

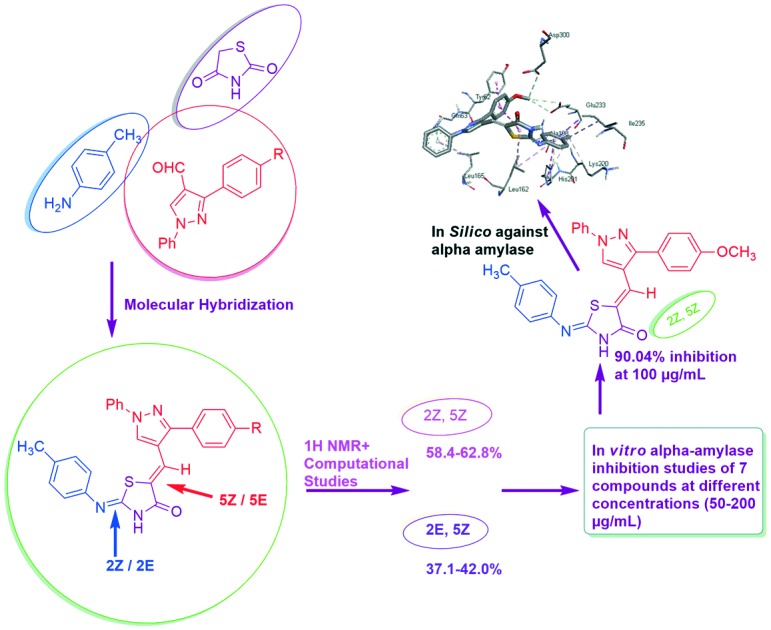
 The results showed that some of the synthesized compounds exhibited significant inhibitory activities. Compound 5a at 100 μg mL–1 concentration showed a remarkable inhibition of 90.04%.
The results showed that some of the synthesized compounds exhibited significant inhibitory activities. Compound 5a at 100 μg mL–1 concentration showed a remarkable inhibition of 90.04%.
Abstract
Postprandial hyperglycemia can be controlled by delaying the absorption of glucose resulting from carbohydrate digestion. α-Amylase is the initiator of the hydrolysis of polysaccharides, and therefore developing α-amylase inhibitors can lead to development of new treatments for metabolic disorders like diabetes mellitus. In the present work, we set out to rationally develop α-amylase inhibitors based on the thiazolidine-4-one scaffold. The structures of all these newly synthesized hybrids were confirmed by spectroscopic analysis (IR, 1H-NMR, MS). The appearance of two sets of signals for some protons in 1H NMR revealed the existence of a mixture of 2E,5Z (37.1–42.0%) and 2Z,5Z isomers (58.4–62.8%), which was further supported by DFT studies. All the newly synthesized compounds have potential inhibitory properties as revealed through in vitro α-amylase inhibition activity. Compound 5a at 100 μg mL–1 concentration showed a remarkable inhibition of 90.04%. In vitro α-amylase inhibition was further supported by docking studies of compound 5a against the active site of human pancreatic α-amylase (PDB ID: ; 2QV4). The docking studies revealed that the bonding interactions found between 5a and human pancreatic α-amylase are similar to those responsible for α-amylase inhibition by acarbose.
Introduction
DM (Diabetes mellitus) is a metabolic disorder characterized by chronic hyperglycemia or increased blood glucose levels with disturbances in carbohydrate, fat and protein metabolism resulting from absolute or relative lack of insulin secretion.1 Diabetes, being one of the most common global diseases, affects approximately 200 million individuals worldwide.2 The management of the blood glucose level as close to normal as possible in patients with diabetes mellitus is the most challenging task. Therefore, enzymes that regulate gluconeogenic or glycogenolytic pathways are key biological targets for therapeutic interventions. Out of several enzymes known, α-amylase is an important key enzyme responsible for carbohydrate digestion. So inhibitors of α-amylase can effectively retard the digestion and assimilation at the early steps of starch digestion, and thus succeed in the significant delay of postprandial hyperglycemia and have a beneficial effect on insulin resistance.3 α-Amylase is considered to be one of the best targets for the development of type II diabetes therapeutic agents due to its ability to catalyze the hydrolysis of α-(1,4)-glycosidic linkages in starch.4,5 Acarbose and voglibose are two known inhibitors of α-amylase which are in clinical use nowadays. However, they often cause severe gastrointestinal side effects such as abdominal pain, flatulence, and diarrhea.6 In recent years, α-amylase has been a point of interest for the development of novel anti-obesity and antidiabetic drugs.7
Thiazolidine-4-one represents one important class of compounds incorporated in several anti-diabetic drugs.8 This compound exhibits a wide range of pharmacological activities such as anti-microbial,9 anti-tubercular activity,10 anti-inflammatory,11 anti-cancer,12 anti-oxidant,13 anti-hyperglycemic agents14,15 and anti-viral.16 There is a wide scope for the different pharmacological properties associated with this molecule due to the presence of an active methylene position in this compound. Similarly, pyrazole also represents an important class of compounds not only for being of theoretical interest but also for its anti-inflammatory, analgesic, anti-tumor, anti-hypertensive, anti-pyretic, sedatives, anti-bacterial and anti-diabetic activities.17–23
In the light of these facts and in continuation of our interest in the synthesis of heterocycles containing a multi-structure for biological activity, we herein report a new class of thiazolidin-4-one linked pyrazoles to see the additive effect of these rings towards α-amylase inhibition.
