP.71,72 In order to study the structure–activity relationship and improve the potency and selectivity, multiple regions of the quinazoline template were investigated. This resulted in the discovery of compound 18 (UNC0224) (Fig. 4) as a potent G9a inhibitor with excellent selectivity.73 Additionally, high resolution X-ray crystal structural analysis of the G9a-18 complex was carried out and depending upon the structural insight discovered in G9a-18 co-crystal structure, F. Liu et al. in 2010 optimized the 7-dimethylaminopropoxy side chain of the quinazoline scaffold. This led to the discovery of 19 (UNC0321) (Fig. 4), which was the most promising G9a inhibitor with picomolar potency.74 Investigation of the 2- and 4-amino and 7-aminoalkoxy regions of this quinazoline scaffold provided valued structure–activity relationship information. Compound 19 exhibited excellent in vitro potency; however it had lower cellular potency. In addition, to improve the cellular potency of the quinazoline series, several novel analogs were designed and synthesized.75 The focus was to improve cell membrane permeability while maintaining in vitro potency. Structure–activity relationship studies were performed on these compounds with G9a biochemical (SAHH-coupled), cell-based functional (anti-H3K9me2 ICW), and cell toxicity (MTT) assays. The results led to the discovery of compounds 20 and 21 (Fig. 4), which exhibited high cellular potency and excellent separation of functional potency versus cell toxicity in a variety of cell lines.
Fig. 4. Quinazoline derivatives 17–21 as protein lysine methyltransferase inhibitors.

2.4. Quinazoline derivatives as topoisomerase I inhibitors
DNA topoisomerases (Top) are very important enzymes involved in DNA modification during cellular processes such as replication, transcription, and repair. The DNA topoisomerases are divided into two major families based on whether they cleave only one or two DNA strands: 1.) Top: Type I (Top1) and 2.) Type II (Top2).76,77 Top1 enzyme inhibitors are considered a new group of anticancer agents with a wide range of activity in hematological and solid tumors. Camptothecin (CPT) 22 (Fig. 5) was identified as the first Top1 inhibitor, but CPTs display a number of limitations, including chemical instability and potential induction of cellular resistance; therefore, they were not considered ideal drugs.78,79 To overcome the main drawbacks of CPTs, several chemical classes of non-CPT Top1 inhibitors (such as phenanthridines and the indenoisoquinolines) were developed as promising antitumor drugs.80,81 In 2013, S. Taliani reported a series of pyrazolo[1,5-a]quinazolines, which were structurally related to the indenoisoquinoline nucleus, as novel non-CPT Top1 inhibitors.82 It was observed that Top1 inhibitory activity was dependent on the nature of the chain at position 5, which is a type –O– or –NH– only, the length of the linker, and the terminal nitrogen containing group. The most active compounds contained dimethylaminoethylamino (23), dimethylaminopropylamino (24), and diethylaminoethylamino (25) chains (Fig. 5). However, it was observed that the presence of an imidazole-containing chain exhibited detrimental effects on cells. The theoretical model for the 23/Top1/DNA ternary complex obtained by hydration docking calculations provided the necessary structural requirement for Top1 inhibitory activity; it was revealed that a properly substituted phenyl ring at position 3 and a protonable dialkylaminoalkylamino chain at position 5 were favorable for Top1 inhibition.
Fig. 5. Quinazoline derivatives 22–25 as topoisomerase I inhibitors.

2.5. Quinazoline derivatives as PI3K/Akt/mTOR inhibitors
The PI3K/Akt/mTOR pathway plays important regulatory roles in various cellular functions, including cell proliferation, differentiation, migration, survival, and angiogenesis.83 PI3K is the key regulator in the PI3K/AKT/mTOR pathway and controls phosphatidylinositol phosphorylation. PI3K dysregulation contributes to uncontrolled cell growth and leads to malignant transformation. Consequently, for malignant tumor treatment, PI3K is considered as one of the most promising targets, and efforts are being undertaken to develop small molecules as PI3K inhibitors.84 The two major classes of promising structural scaffolds with the PI3K inhibition effect are quinolines and 1,3-dihydro-2H-imidazo[4,5-c] quinolin-2-ones.85 Considering the anticancer properties of the quinazoline framework in addition to its bioisosteric behavior toward the quinoline ring, Y.-Y. Hei et al. designed a new series of 4,6-disubstituted quinazoline derivatives as PI3K inhibitors.86 The new series was synthesized by replacing the 1,3-dihydro-2H-imidazo[4,5-c]quinolin-2-one skeleton of PF-04979064 with a quinazoline moiety and by opening the piperidine ring. The in vitro and in vivo anticancer activities of the synthesized compounds were evaluated against HCT-116 and MCF-7 cell lines. All the compounds showed substantial antiproliferative activity and compound 26 (Fig. 6) displayed the most potent anti-proliferative and PI3K inhibitory activity. Therefore, in vivo activity of compound 26 was evaluated and exhibited significant tumor growth inhibition. Structure–activity relationship studies suggested that the presence of a hydrogen-bond receptor group (such as a cyano or a nitro group) at position 5 of the pyridine ring attached at position 6 of quinazoline is important for improving the anti-proliferative activity.
Fig. 6. Quinazoline derivatives 26–28 as PI3K/Akt/mTOR inhibitors.

Substituted 4-morpholine-quinazolines have been previously shown as potent anticancer agents and resemble typical PI3K inhibitor scaffolds.87 W. Peng et al. in 2016 expected that the replacement of a hydrogen atom on the quinazoline scaffold with different substituents such as 6-amino-4-(trifluoromethyl) pyridin-3-yl, 2-amino-pyrimdin-5-yl, and 3-hydroxyphenyl would result in good kinase inhibitory activity.88 Therefore, a novel series of 7- or 8-substituted-4-morpholine-quinazoline derivatives were designed, synthesized, and tested for kinase inhibitory and in vitro anti-cancer activities. Compound 27 (Fig. 6) exhibited significant activity in the micromolar range for both PI3Kα inhibition and antiproliferative activities. Additionally, compound 27 was also screened for its inhibitory activity against other kinases (including PI3Kβ, PI3Kγ, PI3Kδ, and mTOR), its effects on p-Akt (S473), and cell cycle. The results indicated that compound 27 acts as a potent PI3K inhibitor and an anticancer agent, which significantly inhibited the PI3K/Akt/mTOR pathway.
