

Abstract
Streptomycetes are multicellular bacteria with complex developmental cycles. They are of biotechnological importance as they produce most bioactive compounds used in biomedicine, e.g. antibiotic, antitumoral and immunosupressor compounds. Streptomyces genomes encode many Ser/Thr/Tyr kinases, making this genus an outstanding model for the study of bacterial protein phosphorylation events. We used mass spectrometry based quantitative proteomics and phosphoproteomics to characterize bacterial differentiation and activation of secondary metabolism of Streptomyces coelicolor. We identified and quantified 3461 proteins corresponding to 44.3% of the S. coelicolor proteome across three developmental stages: vegetative hypha (first mycelium); secondary metabolite producing hyphae (second mycelium); and sporulating hyphae. A total of 1350 proteins exhibited more than 2-fold expression changes during the bacterial differentiation process. These proteins include 136 regulators (transcriptional regulators, transducers, Ser/Thr/Tyr kinases, signaling proteins), as well as 542 putative proteins with no clear homology to known proteins which are likely to play a role in differentiation and secondary metabolism. Phosphoproteomics revealed 85 unique protein phosphorylation sites, 58 of them differentially phosphorylated during differentiation. Computational analysis suggested that these regulated protein phosphorylation events are implicated in important cellular processes, including cell division, differentiation, regulation of secondary metabolism, transcription, protein synthesis, protein folding and stress responses. We discovered a novel regulated phosphorylation site in the key bacterial cell division protein FtsZ (pSer319) that modulates sporulation and regulates actinorhodin antibiotic production. We conclude that manipulation of distinct protein phosphorylation events may improve secondary metabolite production in industrial streptomycetes, including the activation of cryptic pathways during the screening for new secondary metabolites from streptomycetes.
Keywords: Phosphoproteome, Differentiation, Quantification, Microbiology, Bacteria, antibiotics, sporulation, Streptomyces
Streptomyces is a genus of Gram-positive soil bacterium of great importance for biotechnology given their ability to produce a large array of bioactive compounds, including antibiotics, anticancer agents, immunosuppressants and industrial enzymes (1). Streptomyces has a complex morphogenesis which modulates secondary metabolism (2). After spore germination, a fully compartmentalized mycelium (first mycelium, MI) 1 initiates development (3) until it undergoes an ordered programmed cell death (PCD) (4) and differentiates to a second multinucleated mycelium (substrate mycelium, early second mycelium, MII) which activates secondary metabolism (2) before it starts to express the chaplin and rodlin genes encoding the proteins constituting the hydrophobic coats necessary for growth into the air (aerial mycelium; late MII) (5). At the end of the cycle, there is hyphal septation and sporulation (6).
Reversible protein phosphorylation at serine, threonine and tyrosine is a well-known dynamic post-translational modification with stunning regulatory and signaling potential in eukaryotes (7). In contrast, the extent and biological function of Ser/Thr/Tyr protein phosphorylation in bacteria are poorly defined. Phosphorylation in bacteria is dramatically lower than in eukaryotes, making bacterial phosphoproteomics challenging. Several large-scale Ser/Thr/Tyr phosphoproteome studies in bacteria have been reported (8–24) (summarized in Table I), using, in most cases, large amounts of protein (milligrams) obtained during the vegetative growth to detect the relatively low number of phosphopeptides (Table I). This precludes application of isobaric mass tagging quantitation strategies, because labeling such large amount of peptides is prohibitively expensive. This makes quantitative phosphoproteomics more difficult and to our knowledge, there are only six quantitative phosphoproteomic studies reported for bacteria. Some studies used stable isotope labeling by amino acids in cell culture (SILAC) (25–27), others used the scheduled reaction monitoring analyses (28, 29) and we performed a phosphopeptide profiling (label-free) quantitative proteomics study (16) (see Table I).
Table I. Comparison of the S. coelicolor phosphoproteome with other published prokaryotic and human phosphoproteomes. Quantitative phosphoproteomic studies are indicated by an asterisk. N.r. Not reported.
| Bacterium | Protein (mg)a | Phosphoproteins | Phosphopeptides | Phosphorylation sites | Reference |
|---|---|---|---|---|---|
| Gram + | |||||
| S. coelicolor | 0.1 | 48 | 92 | 85 | This work |
| S. coelicolor | 0.3 | 127 | 260 | 289 | (16)* |
| S. coelicolor | 50 | 40 | 44 | 46 | (18) |
| S. erythraea | 10 | 88 | 109 | n.r. | (28)* |
| B. subtilis | 10 | 78 | 103 | 78 | (15) |
| B. subtilis | 12 | 139 | 177 | 144 | (31)* |
| C. acetobutylicum | 2 | 61 | 82 | 107 | (9) |
| L. lactis | 20 | 63 | 102 | 79 | (22) |
| M. tuberculosis | n.r. | 301 | 380 | 500 | (19) |
| S. pneumoniae | 1 | 84 | 102 | 163 | (23) |
| L. monocytogenes | 10 | 112 | 155 | 143 | (17) |
| L. monocytogenes | 8 | 191 | 256 | 242 | (27)* |
| S. aureus | 50 | 108 | n.r. | 76 | (10) |
| Gram − | |||||
| E. coli | 20 | 79 | 105 | 81 | (14) |
| K. pneumoniae | 30 | 81 | 117 | 93 | (13) |
| P. aeruginosa | 1.2 | 39 | 57 | 61 | (20) |
| P. putida | 1.2 | 59 | 56 | 55 | (20) |
| H. pylori | n.r. | 67 | 82 | 126 | (11) |
| R. palustris (Ch) | 2 | 54 | 100 | 63 | (12) |
| R. palustris (Ph) | 2 | 42 | 74 | 59 | (12) |
| T. thermophilus | 100 | 48 | 52 | 46 | (24) |
| E. coli | 9.8 | 133 | n.r. | 108 | (25)* |
| E. coli | 10 | n.r | 34 | n.r. | (29)* |
| A. baumanii Abh12O-A2 | 9 | 70 | n.r. | 80 | (21) |
| A. baumanii ATCC 17879 | 9 | 41 | n.r. | 48 | (21) |
| Archaea | |||||
| H. salinarum | 20 | 26 | 42 | 31 | (8) |
| Eukarya | |||||
| H. sapiens | 6 | 7832 | >50000 | 38229 | (108) |
aThe total amount of protein used for all the MS/MS or 2D gel experiments is indicated.
Streptomyces coelicolor, the model Streptomyces strain (30), has 47 predicted eukaryotic-like protein kinases, twice the number predicted from genomes of other well characterized bacteria, including E. coli and B. subtilis, but 10-times less than human cells (31). S. coelicolor constitutes an outstanding model for the study of bacterial Ser/Thr/Tyr phosphorylation (16). The S. coelicolor chromosome was predicted to encode for 30 secondary metabolite biosynthetic pathways, 14 of them not yet experimentally observed (cryptic pathways) (32). None of these secondary metabolites has clinical applications, but S. coelicolor still is one of the best models to search for conserved pleiotropic secondary metabolism regulators in streptomycetes.
Here we investigate dynamic regulation of the S. coelicolor proteome and phosphoproteome during differentiation in solid sporulating cultures at three developmental time points, 16-, 30-, and 65-hours, corresponding to vegetative hyphae (MI), antibiotic producing hyphae (MII) and sporulating hyphae, respectively. This study reports the first isobaric-tag labeling quantitative phosphoproteomic study to identify Ser/Thr/Tyr phosphorylation implicated in secondary metabolism and differentiation in Streptomyces. Many of theproteins and phosphorylation sites differentially regulated during the vegetative (MI) and secondary metabolite producing mycelia (MII) are uncharacterised, but conserved in the Streptomyces genus. They are potential modulators of secondary metabolism and differentiation. Genetic manipulation of the genes encoding these biomolecules might improve bioactive compound production in streptomycetes.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Media
The S. coelicolor M145 strain was used in this study (30). Solid cultures were grown in Petri dishes (85 mm diameter) with 25 ml of solid GYM medium (glucose, yeast/malt extract) (33) that were covered with cellophane disks, inoculated with 100 μl of a spore suspension (1 × 107 viable spores/ml) and incubated at 30 °C. FtsZ mutants were grown in solid SFM and MM (supplemented with mannitol) media (30). Liquid cultures were performed in sucrose-free R5A (34) in 100 ml flasks, containing 20 ml media, at 200 rpm and 30 °C).
Experimental Design and Statistical Rationale
Three key developmental stages were analyzed: vegetative hypha (MI16h), secondary metabolite producing hyphae (MII30h) and sporulating hyphae (MII65h) (Fig. 1). Quantitative proteomics and phosphoproteomics were performed by means of TMT-10-plex using 4:3:3 biological replicates from MI16h, MII30h, and MII65h respectively (Fig. 1). In the case of the FtsZ mutants, samples were collected from sucrose-free R5A liquid cultures at 93-hours, the developmental time point preceding the maximum phenotypical differences (see results), and TMT-11-plex was used (4:3:4 biological replicates from Ser-Ser, Glu-Glu, and Ala-Ala FtsZ alleles respectively). As detailed below, abundance changes were statistically significant if they passed the differential expression test between the different developmental stages (q-value less than 0.01).
Fig. 1.
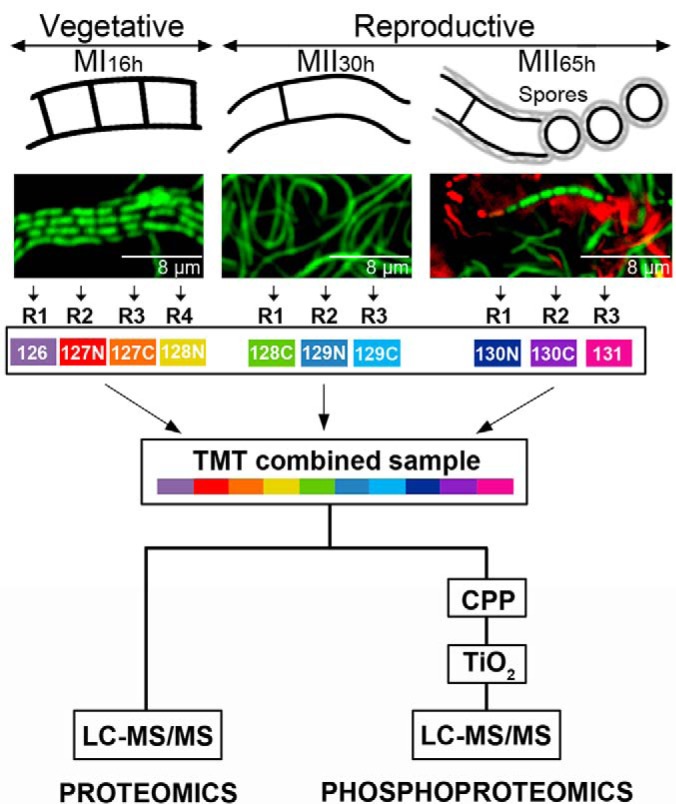
Sample preparation. S. coelicolor hyphae stained with SYTO 9 and propidium iodide observed under the confocal microscope at the compartmentalised MI (16 h), multinucleated substrate mycelium (MII30h) and sporulating aerial mycelium (MII65h) stages are shown (upper panels). TMT labeling and the phosphopeptide enrichment workflows are outlined.
Sampling of S. coelicolor Cells Throughout the Differentiation Cycle
The mycelial lawns of S. coelicolor M145 grown on cellophane disks were scraped off at different time points (16-, 30-, and 65-h; MI, MII, and sporulating MII) (Fig. 1) using a plain spatula. In the case of the FtsZ mutants, samples were collected from sucrose-free R5A liquid cultures at 93-hours by centrifugation at 20,000 × g for 5 min. Samples were resuspended in 2% SDS, 50 mm pH 7 Tris-HCl, 150 mm NaCl, 10 mm MgCl2, 1 mm EDTA, 7 mm β-mercaptoethanol, EDTA-free Protease Inhibitor Mixture Tablets from Roche (Basel, Switzerland) and 1% phosphatase inhibitor mixtures 1 and 2 (Sigma-Aldrich, San Luis, MI) and lysed by boiling for 10 min; sample viscosity was reduced by sonication (MSE soniprep 150, in four cycles of 10 s, on ice); cell debris were discarded after centrifugation at 20,000 × g for 10 min; the sample was cleaned by precipitation with acetone/ethanol (sample/EtOH/acetone 1:4:4 [v/v/v] overnight at −20 °C; washed with EtOH/acetone/H2O 2:2:1 [v/v/v]); resuspended in water; dialyzed against large volumes of water (1 h at 4 °C with four water changes); quantified as previously described (35); lyophilized in aliquots of 100 μg; and stored at −80 °C.
