

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
Shear-induced, Gα13-mediated integrin outside-in signaling facilitates platelet PS exposure, MV release, and coagulation.
An inhibitor of outside-in signaling inhibits not only occlusive platelet thrombus formation but also intravascular coagulation in vivo.
Abstract
It is currently unclear why agonist-stimulated platelets require shear force to efficiently externalize the procoagulant phospholipid phosphatidylserine (PS) and release PS-exposed microvesicles (MVs). We reveal that integrin outside-in signaling is an important mechanism for this requirement. PS exposure and MV release were inhibited in β3−/− platelets or by integrin antagonists. The impaired MV release and PS exposure in β3−/− platelets were rescued by expression of wild-type β3 but not a Gα13 binding–deficient β3 mutant (E733EE to AAA), which blocks outside-in signaling but not ligand binding. Inhibition of Gα13 or Src also diminished agonist/shear-dependent PS exposure and MV release, further indicating a role for integrin outside-in signaling. PS exposure in activated platelets was induced by application of pulling force via an integrin ligand, which was abolished by inhibiting Gα13-integrin interaction, suggesting that Gα13-dependent transmission of mechanical signals by integrins induces PS exposure. Inhibition of Gα13 delayed coagulation in vitro. Furthermore, inhibition or platelet-specific knockout of Gα13 diminished laser-induced intravascular fibrin formation in arterioles in vivo. Thus, β3 integrins serve as a shear sensor activating the Gα13-dependent outside-in signaling pathway to facilitate platelet procoagulant function. Pharmacological targeting of Gα13-integrin interaction prevents occlusive thrombosis in vivo by inhibiting both coagulation and platelet thrombus formation.
Visual Abstract
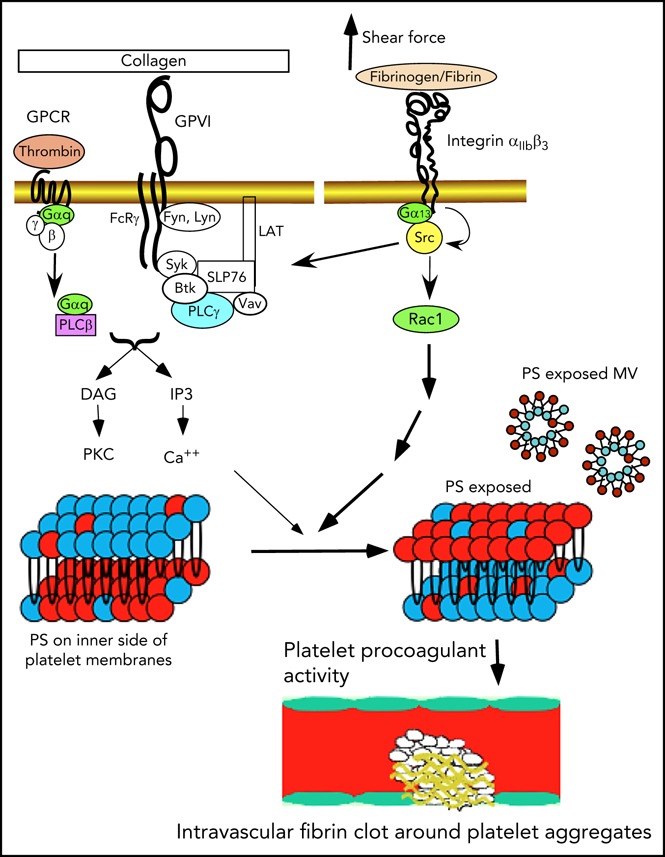
Introduction
Activated platelets facilitate coagulation (platelet procoagulant activity [PPA]) mainly by exposing the procoagulant phospholipid phosphatidylserine (PS) on the outer membrane surfaces and releasing PS-expressed microvesicles (MVs).1-6 Exposure of PS allows binding of important coagulation factors, greatly enhancing the catalytic efficiencies of coagulation enzymes.5 However, under static conditions in vitro, platelet agonists are inefficient in inducing PS exposure, even at high concentrations that robustly cause platelet activation.5 Even the combination of unusually high concentrations of collagen and thrombin together only induces partial platelet PS exposure.5,7 To resolve this discrepancy, we recently demonstrated that efficient PS exposure and MV release not only require agonist stimulation of platelets, but also require flow shear force.8 However, the mechanism responsible for the shear dependence remains unknown. Furthermore, some recent reports that partial inhibition of platelet thrombus formation using PAR4 antagonists or integrin antagonists had minimal effects on fibrin deposition in vivo raised controversy about whether platelets are important in intravascular coagulation in vivo.9,10 In this study, we demonstrate that platelet integrin outside-in signaling is the mechanism responsible for the role of shear force in PPA and is important in facilitating coagulation in vitro and in vivo.