Results and discussion
Chemistry
The target compounds 5-((3-(aryl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino) thiazolidin-4-one (5a–g) were synthesized by the following synthetic protocol. Firstly, reaction of p-tolyl thiourea (2) and ethyl bromoacetate was carried out to synthesise 2-(p-tolylimino) thiazolidin-4-one (3) which is a key reactant for the development of novel hybrids. Compound 3 was further subjected to Knoevenagel condensation with 3-(aryl)-1-phenyl-1H-pyrazole-4-carbaldehyde24 (4a–g) using a catalytic amount of piperidine in refluxing ethanol for 10–12 h. The progress of the reaction was monitored by TLC using petroleum ether : ethylacetate (60 : 40, v/v). After completion of the reaction, the reaction mixture was cooled down at room temperature and the solid so obtained was filtered to yield 5-((3-(aryl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino) thiazolidin-4-one (5a–g) in 72–85% yield (Scheme 1, Table 1). The formation of the compounds (5a–g) was confirmed by using IR, 1H NMR, and mass and elemental analysis.
Scheme 1. Synthesis of 5-((3-(aryl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino) thiazolidin-4-one 5a–g.

Table 1. Synthesis of 5-((3-(aryl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino) thiazolidin-4-one 5a–g.
| S. No. | Compound | –R | Yield (%) | M.pt. °C |
| 1 | 5a | –OCH3 | 85 | 298–300 |
| 2 | 5b | –CH3 | 76 | 270–275 |
| 3 | 5c | –H | 83 | 263–265 |
| 4 | 5d | –Cl | 78 | 292–295 |
| 5 | 5e | –F | 72 | 288–290 |
| 6 | 5f | –Br | 74 | 290–292 |
| 7 | 5g | –NO2 | 85 | 288–290 |
Characterization of compounds and their configurational studies
The structures of these hybrids were ascertained by means of IR, 1H NMR and mass spectral data. The absorption signals corresponding to N–H stretching appeared at 3050–3127 cm–1 and C O stretching appeared in the region of 1700–1720 cm–1 of the thiazolidinone ring, respectively. The presence of a band in the range 1646–1691 cm–1 reveals the formation of an exocyclic C C bond at the 5th position. It is assumed that for compound 5, the restricted rotation about the (C C) linkage as well as the imine (C N) linkage led to the formation of 4 possible configurations which are 2Z,5E; 2E,5E; 2E,5Z and 2Z,5Z, as shown in Fig. 1 (1 to 4).
Fig. 1. Four possible isomers of 5.

A Literature survey reveals that 2-arylimino-4-thiazolidinone 3 exists as a mixture of E and Z isomers of the imino form in dimethylsulphoxide-d6 but predominantly in the Z form.25 The 1H and 13C NMR data of compound 3 also contain two set of signals for N–H, –CH2, and aromatic protons, as shown in Fig. 2.
Fig. 2. Characteristic peaks assigned in the 1H NMR spectrum of compound 3.

In the 1H NMR spectra of compounds 5a–g, the signal for the olefinic proton (H7) was detected at δ 7.24–7.57 ppm, being deshielded by the adjacent C O, which is a higher chemical shift value than is expected for E isomers.26 In the E isomer, due to the lesser deshielding effect of sulphur, such a proton should resonate at lower delta values.27 Therefore we omit the probability of 2Z,5E and 2E,5E isomers. The NMR (1H) spectra of compounds 5a–g also gave two sets of signals which confirmed the existence of two configurational isomers in dimethylsulphoxide-d6 (2E,5Z and 2Z,5Z) and according to the literature25–27 the predominance was assigned to the 2Z,5Z isomer.
Compound 5a was taken as a test sample to study the configurational isomers by means of IR, 1H-NMR, mass, and 1H–1H COSY (Fig. 3 and 4). The disappearance of the signal of the methylene proton at δ 3.80 and the appearance of a signal in the range of δ 7.24–7.57 due to the C C–H olefinic hydrogen confirmed the formation of an exocyclic C C bond. The numeration of the structure is given specifically for NMR analysis only. The amide proton (O C–N–H) observed at δ 12.00–12.53 ppm showed that substitution is on the 2nd position of thiazolidin-4-one which is in agreement with a lactam proton, since an aryl amine proton appears at much higher field.24 Two singlets at δ 2.30 (39.94%) and δ 2.31 (60.55%) were observed due to the methyl protons of the aryl ring of imine. Signals at δ 8.52 (61.72%) and δ 8.61 (39.21%) in the form of two sharp singlets were observed for the proton of pyr-H. Duplication of signals has also been observed for protons showing resonance in the aromatic region associated with the proton of the E and Z geometrical isomers of the compound. Two protons, H28/32, exhibit two doublets at δ 7.92 (59.31%) and δ 7.96 (40.74%) with a coupling constant 7.6 and 8.0 Hz, respectively. Two doublets δ 6.97 (J = 8.0 Hz) (40.82%) and δ 7.19–7.21 (59.17%) are due to two protons, H10/12. In the ESI-MS mass spectra of compound 5a, the m/z value was observed at 466.85. In order to understand the effect of solvent on configurational isomer distribution, the NMR of compound 5a was obtained in TFA. Interestingly the % of the 2Z,5Z isomer has been increased compared to the 2E,5Z isomer in a ratio of 95 : 5 (ESI† file). This may be due to the solvation and stability of different configurations in different solvents. The chemical shift of olefinic protons increased from δ 7.43 to δ 8.06. Moreover, the protons of OCH3 and CH3 were found downfield. This may be due to the protonation of nitrogen and oxygen.
Fig. 3. Characteristic peaks for isomers and H–H COSY correlation of 5a.

Fig. 4. Characteristic peaks and comparative 1H NMR of compound 5a in DMSO and TFA.