In recent years, the development of PI3K inhibitors and especially PI3Kα-selective inhibitors has attracted much attention due to the significance and high frequency of the PIK3CA mutation in most solid tumors. One of the most important approaches for discovering potent anticancer agents is to prepare isoform-selective PI3K-α inhibitors. With this in mind, R. R. Yadav et al. designed and synthesized a series of 6-aryl substituted 4-(4-cyanomethyl) phenylamino quinazolines and screened these for PI3K-α/mTOR inhibitory activity and cytotoxicity in a panel of cancer cell lines.89 The 6-indolyl substituted quinazoline 28 (Fig. 6) exhibited PI3K-α inhibition activity with excellent fold selectivity toward the α-isoform with respect to PI3Kβ and PI3K-δ isoforms and moderate selectivity over PI3K-γ isoforms in both cell-free enzymatic and cell-based gene expression assays. Compound 28 was also investigated for its in vivo anticancer activity in murine tumor models and demonstrated good tumor growth inhibition (62%) at a 25 mg kg–1 dose.
2.6. Quinazoline derivatives as poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors
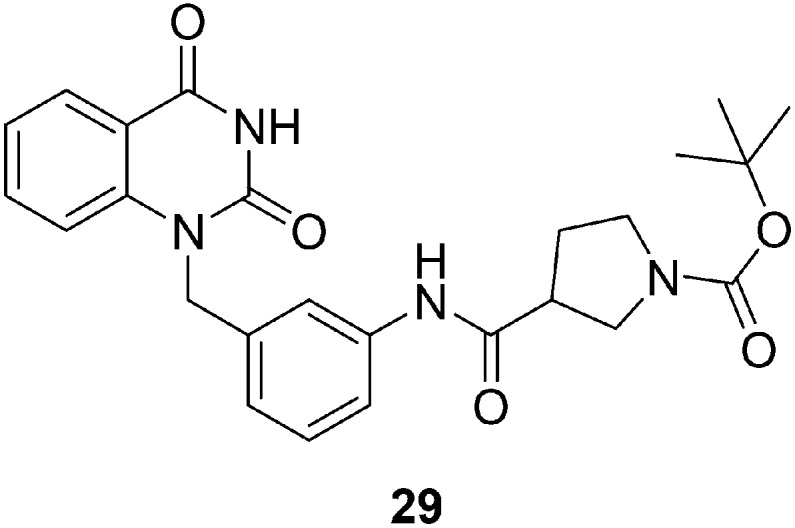
Poly(ADP-ribose)polymerase-1 (PARP-1) plays an important role in the DNA repair process, and it is therefore considered a promising anticancer drug target. H. Yao et al. designed and synthesized a series of quinazoline-2,4(1H,3H)-dione derivatives by employing a range of amino acid building blocks as key pharmacophoric groups and evaluating their PARP inhibition activity and binding features at the PARP active site.90 Several compounds in this series exhibited promising PARP-1 inhibitor activity with IC50 values in the nanomolar range. The structure–activity relationship studies suggested that compounds bearing β-proline and piperidine-4-carboxylic acid groups are good PARP-1 inhibitors. Compound 29 (Fig. 7) having an (S)-N-Boc-pyrrolidin-3-yl substituent exhibited maximum activity as a PARP-1 inhibitor; therefore, it was selected for further evaluation for PARP-2 inhibitory activities, growth inhibition, and temozolomide (TMZ) potentiation effects in cancer cells. The results were very satisfactory, and at both enzymatic and cellular levels, compound 29 displayed high inhibitory activity.
Fig. 7. Quinazoline derivative 29 as a poly(ADP-ribose)polymerase-1 (PARP-1) inhibitor.
2.7. Quinazoline derivatives with apoptotic activity
Quinazolinones and their derivatives are well known as biologically active molecules and are especially used in anti-cancer therapies for breast and other forms of cancer treatment. In 2015, M. Zahedifard et al. synthesized two quinazolinone Schiff base derivatives, which were 3-(5-chloro-2 hydroxybenzylideneamino)-2-(5-chloro-2-hydroxyphenyl)-2,3-dihydroquinazolin-4(1H)-one (30) and 3-(5-nitro-2-hydroxybenzylideneamino)-2-(5-nitro-2-hydroxyphenyl)-2,3-dihydroquinazolin-4(1H)-one (31) (Fig. 8). Their cytotoxic activities were measured on MCF-7, MDA-MB-231, MCF-10A, and WRL-68 cells.91 The colorimetric MTT assay results revealed that both the compounds exhibited substantial inhibition of MCF-7 cells but these compounds did not have cytotoxic effects toward MDA-MB-231, WRL-68, or MCF-10A cells. In addition, the mechanism of compounds A and B on human breast cancer cell lines (such as MCF-7 cells) were also studied. These compounds induced apoptotic cell death in tumor cells by causing cytochrome c release from the mitochondria to the cytosol, which then activates caspase 9 and downstream inhibitor caspase-3/7, which leads to apoptotic changes. Additionally, these compounds induced apoptosis through the extrinsic pathway, which involves caspase-8 and inhibition of NF-κ B translocation from the cytoplasm to the nucleus.
Fig. 8. Quinazoline derivatives 30–31 with apoptotic activity.

2.8. Quinazoline derivatives with anticancer activity
In view of the importance of quinazoline derivatives plus the tendency of some of these molecules to develop resistance, P. M. Chandrika et al. in 2008 designed and synthesized novel quinazoline derivatives with the purpose of finding a more potent compound.92 A series of new 4,6-di-substituted quinazoline derivatives were synthesized and screened for anti-inflammatory and anticancer activities against U937 leukemia cell lines. The anti-inflammatory activity of these compounds was moderate to poor. However, all of the compounds in this series exhibited a noteworthy decrease in cell viability with reference to concentration. The cytotoxic activity of compound 32 (Fig. 9) was in the micromolar range, very close to that of the standard positive control drug (etoposide), and exhibited maximum activity. It was assumed that the maximum activity of 32 was a result of the presence of iodine and l-phenylalanine at positions 6 and 4 of the quinazoline nucleus, respectively.