Integrins mediate cell adhesion11-13 and bind their ligands with a catch bond,14,15 which is resistant to pulling force associated with shear. Integrins also transmit signals bidirectionally.11,12,16 Inside-out signaling activates the ligand binding function of integrins.17-20 Conversely, ligand binding to integrins induces outside-in signaling, leading to a series of cellular responses, including cell spreading, retraction, and migration.16,21 In vascular endothelial cells, it has been suggested that shear stress regulates integrin activity, leading to high-affinity ligand binding, which conversely transmits outside-in signals important in regulation of vascular integrity and tonicity and development of atherosclerosis.22-24 Thus, integrins are proposed as mechanical sensors in vascular endothelial cells, although it remains unclear whether shear stress activates ligand binding to integrins, indirectly leading to outside-in signaling, or whether shear stress directly induces outside-in signaling. In platelets, inside-out signaling activates the integrin αIIbβ3, which mediates platelet adhesion and aggregation. Conversely, outside-in signaling mediates platelet spreading and also greatly amplifies platelet thrombi.17,25-27 We recently demonstrated that a G protein subunit, Gα13, directly binds to integrin β3 cytoplasmic domain,28,29 and this interaction is important for the activation of c-Src and Src-dependent integrin outside-in signaling.28-33 Here we demonstrate that Gα13-dependent integrin outside-in signaling is important for shear-dependent platelet PS exposure and MV release induced by platelet agonists. Furthermore, we demonstrate that the integrin αIIbβ3 serves as a shear sensor that, via Gα13-dependent outside-in signaling, induces PS exposure and facilitates coagulation in vitro and in vivo. Importantly, pharmacologically targeting outside-in signaling potently inhibits both intravascular coagulation and platelet thrombi.
Methods
Animals
Integrin β3−/− mice, PF4-Cre mice, and C57BL/6 mice were obtained from the Jackson Laboratory. Gα13fl/fl mice were gifts from Stefan Offermanns, Max Planck Institute for Heart and Lung Research (Bad Nauheim, Germany).34 Platelet-specific Gα13 knockout mice were generated by breeding Gα13fl/fl with PF4-Cre mice. Animal usage and protocol were approved by the Institutional Animal Care Committee, University of Illinois at Chicago. Mice with similar age, weight, and sex ratios (1:1) were used. For cremaster arteriolar thrombosis model, only male mice were used.
Preparation of blood cells
Details are provided in supplemental Data (available on the Blood Web site).35,36 Platelets were resuspended in modified Tyrode’s buffer37 and allowed to rest for at least 1 hour at 22°C before use. Human subject study was approved by the institutional review boards at the University of Illinois at Chicago and Georgia Institute of Technology. Informed consent was provided by all donors according to the Declaration of Helsinki. Donors had not taken medication within 2 weeks before donation.
Flow cytometric analysis of platelet MV release and PS exposure
Platelet MV release and PS exposure were analyzed by flow cytometry as described previously.8 The washed platelets were stimulated with thrombin, collagen-related peptide (CRP), ADP, or TXA2 mimetic (U46619) for 8 minutes. Flow at defined shear rates was introduced for 8 minutes using a cone-and-plate rheometer (CT35 cone: 0.5°, 35 mm; Thermo Scientific Haake). The samples were then incubated with Alexa Fluor 488–conjugated annexin V (Invitrogen) for PS detection and analyzed using a flow cytometer (AccuriTM C6 flow cytometer; BD Biosciences) and FlowJo software (Tree Star Inc.) as previously described.38 The platelet MVs and platelets were distinguished according to their light scattering pattern as previously described39,40 and verified with anti-αIIb antibody staining.
Peptide inhibitors
Myristoylated peptides mP6 (Myr-FEEERA) and the corresponding control peptides, mP6Scr (Myr-ERAFEE) and AAA (Myr-FAAARA), were synthesized and purified at the Research Resource Center at the University of Illinois at Chicago or provided by Dupage Medical Technology, Inc. (Willowbrook, IL). The peptides were formulated as micellar nanoparticles as previously described29; details provided in supplemental Data.
