

 This article reviews the recent progress in drug development against the African sleeping sickness.
This article reviews the recent progress in drug development against the African sleeping sickness.
Abstract
Human African trypanosomiasis (HAT), also known as African sleeping sickness, is caused by parasitic protozoa of the genus Trypanosoma. As the disease progresses, the parasites cross the blood brain barrier and are lethal for the patients if the disease is left untreated. Current therapies suffer from several drawbacks due to e.g. toxicity of the respective compounds or resistance to approved antitrypanosomal drugs. In this review, the different strategies of drug development against HAT are considered, namely the target-based approach, the phenotypic high throughput screening and the drug repurposing strategy. The most promising compounds emerging from these approaches entering an in vivo evaluation are mentioned herein. Of note, it may turn out to be difficult to confirm in vitro activity in an animal model of infection; however, possible reasons for the missing efficacy in unsuccessful in vivo studies are discussed.
1. Introduction
Human African trypanosomiasis (HAT) is also called sleeping sickness. The disease is mainly restricted to Africa, and depending on the affected region two types of human illness can be found: East and West African trypanosomiasis. Two species of trypanosomatides which are transmitted by the tsetse fly, Trypanosoma brucei rhodesiense and Trypanosoma brucei gambiense, cause the ailment. The number of cases in Africa has dropped drastically, however, approximately 3000 new infections of both East and West African trypanosomiasis have been reported to the World Health Organization in 2015.1 However, underestimating the actual number of cases is a common problem due to weak health infrastructures and restricted access to medical facilities.2
HAT is mainly being spread through the bite of infected tsetse flies; infection seldomly occurs by blood transfusion or through placental transmission. Of note, the disease causes infertility and abortion in pregnant women or of child-bearing age, respectively. This disease is invariably fatal if left untreated. In Africa, HAT-affected zones cover an area of about 8 million square kilometers between 14 degrees north latitude and 20 degrees south latitude.3 In the 20th century three major HAT epidemics occurred, the first one between 1896 and 1906, the second in 1920, and the most recent one in 1970 lasting until the 1990s. Between these epidemics the number of cases constantly decreased to the currently known magnitiude.4
T. brucei rhodesiense causes the more virulent form of the disease which is called East African or Rhodesian African sleeping sickness. It is zoonotic, rare, and extremely virulent; patients usually die within a few months. T. brucei gambiense causes the West African or Gambian African sleeping sickness; the gambiense type shows both long latency and chronicity, whereas in the gambiense HAT, humans only constitute the main reservoir and transmission agent within the life cycle of the parasite.1
Two clinical stages define the status of the disease; i.e., a haemolymphatic initial systemic stage and the meningoencephalytic stage which is characterized by the invasion of the brain by parasites. The latter produces sensory, motor, and psychiatric disturbances, as well as typical and name-giving sleep alteration. The painful bite of an infected tsetse fly produces a distinctive local erythema, heat, edema, and tenderness leading to the appearance of the chancre, an ulcer that lasts for two or three weeks and appears where the parasites are present. The disease subsequently evolves into the two typical phases which have already been mentioned.5,6
The tsetse bite conduces the patient into the haemolymphatic stage I which is characterized by painful lymph nodes; eventually, intermittent fever episodes may occur as a result of the successive waves of trypanosome invasion within the blood stream. Adenopathy, splenomegaly, and liver disturbances are typical signals of the invasion of the reticulo-endothelial system. Finally, skin rashes and pruritus with scratching skin lesions develop which become unbearable for the patient.5–7
Evolving into the meningoencephalitic stage II may take several months or even years. The parasites cross the blood–brain barrier (BBB), infect the central nervous system (CNS), and cause serious changes to the patient's sleep pattern which may be accompanied by confusion, motor, and mental coordination disorders. The cerebrospinal fluid is invaded by enormous quantities of macrophages; non-specific perivascular inflammatory cell infiltrates occur in the leptomeninges and the white matter. Finally, a pronounced activation of microglia and astrocytes occurs.5–7 The terminal phase of the disease is characterized by disturbances in consciousness, the development of dementia including incoherence, double incontinence, and seizures. The patient either dies from heart failure or – in the late stage – from encephalitis in a state of cachexia and physiological collapse.5–7
The Animal trypanosomiasis is known as Nagana. Two African trypanosome subgenera, Nannomona and Duttonella (Trypanosoma congolense and Trypanosoma vivax, respectively), as well as Trypanosoma brucei brucei cause the cattle sickness. From the veterinary point of view this is economically threatening as serious losses in pigs, camelids, goats, and sheep may vastly affect breeding within the affected rural areas.8,9 Trypanosoma evansi (subgenus Trypanozoon) mostly infects mammals including horses, mules, camels, buffalo, cattle, and deer, however, cows, goats, sheep, dogs, and cats can also be affected. The parasite produces a disease called Surra, hip disease, Murrina, or Derrengadera (mal de cadeiras) of great economic impact for many geographic areas: morbidity and mortality rates are usually very high in tropical and semi-tropical regions such as North Africa, China, the Philippines, India, Indonesia, Malaysia, Russia, Central America, and South America.10,11 In fact, in Africa, Asia and South America thousands of animals die from these diseases every year causing tremendous economic losses within these regions.10,11 Besides the huge losses of working animals and deficiencies in milk production the reproductive deficiencies due to infertility also represent a major challenge. In addition, treatment costs are very high.10,11 Of note, Trypanosoma equiperdum (subgenus Trypanozoon) infects equines under natural conditions; by venereal transmission it may also cause an equine disease called dourine.12 Finally, when thinking of the optical identification of the parasites e.g. by microscopy it should be emphasized that Trypanosoma evansi (monomorphic) and Trypanosoma equiperdum (monomorphic but occasionally pleomorphic) are morphologically similar to the slender forms of Trypanosoma brucei brucei, as well as of Trypanosoma brucei rhodesiense and Trypanosoma brucei gambiense.13,14 This makes a valid diagnostic very difficult.
2. Compounds in clinical stages
Only very few active compounds are available for the treatment of the disease, most of them only effective against one stage of the HAT or against one parasite species, i.e. either T. b. gambiense or T. b. rhodesiense. The therapy is far away from being considered optimal and currently only two drugs, fexinidazole and the benzoxaborole SCYX-7158, have reached clinical trials. In addition, the development of resistance to clinically used drugs may aggravate the situation. No vaccines are available.1 Taken together, new antitrypanosomal agents are urgently needed.4 Hence, this review gives an insight in recent trypanocidal drug development. We applied strict selection criteria; in particular, at least in vivo studies and toxicological data should be available (Table 1). Especially in the last three years encouraging research has been performed in the field of antitrypanosomal agents, hence markedly plenty compounds with various scaffolds were tested for in vivo activity recently. Since the vast majority of compounds fails in subsequent clinical stages, a continuous refilling of the pipeline with innovative drug candidates is necessary.
Table 1. In vitro and in vivo data from compounds mentioned herein.
| Compd | T. brucei IC50 [μM| | Cytotoxicity CC50 [μM] | SI | in vivo infection | Treatment regime | Curation | Lit. |
| 6 | 0.89 a | >213 c | >240 | T. b. rhodesiense STIB900 | 20 mg kg–1, ip, for 4 days, begin 3 days pi | 4/4 | 28 |
| T. b. brucei GVR35 | 50 mg kg–1, ip, for 5 days, begin 17 days pi | 1/5 | |||||
| 7 | 0.009 a | 69 c | 7667 | T. b. brucei Lab 110 EATRO | 10 mg kg–1, ip, for 3 days, begin 1 day pi | 3/3 | 40 |
| 0.096 b | |||||||
| T. b. rhodesiense Ketri 2538 | 3/3 | ||||||
| 8 | 0.003 a | 100 c | 33 333 | T. b. brucei Lab 110 EATRO | 5 mg kg–1, ip, for 3 days, begin 1 day pi | 3/3 | 40 |
| 0.002 b | |||||||
| T. b. rhodesiense Ketri 2538 | 3/3 | ||||||
| 9 | 0.002 a | 43 c | 21 500 | T. b. brucei Lab 110 EATRO | 1 mg kg–1, ip, for 3 days, begin 1 day pi | 3/3 | 40 |
| 0.002 b | |||||||
| T. b. rhodesiense Ketri 2002 | 3/3 | ||||||
| 10 | 0.0077 a | >100 d | >13 000 | T. b. brucei Lister 427 | 3 mg kg–1, ip, qd, for 4 days, | 5/5 | 41 |
| >100 e | |||||||
| 11 | 0.002 a | 0.3 f | 150 | T. b. brucei S427 | 12.5 mg kg–1, po, bid, for 4 days, begin 3 days pi | 5/5 | 46 |
| T. b. rhodesiense STIB900 | 50 mg kg–1, po, bid, for 2 days, begin 3 pi | 5/5 | |||||
| T. b. brucei GVR35 | 100 mg kg–1, po, bid, for 5 days, begin 21 pi | 0/5 | 45 | ||||
| 12 | 0.001 a | 1 f | 1000 | T. b. brucei S427 | 50 mg kg–1, po, bid, for 4 days, begin 3 days pi | 5/5 | 47 |
| T. b. brucei GVR35 | 100 mg kg–1, po, bid, for 5 days | 0/5 | |||||
| 50 mg kg–1, po, bid, for 10 days | 0/5 | ||||||
| 100 mg kg–1, po, bid, for 8 days | 1/5 | ||||||
| 100 mg kg–1, po, bid, for 3 rounds of 3 day treatment with a two day hiatus between rounds | 1/5 | ||||||
| 14 | 0.002 a | 30 e | >15 000 | T. b. rhodesiense STIB9000 | 5 mg kg–1, po, bid, for 5 days, begin 2 days pi | 5/5 | 52 |
| 24 g | |||||||
| 15 | 0.07 a | >20 h | >286 | T. b. brucei GVR35-VSL2 | 100 mg kg–1, qd, oral gavage, for 7 days, begin 21 days pi | 6/6 | 53 |
| >50 i | >714 | ||||||
| 16 | 0.191 a | 103 c | 539 | T. b. rhodesiense STIB900 | 50 mg kg–1, ip, bid, for 3 days, begin 1 day pi | 0 | 54 |
| 0.632 b | 162 | ||||||
| 17 | 0.0348 a | >50 g | >1437 | T. b. rhodesiense STIB900 | 50 mg kg–1, po, bid, for 4 days, begin 2 days pi | 5/5 | 55 |
| >50 e | |||||||
| >1437 | |||||||
| T. b. brucei TREU667 | 50 mg kg–1, po, bid, for 10 days, begin 21 days pi | 5/5 | 55 | ||||
| 18 | 0.03 a | >50f | >1800 | T. b. brucei S427 | 10 mg kg–1, ip, bid, for 4 days | 2/3 | 56 |
| 19 | 0.06 a | >50 f | >860 | T. b. brucei GVR35 | 12.5 mg kg–1, ip, bid, for 5 days, begin 21 days pi | 1/5 | |
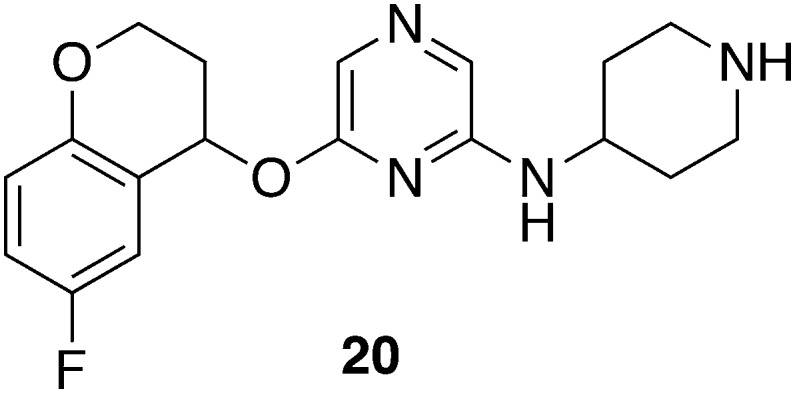
| 20 | 0.120 a | 25 j | 213 | T. b. brucei | 20 mg kg–1, ip, qd, for 5 days, begin 1 day pi | 2/4 | 57 |
| 25 k | |||||||
| 25 mg kg–1, ip, bid, for 10 days, begin 1 day pi | 4/4 | ||||||
| 21 | 0.590 a | 16.8 | Data not shown; according to ref. 21 | 0/4 | 59 | ||
| 0.200 b | |||||||
| 0.01 (Tbg) | |||||||
| 22 | 0.0059 a | T. b. rhodesiense STIB900 | 200 mg kg–1, ip, qd, for 4 days, begin 3 days pi | 0/4 | 61 | ||
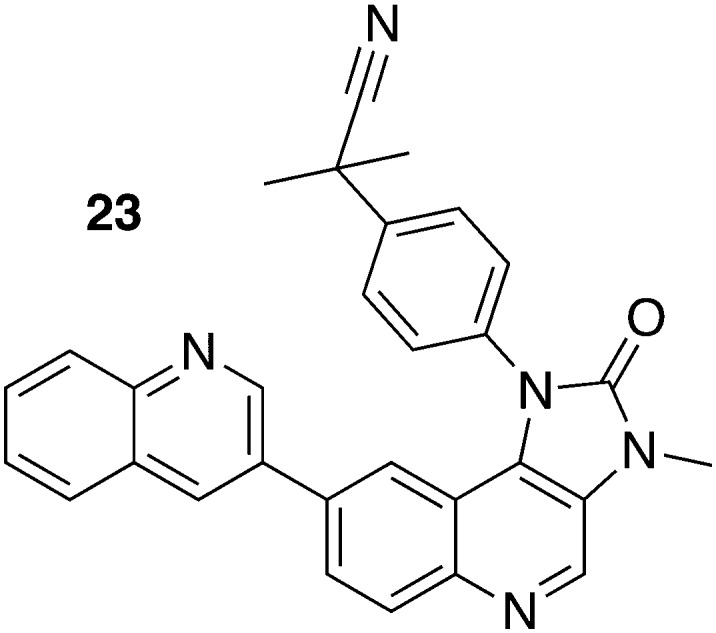
| 23 | 0.0163 a | T. b. rhodesiense EATRO3 | 10 mg kg–1, ip, qd, for 4 days begin 3 days pi | 0/5 | 62 | ||
| 0.00073 b | |||||||
| 24 | 0.00096 a | 12 f | 12 500 | T. b. rhodesiense TREU164 | 10 mg kg–1, ip, bid, for 4 days – 4 days hiatus – 4 days; begin 3 days pi | 4/4 | 66 |
| T. b. brucei Lister 427 | 3/4 | ||||||
| T. b. brucei GVR35 | 20 mg kg–1, ip, bid, for 4 days – 4 days hiatus – 4 days; begin 3 day pi | 0/5 | 67 | ||||
| 25 | 1.5 a | >20 l | >13 | T. b. brucei CA427 | 100 mg kg–1, po, bid, for 14 days, begin 1 day pi | 1/4 | 71 |
| 26 | 1 a | 301 d | 293 | T. b. brucei s427 | 3 mg kg–1, ip, qd, for 4 days | 11 days max survival | 75 |
| 27 | 1 a | 65 d | 62 | 8 days max. survival | |||
| 30 | 6.03 (Tbg) | >30 m | 5 | T. b. brucei CMP | 100 μmol kg–1, ip, qd, | 0/6 | 77 |
| 31 | 0.047 a | 47.2 c | 1004 | T. b. rhodesiense STIB900 | 3.5 mg kg–1, ip, bid, for 4 days, begin 3 days pi | Mean survival 17 days | 79 |
| 0.001 b | 57.0 i | 1212 | |||||
| 26.3 d | 560 | ||||||
| 47.0 e | 1001 | 3.5 mg kg–1, ip, bid, for 4 days, begin 1 day pi, | 3/6 | ||||
| 32 | 0.0023 a | 9.19e | 3916 | T. b. brucei Lister 427 | 20 mg kg–1, iv, for 6 days, begin 3 days pi | 0/4 | 84 |
| 30 mg kg–1, ip, qd, begin 1 day pi, | |||||||
| 33 | 0.82 a | 192 d | 234 | T. b. brucei Lister 427 | 400 mg kg–1, po, qd, 4 days, begin 1 day pi | 0/4 | 87 |
| 34 | 0.68 a | 92.4 d | 136 | 200 mg kg–1, oral gavage, qd, 4 days, begin 1 day pi | 0/4 | 87 | |
| 35 | 0.0031 a | 1 n | 300 | T. b. brucei MiTat1.2 s427 | 30 mg kg–1, ip, qd, begin 1 day pi, | 3/3 | 88 |
| 50 mg kg–1, po, qd, begin 1 day pi | 2/3 | ||||||
| 36 | 0.022 b | 0.146 c | 6 | T. b. rhodesiense STIB900 | 50 mg kg–1, ip, qd, 4 days | Relapse on day 11 | 90 |
| 37 | 0.0018 b | 0.201 c | 11 | Relapse on day 9 | 90 | ||
| 39 | 0.45 a | 15.7 d | 35 | T. b. brucei TC221 | 5 mg kg–1, ip, qd, for 30 days, begin 5 days pi | 92 | |
| 40 | 0.038 a | 12.9 d | 340 | T. b. brucei S427 | 50 mg kg–1, ip, qd, for 4 days, begin 2 days pi | 0/3 | 93 |
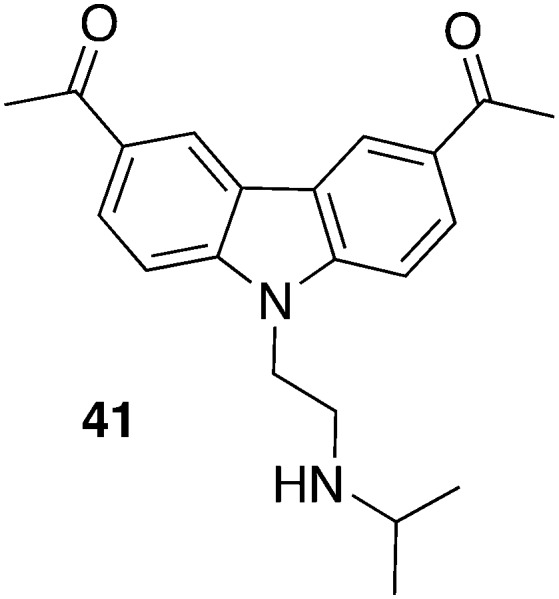
| 41 | 0.055 a | 2.1 e | 38 | T. b. brucei RUMP528 | 40 mg kg–1, po, qd, total 14 doses (4 days on/2 days off), begin 1 day pi | 4/4 | 94 |
| 0.058 b | |||||||
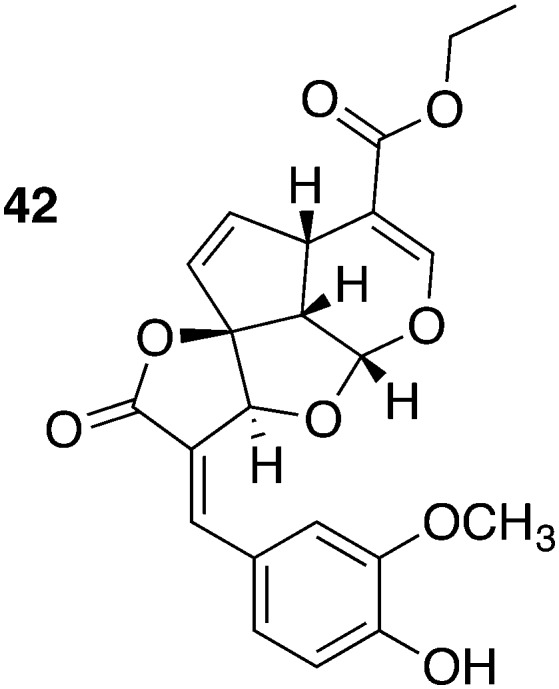
| 42 | 0.430 a | 4.7 o | 11 | T. b. brucei TC-221 | 30 mg kg–1, ip, qd, for 5 days, begin 6 h pi | 5/5 | 97 |
| 10.9 p | 25 |
a T. b. brucei.
b T. b. rhodesiense.
cL6 rat myoblasts.
dHEK293T human embryonic kidney cells.
eHepG2 human hepatocytes.
fMRC5 human lung fibroblasts.
gCRL-8155 human lymphoblasts.
h3T3 fibroblasts.
iMacrophages.
jMOLT4 human T lymphoblasts.
kL929 fibroblasts.
lHeLa cells.
m3T6 fibroblasts.
nL1210 lymphocytic B-cells.
oSkin fibroblasts.
pLung fibroblasts.
Fexinidazole and SCYX-7158
Preclinical details of the current clinical candidates fexinidazole and SCYX-7158 (Fig. 1) have already been reviewed in the literature.15–18
Fig. 1. Clinical candidates fexinidazole (left) and SCYX-7158.