Protein Digestion
Lyophilized proteins were dissolved in 8 m urea in 100 mm TEAB buffer. Protein concentration was measured using bicinchoninic acid assay and adjusted to 2 mg of protein per ml. Samples were reduced with 5 mm DTT at room temperature for 1 h; S-alkylated with 15 mm iodoacetamide (15–30 min at RT in darkness); the alkylation reaction was stopped by adding DTT up to 15 mm; 300 μg of protein were digested using a combined trypsin/LysC digestion protocol (36); samples were desalted using Oasis HLB reverse-phase columns (Waters, Milford, MA).
TMT Labeling
Samples were labeled using TMT-10-plex or TMT-11-plex reagent (Thermo Fisher Scientific, Waltham, MA). The 30- and 65 h samples were analyzed in biological triplicate and the 16 h samples were analyzed in quadruplicate. The samples from the strains harboring the FtsZ Ser-Ser (wild-type) and the Ala-Ala alleles were analyzed in quadruplicate, whereas the sample from the strain harboring the FtsZ Glu-Glu allele in triplicate. Peptide concentration was quantified by amino acid analysis (37). For each condition, 60 μg of peptides was labeled with 0.5 mg of TMT-10-plex reagent following the manufacturer's protocol. The samples were then combined (Fig. 1).
Phosphoenrichment
Phosphopeptides were pre-enriched using calcium phosphate precipitation (CPP) as previously described (16, 38). One-hundred micrograms of TMT labeled peptides were precipitated using CPP and desalted using reverse phase chromatography (POROS R3 resin), before TiO2 enrichment (16) (Fig. 1). The optimal proportion between TiO2 beads and peptides was tested using 100 μg of TMT labeled peptides (before CPP enrichment) and different amounts of TiO2 beads. The best phosphopeptide enrichment efficiency, producing an enriched sample containing 48 ± 1.8% phosphopeptide identifications, was obtained for 0.15 mg TiO2 beads per 100 μg of TMT labeled peptide sample, versus the 26 ± 2.3% and 20 ± 2.6% obtained with 0.6 and 1.2 mg TiO2 beads of TMT labeled peptides.
LC-MS/MS Analysis of the Proteome and Phosphoproteome Temporal Dynamics
Fifty micrograms of TMT-labeled peptides were fractionated by hydrophilic interaction liquid chromatography (HILIC) to generate twelve fractions. Each of the fractions was analyzed by LC-MS/MS on EasyLC system Thermo Fisher Scientific coupled to Orbitrap-Fusion-LUMOS. The LC aqueous mobile phase contained 0.1% (v/v) formic acid in water and the organic mobile phase contained 0.1% (v/v) formic acid in 95% (v/v) acetonitrile. Before injection the trap and analytical columns were pre-equilibrated with 15 and 3.5 μl buffer A, respectively. The samples were injected on a custom 3-cm trap column (100-μm internal diameter silica tubing packed with Reprosil 120 C18 5-μm particles) and desalted with 18 μl of buffer A. Separation was performed on a home-made 20-cm column (75-μm internal diameter silica tubing packed with Reprosil 120 C18 3-μm particles) with a pulled emitter at 250 nL min-1. For total proteome analysis, separation was performed with a 1 to 8% gradient over 3 min, 8 to 28% over 80 min, 28 to 40% over 10 min and 40 to 100% over 5 min, the column was then kept at 100% buffer B for 8 min. For phosphopeptide analysis, peptides were separated with 1 to 34% buffer B over 60 min and 34 to 100% over 5 min and the column was then kept at 100% for 8 min.
The eluted peptides were analyzed on an Orbitrap Fusion mass spectrometer in data-dependent mode. The MS1 spectrum was acquired on an Orbitrap mass analyzer at the 400–1600 mass range 120,000 resolution with an AGC target of 5e5 (60 ms maximum injection time). For MS2 scans, peptides were isolated with a quadrupole using a 1.2-Da isolation window and fragmented at 40% normalized collision energy. AGC was set at 5e4 (maximum injection time 120 ms) and dynamic exclusion was 20 s.
Raw data are available via ProteomeXchange (http://www.proteomexchange.org/) with identifier PXD005558.
LC-MS/MS of the S. coelicolor FtsZ Mutants
Fifty μg of TMT labeled peptides were fractionated by high pH LC into thirty fractions. Each of the fractions was analyzed by LC-MS/MS using a Dionex Ultimate 3000 system (Thermo Fisher Scientific) interfaced to a Thermo Orbitrap Fusion Lumos electrospray ionization (ESI) tandem mass spectrometer. The LC mobile phases contained 0.1% (v/v) formic acid in water (A) and 0.1% (v/v) formic acid in 100% (v/v) acetonitrile (B). The samples were injected on a commercial trap column (300 μm × 5 mm C18 PepMap 100) and desalted with 18 μl of solvent A. Separation was performed at 1.1 μl/min on a home-made 50-cm column (150-μm internal diameter silica tubing packed with Inertsil C18 1.9-μm particles) at 35 °C, equipped with 10 μm ID emitter from PepSep (PSFSE10). The gradient was run from 2 to 10% B in 1 min, from 10 to 12% in 2 min, from 12 to 28% in 44 min, from 28 to 35% in 10 min, from 35 to 47% in 3 min. The column was then washed with 95% B for 3 min.
The Lumos MS instrument was operated with the recommended settings for SPS analysis in data-dependent MS/MS mode. The MS1 spectrum was acquired using the Orbitrap mass analyzer at m/z 375–1500 at mass resolution 120,000, AGC target of 4e5 (50 ms maximum injection time). For MS2 scans, peptides were isolated with the quadrupole using a 0.7 Da isolation window, fragmented at 35% normalized collision energy. Centroid spectra were collected in the ion trap in turbo mode. For MS3 scans, 10 notches were selected in a 2 Da isolation window and fragmented at 65 NCE by HCD and reporter ions were detected at mass resolution 50,000 in the m/z 100–500 range.
Raw data are available via ProteomeXchange (http://www.proteomexchange.org/) with identifier PXD005558.
Data Analysis
Data were analyzed in Proteome Discoverer 2.1 software. A database search was performed with Mascot 2.3.2 using Streptomyces coelicolor UniProt database (retrieved on June 3, 2015, 8038 entries). Methionine oxidation was set as variable modification, whereas Cys carbamidomethylation, TMT6plex (K) and TMT6plex (N-term) were specified as fixed modifications. A maximum of two trypsin missed cleavages were permitted. The precursor and fragment mass tolerances were 10 ppm and 0.02 Da respectively. Contaminants from maxQuant website (retrieved May 2015) were excluded from the final analysis. Peptides were validated by Mascot Percolator with a 0.01 posterior error probability (PEP) threshold. For phosphopeptide analysis, the phosphorylation position was validated using the ptmRS algorithm. Peptide scores, precursor charge, peptide m/z, ptmRS scores, annotated mass labeled spectra for all phosphopeptides and other details are available via ProteomeXchange (http://www.proteomexchange.org/) with identifier PXD005558. Peptide-spectrum matches, peptides per protein and protein coverage are indicated in supplemental Table S1. For phosphopeptides, we report the charge, m/z and mascot score for the highest confident PSM (that showing the highest Mascot score in our data set) in supplemental Table S2. Peptide spectrum matches with total summed reporter intensities of less than 4e5 were not considered for quantitation because of the high level of noise in the quantitation data. The quantification results of peptide spectrum matches were converted to peptide-level quantitation, which in turn was converted into protein quantitation using an R script (39). Only proteins with two or more quantified peptides were considered. Proteins with similar temporal abundance profiles were clustered using a fuzzy c-means algorithm with a Euclidean distance matrix (40).
Protein and phosphopeptide abundances of the reproductive stages (MII) were normalized against the vegetative stage (MI). The fold change of the MII stages (30- and 65- h) compared with the MI was estimated using the average TMT abundances from three or four biological replicates. Abundances were analyzed for differential expression using the limma package in R (41). p values for differential expression were adjusted for multiple comparisons using the R stats package. Phosphopeptide abundances were further normalized against the abundance of the non-phosphorylated proteins. Three thousand and eighty-three proteins exhibited significant expression changes (q-value less than 0.01) at least in one of the two MII stages compared with MI (supplemental Table S1); 78 phosphopeptides passed the abundance expression test (q-value < 0.01) at least in one of the MII stages analyzed compared with MI (supplemental Table S2).
Analysis of Protein Functions
Proteins were classified into functional categories according to their annotated functions in the Gene Bank database, publications and by homology/functions according to the Gene Ontology (42), the Conserved Domain (https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) and the KEGG Pathway (http://www.genome.jp/kegg-bin/show_organism?org=sco) databases. When a protein was involved in the synthesis of a specific secondary metabolite, it was classified in the secondary metabolism group, even if it matched additional categories. When a protein was involved in cell division regulation, it was included in cell division, instead of the category of regulatory proteins.
FtsZ site-directed mutagenesis - Ser 319 (supplemental Table S2) and Ser 387 (16) were replaced by Glu (to mimic phosphate group) and by Ala (to mimic nonphosphorylation). The synthesis of FtsZ mutated alleles was ordered to GeneCust. They included the FtsZ ORF and an upstream region (380 nucleotides) large enough to contain the three promoter regions described by Flärdh et al. (43). Genes were cloned in the integrative plasmid pNG3 (44) and introduced into S. coelicolor by conjugation (30). The native FtsZ ORF was mutated using the CRISPR-Cas9 system developed by Tong et al. (45). The 20-nt target sequence was selected inside the FtsZ gene and amplified using primers sgRNA-F (CATGCCATGGCGATGACTTTGATGACTGCGGTTTTAGAGCTAGAAATAGC) and sgRNA-R (ACGCCTACGTAAAAAAAGCACCGACTCGGTGCC). The product of 110 bps was digested with NcoI/SnaBI and cloned in NcoI/SnaBI digested pCRISPR-Cas9. The FtsZ surrounding regions were amplified by PCR with 2082LeftF (AGGCCTAGACCGACCACCGCCGAG)/2082LeftR (CCTATCACTTCAGGAAGTCCGTGATGACTGCGAGGTAGTTCTG) and 2082RightF (CAGAACTACCTCGCAGTCATCACGGACTTCCTGAAGTGATAGG)/2082RightR (AGGCCTAGTAACCGACCACGGAACGCA) primers. The two DNA fragments were combined by overlap extension PCR (46) using primers 2082LeftR and 2082RightF. The PCR product was cloned and sequenced in pCR™-Blunt II-TOPO®. The insert was released with EcoRI, filled with the Klenow enzyme and cloned into pCRISPR-Cas9 (harboring the 20-nt target sequence) digested with StuI. Mutations were confirmed by PCR using primers 2082LeftF and 2082RightR after plasmid clearance (45).
Antibiotic Quantification
Undecylprodigiosin and actinorhodin were quantified according to the spectrophotometric assays described by Tsao et al. (47) and Bystrykh et al. (48). Cells were ruptured in the culture medium by adding 0.1 N KOH. After vortexing and centrifugation, actinorhodin was quantified in the supernatant (ε640 25,320). Undecylprodigiosin was measured after vacuum drying of the mycelium, followed by extraction with methanol and acidification with HCl (to 0.5 m) (ε530 100,500). Analyses were performed in three biological replicas.
Hypha and Cell Wall Staining
Hypha and cell wall were stained using the LIVE/DEAD BacLight Bacterial Viability Kit (Invitrogen, l-13152) or Texas Red WGA (Invitrogen W21405) respectively as previously reported (49). Samples were observed under a Leica TCS SP8 laser scanning microscope as described before (3).