In the 1H–NMR spectrum of 2-(p-tolylimino)-5-(((3-aryl)-1-phenyl-1H-pyrazol-4-yl)methylene)thiazolidin-4-one (5a–g), two singlets were observed for –CH3(δ 2.11–2.31) and pyr-H (δ 8.18–8.64) whereas other protons of aromatic rings also showed two sets of signals associated with the protons of the 2E,5Z and 2Z,5Z isomers of the compounds. The ratio of 2E,5Z to 2Z,5Z isomers was calculated by means of the NMR integration ratio of respective protons in proton NMR analysis. The integration of the two singlets of methyl (–CH3) and pyrazole protons were used to calculate the relative abundance of each isomer at 25 °C. The percentage of 2E,5Z and 2Z,5Z isomers was found in the range of 37.1–42.0% and 58.4–62.8%, respectively, as shown in Table S1.†
Computational studies
5-((3-(4-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino)thiazolidin-4-one (5a) can exist in 4 possible configurations which are 2Z,5E; 2E,5E; 2E,5Z and 2Z,5Z, as shown in Fig. 1 (1 to 4). In this work, the 3-dimensional structures of the 2-(p-tolylimino)-5-(((3-aryl)-1-phenyl-1H-pyrazol-4-yl)methylene)thiazolidin-4-one configurational isomer in their neutral state were obtained by the DFT28 approach utilizing Becke's three-parameter functional29 with the Vosko et al.30 local and Lee et al.31 non-local correlation, abbreviated as B3LYP1(VWN formula 1 RPA in B3LYP). The rotations about the C2–N6 (Fig. 1) (1st and 2nd) and C5–C7 (Fig. 1) (3rd and 4th) bonds, respectively, were taken into account. We have calculated the rotational energy barrier by steps of 10°. All four isomers presented in Fig. 1 were considered in our study. All the molecules were geometry-optimized until the root-mean square (RMS) gradient value was smaller than 10–6 a.u. To structurally characterize the molecule in detail, a systematic investigation of its potential energy surface was undertaken at the DFT (B3LYP1)/6-311G level of approximation.32 Later, using the surface data generated from Firefly software, the distribution of charge in a molecule was calculated. To obtain a 3D plot of the MEP, the electrostatic potential cube file was calculated from the total SCF density. The contour maps of the electrostatic potential were then drawn using a distance between grid points of 0.02 Å and an iso value of 0.0004. The Firefly software was used to calculate the electrostatic potential maps and surfaces as the distribution of the potential energy of a unit positive charge in a given molecular space, with the resolution controlled by the grid density. The vibrational wave numbers were calculated at the DFT (B3LYP1)/6-311G level of approximation. It is well known in the quantum chemical literature that among the available functionals, the B3LYP1 functional yields a good description of harmonic vibrational wave numbers for small and medium-sized molecules. All calculations were performed using the Firefly software. The visualizations were prepared by the use of the MASK.33 By considering the calculated relative energy barriers (DFT) for the 2Z,5E; 2E,5E; 2E,5Z and 2Z,5Z configurations, the most stable isomers are 2Z,5Z and 2E,5Z, due to the highest energy difference (Table S2†). However, the minimum difference of energy was observed in 2Z,5Z and 2E,5Z isomers i.e. (2.8 kcal per mole), which suggests that these two configurations, 2Z,5Z and 2E,5Z, may not be experimentally accessible as isolate species.
Biological studies
In vitro α-amylase inhibition
All the synthesized compounds (5a–g) and the standard drug were explored for their in vitro α-amylase inhibition at different concentrations (50–200 μg mL–1), as shown in Table 2. All the compounds showed good % inhibition of α-amylase when compared with the standard drug acarbose. Compounds 5b and 5e were found to be more potent among all the synthesized compounds when explored at the concentration of 50 μg mL–1. Compound 5e shows 77.12% inhibition followed by 5b with 79.24% inhibition. There was a significant rise in % inhibition when the concentration has been changed to 100 μg ml–1 from 50 μg mL–1. Among all the compounds, 5b shows 83.72% inhibition followed by 5a which showed 90.04% inhibition at 100 μg ml–1. Apart from this, 5a shows a sudden rise in % inhibition from 75.94 to 90.04% on increasing concentration to 100 μg mL–1 from 50 μg mL–1. Inspired by the results obtained at 100 μg mL–1 concentration, all the synthesized compounds were further screened for in vitro α-amylase inhibition at 200 μg mL–1. Compounds 5a, 5b, 5c and 5f exhibited a linear rise in % inhibition, but a reduction in % inhibition has been observed for compounds 5d, 5e and 5g owing to their lower solubility at a concentration of 200 μg mL–1. Among compounds showing enhanced % inhibition, compound 5a showed % inhibition of 92.5% followed by 5f with % inhibition of 97.4% at a concentration of 200 μg mL–1. A comparative graph of % inhibition of compounds 5a–g with the standard drug acarbose is shown in Fig. 5.