Fexinidazole initially was a designated antimicrobial agent against T. brucei, Trichomonas, T. cruzi, and Entamoeba histolytica by Sanofi-Aventis in 1978. Despite the promising activities of fexinidazole further progression was abandoned primarily due to the common prejudice against nitroaromatics and their toxicological profile.19 A screening approach by the drugs for neglected diseases initiative (DNDi) of 700 already known nitroheterocyclics – mainly nitroimidazoles – against T. brucei rediscovered fexinidazole.20 The convincing in vivo results particularly in the GVR35 mouse model (50 mg kg–1, bid, ip, 4 days) demonstrated full cure (5/5).21 In clinical phase I trials fexinidazole proved good tolerance in oral doses of 100 to 3600 mg.22 Efficacy and safety of fexinidazole, currently being in clinical phase II/III, is tested in 394 acute stage patients in comparison to the nifurtimox-eflornithine combination therapy (NECT).23 Moreover, the recruitment of patients for the phase III trial is ongoing, assessing the therapy success under real life conditions.24 The DNDi expects a hopefully positive result from the regulatory authorities in late 2017, having “only” $45 million spent on the approval fexinidazole so far. This number reflects the money that was spent on drug research, early safety as well as proof-of-concept trials, and efficacy trials.25
The oxaborole SCYX-7158, also derived from phenotype screening, effectively cured (5/5) stage II trypanosomiasis in mice in tolerable doses (25 mg kg–1, qd, orally, 7 days).26 In the following Phase I trial, SCYX-7158 was administered to 128 healthy humans to find the tolerated dose of 960 mg, proceeding with a phase II trial in the Democratic Republic of Congo (DRC) in 2016.27 However, no data are reported yet.
Diamidines: from abandoned clinical trial to promising candidates again
In recent years several diamidines were evaluated as antitrypanosomal drug candidates due to their promising in vitro and in vivo activity against Trypanosoma. Especially the effective cure of stage I of HAT in mice was well established.28,29 For example, pafuramidin (DB289, not shown) successfully completed phase III clinical trials. Disappointingly the progress to market authorization was terminated when unexpected severe nephrotoxic adverse effects emerged.30,31 Although the side effects only appeared in a population in South Africa during the clinical trials, it was not possible to predicted them by preclinical safety models. Only a mouse diversity panel, i.e. well-characterized genetic variants of inbred mice, revealed the kidney safety liability of pafuramidin retrospectively.32 However, the renal toxicity is not supposed to be a general adverse effect of the entire substance group, thus pursuing research on potent diamidines appears reasonable.33 The final aim is to be able to cure both stages of HAT solely by oral administration. Therefore, recently discovered diamidines penetrating the CNS are prominent candidates for supplementing the drug pipeline.
At physiologic pH the diamidines are protonated twice. Since dicationic agents were thought to be unable to cross the BBB, it was even more unexpected that certain compounds like 1 (DB820) and 2 (DB829) (Fig. 2) were active in a stage II mouse model. In mice which had been infected with trypanosomes GVR35, compound 2 was superior to compound 1 (1/5 versus 5/5 cure rate) when 20 mg kg–1 were administered ip over 10 days. As a passive transport mechanism seems to be unlikely for ionized compounds, uptake transporters were suggested channeling these diamidines through the BBB.34 Moreover, it was possible to make the compounds orally bioavailable by masking the amidine structure via a prodrug approach:35 a methoxy group was attached to the amidine. This lowered the diamidines' pKa value in an extend that the molecules are largely unprotonated at physiological pH and warranted a rapid absorption in the gastrointestinal tract. In human liver microsomes these compounds were metabolized via O-demethylation and N-dehydroxylation into the respective active compounds.36 Finally, the corresponding prodrugs 3 (DB844) and 4 (DB868) (Fig. 2) effectively cured all (5/5) mice in a CNS model, administering 100 mg kg–1, po over 5 days.29 In subsequent studies, vervet monkeys were infected with stage II developing strain T. b. rhodesiense KETRI 2537. After determining the tolerable dose of 5 mg kg–1 per day, 8 monkeys were administered 5 mg kg–1, po for 10 days and 6 mg kg–1, po for 14 days, respectively, starting on day 28 pi. Following the monitoring time of 300 days, 3 partially cured 37.5% of the infected animals in lower doses and 42.5% in the elevated dosage regime, respectively.37 Additionally, the pharmacokinetic and safety profiles of 4 were also tested in vervet monkeys for stage I of HAT. Herein, treating the T. b. rhodesiense KETRI 2537 infected animals started 7 days pi, and all animals were cured applying a dose of 3 mg kg–1 per day for 7 days. In this study, biomarkers like creatinine and urea concentration were monitored in order to exclude previously proclaimed nephrotoxicity and kidney liability.33 Further pharmacokinetic investigation was arranged, particularly concerning the CNS exposure by different diamidines. For understanding the outstanding activity of 2 in stage II of HAT compared to other diamidines, Yang et al. proposed a higher unbound fraction of 2 both in plasma and brain (1fu,plasma = 21%/fu,brain = 0.2%; 2fu,plasma = 40%/fu,brain = 1.5%). Thus, high AUC in plasma (7.6 μmol L–1 h–1) and brain (508 μmol L–1 h–1 within 72 h) could be achieved. The CNS permeation was presumed to occur by preferential active transporter; likely the position of the nitrogen played a crucial role.34,38 Finally, the mechanism of the traverse through the BBB should be fully elucidated before proceeding with clinical phases.
Fig. 2. Diamidine compounds being effective in CNS mouse model.