RESULTS
MI and MII Show Distinctive Proteomes and Phosphoproteomes
A total of 3461 proteins (44.3% of S. coelicolor proteome) were identified and quantified (supplemental Table S1). Protein abundances were highly consistent across the biological replicates analyzed (average correlation coefficient of 0.95; median coefficient of variation of 6.5%) (Fig. 2 and supplemental Table S1). Five clusters of proteins with similar temporal abundance profiles were identified (Fig. 3, supplemental Table S1): cluster 1 includes 345 proteins upregulated at the MI stage; cluster 2 includes 241 proteins upregulated at the MI and MII30h stages; clusters 3–5 include 974 proteins upregulated at the MII stages. Most proteins demonstrating significant changes (q-value less than 0.01) in the MII30h and MII65h stages (compared with MI) showed similar trends i.e. they were up- or downregulated in both (see heat maps in Figs. 4A and Fig. 5A) (supplemental Table S1). To focus on the most outstanding changes, we focused on the function of the 1350 proteins with significant expression changes and at least 2-fold change (log 2 ratio MII/MI lower than −1 or higher than 1). The abundances of key proteins are presented as histograms (Figs. 4B and 5B), detailed below (Table II) and outlined in Fig. 6.
Fig. 2.

TMT abundance values distribution and correlation between biological replicates. The distribution of the TMT abundances of two biological replicates of each developmental stage analyzed (MI16h, MII30h and MII65h) (panels in the diagonal), the correlation and coefficients of regressions of TMT abundances between biological replicates of the same developmental stage (marked in black) and the correlation and coefficients of regressions of TMT abundances between different developmental stages (marked in red), are shown. Notice the good correlation between replicates of the same developmental stage and the poor correlation between replicates of different developmental stages.
Fig. 3.

Clusters of the proteins with similar temporal abundance profiles. Cluster 1, proteins upregulated at the MI16h stage; Cluster 2, proteins upregulated at MI16h and MII30h; Clusters 3–5, proteins upregulated at the MII stages.
Fig. 4.

Abundances (logarithm of TMT MII/MI) of proteins related with secondary metabolism, aerial mycelium formation, sporulation, cell division and regulation of cellular processes (transcriptional regulators, transducers Ser/Thr/Tyr kinases and signaling proteins). Proteins involved in the synthesis of specific secondary metabolites, were classified in the secondary metabolism group, even if they fitted additional categories; proteins involved in cell division regulation, were included in cell division (see Methods). A, Heat maps outlining the TMT abundances of proteins with significant variations in both MII stages with respect to MI. Asterisks indicate the few proteins showing opposite differences in both MII (i.e. upregulated at MII30h and downregulated at MII65h or vice versa). B, TMT abundances of the key proteins described in the text. Abundances have significant differences (q-value < 0.01) and passed the 2-fold threshold in at least one of the MII stages, except for cell division proteins, in which proteins below the 2-fold threshold, but showing significant differences, are also shown. The SCO numbers are indicated. Dashed lines indicate the 2-fold threshold. “K” indicates Ser/Thr/Tyr kinases.
Fig. 5.

Abundances (logarithm of TMT MII/MI) of proteins related with central (primary) metabolism (aerobic energy production, catabolism, translation/protein folding). A, heat maps. B, TMT abundances of the key proteins described in the text. Statistics and labeling as in Fig. 4.
Table II. Averaged TMT protein abundances (from at least three biological replicates) of the substrate mycelium (MII30h) and sporulating aerial mycelium (MII65h) compared to the MI stage (16 h). The MII/MI ratio is shown in logarithm (log 2) and linear forms. Cl, clusters of proteins with similar abundance profiles. N.S. Not significant (q-value higher than 0.01). (-) The significant value that did not pass the 2-fold threshold. The proteins discussed in the text are indicated. Only changes of more than 2-fold up or down-regulated are shown, except for cell division proteins.
| Category | SCO n° | Cl | Function | Log 2 (MII30h/MI) | Log 2 (MII65h/MI) | Fold-change (MII30h/MI) | Fold-change (MII65h/MI) |
|---|---|---|---|---|---|---|---|
| Secondary metabolism | SCO6286 | 4 | CPK repressor | 1 | 1.8 | 1.9 | 3.5 |
| SCO6276 | 3 | CPK biosynthesis | 2 | n.s. | 4 | n.s. | |
| SCO6277 | 3 | 1.9 | - | 3.8 | - | ||
| SCO6278 | 3 | 1.4 | n.s. | 2.7 | n.s. | ||
| SCO6279 | n.s. | 2.6 | 1.6 | 6 | 3 | ||
| SCO6282 | 3 | 2.7 | - | 6.5 | - | ||
| SCO6283 | 3 | 2.6 | - | 5,9 | - | ||
| SCO0489 | Coelichelin biosynthesis | 1 | −1.5 | 2 | 0.3 | ||
| SCO0492 | 3 | 1.1 | n.s. | 2.1 | n.s. | ||
| SCO0498 | 3 | 1.6 | - | 3 | - | ||
| SCO0499 | 3 | 1.2 | n.s. | 2.3 | n.s. | ||
| SCO5072 | 5 | ACT biosynthesis | n.s. | 3.6 | n.s. | 12.1 | |
| SCO5073 | 5 | n.s. | 2.7 | n.s. | 6.5 | ||
| SCO5074 | 5 | n.s. | 4.6 | n.s. | 24.2 | ||
| SCO5075 | 5 | n.s. | 2.2 | n.s. | 4.6 | ||
| SCO5077 | n.s. | - | 2.1 | - | 4.3 | ||
| SCO5078 | 5 | n.s. | 3 | n.s. | 8 | ||
| SCO5079 | 5 | - | 3.4 | - | 10.5 | ||
| SCO5080 | 5 | n.s. | 3 | n.s. | 8 | ||
| SCO5083 | 5 | n.s. | 1.9 | n.s. | 3.7 | ||
| SCO5084 | 5 | n.s. | 3 | n.s. | 8 | ||
| SCO5086 | n.s. | n.s. | 1.6 | n.s. | 3 | ||
| SCO5087 | 5 | n.s. | 1.9 | n.s. | 3.7 | ||
| SCO5088 | 5 | n.s. | 1.6 | n.s. | 3 | ||
| SCO5089 | n.s. | - | 2.1 | - | 4.3 | ||
| SCO5090 | 5 | n.s. | 2.4 | n.s. | 5.3 | ||
| SCO5091 | 4 | - | 1.6 | - | 3 | ||
| SCO0381 | 5 | Deoxysugar synthases | n.s. | 2.5 | n.s. | 5.8 | |
| SCO0382 | n.s. | - | 2.5 | - | 5.8 | ||
| SCO0383 | 5 | - | 2.7 | - | 6.3 | ||
| SCO0384 | 5 | n.s. | 2.4 | n.s. | 5.4 | ||
| SCO0385 | 5 | n.s. | 2.1 | n.s. | 4.3 | ||
| SCO0386 | 5 | - | 2.6 | - | 5.9 | ||
| SCO0387 | n.s. | - | 2.3 | - | 5 | ||
| SCO0388 | 5 | - | 2.8 | - | 7 | ||
| SCO0389 | 5 | - | 2.8 | - | 7 | ||
| SCO0391 | 5 | n.s. | 2.1 | n.s. | 4.2 | ||
| SCO0392 | 4 | - | 1.9 | - | 3.7 | ||
| SCO0393 | n.s. | 1 | 3.1 | 2 | 8.2 | ||
| SCO0394 | 5 | - | 3.3 | - | 9.6 | ||
| SCO0395 | n.s. | 1 | 3.1 | 2 | 8.7 | ||
| SCO0396 | 5 | - | 3.3 | - | 9.9 | ||
| SCO0398 | 5 | - | 2.7 | - | 6.4 | ||
| SCO0399 | 4 | 1.2 | 2.8 | 2.3 | 7 | ||
| SCO0400 | n.s. | - | 2.6 | - | 6.7 | ||
| SCO0401 | n.s. | - | 2 | - | 4 | ||
| Bald/whi Sap | SCO0409 | 5 | SapA | - | 1.7 | - | 3.2 |
| SCO1489 | n.s. | BldD | - | -1.5 | - | 0.3 | |
| SCO3323 | n.s. | BldN | 1.5 | 1.9 | 2.8 | 3.7 | |
| SCO3424 | 1 | BldB homologue | −1.8 | −1.8 | 0.3 | 0.3 | |
| SCO3579 | n.s. | WblA | 1.1 | 1.6 | 2.1 | 3 | |
| SCO4091 | n.s. | BldC | - | −1.8 | - | 0.3 | |
| SCO5112 | n.s. | BldKA | −1.1 | n.s. | 0.5 | n.s. | |
| SCO5240 | n.s. | WblE | - | −2.6 | - | 0.2 | |
| SCO5315 | 5 | WhiE ORFVI | n.s. | 1.6 | n.s. | 3 | |
| SCO5321 | 5 | WhiE ORFVIII | n.s. | 1.8 | n.s. | 3.5 | |
| SCO5723 | n.s. | BldB | - | 1.5 | - | 2.8 | |
| SCO5819 | n.s. | WhiH | - | 1.9 | - | 3.7 | |
| SCO3897 | 1 | Possible targets for bldA regulation | −1.4 | −2.7 | 0.4 | 0.2 | |
| SCO4301 | 4 | - | 1.5 | - | 2.8 | ||
| SCO6476 | n.s. | - | 1.4 | - | 2.6 | ||
| SCO6638 | n.s. | 1 | 1.3 | 2 | 2.5 | ||
| SCO6717 | n.s. | - | −1.4 | - | 0.4 | ||
| Cell divisiona | SCO1416 | 5 | SffA | n.s. | 0.8 | n.s. | 1.7 |
| SCO1662 | n.s. | ParJ | 0.33 | 0.55 | 1.3 | 1.5 | |
| SCO2077 | n.s. | DivIVA | n.s. | −0.9 | n.s. | 0.5 | |
| SCO2082 | 2 | FtsZ | −0.2 | −0.8 | 0.9 | 0.57 | |
| SCO2610 | 2 | MreC | n.s. | −0.7 | n.s. | 0.6 | |
| SCO3854 | 5 | CrgA | n.s. | 1.4 | n.s. | 2.6 | |
| SCO3886 | n.s. | ParA | −0.3 | −0.6 | 0.8 | 0.7 | |
| SCO3887 | 2 | ParB | n.s. | −0.3 | n.s. | 0.8 | |
| SCO3904 | 4 | FemX | n.s. | 0.57 | n.s. | 1.48 | |
| SCO4114 | n.s. | Sporulation associated protein | 0.4 | 0.2 | 1.3 | 1.1 | |
| SCO4439 | n.s. | DD-CPase | −0.4 | −0.7 | 0.8 | 0.6 | |
| SCO5556 | 1 | HupS | −2 | −1.7 | 0.25 | 0.3 | |
| SCO5577 | n.s. | SmC | n.s. | 0.6 | n.s. | 1.5 | |
| Regulatory | SCO1468 | 5 | Putative Serine/Threonine protein kinase | n.s. | 1.9 | n.s | 3.8 |
| SCO1596 | n.s. | OhkA | - | 1.5 | - | 2.8 | |
| SCO1628 | 4 | RarC | 1.6 | 3.1 | 3 | 8.6 | |
| SCO1629 | n.s. | RarB | - | 2.6 | - | 6.1 | |
| SCO1630 | 5 | RarA | - | 3 | - | 8 | |
| SCO2666 | n.s. | Putative Serine/Threonine protein kinase | n.s. | 1.3 | n.s. | 2.5 | |
| SCO3102 | 5 | PkaE | n.s. | 1.5 | n.s. | 2.8 | |
| SCO3360 | n.s. | Putative Serine/Threonine protein kinase | −1.1 | n.s. | 0.4 | n.s. | |
| SCO3621 | 5 | Putative Serine/Threonine protein kinase | n.s. | 1.1 | n.s. | 2.1 | |
| SCO3821 | n.s. | Putative Serine/Threonine protein kinase | −1.2 | −0.9 | 0.4 | 0.5 | |
| SCO4776 | n.s. | Putative Serine/Threonine protein kinase | 0.4 | 1.5 | 1.3 | 2.9 | |
| SCO4778 | n.s. | PkaI | 0.9 | 1.5 | 1.9 | 2.8 | |
| SCO4069 | n.s. | SarA | - | 1.5 | - | 2.8 | |
| SCO4118 | 4 | AtrA | - | 1.1 | - | 2.1 | |
| SCO4190 | 5 | DevA | n.s. | 1.3 | n.s. | 2.5 | |
| SCO4325 | n.s. | CspB | - | −2.2 | - | 0.2 | |
| SCO4647 | n.s. | NusG | - | -1.9 | - | 0.3 | |
| SCO5582 | 4 | NsdA | - | 1.1 | - | 2.1 | |
| SCO6265 | n.s. | ScbR | 1.6 | 1.5 | 3 | 2.8 | |
| Unknown | SCO5191 | 1 | Hypothetical protein | −3.4 | −3 | 0.09 | 0.12 |
| SCO6650 | 5 | Hypothetical protein | n.s. | 4.8 | n.s. | 27.8 | |
| Translation/protein folding | SCO1491 | n.s. | Elongation factor | - | −2.3 | - | 0.2 |
| SCO1804 | 5 | tRNA ribosyltransferase-isomerase | n.s. | 2.9 | n.s. | 7.4 | |
| SCO2620 | 2 | Trigger factor chaperone | - | −1.8 | - | 0.8 | |
| SCO4648 | 1 | Ribosomal protein | −1.3 | −2.2 | 0.4 | 0.2 | |
| SCO4725 | 2 | Translation initiation | - | −1.9 | - | 0.3 | |
| SCO4734 | 1 | Ribosomal protein | −1.2 | −2.2 | 0.4 | 0.2 | |
| Energy production | SCO0922 | 3 | Succinate deshydrogenase | 2.1 | n.s. | 4.2 | n.s. |
| SCO0923 | 3 | Succinate deshydrogenase | 2.5 | n.s. | 5.5 | n.s. | |
| SCO4563 | 5 | NADH dehydrogenase | n.s. | 2.4 | n.s. | 5.1 | |
| SCO4564 | 5 | NADH dehydrogenase | n.s. | 3.2 | n.s. | 9.4 | |
| SCO4566 | 5 | NADH dehydrogenase | n.s. | 2.6 | n.s. | 6.1 | |
| SCO5107 | 5 | Succinate deshydrogenase | n.s. | 2.6 | n.s. | 6.2 | |
| Catabolism | SCO1640 | 4 | Proteasome protein | 1.2 | 2.8 | 2.6 | 7 |
| SCO1643 | 4 | Proteasome protein | - | 1.9 | - | 3.7 | |
| SCO1646 | n.s. | Proteasome protein | - | 2.2 | - | 4.7 | |
| SCO5443 | 5 | Alpha-amylase | n.s. | 2.2 | n.s. | 4.7 | |
| SCO6411 | 5 | Hydrolase | - | 2 | - | 4 | |
| SCO6414 | 4 | Hydrolase | 1.2 | 2.3 | 2.3 | 4.9 |
aFor cell division proteins, variations inside the ±1 interval showing significant variations (q-value < 0.01), are shown.