Fig. 9. Quinazoline derivatives 32–49 with anticancer activity.

To find new parent compounds with promising chemotherapeutic activities, M. N. Noolvi in 2011 synthesized a new series of 2,3-disubstituted quinazolinones and quinoxalines that are structurally correlated to erlotinib and lapatinib and evaluated them for in vitro anti-cancer activities.93 The most active compound of the series was 3-(2-chloro benzylideneamine)-2-(furan-2-yl) quinazoline-4(3H)-one 33 (Fig. 9). The rational approach and quantitative structure–activity relationship studies provided the structural features for quinazoline and quinoxaline derivatives needed for enhanced anticancer activity. It was observed that the presence of the quinazoline ring as the backbone, the 2-chloro benzylideneamine group at position 3 of quinazoline, and chalcone on the benzamide amide, sulphonamide, and plane styryl groups at position 3 of the quinoxaline improves the potency and broad spectrum characteristics of these compounds.
In 2011, M. Alafeefy designed and synthesized a series of 2,3-disubstituted-6-iodo-3H-quinazolin-4-one derivatives and screened them for antitumor activities against different cancer cell lines.94 Alkyl substituents (such as allyl, benzyl, or phenacyl) attached to an electron-rich atom are anticipated to be reasonably stable leaving groups. Therefore, the series was designed with benzyl, allyl and/or phenacyl groups attached to the sulfur atom at position 2, phenyl and/or benzyl groups attached at position 3, and an iodine atom at position 6. The most active compounds had allyl and/or benzyl groups at positions 2 and/or 3 of the quinazoline nucleus. Compound 34 (Fig. 9) was the most potent compound of the series and showed in vitro antitumor activity in the micromolar range against the tested cell lines. It was also observed that an electron withdrawing group at position 4 of the phenacyl group linked to 2-mercapto-3H-quinazolin-4one is favorable for antitumor activity. It is expected that these compounds will be effective as double alkylating agents.
In 2012, E. Moreno et al. synthesized a novel series of sulfur and selenium quinazoline and pyrido[2,3-d]pyrimidine compounds and evaluated them for in vitro antiproliferative activity against different cell lines.95 Several compounds showed significant antiproliferative activity and the most potent and selective compounds against MCF-7 cells were compounds 35–37 (Fig. 9), which showed promising antiproliferative activities against all the tested cell lines. The structure–activity relationship studies suggested that the pyridopyrimidines and quinazolines with SeH groups at positions 2 and 4 exhibited enhanced antiproliferative activity than the corresponding analogs with selenoalkyl functionality. However, no significant correlation was observed between the activity and chain length in either the central nucleus or the phenyl ring. In addition, in order to understand the mechanism of action, the cytotoxicity cell death status and cell cycle distribution were tested with compounds 35–37 in MCF-7 cells. It was observed that these compounds induced cell death in a time- and dose-dependent manner without perturbing the cell cycle.
In 2012, S. I. Kovalenko and his coworkers synthesized a series of quinazoline derivatives by introducing dialkylamino(heterocyclyl)alkyl as a substituent on the 6-thio-3-R-2-oxo-2H-[1,2,4] triazino[2,3-c]quinazoline pharmacophore and examined their anticancer activity.96 Six of them demonstrated promising activity and inhibited the growth of leukemia, melanoma, lung, colon, CNS, ovarian, renal, prostate, and breast cancer cell lines. The compound 6-{[2-(diethylamino)ethyl]thio}-3-(4-methoxyphenyl)-2H-[1,2,4]triazino[2,3-c]quinazolin-2-one (38) (Fig. 9) was considered a promising antitumor agent with selective effects on the colon cancer cell lines. The activity order of the substituents at position 6 such as the dialkylamino(heterocyclic)alkyl fragment connected to the heterocyclic system with sulphur was Me < i-Pr < Pyr < Et. Next, a series of novel N-R-2-[(3-R-2-oxo-2H-[1,2,4]triazino[2,3-c]quinazolin-6-yl)thio]acetamides were prepared by introducing thiazole and thiadiazole fragments.97 Thirteen of the synthesized compounds were evaluated for anticancer activity in NCI against 60 cell lines and the majority of them showed antitumor activities against leukemia, melanoma, lung, colon, CNS, ovarian, renal, prostate, and breast cancer cell lines. The most active compound of the series was 2-[(3-methyl-2-oxo-2H-[1,2,4]triazino[2,3-c]quinazolin-6-yl)thio]-N-(1,3-thiazol-2-yl)acetamide (39) (Fig. 9), which exhibited the maximum anticancer activity against the cell lines of colon, melanoma, and ovarian cancer cells with GI50 in the micromolar range. The significant loss in antitumor activity was observed by replacement of thiazolamide by benzothiazole or thiadiazolamide. Later, a new class of the combinatorial library of novel potential anticancer agents was prepared by fusion of the quinazoline and triazole rings and preparing variations of their derivatives.98 The synthesized compounds were screened for antitumor activity against leukemia, melanoma, lung, colon, CNS, ovarian, renal, prostate, and breast cancer cell lines. The most active compound of this series was 3,4,5-trimethoxy-N′-(quinazolin-4(3H)-ylidene)benzohydrazide (40) (Fig. 9), which exhibited GI50 in the micromolar range.