Furthermore, Patrick et al. described diamidines derived from a m-terphenyl scaffold which cured 4/4 mice infected with T. b. rhodesiense STIB900 in doses of 5 mg kg–1, ip, for 4 days.39 The most promising candidates resulted by the replacement of the furan linker by a phenyl moiety. Hence, compound 5 (Fig. 2) – which is chemically closely related to 2 – was assessed in a CNS mouse model. Additionally, detailed pharmacokinetic parameters were examined. 5 (28DAP010) cured all infected mice (5/5) in this stage I model with 20 mg kg–1, ip, for 10 days. The extent of its binding to mouse plasma proteins and brain tissue (fu,plasma = 26%/fu,brain = 0.69%) was slightly stronger when compared to 2.38 Nevertheless, similar AUC values were accomplished in brain tissue (469 μmol L–1 h–1 within 72 h), finally making it a backup compound in case of clinical failure of the diamidine 2.
Besides the prodrug approach, Martinez et al. tried to enhance compound permeability through the BBB by reducing the ionization rate.28 Compound 6 (Fig. 3) carrying N-hydroxy-derivatized imidazoline rings exhibited a pKa of 7.43 and therefore, a less ionized ratio (51.8%) at physiological pH. In a stage I mouse model 6 was fully curative in doses of 20 mg kg–1, ip, for 4 days, hence a model of stage II infection was designed afterwards. In this case, 6 was administered at a dose of 50 mg kg–1, ip, bid, which – in comparison to the control – extended the mean days of relapse to 52. Furthermore, metabolic studies suggested that 6 possessed an intrinsic activity against the parasites without the need of previous bio-activation.28
Fig. 3. Diamidines with varied linker.

According to Vanden Eynde et al. the toxicity of pentamidine could be reduced by replacing the dioxypentyl linker by a diamidealkyl linker.40 The most promising derivatives 7, 8, and 9 (Fig. 3) (IC50 = 2–9 nM, T. b. brucei; CC50 = 43.0 ≥ 100 μM, L6 cells) were tested in mice infected with drug-sensitive and drug-resistant strains of T. b. brucei. One daily dose was administered ip for 3 days, beginning 24 h pi. The three compounds cured all mice (3/3) infected with the drug-sensitive strains (Lab 110 EATRO and KETRI 2002) in a dose of 10 mg kg–1. The most effective compound, 8, cured even in doses lower than 1.0 and 2.5 mg kg–1, respectively. For mice infected with the resistant strain KETRI2538 (refractory to eflornithine and arsenical drugs) a survival of over 30 days could be observed when treated with 7 and 8 (doses between 5 and 25 mg kg–1), and all mice were considered as cured. However, all compounds tested including pentamidine were ineffective against strain Ketri 1992 (refractory to diamidines).40
Yang et al. focused on potent diamidines with 10 (Fig. 3) being the most selective scaffold. 10 exhibited an IC50 value of 7.7 nM against T. B. brucei and a CC50 value greater than 200 μM against both hepatocytes and kidney cells, resulting in a SI > 13 000. Hence, 10 proceeded to in vivo studies for the stage I of HAT. Since the administration of both 3 and 10 mg kg–1, ip, for 4 days effectively cured all mice (5/5), reduced doses of 1 and 0.3 mg kg–1, ip, were applied for 4 days. However, the lower doses were not effective anymore. In this case, A/T-rich DNA motifs (especially in the kinetoplast DNA) could be designated as drug target of the investigated diamidines. Moreover, two additional targets for 10 were identified, particularly the trypanosomal farnesyl diphosphate synthase (FPPS) (more selective to TbFPPS then to human FPPS, representing an improvement towards 2) and the uncoupling of the proton pump in the respiratory chain which is responsible for ATP synthesis.41
3. Target-based approach
Drug discovery programs for neglected tropical diseases rely both on target-based approaches and phenotypic screening. In the target-based approaches compound libraries are screened against a certain molecular target, e.g. enzymes. In further steps, the initial hit is optimized regarding potency, selectivity, and pharmacokinetic profile. Thus, a preliminary validation process is required to ensure that the compound actually binds to the supposed target.42,43 Unfortunately, in the case of African sleeping sickness there are only few validated molecular targets. In the following sections the most distinguished target-based approach was taken into consideration.
N-Myristoyltransferase inhibitors
RNA interference assays in T. brucei confirmed N-myristoyltransferase (NMT) as an essential protein in both life cycle stages of trypanosomes producing the diseases discussed herein, namely in the procyclic vector and the mammalian bloodstream form. Therefore, this enzyme was suggested to be an appropriate target for the development of anti-parasitic agents. The NMT catalyzes the cotranslational myristoylation of the N-terminal glycine of approximately 60 proteins which are predicted to be essential for the parasite growth. Which downstream peptides exactly inhibit the growth of parasites has not been elucidated yet, but pleiotropic effects are assumed.44
Brand et al. optimized the HTS hit which resulted in the very potent NMT-inhibitor 11 (DDD85646) (Fig. 4) (IC50 = 2 nM, TbNMT).45 For pharmacokinetic studies, female NMRI mice were treated with 3 mg kg–1, iv and 10 mg kg–1, po, respectively. It showed a low blood clearance (Clb = 6 mL min–1 kg–1) and a small volume of distribution (Vd = 0.4 L kg–1) with a moderate half-life value (t1/2 = 1.2 h) and a favorable oral bioavailability (F = 19%).46 For the in vivo efficacy studies, female NMRI mice were infected with T. b. brucei S427. The administration of 11 in a dose of 12.5 mg kg–1, po, bid for 4 days could be considered an effective dose for this compound as no parasites were detectable after 30 days.46 Notably, the minimal orally efficacious dose was 50 mg kg–1 for the more clinical relevant T. b. rhodesiense subspecies. A disadvantage was the low brain penetration of this compound (brain–blood-ratio <0.1). Due to lacking efficacy, a stage II HAT efficacy study at the maximum tolerated dose of 100 mg kg–1, po, bid failed.45 All in all, this study could prove NMT as a promising target against HAT, but the examined compound has some limitations, namely the lack of selectivity against the human isoform of NMT (IC50 = 3 nM, HsNMT; IC50 = 2 nM, TbNMT) and its low CNS exposure.45
Fig. 4. TbNMT inhibitors identified through target-based approach.

The Drug Discovery Unit in Dundee (DDU) consequentially guided compound 11 through lead optimization referring to the related crystal structures of NMT of Aspergillus fumigator and Leishmania major.47 The replacement of the pyridine ring by a flexible alkyl linker achieved increased selectivity which was further refined addressing the CNS penetration. Therefore, the sulfonamide group which was suggested to be deprotonated at physiological pH was capped. Since simple alkyl residues like methyl or ethyl groups were metabolized rapidly, a difluorinated methyl group was attached to increase both stability and CNS exposure. Finally, the improved compound 12 (DDD100097) was obtained. The selectivity against HsNMT was increased (IC50 = 12 nM, HsNMT versus 2 nM, TbNMT) which was reflected in the advanced selectivity against MRC5 fibroblasts (CC50 = 0.3 μM versus 1 μM). Moreover, 12 exhibited an advanced permeability through the BBB (blood–brain-ratio = 1.6), contrary to 11. After proof of full cure by using 12 in a stage I mouse model (T. b. brucei S427, 50 mg kg–1, po, bid for 4 days) an evaluation of a stage II mouse model was performed. T. b. brucei GVR35 infected mice were treated with the maximum tolerated dose (100 mg kg–1, bid, po) for 5 days but signs of toxicity occurred. Nevertheless, in order to achieve the proof-of-concept in vivo for stage II, other dosing schedules (e.g. extension of the treatment days or pulse dosing) were tested. Both a prolonged administration of 100 mg kg–1 bid for 8 days and three rounds of 3 days treatment with a two-day drug-free interval each partially cured mice (1/5). Unexpectedly, no absolute efficacy was obtained, though a dose of 20 mg kg–1, bid, po was predicted to be curative. The observed treatment failure might result from the discrepancy in the susceptibility of T. b. brucei strain GVR35 and T. b. brucei S427 towards 12. Previous NMT inhibitors exhibited similar differences in activities (4–5-fold less active against T. b. brucei GVR35). Since the dosing regime was set on the basis of the efficacy against T. b. brucei S427, it was supposed that treatment failure originated from subtherapeutic doses. Unfortunately, the dose was at the limit of therapeutic tolerance, thus no further increase could be performed.47
4. Phenotypic HTS
The whole-cell screening of large compound libraries became feasible due to the application of the inexpensive, robust, and simple resazurin-based assay including 384- and 96-well formats. The phenotype screening hits provide a guaranteed impact on living organisms and hence possess better drug-like properties which is in contrast to compounds being obtained from the target-based approach. Consequently, a cytotoxicity assay against mammalian cells is performed to exclude general toxic molecules and to estimate selectivity.42 Moreover, HTS of diverse libraries could identify novel and unknown drug targets, nevertheless it may be challenging to subsequently elucidate the accurate mode of action.48–51 Of note, when looking at currently approved drugs against sleeping sickness only for eflornithine the mechanism of action could be elucidated, yet.
Oxazolopyridine derived lead compounds
Tatipaka et al. performed a phenotypic screening of 700 000 compounds in cooperation with the Genomics Institution of the Novartis Research Foundation (GNF) to find new starting points for the lead optimization of compounds against HAT.52 Scaffold 13 (Fig. 5), a substituted 2-(3-aminophenyl)oxazolopyridine, was chosen for subsequent hit-to-lead optimization because it lacks stereogenic carbon atoms and possesses desirable properties such as low molecular weight and compliance to the Lipinski's rule of five. In the next step, 110 compounds were synthesized, revealing the most selective 2-(3-aminophenyl)imidazopyridine (14), exhibiting an IC50 of 2 nM in vitro against T. brucei S427 (Fig. 5). An enhanced potency arose from replacing chlorine with fluorine and substituting the furanoyl group by an urea group. In terms of potency, varying the oxazolopyridine core was well tolerated when replacing the oxygen with a nitrogen which resulted in an imidazopyridine scaffold. However, the leap in activity was achieved by attaching an aromatic moiety to C6. Finally, a moderate CC50 towards human lymphoblasts and hepatocytes (24 μM and 30 μM, respectively) suggested a SI value greater than 1000. The analysis of pharmacokinetic parameters indicated that 14 showed an oral bioavailability with sufficient plasma levels (Cmax = 4.3 μM) for parasite inhibition. Additionally, CNS exposure was firstly demonstrated by permeability studies across MDCK-MDR1 cells without the compound being substrate of the efflux pump MDR1. Secondly, the level of brain penetration was proven as brain-to-blood concentration ratio (0.5 and 1.1, determined by two independent experiments) after 60 min of ip dosing. The metabolism studies demonstrated a relatively short half-life of 31 min when incubated utilizing human hepatic microsomes. The plasma clearance was determined to be 14.8 ml min–1 kg–1 and the half-life for plasma elimination was 204 min. For the in vivo studies mice were infected with T. b. rhodesiense. Compound 14 was administered orally 48 h pi. The lowest potent dose of 5 mg kg–1, bid for 5 consecutive days, was able to cure all mice (5/5) after 60 days. Diminishing the dose to 2.5 mg kg–1 or reducing the administration to a qd regime cured 2 of 5 mice only. During a high dose study with 50 mg kg–1, bid, po for 14 consecutive days neither a change in weight compared to mice treated with the vehicle, nor clinical toxicity could be observed. Altogether, these results indicated the high potential of 14 as a representative of a novel drug class against stage I of HAT. Further efforts to examine the potential for stage II are in progress.
Fig. 5. Lead compounds derived from oxazolopyridine 13.