Fig. 6.

Integrated S. coelicolor proteome variations during development (MI16h, MII30h and MII65h). The key proteins discussed in the text are shown at the developmental stage in which they display significant upregulation (q-value < 0.01).
Eighty-five unique phosphorylation sites (48% Ser, 52% Thr) from 92 phosphopeptides and 48 phosphoproteins were identified (supplemental Table S2). All the phosphoproteins detected, were also identified and quantified in the proteomics set, which allowed us to normalize the change in phosphopeptide abundance against the change in protein, avoiding fluctuations because of variations in protein amount. To prevent artificial changes because of ratio suppression (underestimation of the relative change because of co-fragmentation of co-selected ions with similar m/z) (50) we focused on phosphopeptides that passed the abundance expression test (q-value < 0.01) and showed at least 2-fold increase or decrease in abundance (MII versus MI) (58 phosphopeptides) (Figs. 7 and 8 and Table III). There is a clear and striking upregulation of phosphorylation levels at MII, especially at 65 h (red bars in Fig. 7).
Fig. 7.

S. coelicolor phosphoproteome. A–B, Thirty-five peptides differentially phosphorylated during development (q-value < 0.01) that passed the 2-fold threshold at least in one of the MII stages grouped into functional categories. The SCO numbers are indicated. Dashed lines indicate the 2-fold threshold. The 23 peptides differentially phosphorylated, belonging to proteins with unknown function, are not shown. C, Outline indicating the phosphorylation sites and conserved protein domains of two multiphosphorylated proteins with unknown function.
Fig. 8.

S. coelicolor integrated phosphoproteome variations during development (MI16h, MII30h and MII65h). The key proteins discussed in the text are shown at the developmental stage in which they display significant phosphorylation site upregulation (q-value < 0.01).
Table III. Averaged TMT phosphopeptide abundances (from at least three biological replicates) of the substrate mycelium (MII30h) and sporulating aerial mycelium (MII65h) compared to the MI stage (16 h), of the phosphopeptides and phosphoproteins discussed in the manuscript. The MII/MI ratio is shown in the logarithm (log 2) and linear forms. N.S. Not significant (q-value higher than 0.01). (-) significant abundance values that did not pass the 2-fold threshold. Phosphorylation sites are highlighted in bold.
| Category | PROTEIN | Phosphopeptide | Log 2 (MII30h/MI) | Log 2 (MII65h/MI) | Fold-change (MII30h/MI) | Fold-change (MII65h/MI) |
|---|---|---|---|---|---|---|
| Regulatory | SCO0204 | AHpSEQGDNTGSPVR | n.s. | 3.1 | n.s. | 8.5 |
| DevA (SCO4190) | ALQEDGLLTNVpSK | n.s. | 2.1 | n.s. | 4.2 | |
| DasR (SCO5231) | STDVSSAENEGGApTVR | n.s. | 2.1 | n.s. | 4.3 | |
| SCO5357 | VNASAEQAAPADDAPpSER | n.s. | 1.6 | n.s. | 3 | |
| SCO5544 | GHDEPDpSSRTDR | n.s. | 3.2 | n.s. | 9.2 | |
| SCO5544 | GHDEPDSpSRTDRTPR | n.s. | 1.9 | n.s. | 3.7 | |
| SCO5544 | GHDEPDpSpSRTDRTPR | n.s. | 3.6 | n.s. | 12 | |
| SCO5704 | IDIRPDTEQPSDASPEQSpSGGRGE | n.s. | 4.8 | n.s. | 27.8 | |
| SCO7463 | TSDTPGASSEGHEVpS | n.s. | 2.8 | n.s. | 6.8 | |
| Cell division | DivIVA (SCO2077) | QLETQADDpSLAPPR | n.s | 1.6 | n.s | 3.1 |
| SepF (SCO2079) | IAEGGFFNQpS | n.s. | 1.1 | n.s. | 2.1 | |
| SCO2082 (FtsZ) | VTVIAAGFDGGQPPpSK | n.s. | 2 | n.s. | 4 | |
| SCO4439 | DGDADGYDGPPPVDQPpTAVFK | 1.3 | 1.9 | 2.4 | 3.7 | |
| Chaperones, thioredoxin and stress | SCO1836a | SGQDTIETpSGpTAK | n.s. | −1 | n.s. | 0.49 |
| PspA (SCO2168a) | QAIEGGQGQGEApSpSQSQQPQDTPR | n.s. | 1.72 | n.s. | 3.3 | |
| GroES (SCO4761) | IVVQPLDAEQpTTASGLVIPDTAK | - | 2.2 | - | 4.5 | |
| GroES (SCO4761) | IVVQPLDAEQTTApSGLVIPDTAK | n.s. | 2.5 | n.s. | 5.5 | |
| GroES (SCO4761) | IVVQPLDAEQTpTASGLVIPDTAK | n.s. | 2.6 | n.s. | 5.9 | |
| SCO5419 | NVLAEEPGNpTEAK | n.s. | 1.6 | n.s. | 3.1 | |
| Protein synthesis (translation) | RspA (SCO1998) | AAAEGVDTAGAAPAASGGGGGGSYSSEGGDNpSGALASDEALAALR | n.s. | 2.1 | n.s. | 4.3 |
| RspA (SCO1998) | AAAEGVDpTAGAAPAASGGGGGGSYSSEGGDNSGALASDEALAALR | n.s. | 2.4 | n.s. | 5.4 | |
| SCO4703 | AVDpTEGSEA | n.s. | 3.3 | n.s. | 9.6 | |
| SCO4711 | S.E.pSNVTEETK | n.s. | 3.3 | n.s. | 10.2 | |
| Catabolism | SCO1646a | ApTRpSpTEEVEEQAQDAQASEDLK | n.s. | 1.1 | n.s. | 2.2 |
| SCO1646 | pSTEEVEEQAQDAQASEDLK | n.s. | 1.2 | n.s. | 2.3 | |
| SCO1646 | ApTRSTEEVEEQAQDAQASEDLK | n.s. | 1.6 | n.s. | 3 | |
| SCO1646a | ApTRpSTEEVEEQAQDAQASEDLK | n.s. | 3.1 | n.s. | 8.5 | |
| SCO5864 | KTDDDVDSDpSLEELK | n.s. | 3.3 | n.s. | 9.9 | |
| SCO5864 | TDDDVDpSDSLEELK | n.s. | 3 | n.s. | 8 | |
| SCO5864 | KTDDDVDpSDSLEELK | n.s. | 4.4 | n.s. | 21.1 | |
| Nucleotide metabolism | PurE (SCO3059) | EFQQDLNDQApTEK | n.s. | 3.2 | n.s. | 9 |
| SCO3542 | ELPQIDPDQAPPpSR | n.s. | 2 | n.s. | 4 | |
| SCO3542 | ELPQIDPDQAPPpSRR | n.s. | 2.2 | n.s. | 4.5 | |
| Folate biosynthesis | SCO3401 | SAPFAQGPSDPTVQPVPASVIEQVDAADTpTLSNPK | n.s. | −1.5 | n.s. | 0.3 |
| Glycoslisis | SCO7443 | TSGLADGVVVTPpSHNPPADGGFK | n.s. | 1.4 | n.s. | 2.7 |
| Unknown | SCO2668 | AGDIDGApTAK | n.s. | 1.1 | n.s. | 2.1 |
| SCO2668 | VEEADEMTLETRpSpTKpTVR | n.s. | 1.9 | n.s. | 3.8 | |
| SCO2668 | AVQLAGVSGNADpTAK | 1.62 | 1.3 | 3.1 | 2.5 | |
| SCO3859 | SQGGSVLSNTTpTTTpSpSSGAPTVK | n.s. | 3 | n.s. | 8.2 | |
| SCO3859 | SQGGSVLSNTTTpTTpSpSSGAPTVK | n.s. | 3.4 | n.s. | 10.4 | |
| SCO3859 | SQGGSVLSNTTTpTpTpSpSSGAPTVK | n.s. | 3.3 | n.s. | 9.6 | |
| SCO3859 | SQGGSVLSNTTTTpTpSpSSGAPTVK | n.s. | 3.4 | n.s. | 10.4 | |
| SCO3859 | SQGGSVLSNpTTTTTSpSSGAPTVK | n.s. | 3.2 | n.s. | 9.3 | |
| SCO3859 | SQGGpSVLSNTTTTTSpSSGAPTVK | n.s. | 3.5 | n.s. | 11.2 | |
| SCO3859 | SQGGSVLSNTTTTTpSpSSGAPTVK | n.s. | 3.9 | n.s. | 14.8 | |
| SCO3859 | SQGGSVLSNTTTTpTSpSSGAPTVK | n.s. | 5.3 | n.s. | 40.2 | |
| SCO3859 | SQGGSVLSNTpTTTTSpSSGAPTVK | n.s. | 5.9 | n.s. | 61.1 | |
| SCO3859 | SQGGSVLSNTTTTpTSpSpSGAPTVK | n.s. | 3.4 | n.s. | 10.9 | |
| SCO3859 | SQGGSVLSNTTpTpTTpSpSSGAPTVK | n.s. | 3 | n.s. | 8 | |
| SCO3859 | SQGGSVLSNpTpTpTTTSpSSGAPTVK | n.s. | 4.1 | n.s. | 17 |
aOnly one phosphorylation site, at one of the positions marked, exists. See supplemental Table S2.
Proteins Responsible for Biosynthesis of Secondary Metabolites Are Upregulated at MII Stages
Proteins involved in secondary metabolite, such as actinorhodin, CPK, deoxysugar and coelichelin biosynthetic proteins, were upregulated at both MII stages (clusters 3–5 in Fig. 3) (Figs. 4B and 6, Table II).
We did not detect any Ser/Thr/Tyr phosphorylation in proteins involved in secondary metabolite biosynthesis.
Proteins Controlling Aerial Mycelium Formation and Sporulation Showed Different Abundances at the MI and MII Stages
Proteins involved in the regulation of the aerial mycelium formation and sporulation (Bld, Whi, Wbl, Sap proteins) showed different abundances between MI and MII (Figs. 4B and 6, Table II). Most of them were upregulated at the MII stages as SapA (51), WhiE ORFs VI and VIII (52), BldB (53), WhiH (51), SCO4301/SCO6476/SCO6638 (potential BldA targets; they harbor Leu encoded by TTA) (54), BldN (55) and WblA (56). Others were downregulated at the MII stages as BldD/BldC/BldKA (57), WblE (58), SCO3897/SCO6717 potential BldA targets and SCO3424, an uncharacterized BldB homologue.