Table 2. α-Amylase inhibition activity of compounds 5a–g.
| Compound | Concentration (μg mL–1) | OD at 595 nm | Residual activity | % inhibition |
| 5a | 50 | 0.102 | 24.05 | 75.94 |
| 100 | 0.042 | 9.90 | 90.04 | |
| 200 | 0.032 | 7.54 | 92.15 | |
| 5b | 50 | 0.088 | 20.75 | 79.24 |
| 100 | 0.069 | 16.27 | 83.72 | |
| 200 | 0.068 | 16.04 | 83.97 | |
| 5c | 50 | 0.211 | 49.76 | 48.08 |
| 100 | 0.078 | 18.39 | 81.60 | |
| 200 | 0.066 | 15.73 | 84.27 | |
| 5d | 50 | 0.208 | 49.05 | 50.94 |
| 100 | 0.080 | 18.87 | 81.13 | |
| 200 | 0.106 | 24.97 | 75.03 | |
| 5e | 50 | 0.097 | 28.87 | 77.12 |
| 100 | 0.078 | 18.39 | 81.60 | |
| 200 | 0.093 | 21.95 | 78.05 | |
| 5f | 50 | 0.203 | 47.87 | 52.12 |
| 100 | 0.115 | 27.12 | 72.87 | |
| 200 | 0.011 | 2.59 | 97.40 | |
| 5g | 50 | 0.148 | 11.79 | 65.09 |
| 100 | 0.050 | 34.90 | 88.20 | |
| 200 | 0.178 | 41.98 | 58.00 | |
| Acarbose | 50 | 0.218 | 51.52 | 48.58 |
| 100 | 0.196 | 46.23 | 53.77 | |
| 200 | 0.154 | 36.32 | 63.68 |
Fig. 5. Comparative analysis of % inhibition of compounds 5a–g.

Molecular docking studies
The aim of specific structural modeling and accurate prediction of activity can be achieved only by molecular docking studies.34 Interactions between inhibitors and the active site of the target protein can be explored using molecular docking studies. The above results showed that all the synthesized molecules were stronger inhibitors of alpha-amylase compared to acarbose. Although compound 5f showed maximum inhibition at 200 μg mL–1, its % inhibition at 100 μg mL–1 was less than that of compound 5a which showed a maximum inhibition of 90.04% at a concentration of 100 μg mL–1. After comparison of their molar masses, it has been revealed that a lesser dose of 5a was required for maximum inhibition of α-amylase owing to its lower molar mass. Therefore, to ascertain the binding conformation and interactions responsible for the activity, docking simulation of compound 5a was performed against the active site of human pancreatic alpha-amylase (PDB ID: ; 2QV4). The docked conformation of 5a with the highest binding affinity is shown in Fig. 6.
Fig. 6. Interactions (dashed lines) of compound 5a with active site residues of human pancreatic alpha-amylase.

It can be noticed from Fig. 6 that mainly hydrophobic interactions are responsible for anchoring of the compound 5a. The methoxy phenyl ring of the compound was stacked against the hydroxyl phenyl ring of Tyr62 by pi–pi interactions. The methyl phenyl ring attached to the thiazolidinone moiety created T-shaped Pi–Pi interactions with His201. The methyl group was also involved in pi–alkyl interactions with His201. Thiazolidinone, methyl phenyl and phenyl rings were also involved in pi–alkyl interactions with Leu162, Ala188 and Leu165. Gln63 and His201 formed pi–hydrogen bonds with phenyl and methyl phenyl rings, respectively, by acting as hydrogen bond donors. The methoxy group formed carbon hydrogen bonds with Glu233 and Asp300. These two residues (Glu233 and Asp300) are reported to act as catalytic residues in hydrolytic reactions of alpha-amylase.35 Further, Tyr62 has been shown as an important residue for binding of some chalcones to the active site of alpha-amylase.36 Also, Tyr62, Gln63, Glu233 and Asp300 form hydrogen bonding interactions with acarbose (Fig. 7). All these facts show that binding of compound 5a to these active site residues might be the cause of alpha-amylase inhibitory activity. In Fig. 8, compound 5a is shown along with the co-crystalized ligand acarbose in the active site of alpha-amylase.
Fig. 7. Conformation view image of QV4 in 2QV4 .

Fig. 8. Compound 5a along with the co-crystalized ligand acarbose in the active site of human pancreatic alpha-amylase (shown as surface).

Experimental
Structures of all the compounds were identified by their spectral data. Silica gel 60 F254 plates (precoated aluminium plates) from Merck were used to monitor the reaction progress. All the melting points were determined in open glass capillary tubes and are uncorrected. IR spectra were obtained using a Cary 660 Agilent IR spectrophotometer and the values are expressed as νmax cm–1. The 1H and 13C spectra were recorded on Bruker (Avance-II) at 400 MHz and 100 MHz in DMSO-d6 and trifluoroacetic acid using tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ) and coupling constants (J) are expressed in ppm and Hz, respectively. All chemicals used were supplied by Sigma–Aldrich and Merck chemical companies. The mass spectrum was recorded using Waters Micromass Q-Tof Micro. 1-Phenyl-3-aryl-1H-pyrazole-4-carbaldehydes and 1-p-tolylthiourea were synthesized using a reported procedure.37,38
Synthesis of 2-(p-tolylimino)thiazolidin-4-one (3)38
It was prepared by a three-component reaction of 1-p-tolylthiourea (1) (1.0 mmol), ethyl bromoacetate (1.0 mmol) and sodium acetate (2.0 mmol) in glacial acetic acid under refluxing conditions for 2 h. After completion of reaction, the reaction was quenched using ice and the solid so formed was filtered under suction and recrystallized from ethanol. Yield 80%; M.pt: 167–170 °C; 1H NMR (400 MHz, DMSO, d6 ppm): δ 2.27 (s, 3H), 3.96 (d, 2H, J = 18.8 Hz), 6.92 (d, J = 7.2, 2H, Ar), 7.18 (d, J = 7.2 Hz, 2H, Ar), 7.59 (d, J = 7.6, 2H, Ar), 11.10 & 11.66 (2s, NH); 13C NMR (400 MHz, DMSO, d6, ppm): 20.9, 21.4, 35.6, 38.8, 120.6, 122.1, 129.8, 130.2, 134.3, 134.5, 136.8, 143.2, 177.6, 178.2, 188.5.