In 2013, A. Sharma et al. synthesized a series of regioisomeric hybrids, (3-allyl-2-methyl-3H-benzimidazol-5-yl)-(2-substituted-quinazolin-4-yl)-amine and (1-allyl-2-methyl-1H-benzimidazol-5-yl)-(2-substituted-quinazolin-4-yl)-amine, by combining the biologically active moieties benzimidazole and quinazoline.99 To study the effect of electron-donor and acceptor nitrogen groups, the hybrids were substituted with secondary amines, and the focus was to do the derivatization on the secondary amines at position 2 of quinazoline. The synthesized compounds were evaluated against 60 tumor cell lines using a panel assay to obtain an active antitumor agent with promising activity and selectivity toward cancer cells. A number of these hybrids showed significant antitumor activity, and the most active compound was 41 (Fig. 9), bearing a pyrrolidine moiety at position 2 of quinazoline. Compound 41 was considered a broad spectrum antitumor agent and showed efficacy toward several cell lines belonging to different tumor subpanels. Keeping in mind the in vitro anticancer activity of compound 41 toward leukemia cancer cell lines, docking studies were performed with RNR and Topo I and II. The results showed H-bond interactions between the N atom of the quinazoline moiety with the amino acid residues of the active site of RNR and Topo I and Topo II enzymes. Next, a new series of quinazoline and benzimidazole hybrids were synthesized by substituting quinazoline with primary amines at position 2.100 The different electron withdrawing groups besides bulky groups (benzimidazole) were used as primary amine substituents. The compounds of this series were screened for their in vitro antitumor activity in complete NCI 60 cell line panel assays, which included nine tumor cell subpanels, namely; leukemia, non-small cell lung, colon, CNS, melanoma, ovarian, renal, prostate and breast cancer. Compound 42 (Fig. 9) was the most active compound and showed almost twenty and twenty-two fold more activity than quinazoline and benzimidazole analogues, respectively. Compound 42 showed promising anticancer activity toward colon cancer and prostate cancer cell lines at five dose concentrations.
Considering the good performances of quinazoline derivatives as anticancer agents, in 2014, A. M. Alanazi et al. designed and synthesized a novel series of 6-chloro-2-p-tolylquinazolinone derivatives bearing different substituents with diverse electronic environments and studied their effect on lipophilicity and the activity of the target compounds through in vitro antitumor activities.101 In the micromolar range, all the screened compounds of compound 43 (3-(benzylideneamino)-6-chloro-2-p-tolylquinazolin-4(3H)-one) (Fig. 9) having a benzylidine group at position 3 exhibited broad spectrum antitumor activity and inhibited the growth of renal, CNS, ovarian, and non-small lung cancers.
Previous studies have reported that in addition to quinazolinone and its derivatives, compounds containing the imidazolone chromophore are known to have anti-cancer and angiotensin II receptor antagonistic activities.102 Motivated by these studies, in 2014, D. Kumar et al. synthesized a series of novel quinazolinone derivatives fused with imidazolone and evaluated their anticancer activities against cervical cancer (HeLa), breast cancer (MCF-7), leukemia (HL-60), and hepatocellular carcinoma (HepG2) cell lines.103 The most active compound of the series was compound 44 (Fig. 9), which contained an electron-donating methoxy group at the para position in the imidazolone phenyl ring. Compound 44 showed a threefold more potent activity against MCF-7 than the standard drug (cisplatin) and a twofold less potent activity than cisplatin against HepG2. However, the IC50 values of compound 44 HeLa and HL-60 were comparable to that of cisplatin. The structure–activity relationship studies suggested that the electron-donating group favored activity, whereas the electron-withdrawing group had a detrimental effect.
Keeping in mind the significance of the fluorine atom in enhancing the lipophilicity, absorption, and bioavailability of several anticancer drugs in addition to the importance of sulphonamides as anticancer agents, M. F. Zayed et al. synthesized a small series of fluorinated quinazolinone–sulphonamide hybrids and evaluated their in vitro cytotoxic activities.104 Most of the compounds showed substantial anticancer activity in the micromolar range. The most active compound of the series was 4-(6-fluoro-2-methyl-4-oxoquinazolin-3(4H)-yl)-N-(4,6-dimethlpyrimidin-2-yl)benzensulphonamide (45) (Fig. 9) containing sulphamethazine and displaying an IC50 value in the micromolar range in the NCI, MCF-7, and HEK-293 cell lines. This has been considered a suitable parent compound for further modification to produce an effective anticancer agent.
In 2015, J.-P. Yong synthesized a series of quinazoline derivatives containing the isoxazole moiety and evaluated their in vitro anticancer activities against A549, HCT116 and MCF-7 cell lines.105 Most of the compounds of the series exhibited good activity, and compounds 46 and 47 (Fig. 9) showed promising activities against all the tested cell lines. Additionally, to explore the anticancer activity of the isoxazole moiety containing the quinazoline core, a new series of compounds were prepared by varying the substituents on the benzene ring of the quinazoline core and on the phenyl ring attached to the isoxazole moiety.106 The series was screened for in vitro anticancer activities against different cell lines using the MTT method. The compounds of the series exhibited good to excellent anticancer activities, and the most promising compound with low micromolar activity against all the tested cell lines was 48 (Fig. 9) containing a 2-methoxy ethoxy substituent at positions 6 and 7 of the quinazoline core.
In 2016, S. Vodnala et al. designed and synthesized hybrid molecules containing the dihydropyrano[c]chromene moiety fused with an oxa-heterocyclic unit at position 2 of the quinazoline ring.107 These compounds were synthesized using the one-pot three-component reaction catalyzed by 1,4-diazabicyclo[2.2.2]octane. The anticancer activities of the prepared compounds were assessed against breast cancer cell lines of MDA-MB 231 (estrogen-independent) and MDA-MB 453 (breast adenocarcinoma cell line). Compound 49 (Fig. 9) containing bromine and chlorine atoms at the phenyl ring of the oxa-heteryl moiety exhibited maximum activity in both breast cancer cell lines (MDA-MB 453 and MDA-MB 231). Additionally, the author performed molecular docking studies with compound 49 at the receptor site of ERα protein. Compound 49, containing bromo and chloro substituents in the phenyl group, oriented toward the hydrophobic pockets and exhibited H bonding, and π–π, hydrophobic and polar interactions with the amino acids of the receptor site of ERα protein.
3. Conclusion
It is evident from the above discussion that quinazoline and its derivatives have immense potential as anticancer agents. In the discovery of cancer drugs, quinazoline is an important pharmacophore, and several research laboratories worldwide are focused on the synthesis of different quinazoline derivatives for the development of novel and more potent anti-cancer drugs. This review article focused on the anticancer activities of synthetic quinazoline derivatives against various types of cancer cells. It is expected that the information presented in this review article will update researchers about the recently reported quinazoline pharmacophore-based potent anticancer agents and will provide support for the development of novel anticancer drugs.