We did not detect any Ser/Thr/Tyr phosphorylation in proteins controlling aerial mycelium formation and sporulation.
Transcriptional Regulators, Transducers, Ser/Thr/Tyr Kinases, and Signaling Proteins Showed Different Abundances and Phosphorylation at the MI and MII Stages
One-hundred-thirty-six regulatory proteins (transcriptional regulators, transducers, Ser/Thr/Tyr kinases and signaling proteins) had different abundances in at least one of the MII stages compared with MI (supplemental Table S1). Well-characterized regulatory proteins were included here (Figs. 4B and 6, Table II). However, the regulatory role of most of these proteins has been inferred from amino acid sequence similarity to previously characterized regulators.
Most of these regulatory proteins were upregulated at the MII stages (most of them grouped into clusters 4 and 5) (Fig. 3): OhkA, a regulator of secondary metabolism and morphological differentiation (59); RarA-C “restoration of aerial mycelium formation” proteins (60); SarA, a protein involved in the regulation of sporulation and secondary metabolism (61); AtrA, an activator of the actinorhodin biosynthetic gene expressions (62); DevA, a GntR-Like transcriptional regulator controlling aerial mycelium development (63); NsdA, a BldD target regulating morphological differentiation and secondary metabolism (64); and ScbR, a γ-butyrolactone binding protein regulating morphological differentiation and secondary metabolism (65). By contrast, some regulatory proteins were downregulated at the MII stages: CspB, a repressor of secondary metabolism (66) and NusG, a transcription antiterminator repressing undecylprodigiosin production (67).
As introduced above, the Streptomyces coelicolor genome encodes 47 Ser/Thr/Tyr kinases. In this work we identified 40 of these kinases. Most of them were upregulated at the MII65h stage (supplemental Table S2). The variation of eight Ser/Thr/Tyr kinases was over the 2-fold threshold (Fig. 4B): PkaE, an inhibitor of actinorhodin production (68), PkaI, one of the proteins involved in spore wall synthesis (69), and six uncharacterized putative kinases (SCO1468, SCO2666, SCO3360, SCO3621, SCO3821, SCO4776).
Seven regulatory proteins were differentially phosphorylated during development, especially at the MII65h stage (Table III) (Figs. 7A and 8): SCO0204, an orphan response regulator whose expression is affected by the sporulation-specific cell division activator SsgA (70); DevA, a GntR-like transcriptional regulator required for aerial mycelium development (63); DasR, a pleiotropic regulator of differentiation (71); SCO5357, rho transcription terminator involved in RNA degradation (KEGG pathway sco03018); SCO5544, part of the cvn1 conservon, a set of genes involved in regulating antibiotic production and aerial mycelium formation (72); SCO5704, a putative NusA transcription elongation factor; and SCO7463, a putative histidine kinase.
Cell Division Proteins Are Differentially Expressed and Phosphorylated in the Compartmentalized MI and the Multinucleated MII Hyphae
Forty-seven cell division proteins showed significant differences in their abundances between the MI and MII (supplemental Table S1). Cell division proteins do not present large differences in abundance during Streptomyces differentiation (3). However, they are crucial in differentiation, controlling the compartmentalization of the MI hyphae (3) and sporulation (73). Hence and only for cell division proteins, we discuss here all the well-characterized proteins showing significant variation, including those that did not pass the 2-fold threshold defined above (Figs. 4 and 6, Table II). Some cell division proteins that were reported to be involved in sporulation were upregulated at the MII stages (clusters 4 and 5 in Fig. 3): SffA (74), ParJ (75), CrgA (76), FemX (77), SCO4114 (78), and SmC (SCO5577) (79). Other well characterized cell division proteins were downregulated at the MII stages: DivIVA (80), FtsZ (81), MreC (69), ParA, ParB (82), the SCO4439 DD-CPase (83), and the nucleoid-associated HupS protein (84).
Four proteins involved in cell division and cell wall synthesis were differentially phosphorylated during development (Table III) (Figs. 7A and 8). Three cell division proteins were phosphorylated at the MII65h stage: DivIVA, one of the proteins regulating apical growth in Streptomyces, which was demonstrated to be regulated by phosphorylation (80); SepF one of the proteins included in the Streptomyces division and cell wall (dcw) cluster (85) and FtsZ, the key tubulin-like bacterial division protein (81). SCO4439, a d-alanyl-d-alanine carboxypeptidase involved in sporulation (83), was phosphorylated at the MII30h and MII65h stages.
Proteins Involved In Primary Metabolism Are Differentially Expressed and Phosphorylated During Development
Most proteins involved in protein synthesis (translation/protein folding) were upregulated at the MI (cluster 1 in Fig. 3) (Figs. 5 and 6, Table II). Proteins involved in energy production (krebs cycle, oxidative phosphorylation), lipid metabolism and catabolism were mostly upregulated at the MII stages (Figs. 5 and 6, Table II). Other proteins involved in primary metabolic pathways were not clearly overrepresented in the MI or MII developmental stages (supplemental Table S1).
Nine proteins involved in primary metabolism (translation, glycolytic enzymes, catabolism, nucleotide metabolism, folate biosynthesis) were differentially phosphorylated during development. Most of them were phosphorylated at the MII65h stage (Table III) (Figs. 7 and 8).
Stress Proteins and Chaperones Are Regulated by Phosphorylation
Stress proteins and chaperones were not clearly overrepresented in the MI or MII developmental stages (supplemental Table S1). However, some of them were differentially phosphorylated during development, mostly at the MII65h (Figs. 7B and 8, Table III): PspA, a phage shock protein (86); chaperonin GroES; SCO5419, a putative thioredoxin; and SCO1836, a putative stress-like protein.
Proteins and Phosphoproteins of Unknown Function Showed Different Abundances at the MI and MII Stages
Five-hundred-forty-two uncharacterized proteins without any clear homology showed different abundances in at least one of the MII stages analyzed compared with MI. As discussed below, these proteins constitute an outstanding database of potential regulators and effectors of differentiation (supplemental Table S1). For example, SCO5191 was upregulated 11- and 8-fold at the MI16h stage compared with the MII30h and MII65h respectively, whereas SCO6650 was upregulated 28-fold at the MII65h (Table II).
Eight proteins with unknown functions were differentially phosphorylated during development. Interestingly, SCO2668 and SCO3859 were multiphosphorylated at eight and nine positions respectively (Fig. 7C) (supplemental Table S2) (Fig. 8) (Table III). SCO2668 has a “von Willebrand factor type A” domain (vWA, conserved domain database accession cd00198) which is involved in a wide variety of important cellular functions (basal membrane formation, cell migration, cell differentiation, adhesion, homeostasis); SCO3859 has a “helix-turn-helix” domain (HTH, conserved domain database accession cl22854) which is present in several transcriptional regulators and other DNA binding proteins. Interestingly, phosphorylation in both proteins do not look to be random, as it is localized into small regions (58 and 13 amino acids respectively), outside the conserved domains (Fig. 7C).
Phosphorylation of FtsZ Affects Sporulation and Secondary Metabolism
As mentioned above, we identified a novel FtsZ phosphorylation at Ser 319 (supplemental Table S2), which is significantly upregulated at the MII30h and MII65h, 1.25- and 4.1-fold respectively. In our previous label-free study, we identified another FtsZ phosphorylation at Ser 387 (16). To study the effect of these phosphorylations, we replaced the native FtsZ gene with two mutated alleles, one mimicking phosphorylation (replacing phosposerines by Glu) and another one mimicking non-phosphorylation (replacing phosphoserines by Ala), a well-established methodology to analyze the effect of Ser phosphorylation (see some examples in (87–89).
FtsZ is crucial for cell division and sporulation (90) and sporulation is affected in the FtsZ mutants. It is highly reduced in the strain harboring the “Glu-Glu” allele and delayed in the strain harboring the “Ala-Ala” allele (Fig. 9A). However, we did not observe differences in hypha septation between the FtsZ mutants and the wild-type strain during the vegetative stage (Fig. 9B). The Ala-Ala mutant shows a delayed growth at the early development compared with the Glu-Glu mutant and the wild-type strains (Fig. 9C). The most surprising phenotype, was the effect in secondary metabolism. Actinorhodin, purple pigmented polyketide antibiotic, was overproduced (2.9-fold) in the Ala-Ala mutant and repressed (0.53-fold the wild-type value) in the “Glu-Glu” mutant (Fig. 9C), in all the culture media (SFM, MM, GYM and R5A) and conditions (solid and liquid cultures) tested (Fig. 9C, 9D). The production of the tripyrrole antibiotic undecylprodigiosin, was slightly diminished (0.77-fold the wild-type value) in the Ala-Ala mutant and highly repressed (0.24-fold the wild-type value) in the Glu-Glu mutant (Fig. 9C).
Fig. 9.

Phenotypes of the S. coelicolor harboring the FtsZ wild-type gene (Ser-Ser), the FtsZ mutant mimicking duble phosphorylation (“Glu-Glu”) and the FtsZ mutant mimicking no phosphorylation (“Ala-Ala”). A, macroscopic view of sporulated cultures 96-hours in SFM medium); the gray color corresponds to spores. B, peptidoglycan staining (WGA staining) of vegetative hyphae (15-hours in GYM cultures). Arrows indicate septa. C, sucrose-free R5A liquid cultures; upper panel, growth curve (protein/ml); middle panel actinorhodin production; lower panel undecylprodigiosin production. D, macroscopic view of solid cultures; SFM and GYM at 72 h, MM at 96 h. Purple color actinorhodin, red color undecylprodigiosin (not visible at the time points shown).
Next we compared the proteomes of the FtsZ mutants and the wild-type strain (supplemental Table S3). Most of the proteins involved in secondary metabolism that showed significant variations, were upregulated in the Ala-Ala mutant and downregulated in the Glu-Glu mutant (Fig. 10A). When we focus on the proteins that passed the 2-fold threshold (Fig. 10B), we observe that all the actinorhodin biosynthetic proteins are upregulated in the Ala-Ala mutant and not in the Glu-Glu mutant, whereas the undecylprodigiosin biosynthetic proteins are downregulated in both mutants compared with the wild-type strain (Ser-Ser allele). These results corroborate that the actinorhodin overproduction observed in the Ala-Ala mutant and the undecylprodigiosin repression detected in the Ala-Ala and the Glu-Glu mutants (Fig. 9C), are regulated at the protein level.
Fig. 10.

Abundances of proteins implicated in secondary metabolism in the FtsZ Glu-Glu and Ala-Ala mutants compared with the wild-type strain (Ser-Ser). A, heat maps. B, TMT abundances of the key proteins described in the text. Statistics and labeling as in Fig. 4.
DISCUSSION
Here we quantified the variation of 3461 proteins during the MI and MII stages in Streptomyces coelicolor. This corresponds to 44.3% of S. coelicolor theoretical proteome and 45% of the transcriptome identified by us in a previous work at the same developmental stages (91). Our results exceed any previous quantitative studies of the S. coelicolor proteome (92–96), quintuplicating the number of proteins quantified in our previous MI and MII quantitative proteomic study (16). We also quantified the variation of 92 phosphopeptides from 48 phosphoproteins during development, using the proteome abundances to normalize the phosphoproteome abundances and reporting the first quantitative data set of Ser/Thr/Tyr phosphorylation variations during Streptomyces differentiation. We used MS2 TMT to profile proteome and phosphoproteome temporal changes, which is a method known to suffer from ratio suppression (97). However, we and others have studied the issue of ratio compression in TMT based quantitative proteomics and have concluded that the effect on the overall outcome of proteomics studies can be neglected, unless high precision is needed (98–100). To reduce the interference of ratio suppression, we focused on the most reliable quantitative data, i.e. proteins and phosphopeptides that changed in abundance by at least 2-fold. Despite this very stringent criterion, we found that 1350 proteins (supplemental Table S1) and 58 phosphopeptides (supplemental Table S2) were changing, indicating significant reorganization of the Streptomyces coelicolor proteome and phosphoproteome throughout its life cycle. As described above, most of the 40 Ser/Thr/Tyr kinases identified, as well as Ser/Thr/Tyr phosphorylations were overrepresented at the sporulating stage (MII65h), indicating a role of Ser/Thr/Tyr phosphorylation in the regulation of differentiation and sporulation.