Surprisingly, Sykes et al. also identified molecule 13 (Fig. 5) as a potential candidate for drug development in an independently performed whole-cell HTS of 87 296 compounds.48
Additionally, while screening 3 million compounds in a proliferation assay against kinetoplastid parasites T. cruzi, T. brucei, and Leishmania donovani, 13 stood out in being active against all three parasites (IC50 = 150 nM, T. brucei) and was advanced to lead compound 15 (GNF6702) (Fig. 5). The implemented modification comprised substituting chlorine with fluorine, as it was done likewise in the group of Tatipaka. The furanoyl residue was replaced by a dimethyloxazole ring due to the toxicity risks associated with furan. The core modification resulted in an imidazopyrimidine scaffold. Moreover, the C6-substitution initiated the substantial increase in activity against all kinetoplastids, whereas the pyrimidine moiety at C6 introduced the best selectivity towards macrophages and fibroblast cells. Conclusively, the lead compound was optimized regarding potency, oral bioavailability, and plasma clearance. Thus, compound 15 (IC50 = 70 nM) was set to a mouse model of stage II of HAT. However, from the chemical point of view, compounds 14 and 15 are quite similar. The CNS infection was demonstrated in mice by luciferase expressing T. brucei parasites and detection of the bioluminescence. Treating the animals started 21 days pi with 100 mg kg–1, po, qd, for 7 days of 15; the compound was fully curative in all mice (6/6). Due to this outstanding result, efforts on target identification were made to start a target-based drug development. The first hint was gathered using cultivated, drug resistant strains of Trypanosoma which possessed mutations in the proteasome encoding genes. Further investigation of resistant parasites overexpressing PSMB4F24L (i.e., proteasome subunit beta type 4), revealed susceptibility towards 15. Additionally, in contrast to the mammalian protease inhibitor bortezomib compound 15 did not affect the human proteasome but selectively blocked any chymotrypsin-like activity. In summary, the T. brucei proteasome was assessed as a validated drug target of 15 through various evidence such as the correlation between the inhibition of the parasite proteasome and the parasite growth, respectively. It is tempting to speculate that compound 14 may address the same target due to its chemical similarity. As a next step the preclinical toxicity of 15 should be evaluated and consideration about preclinical studies for stage II of HAT should be given.53
Thiazole-2-ethylamine derived lead compound
Patrick et al. subjected molecule 16, one initial hit from the GNF library, to lead optimization (Fig. 6).54 The structure modification focused on both ends of the molecule while retaining the thiazol-2-ethylamine core. Interestingly, Sykes et al. again independently confirmed structurally close related phenylthiazol-4-ylethylamides being promising hit scaffolds.48 Thirty-three analogues of 16 having IC50 values below 0.2 μg mL–1 and SI values greater than 120 towards L6 cells were evaluated in vivo with a dose of 40 mg kg–1 administered ip.54 Two mice infected with T. b. rhodesiense were treated for three consecutive days starting 24 h pi. Despite the good in vitro activity (e.g., IC50 < 25 nM) of certain molecules, none of the analogues was able to cure mice. Studies on the metabolic stability revealed half-lives ranging from 0.3 to 11 min, explaining the lack of in vivo efficacy. Since all the metabolically unstable compounds contained the thiazol-2-ethylamine core, this scaffold was considered as a major weakness.54 Although details about the metabolic degradation remain unexplored, only replacing it by fused ring systems could enhance the stability of the compounds. Additionally, the metabolic stability of the molecule could be increased fourfold by fluorination of the pyrrolidine moiety. Finally, only the benzothiazole scaffold 17 exhibited prolonged half-lives (t1/2 > 60 min) with concurrently retained antitrypanosomal activity (IC50 = 0.0348 μM, T. b. brucei). Compound 17 was administered at 50 mg kg–1, po, bid for 4 days, beginning two days pi and cured all mice (5/5) in a stage I mouse model. Moreover, 17 was given in doses of 50 mg kg–1, po for 10 days, beginning 21 days pi in a stage II study with T. b. brucei TREU667 infected mice. Over a period of 180 days pi, all mice (5/5) were cured and free of parasites in tail blood samples. These results are outstanding for an oral treatment option for both stages and will be complemented by target identification in the near future.55
Fig. 6. Lead compound derived from thiazole-2-ethylamine.

Indoline-2-carboxamides
Based on a protease inhibitor library containing 3400 compounds of the DDU in Dundee, hit compound 18 (Fig. 7) was identified (IC50 = 30 nM, T. b. brucei) with a SI greater than 1800 versus MRC-5 fibroblasts (CC50 > 50 μM). Extensive pharmacokinetic research demonstrated a microsomal metabolic instability of 18. Therefore, the following in vivo experiments were conducted with hepatic CYP reductase null (HRN) mice having a suspended CYP450 activity. The HRN mice were infected with T. b. brucei S427 and the administration began three days pi with 3 mg kg–1, ip, bid for 4 days. All mice (3/3) could be cured without a relapse up to 30 days pi. However, when 3 infected NMRI mice were treated with 10 mg kg–1, bid for 4 days, only a partial cure was obtained (one mouse relapsed; two mice were free of parasites up to 30 days pi). Despite the compound blood concentration of 18 quickly decreased below the EC50 within 4 hours, it was at least partially effective. Hence, 18 is considered to selectively accumulate within the parasites or might have a rapid cidality. Thus, Cleghorn et al. made some efforts in order to prolong compound half-lives and substituted the potentially unstable ether group by a methylene linker.56 Furthermore, regarding the antitrypanosomal activity the R configured enantiomer emerged being superior to the racemate. Other variations addressing e.g. the indoline core or the amide was either not tolerated or only little beneficial. Lead compound 19 (Fig. 7) eventually possessed an improved pharmacokinetic profile regarding brain tissue binding, cmax, t1/2, AUC, and brain to blood ratio, and was passed to a stage II mouse model. T. b. brucei (GVR35 strain) infected mice were treated with 12.5 mg kg–1, ip, bid for 5 days beginning 21 days pi. One of five mice showed no parasitemia after 180 days pi what was specified as cured, while the remaining animals gradually relapsed within this period. The future challenge in optimizing the compound will include improving the microsomal stability.56
Fig. 7. HTS hit from protease inhibitor library and its lead compound.

2-Aminopyrazines/2-aminopyridines
A screening of 5500 small molecules identified 2-aminopyrazines and 2-aminopyridines as hit structures.57 Since the most effective compound 20 (Fig. 8) exhibited in vitro activity against Trypanosoma subsp. (IC50 = 0.12 μM) and a SI > 200 towards mammalian cell lines (MOLT4, L929), in vivo efficacy was studied in the stage I of HAT. A first treatment regime was designed to administer 20 mg kg–1, ip, qd, for 5 days; it yielded a 50% curative outcome (2/4). Subsequently, treatment was changed to 25 mg kg–1, bid for 10 days, as 20 was well tolerated previously; this higher dose effectively cured all mice (4/4) without any signs of toxicity. In silico and in vitro evaluation suggested appropriate pharmacokinetics with a high membrane permeability, low protein binding (fu = 0.4), and high metabolic stability. However, more detailed in vivo investigation about bioavailability and other preclinical properties is in progress.57
Fig. 8. 2-Aminopyrazine from HTS.
5. Drug repurposing
In drug repurposing programs approved drugs are evaluated for novel fields of indication. Besides pharmacokinetics, toxicity, and metabolism it can be challenging to bring new chemical entities (NCEs) into clinical application.50 These preclinical investigations are extremely time consuming and cost intensive factors of the drug development. However, in drug repurposing approaches the development can be accelerated since reliable safety and pharmacokinetic data are already available.58 For pursuing this strategy, whole phenotypic screening as described before is applied.
Fingolimod – screening hit derived from approved FDA drugs
According to the well-established HTS protocols against T. brucei, a library of 741 compounds – either in a late clinical stage or with FDA approval – was screened.48 Compound 21 (fingolimod) (Fig. 9) is a registered drug representing an oral treatment option of multiple sclerosis and demonstrated a selective activity against several T. brucei subspecies (IC50 = 0.01–0.59 μM; SI = 16.8 towards HEK293).59 In order to validate the target of the sphingolipid derivative 21, other sphingosine analogues and sphingosine kinase inhibitors (SPHK) were tested for antitrypanosomal activity (IC50 = 0.02–10.79 μM). TbSPHK was validated as a drug target previously.60 Unfortunately, no correlation between TbSPHK inhibition and activity against T. b. brucei was observed. Nevertheless, fingolimod was evaluated in an in vivo acute mouse model where mice were infected with T. b. rhodesiense. Disappointingly, 21 was not effective in curing mice due to unknown reasons.59
Fig. 9. Omnidirectional HTS hits of FDA approved drugs and agrochemicals, respectively.