Dual biomembrane force probe assay
In a chamber filled with modified Tyrode’s buffer, 2 biomembrane force probes (BFPs) were set up in parallel for stimulating integrin αIIbβ3 and reporting PS exposure, respectively (supplemental Data; supplemental Figure 1). The reporter BFP consisted of a micropipette aspirated human red blood cell with a glass bead coated with annexin V attached to its apex via biotin-streptavidin coupling.41-43 An opposing micropipette aspirated a platelet and was driven by a piezoelectric translator to move forward/backward to get in/out of contact with the annexin V bead to test for binding. The horizontal position of the bead was tracked by a high-speed camera, which reflected the red blood cell axial deformation. A tensile force signal after retraction of the piezo reported a binding event, whereas a 0 force indicated no binding. The approach-contact-retraction cycle was repeated 50 times for 4 platelet-bead pairs to render the average binding frequency. The stimulating BFP was similar to the reporter BFP, except that the glass bead was coated with fibronectin (FN). The platelet was first brought into repeated contact and retraction with the FN beads for a period of 5 minutes, with an average adhesion frequency of 80% (1.6 bonds per contact on average). Then the position of the platelet was quickly (<1 minute) realigned from the stimulating BFP to the reporting BFP for measurement of annexin V binding.
Bone marrow transplantation
Details are provided in supplemental Data and our previous publication.28,29
Recalcification time
Recalcification time of citrated platelet-rich plasma (PRP) was the time from addition of 20 mM of CaCl2 to the formation of a fibrin clot, which was monitored either in a cone-and-plate rheometer (Thermo Scientific Haake) or a turbidometric lumi-aggregometer (Chrono-Log) as described previously (supplemental Data).8
Fluorescence intravital microscopy
Laser-induced mouse cremaster arteriolar thrombosis was performed as described previously44; details are provided in supplemental Data. Platelet and fibrin accumulation around the site of injury was visualized by infusion of DyLight 649–labeled anti-mouse GPIbβ and Alexa Fluor 488–labeled monoclonal antifibrin antibody (clone 59D8) after laser-induced injury.45 The laser was adjusted to be sufficient to cause vascular endothelial injury and thrombosis, but not to cause a large penetration wound to the vessel wall. Fluorescence and bright-field images were recorded by an Olympus BX61W microscope with a 603/1.0 NA water immersion objective and a high-speed camera through an intensifier at 26/27/28 injury sites in 3 mice per group. The experiments were double blinded.
Statistics
Data analysis was performed in GraphPad Prism 5. Statistical significance was assessed using the Student t test for comparison of parametric data between 2 groups and using analysis of variance for comparing >2 groups. Comparisons of nonparametric data from recalcification and intravital microscopy experiments were performed using Mann-Whitney test.
Results
Important role for integrin β3 in shear-dependent MV release and PS exposure in thrombin-activated platelets
To determine the role of integrin β3 in shear-dependent MV release and PS exposure, washed wild-type and β3−/− platelets were stimulated with thrombin (0.05 U/mL) and subjected to defined shear rates (250-6000 s−1) induced by a cone-and-plate rheometer and analyzed using flow cytometry. As expected, neither wild-type nor integrin β3−/− platelets generated MVs or exposed PS under various levels of shear in the absence of thrombin (Figure 1A,C). Conversely, thrombin-stimulated platelets showed a shear-dependent increase in MV release, which reached maximal at ∼3000 s− (Figure 1B). These shear rates are well below the shear rates required for GPIb-IX–dependent MV release as reported in the literature.46 Interestingly, shear-dependent microvesiculation was abolished in β3−/− platelets stimulated with thrombin (Figure 1B), indicating that integrin β3 (predominantly αIIbβ3 in platelets) is critical for shear-dependent MV release in agonist-stimulated platelets. This integrin-dependent platelet MV release is different from previously characterized GPIb-IX–dependent platelet MV release, because integrin-mediated, shear-dependent MV release requires the presence of a platelet agonist. In fact, under shear, thrombin dose dependently increased the amount of MV release only in wild-type but not β3−/− platelets (Figure 1E).
Figure 1.