Synthesis of 5-((3-(aryl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino)thiazolidin-4-one (5a–g)38
2-Arylimino-thiazolidin-4-ones 1 (0.5 mmol) and 1-phenyl-3-(p-substituted phenyl)-1H-pyrazole-4-carbaldehydes 2 (0.6 mmol) were dissolved in absolute ethanol. Piperidine (0.5 mmol) was added to the reaction mixture and the reaction mixture was stirred for 8 h at 60 °C until precipitates formed. Then the mixture was cooled to room temperature, and the precipitates formed were filtered and washed with absolute ethanol to yield the final compounds 5a–g in good to excellent yield.
5-((3-(4-Methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino)thiazolidin-4-one (5a)
Yield 85%, M.pt. 298–300 °C; IR (vmax cm–1, KBr): 3070 (N–H lactam), 3032 (C–H str. of aromatic), 1720 (C O lactam), 1655 (C C), 1574 (C N); 1H NMR (400 MHz, DMSO, d6, ppm): δ 2.30 & 2.31 (2s, 3H, –CH3), 3.84 (s, 3H, –OCH3), 6.97 (d, J = 8.0 Hz, 2H, H10/H12), 7.11–7.14 (m, 2H, H24/H26), 7.19–7.22 (m, 4H, H9/H13, H10/H12), 7.35–7.43 (m, 2H, H7, H30), 7.49–7.64 (m, 6H, H9/H13, H23/H27, H29/H31), 7.92 (d, J = 7.6 Hz, 2H, H28/H32), 7.96 (d, J = 8.0 s Hz, 2H, H28/H32), 8.52 & 8.61 (2s, 2H, H17), 11.95 (broad s, 1H, NH); ESI-MS (m/z): 466.85; anal. calc. C27H22N4O2S: C, 69.51; H, 4.75; N, 12.01; O, 6.86; S, 6.87; Found: C, 69.45; H, 4.71; N, 11.97; O, 6.84; S, 6.85.
5-((3-(4-Methylphenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino)thiazolidin-4-one (5b)
Yield 76%; M.pt. 270–275 °C; IR (vmax cm–1, KBr): 3114 (N–H lactam), 3006 (C–H str. of aromatic), 1700 (C O lactam), 1646 (C C), 1596 (C N); 1H NMR (400 MHz, DMSO, d6, ppm): δ 2.30 & 2.31 (2s, 3H, –CH3), 2.40 (s, 1H, –CH3), 6.96 (d, 2H, J = 7.6 Hz, H10/H12), 7.19–7.23 (m, 4H, H9/H13, H10/H12), 7.36–7.43 (m, 4H, H24/H26, H7, H30), 7.50–7.64 (m, 6H, H9/H13, H23/H27, H29/H31), 7.93 (d, J = 8 Hz, 2H, H28/H32), 7.97 (d, 2H, J = 7.6 Hz, H28/H32), 8.54 & 8.64 (2s, 1H, H17), 12.08 (broad s, 1H, NH); Calc. For C27H22N4OS: C, 71.98; H, 4.92; N, 12.44; O, 3.55; S, 7.12. Found: C, 71.89; H, 4.82; N, 12.39; O, 3.45; S, 7.09.
5-((1-phenyl-3-p-tolyl-1H-pyrazol-4-yl)methylene)-2-(p-phenylimino)thiazolidin-4-one (5c)
Yield 83%; M.pt: 263–265 °C; IR (vmax cm–1, KBr): 3050 (N–H lactam), 3031 (C–H str. of aromatic), 1716 (C O lactam), 1650 (C C), 1535 (C N). 1H NMR (400 MHz, DMSO, d6, ppm): δ 2.11 & 2.13 (2s, 3H, –CH3), 6.74 (d, 2H Hz, J = 8.4 Hz, H10/H12), 6.93–6.98 (m, 4H, H9/H13, H10/H12), 7.02–7.19 (m, 2H, H25, H30), 7.24–7.35 (m, 5H, H29/H31, H24/H26, H7), 7.38–7.48 (m, 4H, H9/H13, H23/H27), 7.66 (d, J = 8.0 Hz, 2H, H28/H32), 7.72 (d, 2H, J = 8.4 Hz, H28/H32), 8.180 & 8.309 (2s, 1H, H17); anal. calc. For C26H20N4OS: C, 71.54; H, 4.62; N, 12.83; O, 3.67; S, 7.35. Found: C, 71.45; H, 4.60; N, 12.79; O, 3.60; S, 7.29.
5-((3-(4-Chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino)thiazolidine-4-one (5d)
Yield 78%; M.pt: 295–298 °C; IR (vmax cm–1, KBr): 3072 (N–H lactam), 3029 (C–H str. of aromatic), 1718 (C O lactam), 1656 (C C), 1598 (C N); 1H NMR (400 MHz, DMSO, d6 ppm): δ 2.30 & 2.31 (2s, 3H, –CH3), 6.93–7.66 (m, 12H, H10/H12, H9/H13, H24/H26, H7, H30, H23/H27, H29/H31), 7.88 (d, J = 8.0 Hz, 2H, H28/H32), 7.93 (d, 2H, J = 7.6 Hz, H28/H32), 8.47 & 8.58 (2s, 1H, H17); anal. calc. For C26H19ClN4OS: C, 66.30; H, 4.07; Cl, 7.53; N, 11.90; O, 3.40; S, 6.81. Found: C, 66.21; H, 4.01; Cl, 7.45; N, 11.85; O, 3.35; S, 6.75.