Zoxamide – screening hit derived from agrochemicals
Witschel et al. initially screened 600 agrochemicals supplied by BASF for their antiparasitic activity. Here, registration and admission requirements could be more stringent in certain points than for pharmaceuticals because for such entities entering the food chain no tolerance towards side effects is allowed. Commercialized agrochemicals need to pass broad toxicological assessments including chronic and reprotoxicological studies in different mammalian species. Besides, beneficial features of these substances are a highly-optimized temperature stability, their multi-ton scale production process, and low production costs. According to in vitro studies with T. b. rhodesiense STIB900, 22 (zoxamide) (Fig. 9) showed a similar activity (IC50 = 6 nM) as melarsoprol (IC50 = 6 nM). However, 22 was favored due to its oral LD50 of >5000 mg kg–1 in rats versus an LD50 iv of 44 mg kg–1 of melarsoprol in mice. For this reason, 22 was warranted for in vivo studies with female NMRI mice previously inoculated with T. b. rhodesiense STIB900. The treatment was initiated on day 3 pi and took 4 days with 200 mg kg–1, ip, qd. No parasitemia was observed up to seven days pi. However, ten days pi all mice showed parasites in blood demonstrating that still efforts have to be done in optimizing these substances. Finally, agrochemicals could be considered being an interesting starting point with good prospects and a source of new lead structures against protozoal diseases which has been untapped so far.61
Kinase-inhibitors
PI3K/mTOR inhibitors
The drug repurposing approach uncovered inhibitors of the phosphoinositide 3-kinase (PI3K) and the mammalian target of rapamycin (mTOR) kinase, both representing genetically validated potential drug targets in trypanosomes. Due to a remarkable structural homology between PI3K and mTOR in humans and trypanosomes, eight commercially available compounds were evaluated. 23 (NVP-BEZ235) (Fig. 10) inhibited both PI3Ks and mTOR in the sub-nanomolar concentration range. The in vitro assessment involving various T. brucei subsp. demonstrated an IC50 values from 0.18 to 16.3 nM. The consequent in vivo evaluation in T. b. rhodesiense EATRO3 infected mice which were treated with 10 mg kg–1, qd, ip for 4 days beginning 3 days pi, extended the mean survival to 13.4 days.62,63
Fig. 10. Repurposed PI3K/mTOR inhibitor.
HTS of kinase inhibitors
Further drug repurposing approaches identified other human kinase inhibitors as potent antitrypanosomal agents.60,63–65 A broadly designed kinase-targeted HTS of 42 444 compounds including the published kinase inhibitor set by GlaxoSmithKline could confirm 797 selective hits. Regarding potency, predicted CNS permeability, physicochemical properties, and selectivity over human kinases, the three most attractive hits were assessed during pharmacokinetic studies. Subsequently, compound 24 (Neu-1053) (Fig. 11) was selected for being evaluated in an efficacy stage I mouse model due to appropriate blood exposure after ip administration, rapid onset of the antitrypanosomal effect in vitro (IC50 = 0.96 nM, T. b. brucei), and a high selectivity. The T. b. rhodesiense infected mice were successfully cured (4/4) after the administration of 10 mg kg–1, ip, bid for 4 consecutive days, followed by a four-day treatment pause and a second round of 4 days of treatment. In a supplemental study with T. b. brucei infected mice compound 24, which was given in the same regime, cured 3 of 4 mice for 90 days.66 Subsequently, a structure–activity relationship (SAR) with 26 analogues of 24 was derived.67 The hydroxyethyl moiety at the indole scaffold was suggested to be prone to metabolism, thus replacement with other residues (e.g. methyl-, aminoethyl-, or methoxyethyl residues) was performed. Moreover, the linker between the two aromatic moieties was varied regarding length and rigidity. When replacing the benzimidazole by other scaffolds, the relevance of having a hydrogen bond donor in this region was unravelled. The dichloro substitution at the indole scaffold apparently affected the activity against T. brucei, as the omission of the lipophilic halogens resulted in a loss of potency. Finally, the structure variation of the primal hit compound 24 did not improve the potency of the compound. Therefore, a pharmacokinetic study of 24 determining the brain-to-plasma ratio (0.39) was subsequently conducted. Since sufficient drug levels could be reached (31.6 ng g–1) in murine brain, a murine CNS infection model was designed. Ten mice were infected with T. b. brucei (GVR 35 strain); five of these animals were administered 40 mg kg–1 diminazene aceturate on day 21 as a control. In comparison, 24 was given to the other half of the mice in doses of 20 mg kg–1 on day 21–25. Afterwards, the treatment was interrupted for 2 days and was continued with doses of 20 mg kg–1 on days 28–32. During the second period of treatment no parasitemia was detected in blood on day 35, however all mice relapsed on day 39.67 The lack of stage II efficacy was suggested to be related to a high plasma protein binding (>99.9) and a rapid clearance (Clint = 24.9 μL min–1/106 cells in rat hepatocyte). Target identification of 24 was carried out. Due to the structural similarity to previously reported T. b. methionyl-tRNA synthetase (MetRS) inhibitors, the inhibition of MetRS was confirmed by means of biochemical (T. brucei MetRS inhibition assay), cellular (comparing the reaction of wild-type T. brucei to T. brucei with upregulated MetRS mRNA), and crystallographic techniques. The binding mode of 24 to MetRs was quite similar to that of reported inhibitors. Furthermore, both the methionine substrate pocket and the auxiliary pocket were addressed by 24 as reported before.68,69 Additionally, novel interactions within the TbruMetRS complex were shaped, especially the hydroxyethyl substituent made a hydrogen bond to an asparagine in distance of 2.6 Å.67
Fig. 11. Hit compound from kinase-targeted HTS and its SAR.

Tyrosine kinase inhibitors
The protein tyrosine kinase inhibitor (PTKI) tyrphostin A47 blocks the endocytosis of transferrin and inhibits trypanosome replication. Driven by the fact that the tyrosine kinase inhibitors (TKI) inhibited the growth of T. b. brucei, Katiyar et al. described the potency of 25 (lapatinib, a TKI applied for cancer) (Fig. 12) in trypanosomes. Since the T. brucei genome does not encode for EGFR or HER2, which are considered as actual targets of lapatinib in humans, the mode of action in T. brucei was investigated.70 The addressed targets of lapatinib included three essential tyrosine kinases of T. brucei which prompted to screen further PTKIs against T. brucei. Therefore, Behera et al. referred to other PTKI (e.g., erlotinib, imatinib, axitinib, and sunitinib) which at least were in an advanced phase of drug development and for which non-toxic profiles and a straightforward synthesis was known.71 Three PTKI were selected for in vivo assessment of which compound 25 exclusively cured 1/4 of the trypanosome-infected mice when administering 100 mg kg–1, po, bid, for 14 days.71 A supplementary SAR study by Patel et al. explored analogues of 25 and could even reveal an increased selectivity due to the replacement of the moiety in position C6 with a 3-morpholinophenyl residue (Fig. 12). However, probably due to the high plasma protein binding survival of the animals could only be prolonged for a few days within in vivo experiments.72 Hence, PTKIs can be considered as an important part in the portfolio of new chemical entities against trypanosomes.73,74
Fig. 12. The human tyrosine kinase inhibitor lapatinib (25) and its analogue.

Farnesyl diphosphate synthetase (FPPS) inhibitors
Initially, a library screen of 925 putative prenyl synthase inhibitors against the T. b. brucei farnesyl diphosphate synthase (TbFPPS) resulted in bisphosphonate molecules, particularly representing lipophilic analogues of the approved drug zoledronate. Since the FPPS gene was assumed to be essential for the trypanosomal sterol biosynthesis, target validation started to correlate TbFPPS inhibition with the antitrypanosomal activity. Interestingly, for none of the 56 hit compounds inhibition of TbFPPS correlated with the activity against T. brucei. However, different targets than the TbFPPS like the squalene synthase or the hexokinase might be addressed by the bisphosphonates which could be advantageous due to a hampered development of resistance. Nevertheless, compounds 26 and 27 (Fig. 13) with a C9 and C7 alkyl side chain, respectively, both exhibited an IC50 value of 1.0 μM against T. brucei and a CC50 values of 301 μM and 64 μM against hepatocytes, respectively. These two most promising compounds were administered ip into T. brucei infected mice over a period of 4 days. A treatment with 26 at a dose of 3 mg kg–1, bid led to a prolongation of life for a maximum of 16 days. Applying a treatment once a day survival was extended for 11 days, while the dose of 1 mg kg–1 prolonged life for 6 to 7 days. Compound 27 was not effective as a single dose of 1 mg kg–1, but 3 mg kg–1 could extend the survival by a maximum of 8 days.75
Fig. 13. Lipophilic bisphosphonates derived from FPPS targeted HTS.

Quinolones
Ciprofloxacin (28), an approved fluoroquinolone with broad-spectrum antibacterial activity, showed slight antitrypanosomal effects with an IC50 value of 52 μM against T. b. brucei.76 The esterification of ciprofloxacin resulting in 29 (Fig. 14) exhibited a decreased IC50 value of 26 μM. However, in vivo experiments involving compound 30 (IC50 = 6.03 μM T. b. gambiense; CC50 > 30 μM, 3 T6 fibroblast cells) with a single ip dose of 100 μmol kg–1 extended survival up to 8 days, but mice could not be fully cured.77 Hiltensperger et al. synthesized novel quinolone amides and tested their activity against T. brucei.78 The SAR studies led to the lead structure 31 which exhibited a promising in vitro activity against T. b. brucei (IC50 = 47 nM) and T. b. rhodesiense (IC50 = 9 nM) as well as a notably moderate cytotoxicity (CC50 = 57 μM, J774.1 macrophages), resulting in a SI of 6333. In order to overcome the poor water solubility of 31, an amorphous formulation was developed by spraydrying the compound with Eudragit L100 which was dissolved in a delivery vehicle of glucose (5%), polysorbate 80 (1%), and PBS buffer (pH 7.4) prior to administration. A stringent mouse model was conducted initially starting on day 3 pi with a treatment of 3.5 mg kg–1, bid, ip for 4 days. The resulting mean survival of mice was 17 days. Afterwards, 3/6 of the infected mice were cured by means of an early-treatment model, i.e. begin of treatment one day pi with the same dosage regime, indicating that 31 is a promising lead structure but demands for further improvement.79
Fig. 14. Quinolone derived compounds.

Bisnaphthalimidopropyl
Bisnaphthalimidopropyl compounds have been evaluated as agents against cancer, bacteria, and Leishmania infections.80–83 The application of 32 was extended to the kinetoplast-holding T. b. brucei, showing an IC50 value of 2.35 nM (Fig. 15). Further cytotoxic studies excluded any harmful effects on human cell lines (THP-1 macrophages, hepatocytes and neurons) within therapeutic doses, exhibiting an SI of 2514, 3916, and 878, respectively. Preliminary pharmacokinetic tests demonstrated 95–100% metabolic stability in mouse microsomes, and a 10 mg kg–1, iv injection of 32 achieved plasma levels above the IC90 value for 24 h. The ensuing in vivo efficacy mice studies of stage I of HAT started 3 days pi by administering 10 or 20 mg kg–1, iv, qd for 6 days. Treatment efficacy was monitored by bioluminescence in luciferase-positive Trypanosoma parasites; additionally, parasitemia was assessed. The elevated dose of 20 mg kg–1 demonstrated in vivo activity and could eliminate the parasite burden during treatment, though infection relapsed when the treatment was stopped at day 9 pi. Graca et al. searched for the reason of the lack of efficacy and proposed a potential invasion of parasites into the peritoneal cavity, i.e. into extravascular tissue next to the injection area. Optimization of 32 regarding improved pharmacokinetic and pharmacodynamic properties, especially with efficacy in a mouse model of stage II of HAT, is the future goal.84 Quarternization of the nitrogen atoms in the middle chain revealed highly active bisnaphthalimido analogues with low cytotoxicities, however these compounds still are too toxic for animal studies.85,86
Fig. 15. Bisnaphthalimidopropyl and tubulin inhibitors.

Tubulin inhibitors
Tubulin inhibitors are well established compounds for the treatment of cancer. Tubulin is a crucial molecule in physiological processes involving locomotion and cell division in T. b. brucei. Starting with anti-cancer agents, Nanavaty et al. succeeded in selectively addressing trypanosomal tubulins.87 Compounds 33 and 34 (Fig. 15) exhibited IC50 values of 0.82 μM and 0.68 μM, respectively, against T. b. brucei. The cytotoxic effects were evaluated against macrophages (RAW267.4) and kidney cells (HEK293) and resulted in a SI greater than 102. Therefore, each compound was administered qd to mice with a dose of 400 mg kg–1 and 200 mg kg–1, respectively, using oral gavage. The level of parasitemia was determined by taking daily blood samples and counting the number of T. brucei cells. The mice relapsed on day 4 and day 3, respectively. Conclusively, the reasons for the inefficacy during the in vivo setting were suspected to be a low bioavailability of the compounds which possibly was due to a high first-pass effect or a high binding affinity to serum albumin. However, these highly selective inhibitors of tubulin could be the starting point for more potent inhibitors against T. b. brucei.87
HSP90 inhibitors
The heat shock protein 90 (HSP90) assists with proper protein folding and the stabilizing of client proteins regulating cell cycle, signal transduction, and receptor formation. For this reason, a couple of HSP90 inhibitors entered various stages of clinical trials against cancer.88 Kinetoplastids express the HSP90 homologue HSP83 which is considered as an appropriate drug target.89 Since 35 (17-DMAG) (Fig. 16) showed an IC50 value of 3.1 nM against trypanosomes and a SI of 300 towards L1210 mammalian cells it was advanced into in vivo experiments. Administering 35 as a single dose of 150 mg kg–1, ip, led to drop outs due to acute toxicity effects. This was the reason why the dose of 150 mg kg–1 was split over 5 days (i.e., 30 mg kg–1, ip, qd). Hence, all mice were free of parasites from day 3 to 30; this implied a full cure (3/3). Additionally, administration by oral gavage for 5 days (50 mg kg–1, qd) cured all but one mouse which died from toxicity and/or gavage-induced trauma. Finally, during the development of novel HSP90 inhibitors it might be valuable to add antitrypanosomal investigations.88 Interestingly, the HSP90 inhibitor geldanamycin stood out from an independently applied screening by another research group.59 These outcomes prompted Giannini et al. to investigate indicated HSP90 inhibitors for their antitrypanosomal activity (Fig. 14). In the beginning they pointed out that there is no correlation between HSP90 binding affinity (HSP90alpha enzyme assay) and in vitro activity against T. b. rhodesiense. An in vitro assay with T. b. rhodesiense (IC50 = 4–22 nM) and cytotoxicity tests with L6 cells (CC50 = 146–264 nM) revealed 36, 37, and 38 as promising test candidates with SI between 6 and 66. For 36 and 37 parasitemia could be decreased below the detection limit after day 4 by an ip treatment of 5 mg kg–1 over 4 days in T. b. rhodesiense infected mice. However, all mice relapsed on day 9 and 11, respectively. For compound 38 the effect of toxicity dominated and all mice died on day 2 after the 2nd injection which was not surprising due to the low SI.90
Fig. 16. HSP90 inhibitors.