Thrombin-stimulated platelet MV release and PS exposure are shear dependent and require integrin αIIbβ3. (A) MV release in resting wild-type (WT) and β3 knockout (KO) mouse platelets under different shear rates as analyzed by flow cytometry. (B) MV release in 0.05 U/mL of thrombin-stimulated WT and β3 KO mouse platelets under different shear rates. (C) PS exposure on the surface of resting WT and β3 KO mouse platelets under different shear rates as indicated by annexin V binding. (D) PS exposure on the surface of 0.05 U/mL of thrombin-stimulated WT and β3 KO mouse platelets under different shear rates as indicated by annexin V binding. (E) MV release from WT and β3 KO mouse platelets stimulated with various doses of thrombin (0.0125, 0.025, and 0.05 U/mL) with or without shear (shear rate 3000 s−1). (F) PS exposure on the surfaces of WT and β3 KO mouse platelets stimulated with various doses of thrombin (0.0125, 0.025, and 0.05 U/mL) with or without flow shear. Data are presented as mean ± standard error of the mean (n = 3-4). **P < .01, ***P < .001, ****P < .0001.
Similar to MV release, PS exposure dramatically increased with increasing shear rates in thrombin-stimulated wild-type but not β3−/− platelets (Figure 1D). Furthermore, PS exposure in wild-type platelets, but not β3−/− platelets, also dose dependently increased in response to increasing thrombin concentrations under shear (Figure 1F). These data indicate that β3 plays an important role in thrombin- and shear-stimulated platelet PS exposure and MV release.
Importance of integrin β3 in shear-dependent MV release and PS exposure induced by CRP
To exclude the possibility that the role of integrins in shear-dependent MV release and PS exposure is restricted to the signaling pathways activated by thrombin, we also investigated the role of integrin β3 in shear-dependent MV release and PS exposure in CRP-stimulated platelets. CRP induces platelet activation via the GPVI-ITAM signaling pathway. Similar to thrombin-stimulated platelets, shear-dependent MV release and PS exposure dose dependently increased in response to CRP in wild-type but not β3−/− platelets (Figure 2A-B). These data indicate that integrin αIIbβ3 plays an important role in shear-dependent MV release and PS exposure induced by both the thrombin receptor and GPVI signaling pathways. Interestingly, U46619 and ADP failed to induce observable PS exposure and MV release even with shear (supplemental Figure 2). Thus, it seems that platelet PS exposure and MV release are selective for certain platelet activation pathways. β3 integrins have an important role in agonist/shear-dependent platelet PS exposure and MV release.
Figure 2.

MV release and PS exposure in CRP-activated platelets are shear dependent and require integrin αIIbβ3. (A) MV release from wild-type (WT) or β3 knockout (KO) platelets stimulated with increasing doses of CRP (0.5, 0.75, and 1.0 µg/mL) with or without shear (shear rate 3000 s−1). (B) PS exposure on WT or β3 KO platelets stimulated with increasing doses of CRP (0.5, 0.75, and 1.0 µg/mL) with or without shear. Data are presented as mean ± standard error of the mean (n = 3-5). ****P < .0001.
Role of Gα13-dependent integrin outside-in signaling in MV release and PS exposure
Ligand binding to integrins not only mediates platelet adhesion and aggregation, but also transmits outside-in signaling, which requires the interaction between Gα13 and the cytoplasmic domain of the integrin β3 subunit. To assess whether outside-in signaling is responsible for shear-dependent MV release and PS exposure, bone marrow stem cells from β3−/− mice were infected with lentivirus encoding the complementary DNA sequence of wild-type or a Gα13 binding–deficient mutant (E733EE to AAA) of β3 (which is defective in outside-in signaling without affecting ligand binding29) and then transplanted into recipient integrin β3−/− mice after irradiation. Six weeks after transplantation, platelets from the recipient mice were analyzed for MV release and PS exposure. Thrombin-induced, shear-dependent MV release and PS exposure in β3−/− platelets were rescued by wild-type β3 expression to the level in wild-type mice (Figure 3A-B). In contrast, β3−/− platelets expressing AAA mutant β3 (Figure 3A-B) remained defective in MV release and PS exposure. Similar results were obtained when the recipient mouse platelets were stimulated with CRP (Figure 3C-D). To further determine whether Gα13-dependent outside-in signaling is important in shear-dependent MV release and PS exposure in agonist-stimulated platelets, human and mouse platelets were treated with a peptide inhibitor of Gα13-integrin interaction, mP6, as previously described.29 As compared with the negative control peptide, mP6 potently inhibited shear- and agonist-induced MV release and PS exposure (Figure 4A-D) both in human and mouse platelets.
Figure 3.