The clearest physiological difference between the MI and MII is the activation of secondary metabolism at the MII (91, 95) and most proteins involved in secondary metabolism were highly upregulated at the MII stages (CPK, ACT, deoxysugar synthases, coelichelin). By contrast, most proteins involved in primary metabolism, particularly those involved in protein synthesis, translation and protein folding were highly upregulated in the MI stage, which is not strange, considering that the MI corresponds to the exponential vegetative growth stage, whereas growth is arrested at the MII stage (101). Other primary metabolism proteins involved in energy production (krebs cycle, oxidative phosphorylation), lipid metabolism, amino acid metabolism, and pentose phosphate pathway, were mostly upregulated in MII stages, perhaps contributing the energy and precursors necessary for secondary metabolism. Secondary metabolism is associated to MII (4), but the expression of different secondary metabolites need specific activators (elicitors) or repressors and not all secondary metabolites are produced simultaneously in the MII (5, 102). For instance, AtrA (SCO4118), an activator of actinorhodin biosynthetic genes (62) is slightly upregulated at the MII65h (2.1-fold), activating actinorhodin production, whereas PkaE, a repressor of actinorhodin production was also upregulated at the MII65h (2.8-fold), indicating that actinorhodin production is blocked in sporulating hyphae as reported before (91, 95). SCO6286, a CPK repressor (103), was upregulated at the MII65h, blocking CPK production during the aerial mycelium stage. SCO3225 and SCO3226, a two-component system repressing CDA biosynthesis showed similar abundances at the MI and MII stages (supplemental Table S1), repressing CDA production under the culture conditions used in this work.
The clearest morphological difference between MII30h (substrate mycelium) and MII65h (aerial sporulating mycelium), is the presence of hydrophobic coats in the aerial sporulating hyphae and proteins involved in the regulation of hydrophobic cover formation and sporulation (Bld, Whi, SapA, RarA-C) were upregulated in the MII65h.
None of the enzymes synthesizing secondary metabolites, or the proteins forming the hydrophobic coats were detected as phosphorylated, indicating that modulation of these processes by Ser/Thr/Tyr phosphorylation is not at the final effectors. By contrast, some proteins and enzymes involved in central metabolic pathways (translation, glycolytic enzymes, catabolism, nucleotide metabolism, folate biosynthesis) were differentially phosphorylated in the MI and MII, suggesting that their activities are modulated by phosphorylation.
There are two developmental stages in which Streptomyces hyphae are compartmentalised, the MI and the sporulating MII, whereas the early stages of MII (substrate and aerial hyphae) consist in multinucleated hyphae with sporadic septa. Streptomyces cell division studies focused on sporulation, whereas MI cell division remains poorly known. Recently, our group, was able to identify the existence of a new kind of cell division based on cross-membranes without detectable peptidoglycan in the MI hyphae (3, 104), whose regulation remains basically uncharacterised. Here, we detected several cell division proteins more abundant at the MI stage than at the sporulating MII stage. This result suggests a putative role of these proteins in the MI cross-membrane based cell division, as it happens with FtsZ, the key effector of bacterial cell division (3). These proteins constitute a good starting point to study the mechanisms controlling cross-membrane cell division in Streptomyces.
Cell division proteins such as DivIVA and FtsZ were more phosphorylated during the MII65h stage, suggesting a role of phosphorylation in the regulation of cell-division accompanying sporulation. FtsZ phosphorylation has a direct effect in sporulation, that is highly reduced in the mutant mimicking double phosphorylation (“Glu-Glu” allele). This might indicate that phosphorylation affects FtsZ polymerization, reducing sporulation, as it happens in Mycobacterium tuberculosis (105). Interestingly, we detected a dramatic effect of FtsZ phosphorylation in actinorhodin production. FtsZ is highly conserved in the Streptomyces genus and the “Ala-Ala” allele, might be used in industrial streptomycetes to enhance the production of other antibiotics. Further work will be necessary to fully understand the biological role of FtsZ phosphorylation and its potential industrial application.
One-hundred-thirty-six regulatory proteins (transcriptional regulators, transducers, Ser/Thr/Tyr kinases, and signaling proteins), most of them uncharacterized putative regulators identified by amino acid sequence homology, and 542 putative proteins (without any clear homology) showed differential abundances during MI and MII. Key regulatory proteins were differentially phosphorylated during S. coelicolor development, such as DasR or DevA, two pleiotropic regulators controlling secondary metabolism and aerial mycelium formation. These results suggest a putative role of phosphorylation in the regulation of secondary metabolism, aerial mycelium formation and sporulation to be studied and characterized in the future. Eight proteins without known function were also differentially phosphorylated during development. For instance, SCO2668 is highly multiphosphorylated at the sporulating mycelium (MII65h) and harbors a pleiotropic eukaryotic vWA domain which is involved in a wide variety of important cellular functions in eukaryotic cells (basal membrane formation, cell migration, cell differentiation, adhesion, homeostasis). This suggests a complex regulatory role of SCO2668 in sporulation, resembling eukaryotic regulatory pathways. S. coelicolor is the best characterized streptomycete, despite that it does not produce commercial secondary metabolites. However, many of the phosphorylated proteins, as well as regulatory proteins and proteins with unknown function differentially expressed during the vegetative (MI) and secondary metabolite producing mycelia (MII), are conserved in the Streptomyces genus. These proteins are potential modulators of secondary metabolism and differentiation to be studied and characterized in the future, as it happens with FtsZ. This knowledge can contribute to the optimization of secondary metabolite production in industrial streptomycetes, including the activation of cryptic secondary metabolite pathways (pathways that are not activated in laboratory cultures) during the screening for new secondary metabolites from streptomycetes (106).
DATA AVAILABILITY
Raw data are available via ProteomeXchange (http://www.proteomexchange.org/) with identifier PXD005558.
Supplementary Material
Acknowledgments
We thank Beatriz Gutierrez Magan (Universidad de Oviedo, Dpto. Biología Funcional, Área de Microbiología) for her laboratory assistance.
Footnotes
* This work was supported by the European Research Council (ERC Starting Grant; Strp-differentiation 280304) and the Spanish “Ministerio de Economía y Competitividad” (MINECO; BIO2015-65709-R). The mass spectrometry and proteomics infrastructure used in this study was provided by VILLUM Center for Bioanalytical Sciencesat University of Southern Denmark supported by a generous grant from the VILLUM Foundation (O.N.J). P.S. was supported by a postdoctoral fellowship from the Lundbeck Foundation, Denmark. Nathaly Gonzalez-Quiñonez was funded by a Severo Ochoa fellowship (FICYT, Consejería de Educación y Ciencia, Asturias, Spain).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- MI
- first mycelium
- MII
- second mycelium
- PCD
- programmed cell death
- LC/MS/MS
- liquid chromatography-mass spectrometry-mass spectrometry
- CPP
- calcium phosphate precipitation
- TMT
- tandem mass tag.
REFERENCES
- 1. Hopwood D. A. (2007) Streptomyces in nature and medicine: the antibiotic makers., John Innes Centre, Oxford University Press [Google Scholar]
- 2. Rioseras B., Lopez-Garcia M. T., Yague P., Sanchez J., and Manteca A. (2014) Mycelium differentiation and development of Streptomyces coelicolor in lab-scale bioreactors: programmed cell death, differentiation, and lysis are closely linked to undecylprodigiosin and actinorhodin production. Bioresour. Technol. 151, 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yague P., Willemse J., Koning R. I., Rioseras B., Lopez-Garcia M. T., Gonzalez-Quinonez N., Lopez-Iglesias C., Shliaha P. V., Rogowska-Wrzesinska A., Koster A. J., Jensen O. N., van Wezel G. P., and Manteca A. (2016) Subcompartmentalization by cross-membranes during early growth of Streptomyces hyphae. Nat. Commun. 7, 12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Manteca A., Fernandez M., and Sanchez J. (2006) Cytological and biochemical evidence for an early cell dismantling event in surface cultures of Streptomyces antibioticus. Res. Microbiol. 157, 143–152 [DOI] [PubMed] [Google Scholar]
- 5. Yague P., Lopez-Garcia M. T., Rioseras B., Sanchez J., and Manteca A. (2013) Pre-sporulation stages of Streptomyces differentiation: state-of-the-art and future perspectives. FEMS Microbiol. Lett. 342, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jakimowicz D., and van Wezel G. P. (2012) Cell division and DNA segregation in Streptomyces: how to build a septum in the middle of nowhere? Mol. Microbiol. 85, 393–404 [DOI] [PubMed] [Google Scholar]
- 7. Pawson T., and Scott J. D. (2005) Protein phosphorylation in signaling–50 years and counting. Trends Biochem. Sci. 30, 286–290 [DOI] [PubMed] [Google Scholar]
- 8. Aivaliotis M., Macek B., Gnad F., Reichelt P., Mann M., and Oesterhelt D. (2009) Ser/Thr/Tyr protein phosphorylation in the archaeon Halobacterium salinarum–a representative of the third domain of life. PLoS ONE 4, e4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bai X., and Ji Z. (2012) Phosphoproteomic investigation of a solvent producing bacterium Clostridium acetobutylicum. Appl. Microbiol. Biotechnol. 95, 201–211 [DOI] [PubMed] [Google Scholar]
- 10. Basell K., Otto A., Junker S., Zuhlke D., Rappen G. M., Schmidt S., Hentschker C., Macek B., Ohlsen K., Hecker M., and Becher D. (2014) The phosphoproteome and its physiological dynamics in Staphylococcus aureus. Int. J. Med. Microbiol. 304, 121–132 [DOI] [PubMed] [Google Scholar]
- 11. Ge R., Sun X., Xiao C., Yin X., Shan W., Chen Z., and He Q. Y. (2011) Phosphoproteome analysis of the pathogenic bacterium Helicobacter pylori reveals over-representation of tyrosine phosphorylation and multiply phosphorylated proteins. Proteomics 11, 1449–1461 [DOI] [PubMed] [Google Scholar]
- 12. Hu C. W., Lin M. H., Huang H. C., Ku W. C., Yi T. H., Tsai C. F., Chen Y. J., Sugiyama N., Ishihama Y., Juan H. F., and Wu S. H. (2012) Phosphoproteomic analysis of Rhodopseudomonas palustris reveals the role of pyruvate phosphate dikinase phosphorylation in lipid production. J. Proteome Res. 11, 5362–5375 [DOI] [PubMed] [Google Scholar]
- 13. Lin M. H., Hsu T. L., Lin S. Y., Pan Y. J., Jan J. T., Wang J. T., Khoo K. H., and Wu S. H. (2009) Phosphoproteomics of Klebsiella pneumoniae NTUH-K2044 reveals a tight link between tyrosine phosphorylation and virulence. Mol. Cell. Proteomics 8, 2613–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Macek B., Gnad F., Soufi B., Kumar C., Olsen J. V., Mijakovic I., and Mann M. (2008) Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell. Proteomics 7, 299–307 [DOI] [PubMed] [Google Scholar]
- 15. Macek B., Mijakovic I., Olsen J. V., Gnad F., Kumar C., Jensen P. R., and Mann M. (2007) The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol. Cell. Proteomics 6, 697–707 [DOI] [PubMed] [Google Scholar]