CTP Synthetase inhibitor
It is known that T. brucei has low pools of cytidine triphosphate (CTP) in comparison to mammalian cells.91 Nevertheless, CTP is essential for cell survival rendering the CTP synthetase (CTPS) an excellent target: the parasites are not able to compensate the inhibition of CTPS by the salvage of cytidine. Compound 39 (acivicin) (Fig. 17), primarily developed as an antitumor agent, showed good inhibitory activity on CTPS but failed in in vivo mouse models.92 The mode of action involves a nucleophilic attack by a thiol group with the displacement of the chlorine atom. Therefore, the halogen atom plays an important role for the inhibitory activity. The exchange of the chlorine in 39 by a bromine atom in 40 (3-bromoacivicin) yielded a 12-fold higher in vitro activity against T. brucei (IC50 = 38 nM versus IC50 = 450 nM) while retaining a moderate cytotoxicity (CC50 = 12.91 μM versus 15.65 μM, HEK cells). For the in vivo studies, three female adult mice infected with T. b. brucei S427 were administered 50 mg kg–1 ip, for 4 consecutive days, beginning 2 days pi. Parasites could not be detected throughout the treatment days but re-emerged on day 8, hence the studies were discontinued on day 11. Conclusively, compound 40 failed to completely eradicate protozoa in vivo at its maximum tolerable dose.93
Fig. 17. CTP synthetase inhibitors acivicin and 3-bromoacivicin.

Carbazoles
Most recently, Thomas et al. made use of carbazole derived compounds, so-called curaxins, for their drug repositioning program94 and warranted them to be tested against T. brucei for several reasons: most compounds previously showed oral bioavailability, a promising in vivo toxicity profile, and were accessible by straightforward synthesis. All advantageous aspects of drug repurposing that are mentioned herein could be utilized. Particularly compound 41 (Fig. 18) is trailed in clinic phase I against solid tumors which might speed up the development for an antiprotozoan drug. According to encouraging in vitro data (50% growth inhibitor concentration GI50 = 58 nM, T. b. rhodesiense; GI50 = 55 nM, T. b. brucei; CC50 = 2100 nM, HeLa cells) compound 41 was advanced in an acute mouse model. T. b. brucei infected mice were treated with 40 mg kg–1, po, starting 1 day pi. All mice (4/4) were cured since there was no detectable parasites in peripheral blood after 30 days. The fact that some curaxins intercalate into DNA prompted the authors to investigate their effect on DNA replication and mitosis. Interestingly, 41 blocked the execution of mitosis but did not affect DNA replication, hence other targets like proteins involved in DNA replication, e.g., CRK3, AUK1, and TLK1, were presumed.94
Fig. 18. Carbazole-derived structure.
6. Derived from plants
Medicinal plants are always a rich source for antiinfective drugs. e.g., The methanol extract of Morinda lucida Benth. possessed antitrypanosomal activity in vivo. Tetracyclic iridoids were identified as the causative agents by bioassay-guided fractionation.95,96 The most potent compound, the iridoid 42, (IC50 = 0.43 μM, T. b. brucei; CC50 = 4.7–18.14 μM, various fibroblast cells) was isolated from leaves of M. lucida. Administering 30 mg kg–1, ip, for 5 days effectively cured all T. b. brucei (TC-221 strain) infected mice (5/5). Additionally, Kwofie et al. suggested the induction of apoptosis in trypanosomes to be the mode of action of 42 (Fig. 19) or, more specifically, the inhibition of the expression of paraflagellar rod subunit 2 (PFR-2). The pivotal role of the parasite flagellum both in cell cycle division and the migration is well known. The induced disorder of cell cycle and the flagellum having fragmented nuclei was revealed by means of Nexin assay, Western Blot, and fluorescence-activated cell sorting (FACS) analysis.97
Fig. 19. Tetracyclic iridoid as active compound isolated from Morinda lucida Benth.
7. Adjunct: Nagana treatment options
The treatment of Nagana (Animal African Trypanosomiasis, cf. above) is restricted to three available drugs: diminazene diaceturate, ethidium bromide, and isometamidium chloride. Unfortunately, treatment options suffered a setback as drug-resistant strains evolved due to the application of these three drugs for 50 years.98,99 For this reason, Gillingwater et al. recently tested the in vivo activity of the well-established diamidines against T. congolense and T. vivax. Among other diamidines particularly compounds 1 and 2 (Fig. 2) were both fully curative in a mouse model (4/4) when 2.5 mg kg–1 (ip, in a single bolus dose) and 1.25 mg kg–1, respectively, were administered. However, a potential cross-resistance to the closely related compounds diminazene aceturate (diamidine) and isometamidium chloride (amidine) should be investigated for T. congolense and T. vivax.100
8. Conclusion
There are various starting points to generate hit compounds for the treatment of African sleeping sickness. Especially stage II of HAT which is very hard to treat poses a tough challenge for drug discovery programs as molecules inevitably need to cross the BBB. However, promising compounds (2, 15, and 17) are in the pipeline accomplishing these criteria for CNS mouse models, and in some cases even are orally bioavailable (15 and 17). Especially the large phenotypic screening campaigns performed by the GNF, GlaxoSmithKline, DDU, and Sykes et al. resulted in promising hits discussed herein. Nevertheless, it is not always easy to translate results from in vitro studies into in vivo efficacy like shown in several of the mentioned studies. The reasons for in vivo failures are multilayered and might originate from (I) extensive metabolism, (II) high plasma protein binding, (III) poor water solubility, (IV) efflux transporters, (V) different sensitivity for particular strains, (VI) reduced permeability, and (VII) growth inhibition rather than trypanocidal effects.
Abbreviations
- AUC
Area under the curve
- BBB
blood–brain barrier; bid, “bis in die”, twice a day
- CC50
Half maximal cytotoxic concentration
- CNS
Central nervous system
- CTP
Cytidine triphosphate
- DDU
Drug Discovery Unit
- DNDi
Drugs for neglected diseases initiative
- EGFR
Epidermal growth factor receptor
- GI50
Half maximal growth inhibitor concentration
- GNF
Genomics institute of the Novartis research foundation
- HAT
Human African trypanosomaisis
- HRN
Hepatic cytochrome P450 reductase null
- HSP90
Heat shock protein 90
- HTS
High throughput screening
- IC50
Half maximal inhibitory concentration
- ip
Intraperitoneal
- iv
Intravenous
- MDR1
Multidrug-resistance-protein 1
- mTOR
Mammalian target of rapamycin
- PI3K
Phosphoinositide 3-kinase
- pi
Post infection
- p.o.
per os, orally
- SI
Selectivity index, CC50 (mammalian cell line)/IC50 (T. brucei)
- SAR
Structure–activity relationship
- T. b. b.
T. brucei brucei
- T. b. r.
T. brucei rhodesiense
- T. b. g.
T. brucei gambiense
- TbFPPS
T. b. brucei farnesyl diphosphate synthase
- qd
“Quaque die”, once a day
Conflicts of interest
The authors declare no competing interests.
Biographies

Michael Berninger
Michael Berninger graduated in pharmacy in 2013 at the University of Würzburg, Germany. Since then, he is a PhD student under the guidance of Prof. Dr. Ulrike Holzgrabe (Institute of Pharmacy and Food Chemistry, University of Würzburg). His current research projects are focused on the synthesis of novel quinolone amides against the African trypanosomiasis.

Ines Schmidt
Ines Schmidt holds a diploma in chemistry from the University of Würzburg, Germany. Afterwards she joined the group of Prof. Dr. Ulrike Holzgrabe (Institute of Pharmacy and Food Chemistry, University of Würzburg) for her doctoral research on dimeric tacrine compounds against tropical infectious diseases. She received her PhD degree in Pharmaceutical Chemistry in 2016 from the University of Würzburg.

Alicia Ponte-Sucre
Alicia Ponte-Sucre is Emeritus Full Professor and Head of the Molecular Physiology Laboratory, Faculty of Medicine, UCV, Venezuela. She occupied diverse academic positions during her career. Her scientific work has linked two control research areas, the “Eros and Thanatos” of Pharmacology: the activity and mechanism of action (against Leishmania and Trypanosoma) of compounds belonging to different chemical classes, and the concept of drug-resistance as a dynamic process that encompasses essential changes in the physiology of the parasites. Since 1995 she collaborates with health science laboratories at the University of Würzburg (Germany).