Comparison of wild-type (WT) and Gα13binding–deficient mutant β3in agonist and shear-dependent MV release and PS exposure. (A) MV release from 3000 s−1 of shear/0.05 U/mL of thrombin-stimulated β3−/− platelets expressing WT or AAA733-mutant β3 (Gα13 binding deficient) transplanted using bone marrow stem-cell transplantation technique. (B) PS exposure on platelets expressing WT or AAA733-mutant β3 treated with thrombin and shear. (C) MV release from platelets expressing WT or AAA733-mutant β3 treated with CRP (1 µg/mL) and shear. (D) PS exposure on platelets expressing WT or AAA733-mutant β3 treated with CRP and shear. Data are presented as mean ± standard error of the mean (n = 4). ****P < .0001.
Figure 4.

Inhibition of shear-dependent platelet MV release and PS exposure in mouse and human platelets by mP6, a peptide inhibitor of Gα13-integrin interaction and outside-in signaling. (A) Effects of mP6 or a negative control peptide (AAA mutant, Myr-FAAARA) on MV release from mouse platelets stimulated by 0.05 U/mL of thrombin with shear of 3000 s−1 as analyzed by flow cytometry. (B) Effects of mP6 or a control peptide on PS exposure on mouse platelets stimulated by 0.05 U/mL of thrombin with shear of 3000 s−1 as indicated by annexin V binding. (C) Effects of mP6 or a control peptide on MV release from human platelets stimulated with 0.05 U/mL of thrombin with or without shear. (D) Effects of mP6 (40 µM) or a control peptide on PS exposure on human platelets stimulated with 0.05 U/mL of thrombin with or without shear. (E) MV release from thrombin- and shear-stimulated mouse platelets treated with dimethyl sulfoxide (DMSO; vehicle control), Src inhibitor PP2, integrin antagonist integrilin, or Rac1 inhibitor NSC23766. (F) PS exposure on thrombin- and shear-stimulated platelets treated with DMSO, PP2 (10 µM), integrilin (20 µg/mL), or NSC23766 (100 µM). (G) MV release from CRP- and shear-stimulated platelets treated with DMSO, PP2, integrilin, or NSC23766. (H) PS exposure on CRP- and shear-stimulated platelets treated with DMSO, PP2, integrilin, or NSC23766. Data are presented as mean ± standard error of the mean (n = 3-4). **P < .01, ***P < .001, and ****P < .0001.
We further investigated the effect of inhibitors of Src and Rac1 in integrin-dependent MV release and PS exposure in platelets stimulated with thrombin or CRP under shear. Both the Src inhibitor PP2 and Rac1 inhibitor NSC23766 significantly inhibited platelet MV release and PS exposure (Figure 4E-H) in platelets stimulated with thrombin and CRP under shear, suggesting that the outside-in signaling pathway leading to PS exposure and MV release requires Src and Rac1. Together, these data indicate a Gα13-Src/Rac1 outside-in signaling pathway is important for agonist-induced, shear-dependent MV release and PS exposure both in mouse and human platelets.
Integrin αIIbβ3 serves as a mechanical force sensor in mediating PS exposure in activated platelets
To understand why integrin outside-in signaling is important in agonist- and shear-induced PS exposure and the release of PS-exposed MVs, we used a newly developed dual BFP assay (supplemental Figure 1) to characterize the effect of pulling force applied to integrin αIIbβ3 through the engaged ligand FN on platelet PS exposure on a single-platelet level. The platelet under interrogation was first allowed to repeatedly touch the FN-conjugated force probe to engage and pull on αIIbβ3 at a defined frequency and reoriented to repeatedly contact a second, annexin V–conjugated force probe to detect binding of the exposed PS. The frequency of resting platelet binding to the annexin V probe was near 0 without prestimulation by pulling αIIbβ3 via FN, reflecting the fact that resting platelets minimally expose PS. Thrombin stimulation alone only moderately increased binding frequency (Figure 5A). In contrast, when a low number (1.6 per contact on average) of integrin αIIbβ3-FN bonds was allowed to form on the platelet surface and force was applied to pull on these bonds, a marked further increase in annexin V binding frequency of thrombin-activated platelets was observed, indicating that pulling force applied through integrin-ligand interaction elicited a signal leading to exposure of PS (Figure 5A). This mechanically induced integrin signaling leading to PS exposure is likely the Gα13-dependent outside-in signal, because the annexin V binding was reduced to the level of thrombin-stimulated platelets without the FN interaction when platelets were preincubated with mP6, which inhibited Gα13-integrin interaction and outside-in signaling (Figure 5A), but not the scrambled peptide (Figure 5A). These data mechanistically explain why integrin signaling is important in shear force–dependent PS exposure and suggest that by interacting with its ligands, integrin αIIbβ3 serves as a mechanical force sensor, converting the mechanical force to outside-in signaling, leading to PS exposure.