Acknowledgments
The authors appreciate the financial support provided by the School of Graduate Studies and Research, American University of Ras Al Khaimah through seed grant funded project No. AAS/003/15.
Footnotes
†The authors declare no competing interests.
References
- Cancer facts and figure 2017, https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2017.html.
- Harris C. C., Hollstein M. N. Engl. J. Med. 1993;329:1318–1327. doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- Salerno S., Da Settimo F., Taliani S., Simorini F., La Motta C., Fornaciari G., Marini A. M. Curr. Med. Chem. 2010;17:4270–4290. doi: 10.2174/092986710793361252. [DOI] [PubMed] [Google Scholar]
- Baselga J., Swain S. M. Nat. Rev. Cancer. 2009;9:463–475. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- Brown C. H. J., Lain S., Verma C. H. S., Fersht A. R., Lane D. P. Nat. Rev. Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- Ljungman M. Chem. Rev. 2009;109:2929–2950. doi: 10.1021/cr900047g. [DOI] [PubMed] [Google Scholar]
- Semenza G. L. Cancer Metastasis Rev. 2007;26:223–224. doi: 10.1007/s10555-007-9058-y. [DOI] [PubMed] [Google Scholar]
- Eckford P. D. W., Sharom F. J. Chem. Rev. 2009;109:2989–3011. doi: 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- Ullah M. F. Asian Pac. J. Cancer Prev. 2008;9:1–6. [PubMed] [Google Scholar]
- Shagufta, Ahmad I. Eur. J. Med. Chem. 2016;116:267–280. doi: 10.1016/j.ejmech.2016.03.058. [DOI] [PubMed] [Google Scholar]
- Ahmad I., Shagufta Eur. J. Med. Chem. 2015;102:375–386. doi: 10.1016/j.ejmech.2015.08.010. [DOI] [PubMed] [Google Scholar]
- Ahmad I., Shagufta Int. J. Pharm. Sci. 2015;7:19–27. [Google Scholar]
- Demeunynck M., Baussanne I. Curr. Med. Chem. 2013;20:794–814. [PubMed] [Google Scholar]
- Raghavendra N. M., Thampi P., Gurubasavarajaswamy P. M., Sriram D. Chem. Pharm. Bull. 2007;55:1615–1619. doi: 10.1248/cpb.55.1615. [DOI] [PubMed] [Google Scholar]
- Panneerselvam P., Rather B. A., Reddy D. R. S., Kumar N. R. Eur. J. Med. Chem. 2009;44:2328–2333. doi: 10.1016/j.ejmech.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Verhaeghe P., Azas N., Gasquet M., Hutter S., Ducros C., Laget M., Rault S., Rathelot P., Vanelle P. Bioorg. Med. Chem. Lett. 2008;18:396–401. doi: 10.1016/j.bmcl.2007.10.027. [DOI] [PubMed] [Google Scholar]
- Saravanan G., Pannerselvam P., Prakash C. R. Pharm. Lett. 2010;2:216–226. doi: 10.4103/0110-5558.72426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagarsamy V., Solomon V. R., Sheorey R. V., Jayakumar R. Chem. Biol. Drug Des. 2009;73:471–479. doi: 10.1111/j.1747-0285.2009.00794.x. [DOI] [PubMed] [Google Scholar]
- Smits R. A., Adami M., Istyastono E. P., Zuiderveld O. P., van Dam C. M. E., de Kanter F. J. J., Jongejan A., Coruzzi G., Leurs R., de Esch I. J. J. Med. Chem. 2010;53:2390–2400. doi: 10.1021/jm901379s. [DOI] [PubMed] [Google Scholar]
- Georgey H., Abdel-Gawad N., Abbas S. Molecules. 2008;13:2557–2569. doi: 10.3390/molecules13102557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N. B. Acta Pol. Pharm. 2010;67:267–275. [PubMed] [Google Scholar]
- Ismail M. A. H., Barker S., Abau El Ella D. A., Abouzid K. A. M., Toubar R. A., Todd M. H. J. Med. Chem. 2006;49:1526–1535. doi: 10.1021/jm050232e. [DOI] [PubMed] [Google Scholar]
- Zaranappa, Vagdevi H. M., Lokesh M. R., Gowdarshivannanava B. C. Int. J. ChemTech Res. 2012;4:1527–1533. [Google Scholar]
- Krishnan S. K. Riv. Eur. Sci. Med. Farmacol. 2011;15:673–681. [PubMed] [Google Scholar]
- Pati B., Banerjee S. J. Adv. Pharm. Educ. Res. 2013;3:136–151. [Google Scholar]
- Katrin S. N. Chemother. Res. Pract. 2012;3:22–27. [Google Scholar]
- Manasa A. K., Sidhaye R. V., Radhika G., Nalini C. N. Curr. Pharma Res. 2011;1:101–105. [Google Scholar]
- Nerkar B., Saxena A., Ghone S., Thakeri A. K. E-J. Chem. 2009;6:97–102. [Google Scholar]
- Ahmed M. F., Youns M. Arch. Pharm. 2013;346:610–617. doi: 10.1002/ardp.201300158. [DOI] [PubMed] [Google Scholar]
- Moon D. O., Kim M. O., Heo M. S., Lee J. D., Choi Y. H., Kim G. Y. Arch. Pharmacal Res. 2009;32:1351–1360. doi: 10.1007/s12272-009-2002-7. [DOI] [PubMed] [Google Scholar]
- Sirisoma N., Pervin A., Zhang H., Jiang S., Adam Willardsen J., Anderson M. B., Mather G., Pleiman C. M., Kasibhatla S., Tseng B., Drewe J., Cai S. X. Bioorg. Med. Chem. Lett. 2010;20:2330–2334. doi: 10.1016/j.bmcl.2010.01.155. [DOI] [PubMed] [Google Scholar]
- Font M., Gonzalez A., Palop J. A., Sanmartin C. Eur. J. Med. Chem. 2011;46:3887–3899. doi: 10.1016/j.ejmech.2011.05.060. [DOI] [PubMed] [Google Scholar]