Ulrike Holzgrabe
Ulrike Holzgrabe studied chemistry and pharmacy at the University of Marburg and Kiel; she completed her PhD and habilitation at Kiel. In 1990 she became an associate professor at the University of Bonn where she served as a vice-rector from 1997 to 1999. Since 1999 she has held a full professorship at the University of Würzburg. She was president of the German Pharmaceutical society from 2003 to 2007. Her main research interests are the development of antiinfectives and ligands for muscarinic receptors.
References
- http://www.who.int/mediacentre/factsheets/fs259/en/, (accessed 27.01.17).
- http://www.webmd.com/a-to-z-guides/trypanosomiasis?page=3, (accessed 27.01.17).
- Steverding D. Parasites Vectors. 2008;1:3. doi: 10.1186/1756-3305-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stich A., Ponte-Sucre A., Holzgrabe U. Lancet Infect. Dis. 2013;13:733–734. doi: 10.1016/S1473-3099(13)70191-9. [DOI] [PubMed] [Google Scholar]
- Sternberg J. M. Parasite Immunol. 2004;26:469–476. doi: 10.1111/j.0141-9838.2004.00731.x. [DOI] [PubMed] [Google Scholar]
- Kennedy P. G. E. Lancet Neurol. 2013;12:186–194. doi: 10.1016/S1474-4422(12)70296-X. [DOI] [PubMed] [Google Scholar]
- Franco J. R., Simarro P. P., Diarra A., Jannin J. G. Clin. Epidemiol. 2014;6:257–275. doi: 10.2147/CLEP.S39728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy C. W., Morrison L. J., Black A., Pinchbeck G. L., Christley R. M., Schoenefeld A., Tait A., Turner C. M., MacLeod A. Int. J. Parasitol. 2009;39:1475–1483. doi: 10.1016/j.ijpara.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Morrison L. J. Parasite Immunol. 2011;33:448–455. doi: 10.1111/j.1365-3024.2011.01286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desquesnes M., Dargantes A., Lai D. H., Lun Z. R., Holzmuller P., Jittapalapong S. BioMed Res. Int. 2013;2013:321237. doi: 10.1155/2013/321237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desquesnes M., Livestock trypanosomoses and their vectors in Latin America. OIE (World Organisation for Animal Health), 2004. [Google Scholar]
- Camargo R., Izquier A., Uzcanga G. L., Perrone T., Acosta-Serrano A., Carrasquel L., Arias L. P., Escalona J. L., Cardozo V., Bubis J. Vet. Parasitol. 2015;207:17–33. doi: 10.1016/j.vetpar.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacklock B., Yorke W. Proc. R. Soc. London, Ser. B. 1913;87:89–96. [Google Scholar]
- Brun R., Hecker H., Lun Z. R. Vet. Parasitol. 1998;79:95–107. doi: 10.1016/s0304-4017(98)00146-0. [DOI] [PubMed] [Google Scholar]
- Brun R., Don R., Jacobs R. T., Wang M. Z., Barrett M. P. Future Microbiol. 2011;6:677–691. doi: 10.2217/fmb.11.44. [DOI] [PubMed] [Google Scholar]
- Eperon G., Balasegaram M., Potet J., Mowbray C., Valverde O., Chappuis F. Expert Rev. Anti-Infect. Ther. 2014;12:1407–1417. doi: 10.1586/14787210.2014.959496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle A. S., Khare S., Kumar A. B., Supek F., Buchynskyy A., Mathison C. J., Chennamaneni N. K., Pendem N., Buckner F. S., Gelb M. H., Molteni V. Chem. Rev. 2014;114:11305–11347. doi: 10.1021/cr500365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs R. T., Plattner J. J., Nare B., Wring S. A., Chen D., Freund Y., Gaukel E. G., Orr M. D., Perales J. B., Jenks M., Noe R. A., Sligar J. M., Zhang Y. K., Bacchi C. J., Yarlett N., Don R. Future Med. Chem. 2011;3:1259–1278. doi: 10.4155/fmc.11.80. [DOI] [PubMed] [Google Scholar]
- Patterson S., Wyllie S. Trends Parasitol. 2014;30:289–298. doi: 10.1016/j.pt.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torreele E., Bourdin Trunz B., Tweats D., Kaiser M., Brun R., Mazué G., Bray M. A., Pécoul B. PLoS Neglected Trop. Dis. 2010;4:e923. doi: 10.1371/journal.pntd.0000923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser M., Bray M. A., Cal M., Bourdin Trunz B., Torreele E., Brun R. Antimicrob. Agents Chemother. 2011;55:5602–5608. doi: 10.1128/AAC.00246-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarral A., Blesson S., Mordt O. V., Torreele E., Sassella D., Bray M. A., Hovsepian L., Evene E., Gualano V., Felices M., Strub-Wourgaft N. Clin. Pharmacokinet. 2014;53:565–580. doi: 10.1007/s40262-014-0136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.clinicaltrials.gov/ Identifier: NCT01685827, (accessed 29.01.17).
- http://www.clinicaltrials.gov/ Identifier: NCT03025789, (accessed 30.01.17).
- Maxmen A. Nature. 2016;536:388–390. doi: 10.1038/536388a. [DOI] [PubMed] [Google Scholar]
- Jacobs R. T., Nare B., Wring S. A., Orr M. D., Chen D., Sligar J. M., Jenks M. X., Noe R. A., Bowling T. S., Mercer L. T., Rewerts C., Gaukel E., Owens J., Parham R., Randolph R., Beaudet B., Bacchi C. J., Yarlett N., Plattner J. J., Freund Y., Ding C., Akama T., Zhang Y. K., Brun R., Kaiser M., Scandale I., Don R. PLoS Neglected Trop. Dis. 2011;5:e1151. doi: 10.1371/journal.pntd.0001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.dndi.org/2015/media-centre/press-releases/pr-scyx-7158/, (accessed 28.01.17).
- Rios Martinez C. H., Miller F., Ganeshamoorthy K., Glacial F., Kaiser M., de Koning H. P., Eze A. A., Lagartera L., Herraiz T., Dardonville C. Antimicrob. Agents Chemother. 2015;59:890–904. doi: 10.1128/AAC.03958-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzler T., Boykin D. W., Ismail M. A., Hall J. E., Tidwell R. R., Brun R. Antimicrob. Agents Chemother. 2009;53:4185–4192. doi: 10.1128/AAC.00225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine M. F., Wang M. Z., Generaux C. N., Boykin D. W., Wilson W. D., De Koning H. P., Olson C. A., Pohlig G., Burri C., Brun R., Murilla G. A., Thuita J. K., Barrett M. P., Tidwell R. R. Curr. Opin. Invest. Drugs. 2010;11:876–883. [PubMed] [Google Scholar]
- Pohlig G., Bernhard S. C., Blum J., Burri C., Mpanya A., Lubaki J. P., Mpoto A. M., Munungu B. F., N'Tombe P. M., Deo G. K., Mutantu P. N., Kuikumbi F. M., Mintwo A. F., Munungi A. K., Dala A., Macharia S., Bilenge C. M., Mesu V. K., Franco J. R., Dituvanga N. D., Tidwell R. R., Olson C. A. PLoS Neglected Trop. Dis. 2016;10:e0004363. doi: 10.1371/journal.pntd.0004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrill A. H., Desmet K. D., Wolf K. K., Bridges A. S., Eaddy J. S., Kurtz C. L., Hall J. E., Paine M. F., Tidwell R. R., Watkins P. B. Toxicol. Sci. 2012;130:416–426. doi: 10.1093/toxsci/kfs238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuita J. K., Wolf K. K., Murilla G. A., Liu Q., Mutuku J. N., Chen Y., Bridges A. S., Mdachi R. E., Ismail M. A., Ching S., Boykin D. W., Hall J. E., Tidwell R. R., Paine M. F., Brun R., Wang M. Z. PLoS Neglected Trop. Dis. 2013;7:e2230. doi: 10.1371/journal.pntd.0002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Wenzler T., Miller P. N., Wu H., Boykin D. W., Brun R., Wang M. Z. Antimicrob. Agents Chemother. 2014;58:4064–4074. doi: 10.1128/AAC.02605-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson W. D., Tanious F. A., Mathis A., Tevis D., Hall J. E., Boykin D. W. Biochimie. 2008;90:999–1014. doi: 10.1016/j.biochi.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansede J. H., Voyksner R. D., Ismail M. A., Boykin D. W., Tidwell R. R., Hall J. E. Xenobiotica. 2005;35:211–226. doi: 10.1080/00498250500087671. [DOI] [PubMed] [Google Scholar]
- Thuita J. K., Wang M. Z., Kagira J. M., Denton C. L., Paine M. F., Mdachi R. E., Murilla G. A., Ching S., Boykin D. W., Tidwell R. R., Hall J. E., Brun R. PLoS Neglected Trop. Dis. 2012;6:e1734. doi: 10.1371/journal.pntd.0001734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzler T., Yang S., Patrick D. A., Braissant O., Ismail M. A., Tidwell R. R., Boykin D. W., Wang M. Z., Brun R. Antimicrob. Agents Chemother. 2014;58:4452–4463. doi: 10.1128/AAC.02309-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick D. A., Ismail M. A., Arafa R. K., Wenzler T., Zhu X., Pandharkar T., Jones S. K., Werbovetz K. A., Brun R., Boykin D. W., Tidwell R. R. J. Med. Chem. 2013;56:5473–5494. doi: 10.1021/jm400508e. [DOI] [PubMed] [Google Scholar]
- Vanden Eynde J. J., Mayence A., Mottamal M., Bacchi C. J., Yarlett N., Kaiser M., Brun R., Huang T. L. Pharmaceuticals. 2016;9:20. doi: 10.3390/ph9020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G., Zhu W., Wang Y., Huang G., Byun S. Y., Choi G., Li K., Huang Z., Docampo R., Oldfield E., No J. H. ACS Infect. Dis. 2015;1:388–398. doi: 10.1021/acsinfecdis.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert I. H. J. Med. Chem. 2013;56:7719–7726. doi: 10.1021/jm400362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert I. H. Parasitology. 2014;141:28–36. doi: 10.1017/S0031182013001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price H. P., Menon M. R., Panethymitaki C., Goulding D., McKean P. G., Smith D. F. J. Biol. Chem. 2003;278:7206–7214. doi: 10.1074/jbc.M211391200. [DOI] [PubMed] [Google Scholar]
- Brand S., Cleghorn L. A., McElroy S. P., Robinson D. A., Smith V. C., Hallyburton I., Harrison J. R., Norcross N. R., Spinks D., Bayliss T., Norval S., Stojanovski L., Torrie L. S., Frearson J. A., Brenk R., Fairlamb A. H., Ferguson M. A., Read K. D., Wyatt P. G., Gilbert I. H. J. Med. Chem. 2012;55:140–152. doi: 10.1021/jm201091t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frearson J. A., Brand S., McElroy S. P., Cleghorn L. A., Smid O., Stojanovski L., Price H. P., Guther M. L., Torrie L. S., Robinson D. A., Hallyburton I., Mpamhanga C. P., Brannigan J. A., Wilkinson A. J., Hodgkinson M., Hui R., Qiu W., Raimi O. G., van Aalten D. M., Brenk R., Gilbert I. H., Read K. D., Fairlamb A. H., Ferguson M. A., Smith D. F., Wyatt P. G. Nature. 2010;464:728–732. doi: 10.1038/nature08893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand S., Norcross N. R., Thompson S., Harrison J. R., Smith V. C., Robinson D. A., Torrie L. S., McElroy S. P., Hallyburton I., Norval S., Scullion P., Stojanovski L., Simeons F. R., van Aalten D., Frearson J. A., Brenk R., Fairlamb A. H., Ferguson M. A., Wyatt P. G., Gilbert I. H., Read K. D. J. Med. Chem. 2014;57:9855–9869. doi: 10.1021/jm500809c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes M. L., Baell J. B., Kaiser M., Chatelain E., Moawad S. R., Ganame D., Ioset J. R., Avery V. M. PLoS Neglected Trop. Dis. 2012;6:e1896. doi: 10.1371/journal.pntd.0001896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling T., Mercer L., Don R., Jacobs R., Nare B. Int. J. Parasitol.: Drugs Drug Resist. 2012;2:262–270. doi: 10.1016/j.ijpddr.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njoroge M., Njuguna N. M., Mutai P., Ongarora D. S., Smith P. W., Chibale K. Chem. Rev. 2014;114:11138–11163. doi: 10.1021/cr500098f. [DOI] [PubMed] [Google Scholar]
- Sykes M. L., Avery V. M. J. Med. Chem. 2013;56:7727–7740. doi: 10.1021/jm4004279. [DOI] [PubMed] [Google Scholar]