Figure 5.

BFP analysis of integrin-dependent transduction of mechanical force leading to PS exposure and in vitro analysis of the role of integrin outside-in signaling in PPA. (A) Integrin ligand FN-conjugated BFP was allowed to repeatedly touch and pull the membrane surface of a resting or thrombin-stimulated platelet treated with or without mP6 or a scrambled control peptide (mP6Scr). Annexin V binding frequency to the same platelet was then assessed using a second BFP conjugated with annexin V (supplemental Figure 1). Data are shown as median (center bars) ± interquartile range (boxes) and maximal/minimal range (range bars). Note that annexin V binding to thrombin-stimulated platelets was significantly enhanced by frequent pulling applied through FN-coated BFP, which was inhibited by mP6 but not by mP6Scr. (B) The recalcification-induced clotting time (mean ± standard error of the mean [SEM]) of citrated human platelet-depleted plasma reconstituted with washed wild-type (WT) or β3−/− mouse platelets (WT, n = 6; β3−/−−, n = 8) under stirring conditions. (C-D) The recalcification-induced clotting time (mean ± SEM) of human citrated platelet-rich plasma treated with 40 µM of mP6 or a control peptide was monitored under stirring conditions in a turbidometric platelet aggregometer (C) (n = 9) or detected using a cone-and-plate rheometer under shear rate of 6000 s−1 (D) (n = 5). *P < .05, **P < .01, and ****P < .0001. N.S., not significant.
Role of integrin outside-in signaling in promoting coagulation in vitro
To determine whether outside-in signaling-mediated PS exposure and MV release is important in coagulation, we measured the clotting time of citrated PRP after recalcification under the flow induced by stirring in vitro. Integrin β3−/− platelets suspended in human platelet-poor plasma showed significantly longer clotting time than wild-type platelets suspended in human platelet-poor plasma (Figure 5B). Strikingly, incubation of citrated human PRP or mouse platelet-containing plasma with mP6, but not its negative control peptide mP6Scr, also delayed the occurrence of coagulation under flow conditions induced by stirring or under high shear (6000 s−1; Figure 5C-D). Thus, integrin outside-in signaling is important in promoting coagulation.
Role of integrin outside-in signaling in promoting fibrin formation in vivo
To assess the role of outside-in signaling in fibrin formation in vivo, we investigated the effect of an outside-in signal inhibitor, mP6,29 on thrombus formation in a laser-induced cremaster arteriolar thrombosis model. Under our experimental conditions (laser intensity is sufficient for endothelial injury, but not for burning large holes in blood vessels), platelet thrombi formed rapidly at the site of arteriolar wall injury, but the thrombus size rapidly decreased after peaking at ∼2 minutes. Fibrin formation, however, showed a significant delay and followed platelet thrombus formation. In addition to the major intravascular fibrin formation at the site of laser-induced injury (Figure 6) that expanded into the lumen, we also observed occasional fibrin formation located outside the vessel lumen near the injury sites. The extravascular fibrin formation seemed to be independent of the bulk of the intravascular platelet thrombus (see Movie 2), which is consistent with the knowledge that tissue factor and PS can be provided by many types of cells/tissues.47,48 However, the intravascular fibrin formed was associated with platelet thrombi and stabilization of platelet thrombi (Figure 6A-B). The sizes of the fibrin clot and platelet thrombus were both inhibited in mice treated with mP6 but not scrambled control peptide (Figure 6C-D). To exclude possible off-target effects of mP6, we also examined intravascular platelet thrombus and fibrin formation in platelet-specific Gα13 knockout mice (Gα13fl/fl–PF4-Cre) as compared with wild-type control from the same genetic background (Gα13fl/fl) using the laser-induced cremaster arteriolar thrombosis model. Mice deficient in platelet Gα13 showed a dramatic inhibition in both fibrin formation and platelet thrombus size as compared with control mice (Figure 7), which mirrored the effect of mP6. Thus, our data suggest that Gα13-dependent outside-in signaling of integrin αIIbβ3 is important for intravascular fibrin clot formation in vivo and, importantly, that selective inhibition of integrin outside-in signaling inhibits not only platelet thrombus formation but also intravascular fibrin clot formation in vivo.