5-((3-(4-Fluorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino)thiazolidine-4-one (5e)
Yield 72%; M.pt: 288–290 °C; IR (vmax cm–1, KBr): 3127 (N–H lactam), 3006 (C–H str. of aromatic), 1706 (C O lactam), 1664 (C C), 1598 (C N); 1H NMR (400 MHz, DMSO, d6, ppm): δ 2.30 & 2.31 (2s, 3H, –CH3), 6.93–7.88 (m, 14H, H10/H12, H9/H13, H24/H26, H7, H30, H28/H32, H23/H27, H29/H31), 8.42 & 8.54 (2s, 1H, H17); anal. calcd. For C26H19FN4OS: C, 68.71; H, 4.21; F, 4.18; N, 12.33; O, 3.52; S, 7.05. Found: C, 68.69; H, 4.14; F, 4.08; N, 12.30; O, 3.45; S, 6.98.
5-((3-(4-Bromophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino)thiazolidine-4-one (5f)
Yield 74%; M.pt: 290–292 °C; IR (vmax cm–1, KBr): 3064 (N–H lactam), 3035 (C–H str. of aromatic), 1720 (C O lactam), 1654 (C C), 1596 (C N); 1H NMR (400 MHz, DMSO, d6, ppm): δ 2.30 & 2.31 (2s, 3H, –CH3), 6.93 (d, J = 8.0 Hz, 2H, H10/H12), 7.11–7.18 (m, 4H, H9/H13, H10/H12), 7.32–7.43 (m, 2H, H30, H7), 7.46–7.72 (m, 8H, H24/H26, H9/H13, H23/H27, H29/H31), 7.87 (d, J = 8.0 Hz, 2H, H28/H32), 7.92 (d, J = 8.0 Hz, 2H, H28/H32), 8.44 & 8.56 (2s, 1H, H17); anal. calc. For C26H19BrN4OS: C, 60.59; H, 3.72; Br, 15.50; N, 10.87; O, 3.10; S, 6.22. Found: C, 60.55; H, 3.69; Br, 15.45; N, 10.81; O, 3.01; S, 6.19.
5-((3-(4-Nitrophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino)thiazolidine-4-one (5g)
Yield 85%; M.pt: 288–290 °C; IR (vmax cm–1, KBr): 3072 (N–H lactam), 3037 (C–H str. of aromatic), 1716 (C O lactam), 1691 (C C), 1600 (C N); 1H NMR (400 MHz, DMSO, d6, ppm): δ 2.30 & 2.31 (s, 3H, –CH3), 6.91 (d, J = 8.0 Hz, 2H, H10/H12), 7.16 (d, J = 8.0 Hz, 2H, H9/H13, H10/H12), 7.34–7.41 (m, 1H, H30), 7.48–7.57 (m, 3H, H29/H31, H7), 7.62–7.64 (d, 2H, H9/H13, J = 8.4 Hz), 7.87–7.91 (m, 2H, H27/H31), 7.93–7.96 (m, 2H, H28/H32), 8.35–8.39 (m, 2H, H24/H26); 8.49 & 8.61 (2s, 1H, H17); anal. calc. For C26H19N5O3S: C, 64.85; H, 3.98; N, 14.54; O, 9.97; S, 6.66. Found: C, 64.81; H, 3.87; N, 14.43; O, 9.91; S, 6.60.
Inhibition assay for α-amylase activity
A stock solution of 10 mg/10 mL concentration was prepared by using DMSO solvent. The activity of amylase was assayed with different concentrations (50, 100, 200 μg mL–1) of the sample with control and a reagent solution without the test sample was used as the control. Starch solution (1% w/v) or (0.5% w/v) was prepared by stirring and boiling 0.5 g of soluble potato starch in 50 mL of deionized water for 15 minutes. The enzyme solution (1 unit per mL) was prepared by mixing 100 mg in 100 mL of 20 mM sodium phosphate buffer (pH 6.9). The color reagent was a solution containing 96 mM 3,5-dinitrosalicylic acid (DNSA) (20 mL), 5.31 M sodium potassium tartrate in 2 M NaOH (8 mL) and deionized water (12 mL). Acarbose was used as a standard at the concentration of 1 mg mL–1. 100 μl of test solution and 100 μL of enzyme solution were mixed in vials and incubated at 25 °C for 30 min. To this mixture 100 μL of the color reagent was added and the mixture was heated in a water bath at 85 °C for 15 min. After that, the reaction mixture was removed from the water bath, cooled and the absorbance value determined at 595 nm. Individual blanks were prepared for correcting the background absorbance. A control experiment was conducted in the same manner by replacing the drug sample with 1 mL DMSO. The inhibition percentage of α-amylase was calculated using a formula.39 The enzyme activity was calculated and the percentage of inhibition shown in the table.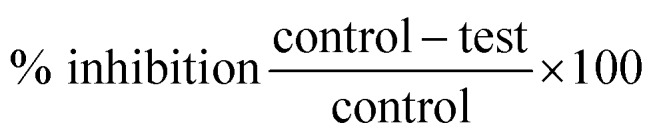
Docking simulations
The 3D structure of compound 5a was obtained and optimized with MarvinSketch.40 The enzyme alpha-amylase (PDB ID:; 2QV4) was downloaded from the Brookhaven Protein Data Bank (; http://www.rcsb.org/pdb). The preparation of protein was performed with UCSF Chimera41 and AutoDock Tools42 using a reported procedure.43 The docking simulation was performed using the well-established AutoDock Vina program.44 The validation of the docking protocols was done following a reported technique45 and these protocols were adopted for carrying out docking studies. The search space for docking was centered at x = 11.0976587428, y = 47.4125222511, and z = 26.0545925088 with size x = 25.0, y = 25.0 and z = 25.0. The exhaustiveness was set to be 100. The results were visualized with Discovery Studio46 and PyMol.47
Conclusion
In conclusion, the present study describes the synthesis of seven novel 5-((3-(aryl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino)thiazolidin-4-one (5a–g) compounds by condensation of 1-phenyl-3-(aryl)-1H-pyrazole-4-carbaldehyde (4a–g) with 2-(p-tolylimino)thiazolidin-4-one (3) and evaluates these compounds for their α-amylase inhibition. The structures of all the newly synthesized compounds were confirmed by elemental and spectroscopic analysis (IR, 1H-NMR, MS). 1H NMR shows that the 2Z,5Z-isomer is present in 58.4–62.8%, which was further supported by DFT studies. The biological potential of the newly synthesized compounds was investigated through in vitro α-amylase inhibition activity. The results showed that some of the synthesized compounds exhibited significant inhibitory activities. Compound 5a at 100 μg mL–1 concentration showed a remarkable inhibition of 90.04%. Docking studies of compound 5a were performed against the active site of human pancreatic alpha-amylase (PDB ID: ; 2QV4). It has been revealed from the docking studies that the bonding interactions found between 5a and human pancreatic α-amylase are similar to those responsible for α-amylase inhibition by acarbose.