- Liu F., Lovejoy D. B., Hassani A. A., He Y., Herold J. M., Chen X., Yates C. M., Frye S. V., Brown P. J., Huang J., Vedadi M., Arrowsmith C. H., Jin J. J. Med. Chem. 2011;54:6139–6150. doi: 10.1021/jm200903z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EI-Azab A. S., Al-Omar M. A., Abdel-Aziz A. A. M., Abdel-Aziz N. I., EI-Sayed M. A.-A., Aleisa A. M., Sayad-Ahmed M. M., Abdel-Hamide S. G. Eur. J. Med. Chem. 2010;45:4188–4198. doi: 10.1016/j.ejmech.2010.06.013. [DOI] [PubMed] [Google Scholar]
- Al-Obaid A. M., Abdel-Hamide S. G. A., El-Kashef H. A., Abdel-Aziz A. A.-M., El-Azad A. S., Al-Khamees H. A., El-Subbagh H. I. Eur. J. Med. Chem. 2009;44:2379–2391. doi: 10.1016/j.ejmech.2008.09.015. [DOI] [PubMed] [Google Scholar]
- Alafeefy A. M., Kadi A. A., El-Azab A. S., Abdel-Hamide S. G., Daba M. H. Arch. Pharm. 2008;341:377–385. doi: 10.1002/ardp.200700271. [DOI] [PubMed] [Google Scholar]
- Kumar A., Sharma S., Archana A., Bajaj K., Sharma S., Panwar H., Singh T., Srivastava V. K. Bioorg. Med. Chem. 2003;11:5293–5299. doi: 10.1016/s0968-0896(03)00501-7. [DOI] [PubMed] [Google Scholar]
- El-Azab A. S., Kamal E. H. Bioorg. Med. Chem. Lett. 2012;22:1879–1885. doi: 10.1016/j.bmcl.2012.01.071. [DOI] [PubMed] [Google Scholar]
- Kashaw S. K., Kashaw V., Mishra P., Jain N. K., Stables J. P. Eur. J. Med. Chem. 2009;44:4335–4343. doi: 10.1016/j.ejmech.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Archana V., Srivastava K., Kumar A. Bioorg. Med. Chem. 2004;12:1257–1264. doi: 10.1016/j.bmc.2003.08.035. [DOI] [PubMed] [Google Scholar]
- Al-Suwaidan I. A., Alanazi A. M., Abdel-Aziz A. A., Mohamed M. A., El-Azab A. S. Bioorg. Med. Chem. Lett. 2013;23:3935–3941. doi: 10.1016/j.bmcl.2013.04.056. [DOI] [PubMed] [Google Scholar]
- Abdel Gawad N. M., Georgey H. H., Youssef R. M., El-Sayed N. A. Eur. J. Med. Chem. 2010;45:6058–6067. doi: 10.1016/j.ejmech.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Ismail R. S. M., Ismail N. S. M., Abuserii S., Abou El Ella D. A., Future Journal of Pharmaceutical Sciences, 2016, 2 , 9 –19 , , and references cited in . [Google Scholar]
- Karamouzis M. V., Grandis J. R., Argiris A. JAMA, J. Am. Med. Assoc. 2007;298:70–82. doi: 10.1001/jama.298.1.70. [DOI] [PubMed] [Google Scholar]
- Carmi C., Mor M., Petronini P. G., Alfieri R. R. Biochem. Pharmacol. 2012;84:1388–1399. doi: 10.1016/j.bcp.2012.07.031. [DOI] [PubMed] [Google Scholar]
- Gotink K. J., Verheul H. M. Angiogenesis. 2010;13:1–14. doi: 10.1007/s10456-009-9160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. C., Adams J. L. Curr. Opin. Biotechnol. 1995;6:657–661. doi: 10.1016/0958-1669(95)80108-1. [DOI] [PubMed] [Google Scholar]
- Lemmon A. M., Schlessinger J. Cell. 2010;7:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantl W. J., Johnson D. E., Williams L. T. Annu. Rev. Biochem. 1993;62:453–481. doi: 10.1146/annurev.bi.62.070193.002321. [DOI] [PubMed] [Google Scholar]
- Kris M. G., Natale R. B., Herbst R. S., Lynch Jr. T. J., Prager D., Belani C. P., Schiller J. H., Kelly K., Spiridonidis H., Sandler A., Albain K. S., Cella D., Wolf M. K., Averbuch S. D., Ochs J. J., Kay A. C. JAMA, J. Am. Med. Assoc. 2003;290:2149–2158. doi: 10.1001/jama.290.16.2149. [DOI] [PubMed] [Google Scholar]
- Fukuoka M., Yano S., Giaccone G., Tamura T., Nakagawa K., Douillard J. Y., Nishiwaki Y., Vansteenkiste J., Kudoh S., Rischin D., Eek R., Horai T., Noda K., Takata I., Smit E., Averbuch S., Macleod A., Feyereislova A., Dong R. P., Baselga J. J. Clin. Oncol. 2003;21:2237–2246. doi: 10.1200/JCO.2003.10.038. [DOI] [PubMed] [Google Scholar]
- Shepherd F. A., Rodrigues Pereira J., Ciuleanu T., Tan E. H., Hirsh V., Thongprasert S., Campos D., Maoleekoonpiroj S., Smylie M., Martins R., van Kooten M., Dediu M., Findlay B., Tu D., Johnston D., Bezjak A., Clark G., Santabarbara P., Seymour L. N. Engl. J. Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- Kobayashi S., Boggon T. J., Dayaram T., Janne P. A., Kocher O., Meyerson M., Johnson B. E., Eck M. J., Tenen D. G., Halmos B. N. Engl. J. Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Wu X., Li M., Qu Y., Tang W., Zheng Y., Lian J., Ji M., Xu L. Bioorg. Med. Chem. 2010;18(11):3812–3822. doi: 10.1016/j.bmc.2010.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Li M., Tang W., Zheng Y., Lian J., Xu L., Ji M. Chem. Biol. Drug Des. 2011;78(6):932–940. doi: 10.1111/j.1747-0285.2011.01234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Jin L. H., Xiang H. M., Wu J., Wang P. Y., Hu D. Y., Xue W., Yang S. Eur. J. Med. Chem. 2013;66:335–344. doi: 10.1016/j.ejmech.2013.05.043. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Huang Y.-J., Xiang H.-M., Wang P.-Y., Hu D.-Y., Xue W., Song B.-A., Yang S. Eur. J. Med. Chem. 2014;78:23–34. doi: 10.1016/j.ejmech.2014.03.036. [DOI] [PubMed] [Google Scholar]