Figure 6.

The effect of mP6 on fibrin generation and platelet thrombus formation in vivo using laser-induced mouse arteriolar thrombosis model. Intravital microscopy was used to monitor fibrin generation and platelet thrombi in vivo after laser-induced cremaster arteriole wall injury in wild-type mice treated with or without selective outside-in signaling inhibitor mP6 (10 µmol/kg) or scrambled control peptide (mP6Scr; 10 µmol/kg). The median integrated (Int.) fluorescence (FL) signals of fibrin (A) and platelets (B) from 28 thrombi in mP6-treated mice, 26 thrombi in mP6Scr-treated mice, or 27 thrombi in untreated mice are shown as a function of time. The median of the total FL detected over time for fibrin (C) or platelets (D) is shown, calculated by integrating the area under the curve. (E) Representative images of fibrin generation (green) and platelet thrombi formation (red) and merged images. ****P < .0001.
Figure 7.

Diminished fibrin generation and platelet thrombus formation in Gα13 fl/fl–PF4-Cre mice in vivo. Intravital microscopy was used to monitor fibrin generation and platelet thrombi in vivo after laser-induced cremaster arteriole wall injury in Gα13fl/fl control and Gα13fl/fl–PF4-Cre mice. (A) Western-blot analysis of Gα13 expression in platelets from Gα13fl/fl and Gα13fl/fl–PF4-Cre mouse. (B-C) The median integrated (Int.) fluorescence (FL) signals of fibrin (B) and platelets (C) from 25 thrombi in Gα13fl/fl control mice and from 27 thrombi in Gα13fl/fl–PF4-Cre mice are shown as a function of time. (D-E) The median of the total FL detected over time for fibrin (D) or platelets (E) is shown, calculated by integrating the area under the curve. (F) Representative images of fibrin generation (green) and platelet thrombi formation (red) and merged images. ***P < .001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
In this study, we made a significant advance in our understanding of the mechanism that regulates the procoagulant response of platelets to shear. We demonstrate that integrin αIIbβ3 serves as a mechanical sensor that transduces mechanical force into outside-in signals, which, via a Gα13-, Src-, and Rac1-dependent signaling pathway, leads to PS exposure and PS-exposed MV release. This integrin outside-in signaling pathway is important for facilitating coagulation in vitro and intravascularly in vivo. Importantly, we show that a selective integrin outside-in signaling inhibitor is potently antithrombotic, because it inhibits not only platelet thrombus expansion but also intravascular coagulation at the site of vascular injury.
Our discovery that integrin outside-in signaling is critically important for platelet PS exposure, MV release, and procoagulant function is consistent with previous knowledge of the important roles of platelets and integrin αIIbβ3 in promoting coagulation. Despite numerous early evidence that platelets play a critical role in promoting coagulation, there have been recent controversies with regard to the roles of platelets and integrin αIIbβ3 in facilitating coagulation in vivo. Studies from 2 laboratories showed significant inhibitory effects of integrin antagonists on platelet thrombus formation but not on fibrin deposition at the site of vascular injury using the laser-induced mouse arteriolar thrombosis model.9,10 However, another study from Cho et al of the Furie group, using the same technology, demonstrated defective platelet thrombus formation and fibrin clot formation in integrin β3 knockout mice.44 In analyzing these apparently contradictory results, we noted 2 major differences. First, the studies by Cho et al44 used limited laser power/duration that was sufficient to injure the endothelial cells causing thrombosis, but insufficient to cause a major penetrating hole in the blood vessel. The laser power used in some other studies, however, caused a more severe penetrating injury, with fibrin located primarily outside of the vascular lumen.9 Thus, platelets are required to provide sufficient PS surface for intralumen coagulation, but coagulation may also be facilitated by PS provided by cells on or outside of the blood vessel wall. Second, the studies showing ineffective inhibition of fibrin clot formation by integrin inhibitors and PAR4 antagonists also showed only partial inhibition of platelet thrombus formation,9,10 whereas studies using the β3 knockout mice completely inhibited platelet thrombus formation as expected with total lack of this receptor.44 Considering the vast reserve capacity of platelets in PPA, it is likely that the remaining platelet activation and integrin ligation after partial inhibition are already sufficient to transmit integrin outside-in signaling, leading to the induction of PPA. When the signaling molecules important for integrin outside-in signaling are inhibited, such as in the case of inhibition of Gα13-integrin interaction reported here, we have observed significantly diminished PPA without inhibition of the ligand binding function of integrins per se. Thus, combining previous controversies and the current data, it is clear that integrin outside-in signaling, but not the ligand binding function of integrin αIIbβ3 or its activation per se, is important for PPA in vivo intravascularly. Furthermore, we demonstrate that integrin αIIbβ3 functions as a mechanical force sensor triggering a Gα13/Src-dependent signaling pathway that cooperates with agonist receptor signaling pathways to induce PS exposure and the release of PS-exposed MVs, which are important for intravascular coagulation. However, we do not exclude the possibility that other conditions of platelet activation might be discovered to replicate or substitute for shear-induced integrin signaling, even though we are not aware of what these conditions are at the present time.