- Tatipaka H. B., Gillespie J. R., Chatterjee A. K., Norcross N. R., Hulverson M. A., Ranade R. M., Nagendar P., Creason S. A., McQueen J., Duster N. A., Nagle A., Supek F., Molteni V., Wenzler T., Brun R., Glynne R., Buckner F. S., Gelb M. H. J. Med. Chem. 2014;57:828–835. doi: 10.1021/jm401178t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare S., Nagle A. S., Biggart A., Lai Y. H., Liang F., Davis L. C., Barnes S. W., Mathison C. J. N., Myburgh E., Gao M.-Y., Gillespie J. R., Liu X., Tan J. L., Stinson M., Rivera I. C., Ballard J., Yeh V., Groessl T., Federe G., Koh H. X. Y., Venable J. D., Bursulaya B., Shapiro M., Mishra P. K., Spraggon G., Brock A., Mottram J. C., Buckner F. S., Rao S. P. S., Wen B. G., Walker J. R., Tuntland T., Molteni V., Glynne R. J., Supek F. Nature. 2016;537:229–233. doi: 10.1038/nature19339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick D. A., Wenzler T., Yang S., Weiser P. T., Wang M. Z., Brun R., Tidwell R. R. Bioorg. Med. Chem. 2016;24:2451–2465. doi: 10.1016/j.bmc.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick D. A., Gillespie J. R., McQueen J., Hulverson M. A., Ranade R. M., Creason S. A., Herbst Z. M., Gelb M. H., Buckner F. S., Tidwell R. R. J. Med. Chem. 2017;60:957–971. doi: 10.1021/acs.jmedchem.6b01163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleghorn L. A., Albrecht S., Stojanovski L., Simeons F. R., Norval S., Kime R., Collie I. T., De Rycker M., Campbell L., Hallyburton I., Frearson J. A., Wyatt P. G., Read K. D., Gilbert I. H. J. Med. Chem. 2015;58:7695–7706. doi: 10.1021/acs.jmedchem.5b00596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodnala S. K., Lundback T., Sjoberg B., Svensson R., Rottenberg M. E., Hammarstrom L. G. Antimicrob. Agents Chemother. 2013;57:1012–1018. doi: 10.1128/AAC.01870-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews K. T., Fisher G., Skinner-Adams T. S. Int. J. Parasitol.: Drugs Drug Resist. 2014;4:95–111. doi: 10.1016/j.ijpddr.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. J., Kaiser M., Avery V. M. Antimicrob. Agents Chemother. 2015;60:1859–1861. doi: 10.1128/AAC.02116-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternack sD. A., Sharma A. I., Olson C. L., Epting C. L., Engman D. M. MBio. 2015;6:e01291-15. doi: 10.1128/mBio.01291-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witschel M., Rottmann M., Kaiser M., Brun R. PLoS Neglected Trop. Dis. 2012;6:e1805. doi: 10.1371/journal.pntd.0001805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Gonzalez R., Kuhlmann F. M., Galan-Rodriguez C., Madeira da Silva L., Saldivia M., Karver C. E., Rodriguez A., Beverley S. M., Navarro M., Pollastri M. P. PLoS Neglected Trop. Dis. 2011;5:e1297. doi: 10.1371/journal.pntd.0001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seixas J. D., Luengo-Arratta S. A., Diaz R., Saldivia M., Rojas-Barros D. I., Manzano P., Gonzalez S., Berlanga M., Smith T. K., Navarro M., Pollastri M. P. J. Med. Chem. 2014;57:4834–4848. doi: 10.1021/jm500361r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urich R., Grimaldi R., Luksch T., Frearson J. A., Brenk R., Wyatt P. G. J. Med. Chem. 2014;57:7536–7549. doi: 10.1021/jm500239b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oduor R. O., Ojo K. K., Williams G. P., Bertelli F., Mills J., Maes L., Pryde D. C., Parkinson T., Van Voorhis W. C., Holler T. P. PLoS Neglected Trop. Dis. 2011;5:e1017. doi: 10.1371/journal.pntd.0001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz R., Luengo-Arratta S. A., Seixas J. D., Amata E., Devine W., Cordon-Obras C., Rojas-Barros D. I., Jimenez E., Ortega F., Crouch S., Colmenarejo G., Fiandor J. M., Martin J. J., Berlanga M., Gonzalez S., Manzano P., Navarro M., Pollastri M. P. PLoS Neglected Trop. Dis. 2014;8:e3253. doi: 10.1371/journal.pntd.0003253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine W. G., Diaz-Gonzalez R., Ceballos-Perez G., Rojas D., Satoh T., Tear W., Ranade R. M., Barros-Alvarez X., Hol W. G., Buckner F. S., Navarro M., Pollastri M. P. ACS Infect. Dis. 2017;3:225–236. doi: 10.1021/acsinfecdis.6b00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S., Gillespie J. R., Kelley A. M., Napuli A. J., Zhang Z., Kovzun K. V., Pefley R. M., Lam J., Zucker F. H., Van Voorhis W. C., Merritt E. A., Hol W. G., Verlinde C. L., Fan E., Buckner F. S. Antimicrob. Agents Chemother. 2011;55:1982–1989. doi: 10.1128/AAC.01796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh C. Y., Kim J. E., Wetzel A. B., de van der Schueren W. J., Shibata S., Ranade R. M., Liu J., Zhang Z., Gillespie J. R., Buckner F. S., Verlinde C. L., Fan E., Hol W. G. PLoS Neglected Trop. Dis. 2014;8:e2775. doi: 10.1371/journal.pntd.0002775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katiyar S., Kufareva I., Behera R., Thomas S. M., Ogata Y., Pollastri M., Abagyan R., Mensa-Wilmot K. PLoS One. 2013;8:e56150. doi: 10.1371/journal.pone.0056150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera R., Thomas S. M., Mensa-Wilmot K. Antimicrob. Agents Chemother. 2014;58:2202–2210. doi: 10.1128/AAC.01691-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel G., Karver C. E., Behera R., Guyett P. J., Sullenberger C., Edwards P., Roncal N. E., Mensa-Wilmot K., Pollastri M. P. J. Med. Chem. 2013;56:3820–3832. doi: 10.1021/jm400349k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine W., Thomas S. M., Erath J., Bachovchin K. A., Lee P. J., Leed S. E., Rodriguez A., Sciotti R. J., Mensa-Wilmot K., Pollastri M. P. ACS Med. Chem. Lett. 2017;8:350–354. doi: 10.1021/acsmedchemlett.7b00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodring J. L., Patel G., Erath J., Behera R., Lee P. J., Leed S. E., Rodriguez A., Sciotti R. J., Mensa-Wilmot K., Pollastri M. P. MedChemComm. 2015;6:339–346. doi: 10.1039/C4MD00441H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G., Zhu W., Kim K., Byun S. Y., Choi G., Wang K., Cha J. S., Cho H. S., Oldfield E., No J. H. Antimicrob. Agents Chemother. 2015;59:7530–7539. doi: 10.1128/AAC.01873-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenortas E., Burri C., Shapiro T. A. Antimicrob. Agents Chemother. 1999;43:2066–2068. doi: 10.1128/aac.43.8.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomel S., Dubar F., Forge D., Loiseau P. M., Biot C. Bioorg. Med. Chem. 2015;23:5168–5174. doi: 10.1016/j.bmc.2015.03.029. [DOI] [PubMed] [Google Scholar]
- Hiltensperger G., Jones N. G., Niedermeier S., Stich A., Kaiser M., Jung J., Puhl S., Damme A., Braunschweig H., Meinel L., Engstler M., Holzgrabe U. J. Med. Chem. 2012;55:2538–2548. doi: 10.1021/jm101439s. [DOI] [PubMed] [Google Scholar]
- Hiltensperger G., Hecht N., Kaiser M., Rybak J. C., Hoerst A., Dannenbauer N., Muller-Buschbaum K., Bruhn H., Esch H., Lehmann L., Meinel L., Holzgrabe U. Antimicrob. Agents Chemother. 2016;60:4442–4452. doi: 10.1128/AAC.01757-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopsida M., Barron G. A., Bermano G., Kong Thoo Lin P., Goua M. Org. Biomol. Chem. 2016;14:9780–9789. doi: 10.1039/c6ob01850e. [DOI] [PubMed] [Google Scholar]
- Menzel T. M., Tischer M., Francois P., Nickel J., Schrenzel J., Bruhn H., Albrecht A., Lehmann L., Holzgrabe U., Ohlsen K. Antimicrob. Agents Chemother. 2011;55:311–320. doi: 10.1128/AAC.00586-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares J., Ouaissi A., Kong Thoo Lin P., Loureiro I., Kaur S., Roy N., Cordeiro-da-Silva A. ChemMedChem. 2010;5:140–147. doi: 10.1002/cmdc.200900367. [DOI] [PubMed] [Google Scholar]
- Oliveira J., Ralton L., Tavares J., Codeiro-da-Silva A., Bestwick C. S., McPherson A., Thoo Lin P. K. Bioorg. Med. Chem. 2007;15:541–545. doi: 10.1016/j.bmc.2006.09.031. [DOI] [PubMed] [Google Scholar]
- Graca N. A., Gaspar L., Costa D. M., Loureiro I., Thoo-Lin P. K., Ramos I., Roura M., Pruvost A., Pemberton I. K., Loukil H., MacDougall J., Tavares J., Cordeiro-da-Silva A. Antimicrob. Agents Chemother. 2016;60:2532–2536. doi: 10.1128/AAC.02490-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth M., Hoerr V., Glaser M., Ponte-Sucre A., Moll H., Stich A., Holzgrabe U. Bioorg. Med. Chem. Lett. 2007;17:1590–1593. doi: 10.1016/j.bmcl.2006.12.088. [DOI] [PubMed] [Google Scholar]
- Ponte-Sucre A., Bruhn H., Schirmeister T., Cecil A., Albert C. R., Buechold C., Tischer M., Schlesinger S., Goebel T., Fuss A., Mathein D., Merget B., Sotriffer C. A., Stich A., Krohne G., Engstler M., Bringmann G., Holzgrabe U. Parasitol. Res. 2015;114:501–512. doi: 10.1007/s00436-014-4210-4. [DOI] [PubMed] [Google Scholar]
- Nanavaty V., Lama R., Sandhu R., Zhong B., Kulman D., Bobba V., Zhao A., Li B., Su B. PLoS One. 2016;11:e0146289. doi: 10.1371/journal.pone.0146289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K. J., Shapiro T. A. J. Infect. Dis. 2013;208:489–499. doi: 10.1093/infdis/jit179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro J. C., Hills T., Senisterra G., Wernimont A. K., Mackenzie C., Norcross N. R., Ferguson M. A., Wyatt P. G., Gilbert I. H., Hui R. PLoS Neglected Trop. Dis. 2013;7:e2492. doi: 10.1371/journal.pntd.0002492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini G., Battistuzzi G. Bioorg. Med. Chem. Lett. 2015;25:462–465. doi: 10.1016/j.bmcl.2014.12.048. [DOI] [PubMed] [Google Scholar]
- Hofer A., Steverding D., Chabes A., Brun R., Thelander L. Proc. Natl. Acad. Sci. U. S. A. 2001;98:6412–6416. doi: 10.1073/pnas.111139498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijolek A., Hofer A., Thelander L. J. Biol. Chem. 2007;282:11858–11865. doi: 10.1074/jbc.M611580200. [DOI] [PubMed] [Google Scholar]
- Conti P., Pinto A., Wong P. E., Major L. L., Tamborini L., Iannuzzi M. C., De Micheli C., Barrett M. P., Smith T. K. ChemMedChem. 2011;6:329–333. doi: 10.1002/cmdc.201000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S. M., Purmal A., Pollastri M., Mensa-Wilmot K. Sci. Rep. 2017;7:40565. doi: 10.1038/srep40565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuzu I. U., Chineme C. N. J. Ethnopharmacol. 1990;30:307–313. doi: 10.1016/0378-8741(90)90109-7. [DOI] [PubMed] [Google Scholar]
- Suzuki M., Tung N. H., Kwofie K. D., Adegle R., Amoa-Bosompem M., Sakyiamah M., Ayertey F., Owusu K. B.-A., Tuffour I., Atchoglo P., Frempong K. K., Anyan W. K., Uto T., Morinaga O., Yamashita T., Aboagye F., Appiah A. A., Appiah-Opong R., Nyarko A. K., Yamaoka S., Yamaguchi Y., Edoh D., Koram K., Ohta N., Boakye D. A., Ayi I., Shoyama Y. Bioorg. Med. Chem. Lett. 2015;25:3030–3033. doi: 10.1016/j.bmcl.2015.05.003. [DOI] [PubMed] [Google Scholar]
- Kwofie K. D., Tung N. H., Suzuki-Ohashi M., Amoa-Bosompem M., Adegle R., Sakyiamah M. M., Ayertey F., Owusu K. B.-A., Tuffour I., Atchoglo P., Frempong K. K., Anyan W. K., Uto T., Morinaga O., Yamashita T., Aboagye F., Appiah A. A., Appiah-Opong R., Nyarko A. K., Yamaguchi Y., Edoh D., Koram K. A., Yamaoka S., Boakye D. A., Ohta N., Shoyama Y., Ayi I. Antimicrob. Agents Chemother. 2016;60:3283–3290. doi: 10.1128/AAC.01916-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matovu E., Seebeck T., Enyaru J. C. K., Kaminsky R. Microbes Infect. 2001;3:763–770. doi: 10.1016/s1286-4579(01)01432-0. [DOI] [PubMed] [Google Scholar]
- Yaro M., Munyard K. A., Stear M. J., Groth D. M. Vet. Parasitol. 2016;225:43–52. doi: 10.1016/j.vetpar.2016.05.003. [DOI] [PubMed] [Google Scholar]
- Gillingwater K., Kunz C., Braghirolia B. C., Boykin D. W., Tidwell R. R., Brun R. Antimicrob. Agents Chemother. 2017;61:e02356-16. doi: 10.1128/AAC.02356-16. [DOI] [PMC free article] [PubMed] [Google Scholar]