Conflict of interest
The authors declare no competing interests.
Supplementary Material
Acknowledgments
Meenakshi Duhan thanks the Haryana State Council of Science and Technology (HSCST) for the award of the research fellowship.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00080d
References
- Kokil G. R., Rewatkar P. V., Verma A., Thareja S., Naik S. R. Curr. Med. Chem. 2010;17:4405–4423. doi: 10.2174/092986710793361225. [DOI] [PubMed] [Google Scholar]
- Mc Cune L. M., Johns T. J. Ethnopharmacol. 2002;82:197–202. doi: 10.1016/s0378-8741(02)00180-0. [DOI] [PubMed] [Google Scholar]
- de Sales P. M., de Souza P. M., Simeoni L. A., Magalhães P. O., Silveira D. J. Pharm. Pharm. Sci. 2012;15:141–183. doi: 10.18433/j35s3k. [DOI] [PubMed] [Google Scholar]
- Qin X., Ren L., Yang X., Bai F., Wang L., Geng P., Bai G., Shen Y. J. Struct. Biol. 2011;174:196–202. doi: 10.1016/j.jsb.2010.11.020. [DOI] [PubMed] [Google Scholar]
- de Sales P. M., de Souza P. M., Simeoni L. A., de O. Magalhães P., Silveira D. J. Pharm. Pharm. Sci. 2012;15:141–183. doi: 10.18433/j35s3k. [DOI] [PubMed] [Google Scholar]
- van de Laar F. A., Lucassen P. L., Akkermans R. P., van de Lisdonk E. H., Rutten G. E., van Weel C. Diabetes Care. 2005;28:166–175. doi: 10.2337/diacare.28.1.154. [DOI] [PubMed] [Google Scholar]
- Kajaria D., Ranjana, Triphati J., Triphati Y. B., Tiwari S. J. Adv. Pharm. Technol. Res. 2013;4:206–209. doi: 10.4103/2231-4040.121415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhosle M. R., Mali J. R., Pal S., Srivastava A. K., Mane R. A. Bioorg. Med. Chem. Lett. 2014;24:2651–2654. doi: 10.1016/j.bmcl.2014.04.064. [DOI] [PubMed] [Google Scholar]
- Pansare D. N., Mulla N. A., Pawar C. D., Shende V. R., Shinde D. B. Bioorg. Med. Chem. Lett. 2014;24:3569–3573. doi: 10.1016/j.bmcl.2014.05.051. [DOI] [PubMed] [Google Scholar]
- Subhedar D. D., Shaikh M. H., Arkile M. A., Yeware A., Sarkar D., Shingate B. B. Bioorg. Med. Chem. Lett. 2016;26:1704–1708. doi: 10.1016/j.bmcl.2016.02.056. [DOI] [PubMed] [Google Scholar]
- Chougala B. M., Samundeeswari S., Holiyachi M., Shastri L. A., Dodamani S., Jalalpure S., Dixit S. R., Joshi S. D., Sunagar V. A. Eur. J. Med. Chem. 2017;125:101–116. doi: 10.1016/j.ejmech.2016.09.021. [DOI] [PubMed] [Google Scholar]
- Rashid M., Husain A., Shaharyar M., Mishra R., Hussain A., Afzal O. Eur. J. Med. Chem. 2014;83:630–645. doi: 10.1016/j.ejmech.2014.06.033. [DOI] [PubMed] [Google Scholar]
- Koppireddi S., Komsani J. R., Avula S., Pombala S., Vasamsetti S., Kotamraju S., Yadla R. Eur. J. Med. Chem. 2013;66:305–313. doi: 10.1016/j.ejmech.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Iqbal A. K. M., Khan A. Y., Kalashetti M. B., Belavagi N. S., Gong Y. D., Khazi I. A. M. Eur. J. Med. Chem. 2012;53:308–315. doi: 10.1016/j.ejmech.2012.04.015. [DOI] [PubMed] [Google Scholar]
- Raza S., Srivastava S. P., Srivastava D. S., Srivastava A. K., Haq W., Katti S. B. Eur. J. Med. Chem. 2013;63:611–620. doi: 10.1016/j.ejmech.2013.01.054. [DOI] [PubMed] [Google Scholar]
- Barreca M. L., Chimirri A., Luca L. D., Monforte A. M., Monforte P., Rao A., Zappala M., Balzarini J., Clercq E. D., Pannecouquec C., Witvrouwc M. Bioorg. Med. Chem. Lett. 2001;11:1793–1796. doi: 10.1016/s0960-894x(01)00304-3. [DOI] [PubMed] [Google Scholar]
- Alam R., Wahi D., Singh R., Sinha D., Tandon V., Rahisuddin A. G. Bioorg. Chem. 2016;69:77–90. doi: 10.1016/j.bioorg.2016.10.001. [DOI] [PubMed] [Google Scholar]
- Alegaon S. G., Alagawadi K. R., Garg M. K., Dushyant K., Vinod D. Bioorg. Chem. 2014;54:51–59. doi: 10.1016/j.bioorg.2014.04.001. [DOI] [PubMed] [Google Scholar]