- Gschwind A., Fischer O. M., Ullrich A. Nat. Rev. Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- Qin X., Lv Y., Liu P., Li Z., Hu L., Zeng C., Yang L. Bioorg. Med. Chem. Lett. 2016;26:1571–1575. doi: 10.1016/j.bmcl.2016.02.009. [DOI] [PubMed] [Google Scholar]
- Chen J.-N., Wang X.-F., Li T., Wu D.-W., Fu X.-B., Zhang G.-J., Shen X.-C., Wang H.-S. Eur. J. Med. Chem. 2016;107:12–25. doi: 10.1016/j.ejmech.2015.10.045. [DOI] [PubMed] [Google Scholar]
- Dumontet C., Jordan M. A. Nat. Rev. Drug Discovery. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemann D. W. Cancer Treat. Rev. 2011;37:63–74. doi: 10.1016/j.ctrv.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conconi M. T., Marzaro G., Urbani L., Zanusso I., Di Liddo R., Castagliuolo I., Brun P., Tonus F., Ferrarese A., Guiotto A., Chilin A. Eur. J. Med. Chem. 2013;67:373–383. doi: 10.1016/j.ejmech.2013.06.057. [DOI] [PubMed] [Google Scholar]
- Marzaro G., Coluccia A., Ferrarese A., Brun P., Castagliuolo I., Conconi M. T., Regina G. L., Bai R., Silvestri R., Hamel E., Chilin A. J. Med. Chem. 2014;57:4598–4605. doi: 10.1021/jm500034j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. F., Wang S. B., Ohkoshi E., Wang L. T., Hamel E., Qian K., Morris-Natschke S. L., Lee K. H., Xie L. Eur. J. Med. Chem. 2013;67:196–207. doi: 10.1016/j.ejmech.2013.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.-F., Guan F., Ohkoshi E., Guo W., Wang L., Zhu D.-Q., Wang S.-B., Wang L.-T., Hamel E., Yang D., Li L., Qian K., Morris-Natschke S. L., Yuan S., Lee K.-H., Xie L. J. Med. Chem. 2014;57:1390–1402. doi: 10.1021/jm4016526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroiwa K., Ishii H., Matsuno K., Asai A., Suzuki Y. ACS Med. Chem. Lett. 2015;6:287–291. doi: 10.1021/ml5004684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarvey K. M., Fahrner J. A., Greene E., Martens J., Jenuwein T., Baylin S. B. Cancer Res. 2006;66:3541–3549. doi: 10.1158/0008-5472.CAN-05-2481. [DOI] [PubMed] [Google Scholar]
- Kondo Y., Shen L., Ahmed S., Boumber Y., Sekido Y., Haddad B. R., Issa J. P. PLoS One. 2008;3:e2037. doi: 10.1371/journal.pone.0002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Dorsey J., Chuikov S., Zhang X., Jenuwein T., Reinberg D., Berger S. L. J. Biol. Chem. 2010;285:9636–9641. doi: 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek S., O'Sullivan R. J., August E. M., Hickey E. R., Zhang Q., Teodoro M. L., Rea S., Mechtler K., Kowalski J. A., Homon C. A., Kelly T. A., Jenuwein T. Mol. Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Chang Y., Zhang X., Horton J. R., Upadhyay A. K., Spannhoff A., Liu J., Snyder J. P., Bedford M. T., Cheng X. Nat. Struct. Mol. Biol. 2009;16:312–317. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Chen X., Allali-Hassani A., Quinn A. M., Wasney G. A., Dong A., Barsyte D., Kozieradzki I., Senisterra G., Chau I., Siarheyeva A., Kireev D. B., Jadhav A., Herold J. M., Frye S. V., Arrowsmith C. H., Brown P. J., Simeonov A., Vedadi M., Jin J. J. Med. Chem. 2009;52:7950–7953. doi: 10.1021/jm901543m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Chen X., Allali-Hassani A., Quinn A. M., Wigle T. J., Wasney G. A., Dong A., Senisterra G., Chau I., Siarheyeva A., Norris J. L., Kireev D. B., Herold J. M., Janzen W. P., Arrowsmith C. H., Frye S. V., Brown P. J., Simeonov A., Vedadi M., Jin J. J. Med. Chem. 2010;53(15):5844–5857. doi: 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Lovejoy D. B., Allali-Hassani A., He Y., Herold J. M., Chen X., Yates C. M., Frye S. V., Brown P. J., Huang J., Vedadi M., Arowsmith C. H., Jin J. J. Med. Chem. 2010;54(17):6139–6150. doi: 10.1021/jm200903z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. C., Nat. Rev. Mol. Cell Biol., 2002, 3 , 430 –440 , . 1,2 and 3,6 and 8,9 . [DOI] [PubMed] [Google Scholar]
- Pommier Y. ACS Chem. Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall M. E., Wani M. C., Cook C. E., Palmer K. H., McPhail A. T., Sim G. A. J. Am. Chem. Soc. 1966;88:3888–3890. [Google Scholar]
- Burke T. G., Mi Z. J. Med. Chem. 1994;37:40–46. doi: 10.1021/jm00027a005. [DOI] [PubMed] [Google Scholar]
- Pommier Y. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- Beretta G. L., Zuco V., Perego P., Zaffaroni N. Curr. Med. Chem. 2012;19:1238–1257. doi: 10.2174/092986712799320529. [DOI] [PubMed] [Google Scholar]