- 16. Manteca A., Ye J., Sanchez J., and Jensen O. N. (2011) Phosphoproteome analysis of Streptomyces development reveals extensive protein phosphorylation accompanying bacterial differentiation. J. Proteome Res. 10, 5481–5492 [DOI] [PubMed] [Google Scholar]
- 17. Misra S. K., Milohanic E., Ake F., Mijakovic I., Deutscher J., Monnet V., and Henry C. (2011) Analysis of the serine/threonine/tyrosine phosphoproteome of the pathogenic bacterium Listeria monocytogenes reveals phosphorylated proteins related to virulence. Proteomics 11, 4155–4165 [DOI] [PubMed] [Google Scholar]
- 18. Parker J. L., Jones A. M., Serazetdinova L., Saalbach G., Bibb M. J., and Naldrett M. J. (2010) Analysis of the phosphoproteome of the multicellular bacterium Streptomyces coelicolor A3(2) by protein/peptide fractionation, phosphopeptide enrichment and high-accuracy mass spectrometry. Proteomics 10, 2486–2497 [DOI] [PubMed] [Google Scholar]
- 19. Prisic S., Dankwa S., Schwartz D., Chou M. F., Locasale J. W., Kang C. M., Bemis G., Church G. M., Steen H., and Husson R. N. (2010) Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U.S.A. 107, 7521–7526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ravichandran A., Sugiyama N., Tomita M., Swarup S., and Ishihama Y. (2009) Ser/Thr/Tyr phosphoproteome analysis of pathogenic and non-pathogenic Pseudomonas species. Proteomics 9, 2764–2775 [DOI] [PubMed] [Google Scholar]
- 21. Soares N. C., Spat P., Mendez J. A., Nakedi K., Aranda J., and Bou G. (2014) Ser/Thr/Tyr phosphoproteome characterization of Acinetobacter baumannii: comparison between a reference strain and a highly invasive multidrug-resistant clinical isolate. J. Proteomics 102, 113–124 [DOI] [PubMed] [Google Scholar]
- 22. Soufi B., Gnad F., Jensen P. R., Petranovic D., Mann M., Mijakovic I., and Macek B. (2008) The Ser/Thr/Tyr phosphoproteome of Lactococcus lactis IL1403 reveals multiply phosphorylated proteins. Proteomics 8, 3486–3493 [DOI] [PubMed] [Google Scholar]
- 23. Sun X., Ge F., Xiao C. L., Yin X. F., Ge R., Zhang L. H., and He Q. Y. (2010) Phosphoproteomic analysis reveals the multiple roles of phosphorylation in pathogenic bacterium Streptococcus pneumoniae. J. Proteome Res 9, 275–282 [DOI] [PubMed] [Google Scholar]
- 24. Takahata Y., Inoue M., Kim K., Iio Y., Miyamoto M., Masui R., Ishihama Y., and Kuramitsu S. (2012) Close proximity of phosphorylation sites to ligand in the phosphoproteome of the extreme thermophile Thermus thermophilus HB8. Proteomics 12, 1414–1430 [DOI] [PubMed] [Google Scholar]
- 25. Soares N. C., Spat P., Krug K., and Macek B. (2013) Global dynamics of the Escherichia coli proteome and phosphoproteome during growth in minimal medium. J. Proteome Res. 12, 2611–2621 [DOI] [PubMed] [Google Scholar]
- 26. Ravikumar V., Shi L., Krug K., Derouiche A., Jers C., Cousin C., Kobir A., Mijakovic I., and Macek B. (2014) Quantitative phosphoproteome analysis of Bacillus subtilis reveals novel substrates of the kinase PrkC and phosphatase PrpC. Mol. Cell. Proteomics 13, 1965–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Misra S. K., Moussan Desiree Ake F., Wu Z., Milohanic E., Cao T. N., Cossart P., Deutscher J., Monnet V., Archambaud C., and Henry C. (2014) Quantitative proteome analyses identify PrfA-responsive proteins and phosphoproteins in Listeria monocytogenes. J. Proteome Res. 13, 6046–6057 [DOI] [PubMed] [Google Scholar]
- 28. Licona-Cassani C., Lim S., Marcellin E., and Nielsen L. K. (2014) Temporal dynamics of the Saccharopolyspora erythraea phosphoproteome. Mol. Cell. Proteomics 13, 1219–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lim S., Marcellin E., Jacob S., and Nielsen L. K. (2015) Global dynamics of Escherichia coli phosphoproteome in central carbon metabolism under changing culture conditions. J. Proteomics 126, 24–33 [DOI] [PubMed] [Google Scholar]
- 30. Kieser T., Bibb M. J., Buttner M. J., Chater K. F., and Hopwood D. A. (2000) Practical Streptomyces genetics., John Innes Foundation, Norwich, England [Google Scholar]
- 31. Milanesi L., Petrillo M., Sepe L., Boccia A., D'Agostino N., Passamano M., Di Nardo S., Tasco G., Casadio R., and Paolella G. (2005) Systematic analysis of human kinase genes: a large number of genes and alternative splicing events result in functional and structural diversity. BMC Bioinformatics 6, S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nett M., Ikeda H., and Moore B. S. (2009) Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep. 26, 1362–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Novella I. S., Barbes C., and Sanchez J. (1992) Sporulation of Streptomyces antibioticus ETHZ 7451 in submerged culture. Can. J. Microbiol. 38, 769–773 [DOI] [PubMed] [Google Scholar]
- 34. Fernandez E., Weissbach U., Sanchez Reillo C., Brana A. F., Mendez C., Rohr J., and Salas J. A. (1998) Identification of two genes from Streptomyces argillaceus encoding glycosyltransferases involved in transfer of a disaccharide during biosynthesis of the antitumor drug mithramycin. J. Bacteriol. 180, 4929–4937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 36. Glatter T., Ludwig C., Ahrne E., Aebersold R., Heck A. J., and Schmidt A. (2012) Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion. J. Proteome Res. 11, 5145–5156 [DOI] [PubMed] [Google Scholar]
- 37. Hojrup P. (2015) Analysis of peptides and conjugates by amino acid analysis. Methods Mol. Biol. 1348, 65–76 [DOI] [PubMed] [Google Scholar]
- 38. Zhang X., Ye J., Jensen O. N., and Roepstorff P. (2007) Highly Efficient Phosphopeptide Enrichment by Calcium Phosphate Precipitation Combined with Subsequent IMAC Enrichment. Mol. Cell. Proteomics 6, 2032–2042 [DOI] [PubMed] [Google Scholar]
- 39. Polpitiya A. D., Qian W. J., Jaitly N., Petyuk V. A., Adkins J. N., Camp D. G. 2nd, Anderson G. A., and Smith R. D. (2008) DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics 24, 1556–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schwammle V., and Jensen O. N. (2010) A simple and fast method to determine the parameters for fuzzy c-means cluster analysis. Bioinformatics 26, 2841–2848 [DOI] [PubMed] [Google Scholar]
- 41. Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., and Smyth G. K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., and Sherlock G. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Flardh K., Leibovitz E., Buttner M. J., and Chater K. F. (2000) Generation of a non-sporulating strain of Streptomyces coelicolor A3(2) by the manipulation of a developmentally controlled ftsZ promoter. Mol. Microbiol. 38, 737–749 [DOI] [PubMed] [Google Scholar]
- 44. Gonzalez-Quinonez N., Lopez-Garcia M. T., Yague P., Rioseras B., Pisciotta A., Alduina R., and Manteca A. (2016) New PhiBT1 site-specific integrative vectors with neutral phenotype in Streptomyces. Appl. Microbiol. Biotechnol. 100, 2797–2808 [DOI] [PubMed] [Google Scholar]
- 45. Tong Y., Charusanti P., Zhang L., Weber T., and Lee S. Y. (2015) CRISPR-Cas9 based engineering of Actinomycetal genomes. ACS Synthetic Biol. 4, 1020–1029 [DOI] [PubMed] [Google Scholar]
- 46. Lee J., Shin M. K., Ryu D. K., Kim S., and Ryu W. S. (2010) Insertion and deletion mutagenesis by overlap extension PCR. Methods Mol. Biol. 634, 137–146 [DOI] [PubMed] [Google Scholar]
- 47. Tsao S. W., Rudd B. A., He X. G., Chang C. J., and Floss H. G. (1985) Identification of a red pigment from Streptomyces coelicolor A3(2) as a mixture of prodigiosin derivatives. The J. Antibiot. 38, 128–131 [DOI] [PubMed] [Google Scholar]
- 48. Bystrykh L. V., Fernandez-Moreno M. A., Herrema J. K., Malpartida F., Hopwood D. A., and Dijkhuizen L. (1996) Production of actinorhodin-related “blue pigments” by Streptomyces coelicolor A3(2). J. Bacteriol. 178, 2238–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Manteca A., Fernandez M., and Sanchez J. (2005) A death round affecting a young compartmentalized mycelium precedes aerial mycelium dismantling in confluent surface cultures of Streptomyces antibioticus. Microbiology 151, 3689–3697 [DOI] [PubMed] [Google Scholar]
- 50. Christoforou A. L., and Lilley K. S. (2012) Isobaric tagging approaches in quantitative proteomics: the ups and downs. Anal. Bioanal. Chem. 404, 1029–1037 [DOI] [PubMed] [Google Scholar]
- 51. Chater K. F. (2001) Regulation of sporulation in Streptomyces coelicolor A3(2): a checkpoint multiplex? Curr. Opin. Microbiol. 4, 667–673 [DOI] [PubMed] [Google Scholar]
- 52. Kelemen G. H., Brian P., Flardh K., Chamberlin L., Chater K. F., and Buttner M. J. (1998) Developmental regulation of transcription of whiE, a locus specifying the polyketide spore pigment in Streptomyces coelicolor A3 (2). J. Bacteriol. 180, 2515–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pope M. K., Green B., and Westpheling J. (1998) The bldB gene encodes a small protein required for morphogenesis, antibiotic production, and catabolite control in Streptomyces coelicolor. J. Bacteriol. 180, 1556–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chandra G., and Chater K. F. (2008) Evolutionary flux of potentially bldA-dependent Streptomyces genes containing the rare leucine codon TTA. Antonie Van Leeuwenhoek 94, 111–126 [DOI] [PubMed] [Google Scholar]
- 55. Bibb M. J., Molle V., and Buttner M. J. (2000) sigma(BldN), an extracytoplasmic function RNA polymerase sigma factor required for aerial mycelium formation in Streptomyces coelicolor A3(2). J. Bacteriol. 182, 4606–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fowler-Goldsworthy K., Gust B., Mouz S., Chandra G., Findlay K. C., and Chater K. F. (2011) The actinobacteria-specific gene wblA controls major developmental transitions in Streptomyces coelicolor A3(2). Microbiology 157, 1312–1328 [DOI] [PubMed] [Google Scholar]
- 57. Chater K. F. (1993) Genetics of differentiation in Streptomyces. Annu. Rev. Microbiol. 47, 685–713 [DOI] [PubMed] [Google Scholar]
- 58. Homerova D., Sevcikova J., and Kormanec J. (2003) Characterization of the Streptomyces coelicolor A3(2) wblE gene, encoding a homologue of the sporulation transcription factor. Folia Microbiol. 48, 489–495 [DOI] [PubMed] [Google Scholar]
- 59. Lu Y., He J., Zhu H., Yu Z., Wang R., Chen Y., Dang F., Zhang W., Yang S., and Jiang W. (2011) An orphan histidine kinase, OhkA, regulates both secondary metabolism and morphological differentiation in Streptomyces coelicolor. J. Bacteriol. 193, 3020–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Komatsu M., Takano H., Hiratsuka T., Ishigaki Y., Shimada K., Beppu T., and Ueda K. (2006) Proteins encoded by the conservon of Streptomyces coelicolor A3(2) comprise a membrane-associated heterocomplex that resembles eukaryotic G protein-coupled regulatory system. Mol. Microbiol. 62, 1534–1546 [DOI] [PubMed] [Google Scholar]
- 61. Ou X., Zhang B., Zhang L., Dong K., Liu C., Zhao G., and Ding X. (2008) SarA influences the sporulation and secondary metabolism in Streptomyces coelicolor M145. Acta Biochim. Biophys. Sin. 40, 877–882 [PubMed] [Google Scholar]
- 62. Uguru G. C., Stephens K. E., Stead J. A., Towle J. E., Baumberg S., and McDowall K. J. (2005) Transcriptional activation of the pathway-specific regulator of the actinorhodin biosynthetic genes in Streptomyces coelicolor. Mol. Microbiol. 58, 131–150 [DOI] [PubMed] [Google Scholar]
- 63. Hoskisson P. A., Rigali S., Fowler K., Findlay K. C., and Buttner M. J. (2006) DevA, a GntR-like transcriptional regulator required for development in Streptomyces coelicolor. J. Bacteriol. 188, 5014–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. den Hengst C. D., Tran N. T., Bibb M. J., Chandra G., Leskiw B. K., and Buttner M. J. (2010) Genes essential for morphological development and antibiotic production in Streptomyces coelicolor are targets of BldD during vegetative growth. Mol. Microbiol. 78, 361–379 [DOI] [PubMed] [Google Scholar]