- Hafez H. N., El-Gazzar A. R. B. A., Al-Hussain S. A. Bioorg. Med. Chem. Lett. 2016;26:2428–2433. doi: 10.1016/j.bmcl.2016.03.117. [DOI] [PubMed] [Google Scholar]
- Lv X. H., Ren Z. L., Zhou B. G., Li Q. S., Chu M. J., Liu D. H., Mo K., Zhang L. S., Yao X. K., Cao H. Q. Bioorg. Med. Chem. 2016;24:4652–4659. doi: 10.1016/j.bmc.2016.08.002. [DOI] [PubMed] [Google Scholar]
- Naidu K. M., Srinivasarao S., Agnieszka N., Ewa A. K., Kumar M. M. K., Sekhar K. V. G. C. Bioorg. Med. Chem. Lett. 2016;26:2245–2250. doi: 10.1016/j.bmcl.2016.03.059. [DOI] [PubMed] [Google Scholar]
- Vázquez E. H., Barrera S. S., Espinosa J. J. R., Soto S. E., Luis F. H. Bioorg. Med. Chem. 2016;24:2298–2306. [Google Scholar]
- Shi W., Hu J., Bao N., Li D., Chen L., Sun J. Bioorg. Med. Chem. Lett. 2017;27:147–151. doi: 10.1016/j.bmcl.2016.11.089. [DOI] [PubMed] [Google Scholar]
- Kumar P., Kumar S., Husain K., Kumar A. Chin. Chem. Lett. 2011;22:37–40. [Google Scholar]
- Ramesh S. M., Smorygo N. A., Khrabrova E. S., Ginak A. I. Chem. Heterocycl. Compd. 1986;22:447–451. [Google Scholar]
- Vicini P., Geronikaki A., Anastasia K., Incertia M., Zania F. Bioorg. Med. Chem. 2006;14:3859–3864. doi: 10.1016/j.bmc.2006.01.043. [DOI] [PubMed] [Google Scholar]
- Ottana R., Maccari R., Barreca M. L., Bruno G., Rotondo A., Rossi A., Chiricosta G., Paola R. D., Sautebin L., Cuzzocread S., Vigoritaa M. G. Bioorg. Med. Chem. 2005;13:4243–4252. doi: 10.1016/j.bmc.2005.04.058. [DOI] [PubMed] [Google Scholar]
- Parr R. G. and Weitao Y., Density-functional theory of atoms and molecules, Oxford University Press, USA, 1994, p. 16. [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- Vosko S. H., Wilk L., Nusair M. Can. J. Phys. 1980;58:1200–1211. [Google Scholar]
- Lee C., Yang W., Parr R. G. Phys. Rev. B: Condens. Matter Mater. Phys. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- Frisch M. J., Pople J. A., Binkley J. S. J. Chem. Phys. 1984;80:3265–3267. [Google Scholar]
- Ostapiuk Y. V., Obushak M. D., Matiychuk V. S., Naskrent M., Gzella A. K. Tetrahedron Lett. 2012;53:543–545. [Google Scholar]
- Kitchen D. B., Decornez H., Furr J. R., Bajorath J. Nat. Rev. Drug. Discovery. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- Piparo E. L., Scheib H., Frei N., Williamson G., Grigorov M., Chou C. J. J. Med. Chem. 2008;51:3555–3561. doi: 10.1021/jm800115x. [DOI] [PubMed] [Google Scholar]
- Najafian M., Habibi A. E., Hezareh N., Yaghmaei P., Parivar K., Larijani B. Mol. Biol. Rep. 2011;38:1617–1620. doi: 10.1007/s11033-010-0271-3. [DOI] [PubMed] [Google Scholar]
- Kumar P., Kumar S., Husain K., Kumar A. Chin. Chem. Lett. 2011;22:37–40. [Google Scholar]
- Zhou H., Wu S., Zhai S., Liu A., Sun Y., Li R., Zhang Y., Ekins S., Swaan P. W., Fang B., Zhang B., Yan B. J. Med. Chem. 2008;51:1242–1251. doi: 10.1021/jm7012024. [DOI] [PubMed] [Google Scholar]
- Nickavar B., Yousefian N. Iran. J. Pharm. Res. 2009;8:53–57. [Google Scholar]
- Mavin Sketch 5.10.1 Chem Axon Ltd. 1998–2012. http://www.chemaxon.com.
- Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- AutoDock Tools (version 1.5.6 rc2), Stefano Forte. Molecular Graphics Laboratory, Department of Molecular Biology, The Scripps Research Institute, 1999–2010. http://mgltools.scripps.edu.
- Lal K., Kaushik C. P., Kumar A. Med. Chem. Res. 2015;24:3258–3271. [Google Scholar]
- Trott O., Olson A. J. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Kumar S., Jain S., Kumar P., Goyal R. Med. Chem. Res. 2013;22:5431–5441. [Google Scholar]
- Discovery Studio Visualizer v4.0.100.13345. Accelrys Software Inc, 2005–2013.
- DeLano W. L., PyMOL(TM) Molecular Graphics System, Version 1.7.5.0. Copyright (c) Schrodinger, LLC.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.