- Taliani S., Pugliesi I., Barresi E., Salerno S., Marchand C., Agama K., Simorini F., Motta C. L., Marini A. M., Saverio Di Leva F., Marinelli L., Cosconati S., Novellino E., Pommier Y., Di Santo R., Da Settimo F. J. Med. Chem. 2013;56:7458–7462. doi: 10.1021/jm400932c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer C. E., Forster J., Lindquist D., Chan S., Romieu C. G., Pienkowski T., Jagiello-Gruszfeld A., Crown J., Chan A., Kaufman B. N. Engl. J. Med. 2006;355:2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- Wu P., Hu Y. Z. Med. Chem. Commun. 2012;3:1337–1355. [Google Scholar]
- Nishimura N., Siegmund A., Liu L., Yang K., Bryan M. C., Andrews K. L., Bo Y. X., Booker S. K., Caenepeel S., Freeman D., Liao H. Y., McCarter J., Mullady E. L., Miguel T. S., Subramanian R., Tamayo N., Wang L., Whittington D. A., Zalameda L., Zhang N., Hughes P. E., Norman M. H. J. Med. Chem. 2011;54:4735–4751. doi: 10.1021/jm200386s. [DOI] [PubMed] [Google Scholar]
- Hei Y.-Y., Xin M., Zhang H., Xie X. X., Mao S., Zhang S.-Q. Bioorg. Med. Chem. Lett. 2016;26:4408–4413. doi: 10.1016/j.bmcl.2016.08.015. [DOI] [PubMed] [Google Scholar]
- Wang X.-M., Xin M.-H., Xu J., Kang B.-R., Li Y., Lu S.-M., Zhang S.-Q. Eur. J. Med. Chem. 2015;96:382–395. doi: 10.1016/j.ejmech.2015.04.037. [DOI] [PubMed] [Google Scholar]
- Peng W., Tu Z.-C., Long Z.-J., Liu Q., Lu G. Eur. J. Med. Chem. 2016;108:644–654. doi: 10.1016/j.ejmech.2015.11.038. [DOI] [PubMed] [Google Scholar]
- Yadav R. R., Guru S. K., Joshi P., Mahajan G., Mintoo M. J., Kumar V., Bharate S. S., Mondhe D. M., Vishwakarma R. A., Bhushan S., Bharate S. B. Eur. J. Med. Chem. 2016;122:731–743. doi: 10.1016/j.ejmech.2016.07.006. [DOI] [PubMed] [Google Scholar]
- Yao H., Ji M., Zhu Z., Zhou J., Cao R., Chen X., Xu B. Bioorg. Med. Chem. 2015;23:681–693. doi: 10.1016/j.bmc.2014.12.071. [DOI] [PubMed] [Google Scholar]
- Zahedifard M., Faraj F. L., Paydar M., Looi C. Y., Hajrezaei M., Hasanpourghadi M., Kamalidehghan B., Majid N. A., Ali H. M., Abdulla M. A., Sci. Rep., 2015, 5 , 11544 , , (1–17) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrika P. M., Yakaiah T., Rao A. R. R., Narsaiah B., Reddy N. C., Sridhar V., Rao J. V. Eur. J. Med. Chem. 2008;43:846–852. doi: 10.1016/j.ejmech.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Noolvi M. N., Patel H. M., Bhardwaj V., Chauhan A. Eur. J. Med. Chem. 2011;46:2327–2346. doi: 10.1016/j.ejmech.2011.03.015. [DOI] [PubMed] [Google Scholar]
- Alafeefy A. M. J. Saudi Chem. Soc. 2011;15:337–343. [Google Scholar]
- Moreno E., Plano D., Lamberto I., Font M., Encío I., Palop J. A., Sanmartín C. Eur. J. Med. Chem. 2012;47:283–298. doi: 10.1016/j.ejmech.2011.10.056. [DOI] [PubMed] [Google Scholar]
- Berest G. G., Voskoboynik O. Y., Kovalenko S. I., Nosulenko I. S., Antypenko L. M., Antypenko O. M., Shvets V. M., Katsev A. M. Sci. Pharm. 2012;80:37–65. doi: 10.3797/scipharm.1111-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko S. I., Nosulenko I. S., Voskoboynik A. Y., Berest G. G., Antypenko L. N., Antypenko A. N., Katsev A. M. Sci. Pharm. 2012;80:837–865. doi: 10.3797/scipharm.1208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko S. I., Antypenko L. M., Bilyi A. K., Kholodnyak S. V., Karpenko O. V., Antypenko O. M., Mykhaylova N. S., Los T. I., Kolomoets O. S. Sci. Pharm. 2013;81:359–391. doi: 10.3797/scipharm.1211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A., Luxami V., Paul K. Bioorg. Med. Chem. Lett. 2013;23:3288–3294. doi: 10.1016/j.bmcl.2013.03.107. [DOI] [PubMed] [Google Scholar]
- Paul K., Sharma A., Luxami V. Bioorg. Med. Chem. Lett. 2014;24:624–629. doi: 10.1016/j.bmcl.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Alanazi A. M., Abdel-Aziz A. A.-M., Al-Suwaidan I. A., Abdel-Hamid S. G., Shawer T. Z., El-Azab A. S. Eur. J. Med. Chem. 2014;79:446–454. doi: 10.1016/j.ejmech.2014.04.029. [DOI] [PubMed] [Google Scholar]
- Siamaki A. R., Black D. A., Arndtsen B. A. J. Org. Chem. 2008;73:1135–1138. doi: 10.1021/jo701875b. [DOI] [PubMed] [Google Scholar]
- Kumar D., Mariappan G., Husain A., Monga J., Kumar S. Arabian J. Chem. 2017;10:344–350. [Google Scholar]
- Zayed M. F., Ahmed H. E. A., Ihmaid S., Omar A. S. M., Abdelrahim A. S. J. Taibah Univ. Med. Sci. 2015;10:333–339. [Google Scholar]
- Yong J.-P., Lu C.-Z., Wu X. Med. Chem. 2015;15:131–136. doi: 10.2174/1871520614666140812105445. [DOI] [PubMed] [Google Scholar]
- Yong J.-P., Lu C.-Z., Wu X. Med. Chem. 2015;15:1326–1332. doi: 10.2174/1871520615666150526115904. [DOI] [PubMed] [Google Scholar]
- Vodnala S., Bhavani A. K. D., Kamutam R., Naidu V. G. M., Prabhakar P. C. Bioorg. Med. Chem. Lett. 2016;26:3973–3977. doi: 10.1016/j.bmcl.2016.07.003. [DOI] [PubMed] [Google Scholar]