- 65. Hsiao N. H., Nakayama S., Merlo M. E., de Vries M., Bunet R., Kitani S., Nihira T., and Takano E. (2009) Analysis of two additional signaling molecules in Streptomyces coelicolor and the development of a butyrolactone-specific reporter system. Chem. Biol. 16, 951–960 [DOI] [PubMed] [Google Scholar]
- 66. Mikulik K., Khanh-Hoang Q., Halada P., Bezouskova S., Benada O., and Behal V. (1999) Expression of the Csp protein family upon cold shock and production of tetracycline in Streptomyces aureofaciens. Biochem. Biophys. Res. Commun. 265, 305–310 [DOI] [PubMed] [Google Scholar]
- 67. Meng L., Yang S. H., Palaniyandi S. A., Lee S. K., Lee I. A., Kim T. J., and Suh J. W. (2011) Phosphoprotein affinity purification identifies proteins involved in S-adenosyl-L-methionine-induced enhancement of antibiotic production in Streptomyces coelicolor. J. Antibiot. 64, 97–101 [DOI] [PubMed] [Google Scholar]
- 68. Urabe H., Ogawara H., and Motojima K. (2015) Expression and characterization of Streptomyces coelicolor serine/threonine protein kinase PkaE. Biosci. Biotechnol. Biochem. 79, 855–862 [DOI] [PubMed] [Google Scholar]
- 69. Kleinschnitz E. M., Heichlinger A., Schirner K., Winkler J., Latus A., Maldener I., Wohlleben W., and Muth G. (2011) Proteins encoded by the mre gene cluster in Streptomyces coelicolor A3(2) cooperate in spore wall synthesis. Mol. Microbiol. 79, 1367–1379 [DOI] [PubMed] [Google Scholar]
- 70. Wang W., Shu D., Chen L., Jiang W., and Lu Y. (2009) Cross-talk between an orphan response regulator and a noncognate histidine kinase in Streptomyces coelicolor. FEMS Microbiol. Lett. 294, 150–156 [DOI] [PubMed] [Google Scholar]
- 71. Rigali S., Titgemeyer F., Barends S., Mulder S., Thomae A. W., Hopwood D. A., and van Wezel G. P. (2008) Feast or famine: the global regulator DasR links nutrient stress to antibiotic production by Streptomyces. EMBO Rep. 9, 670–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Takano H., Hashimoto K., Yamamoto Y., Beppu T., and Ueda K. (2011) Pleiotropic effect of a null mutation in the cvn1 conservon of Streptomyces coelicolor A3(2). Gene 477, 12–18 [DOI] [PubMed] [Google Scholar]
- 73. Zhang L., Willemse J., Claessen D., and van Wezel G. P. (2016) SepG coordinates sporulation-specific cell division and nucleoid organization in Streptomyces coelicolor. Open Biol. 6, 150164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ausmees N., Wahlstedt H., Bagchi S., Elliot M. A., Buttner M. J., and Flardh K. (2007) SmeA, a small membrane protein with multiple functions in Streptomyces sporulation including targeting of a SpoIIIE/FtsK-like protein to cell division septa. Mol. Microbiol. 65, 1458–1473 [DOI] [PubMed] [Google Scholar]
- 75. Ditkowski B., Troc P., Ginda K., Donczew M., Chater K. F., Zakrzewska-Czerwinska J., and Jakimowicz D. (2010) The actinobacterial signature protein ParJ (SCO1662) regulates ParA polymerization and affects chromosome segregation and cell division during Streptomyces sporulation. Mol. Microbiol. 78, 1403–1415 [DOI] [PubMed] [Google Scholar]
- 76. Del Sol R., Mullins J. G., Grantcharova N., Flardh K., and Dyson P. (2006) Influence of CrgA on assembly of the cell division protein FtsZ during development of Streptomyces coelicolor. J. Bacteriol. 188, 1540–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hong H. J., Hutchings M. I., Hill L. M., and Buttner M. J. (2005) The role of the novel Fem protein VanK in vancomycin resistance in Streptomyces coelicolor. J. Biol. Chem. 280, 13055–13061 [DOI] [PubMed] [Google Scholar]
- 78. Li W., Ying X., Guo Y., Yu Z., Zhou X., Deng Z., Kieser H., Chater K. F., and Tao M. (2006) Identification of a gene negatively affecting antibiotic production and morphological differentiation in Streptomyces coelicolor A3(2). J. Bacteriol. 188, 8368–8375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dedrick R. M., Wildschutte H., and McCormick J. R. (2009) Genetic interactions of smc, ftsK, and parB genes in Streptomyces coelicolor and their developmental genome segregation phenotypes. J. Bacteriol. 191, 320–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hempel A. M., Cantlay S., Molle V., Wang S. B., Naldrett M. J., Parker J. L., Richards D. M., Jung Y. G., Buttner M. J., and Flardh K. (2012) The Ser/Thr protein kinase AfsK regulates polar growth and hyphal branching in the filamentous bacteria Streptomyces. Proc. Natl. Acad. Sci. U.S.A. 109, E2371–E2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Flardh K. (2003) Growth polarity and cell division in Streptomyces. Curr. Opin. Microbiol. 6, 564–571 [DOI] [PubMed] [Google Scholar]
- 82. Jakimowicz D., Zydek P., Kois A., Zakrzewska-Czerwinska J., and Chater K. F. (2007) Alignment of multiple chromosomes along helical ParA scaffolding in sporulating Streptomyces hyphae. Mol. Microbiol. 65, 625–641 [DOI] [PubMed] [Google Scholar]
- 83. Rioseras B., Yague P., Lopez-Garcia M. T., Gonzalez-Quinonez N., Binda E., Marinelli F., and Manteca A. (2016) Characterization of SCO4439, a D-alanyl-D-alanine carboxypeptidase involved in spore cell wall maturation, resistance, and germination in Streptomyces coelicolor. Sci. Rep. 6, 21659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Salerno P., Larsson J., Bucca G., Laing E., Smith C. P., and Flardh K. (2009) One of the two genes encoding nucleoid-associated HU proteins in Streptomyces coelicolor is developmentally regulated and specifically involved in spore maturation. J. Bacteriol. 191, 6489–6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Letek M., Fiuza M., Ordonez E., Villadangos A. F., Ramos A., Mateos L. M., and Gil J. A. (2008) Cell growth and cell division in the rod-shaped actinomycete Corynebacterium glutamicum. Antonie Van Leeuwenhoek 94, 99–109 [DOI] [PubMed] [Google Scholar]
- 86. Vrancken K., Van Mellaert L., and Anne J. (2008) Characterization of the Streptomyces lividans PspA response. J. Bacteriol. 190, 3475–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhao Y., Hawes J., Popov K. M., Jaskiewicz J., Shimomura Y., Crabb D. W., and Harris R. A. (1994) Site-directed mutagenesis of phosphorylation sites of the branched chain alpha-ketoacid dehydrogenase complex. J. Biol. Chem. 269, 18583–18587 [PubMed] [Google Scholar]
- 88. Keller-Pinter A., Ughy B., Domoki M., Pettko-Szandtner A., Letoha T., Tovari J., Timar J., and Szilak L. (2017) The phosphomimetic mutation of syndecan-4 binds and inhibits Tiam1 modulating Rac1 activity in PDZ interaction-dependent manner. PloS One 12, e0187094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hewitt S. L., Wong J. B., Lee J. H., Nishana M., Chen H., Coussens M., Arnal S. M., Blumenberg L. M., Roth D. B., Paull T. T., and Skok J. A. (2017) The conserved ATM kinase RAG2-S365 phosphorylation site limits cleavage events in individual cells independent of any repair defect. Cell Reports 21, 979–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. McCormick J. R., Su E. P., Driks A., and Losick R. (1994) Growth and viability of Streptomyces coelicolor mutant for the cell division gene ftsZ. Mol. Microbiol. 14, 243–254 [DOI] [PubMed] [Google Scholar]
- 91. Yague P., Rodriguez-Garcia A., Lopez-Garcia M. T., Martin J. F., Rioseras B., Sanchez J., and Manteca A. (2013) Transcriptomic analysis of Streptomyces coelicolor differentiation in solid sporulating cultures: first compartmentalized and second multinucleated mycelia have different and distinctive transcriptomes. PLoS ONE 8, e60665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Gubbens J., Janus M., Florea B. I., Overkleeft H. S., and van Wezel G. P. (2012) Identification of glucose kinase-dependent and -independent pathways for carbon control of primary metabolism, development and antibiotic production in Streptomyces coelicolor by quantitative proteomics. Mol. Microbiol. 86, 1490–1507 [DOI] [PubMed] [Google Scholar]
- 93. Jayapal K. P., Philp R. J., Kok Y. J., Yap M. G., Sherman D. H., Griffin T. J., and Hu W. S. (2008) Uncovering genes with divergent mRNA-protein dynamics in Streptomyces coelicolor. PLoS ONE 3, e2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Manteca A., Jung H. R., Schwammle V., Jensen O. N., and Sanchez J. (2010) Quantitative proteome analysis of Streptomyces coelicolor Nonsporulating liquid cultures demonstrates a complex differentiation process comparable to that occurring in sporulating solid cultures. J. Proteome Res. 9, 4801–4811 [DOI] [PubMed] [Google Scholar]
- 95. Manteca A., Sanchez J., Jung H. R., Schwammle V., and Jensen O. N. (2010) Quantitative proteomics analysis of Streptomyces coelicolor development demonstrates that onset of secondary metabolism coincides with hypha differentiation. Mol. Cell. Proteomics 9, 1423–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Millan-Oropeza A., Henry C., Blein-Nicolas M., Aubert-Frambourg A., Moussa F., Bleton J., and Virolle M. J. (2017) Quantitative Proteomics Analysis Confirmed Oxidative Metabolism Predominates in Streptomyces coelicolor versus Glycolytic Metabolism in Streptomyces lividans. J. Proteome Res. 16, 2597–2613 [DOI] [PubMed] [Google Scholar]
- 97. Ow S. Y., Salim M., Noirel J., Evans C., Rehman I., and Wright P. C. (2009) iTRAQ underestimation in simple and complex mixtures: “the good, the bad and the ugly”. J. Proteome Res. 8, 5347–5355 [DOI] [PubMed] [Google Scholar]
- 98. Hogrebe A., von Stechow L., Bekker-Jensen D. B., Weinert B. T., Kelstrup C. D., and Olsen J. V. (2018) Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Communs. 9, 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hughes C. S., Spicer V., Krokhin O. V., and Morin G. B. (2017) Investigating Acquisition Performance on the Orbitrap Fusion When Using Tandem MS/MS/MS Scanning with Isobaric Tags. J. Proteome Res. 16, 1839–1846 [DOI] [PubMed] [Google Scholar]
- 100. Williamson J. C., Edwards A. V., Verano-Braga T., Schwammle V., Kjeldsen F., Jensen O. N., and Larsen M. R. (2016) High-performance hybrid Orbitrap mass spectrometers for quantitative proteome analysis: Observations and implications. Proteomics 16, 907–914 [DOI] [PubMed] [Google Scholar]
- 101. Manteca A., Alvarez R., Salazar N., Yague P., and Sanchez J. (2008) Mycelium differentiation and antibiotic production in submerged cultures of Streptomyces coelicolor. Appl. Environ. Microbiol. 74, 3877–3886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Liu G., Chater K. F., Chandra G., Niu G., and Tan H. (2013) Molecular regulation of antibiotic biosynthesis in streptomyces. Microbiol. Mol. Biol. Rev. 77, 112–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Gottelt M., Kol S., Gomez-Escribano J. P., Bibb M., and Takano E. (2010) Deletion of a regulatory gene within the cpk gene cluster reveals novel antibacterial activity in Streptomyces coelicolor A3(2). Microbiology 156, 2343–2353 [DOI] [PubMed] [Google Scholar]
- 104. Celler K., Koning R. I., Willemse J., Koster A. J., and van Wezel G. P. (2016) Cross-membranes orchestrate compartmentalization and morphogenesis in Streptomyces. Nat. Commun. 7, ncomms11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Thakur M., and Chakraborti P. K. (2006) GTPase activity of mycobacterial FtsZ is impaired due to its transphosphorylation by the eukaryotic-type Ser/Thr kinase, PknA. J. Biol. Chem. 281, 40107–40113 [DOI] [PubMed] [Google Scholar]
- 106. Onaka H. (2017) Novel antibiotic screening methods to awaken silent or cryptic secondary metabolic pathways in actinomycetes. J. Antibiot. 70, 865–870 [DOI] [PubMed] [Google Scholar]
- 107. Sharma K., D'Souza R. C., Tyanova S., Schaab C., Wisniewski J. R., Cox J., and Mann M. (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 8, 1583–1594 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are available via ProteomeXchange (http://www.proteomexchange.org/) with identifier PXD005558.