Platelets have 2 distinct adhesion receptor families that are known to be important in platelet adhesion under shear stress: the platelet vWF receptor, GPIb-IX, and integrins. GPIb-IX has been previously reported to mediate platelet microparticle formation induced by shear stress, although at much higher shear stress than we applied, and GPIb-IX–dependent shear stress–induced microparticle (microvesicle) release does not require stimulation by other agonists.49,50 Our study clearly shows that physiological levels of shear stress alone are not sufficient to induce microvesiculation or PS exposure. Instead, agonist stimulation and β3 integrin are both required for platelet PS exposure and MV release. Importantly, we have directly demonstrated, at a molecular level, that integrin ligand-mediated pulling force induces mechanical signal transduction via the Gα13-dependent integrin outside-in signaling pathway, leading to PS exposure. This discovery is significant to elucidate not only the mechanisms of PPA induction but also integrin-mediated mechanical signaling in other types of vascular cells with quite similar integrin outside-in signaling mechanisms. These cells, such as leukocytes and vascular endothelial cells, are exposed to shear stress of blood flow and release PS+ MVs during activation, inflammation, and injury. It would be interesting to further investigate whether similar mechanisms of activation of PS exposure and MV release occur in these cells.
Although we have demonstrated an important role for Gα13-dependent integrin outside-in signaling in shear-dependent platelet PS exposure and MV release, the responsible downstream signaling mechanisms remain unclear. Previous studies have demonstrated the importance of intracellular calcium elevation and the calcium-activated ion channel TMEM16F in PS exposure and MV release.1,5,51 Because integrin ligation is known to induce cytoplasmic calcium elevation,52,53 it is possible that shear/outside-in signaling greatly elevates cytoplasmic calcium to the level that is not otherwise achieved by the agonist receptor signaling pathways. Thus, it is tempting to hypothesize that shear-dependent integrin outside-in signaling serves as physiological calcium ionophore, greatly elevating cytoplasmic calcium after agonist stimulation, inducing PS exposure and MV release. However, PS exposure and MV release seem to be agonist selective. TXA2 analog, U46619, a potent platelet agonist that stimulates robust calcium elevation, does not seem to induce significant PS exposure or microvesiculation. Also, integrin deficiency reportedly only affects calcium influx induced by low-dose thrombin, but not high-dose thrombin.52 Furthermore, shear not only promotes platelet PS exposure and microvesiculation induced by physiological agonists, but also greatly enhances PS exposure and MV release induced by calcium ionophore A23187.8 Thus, it is possible that shear/integrin-dependent PS exposure and microvesiculation may also involve mechanisms other than calcium elevation. In any case, integrin outside-in signaling serves as an important physiological facilitator of platelet PS exposure, microvesiculation, and PPA, and its downstream mechanisms warrant in-depth further investigation.
Not only is the discovery of an important role for integrin outside-in signaling in platelet PS exposure and MV release significant to our understanding of the mechanisms that regulate PPA, but it also has important implications in antiplatelet drug development. Recent studies indicate that selective targeting of integrin outside-in signaling as a new antithrombotic strategy has the advantage of potently inhibiting occlusive platelet thrombus expansion without causing excessive bleeding.29 This study further indicates that selective inhibition of integrin outside-in signaling not only inhibits platelet thrombus expansion, but also inhibits intravascular fibrin clot formation. This is significant, particularly in view of the data showing that partial inhibition of platelet thrombus formation by platelet activation inhibitors or by integrin inhibitors is not effective in inhibiting fibrin clot formation,9,10 whereas complete inhibition of integrin ligand binding function is likely to cause excessive bleeding. Our work suggests that by targeting integrin outside-in signaling without blocking platelet adhesion and primary aggregation, it is possible to diminish platelet-dependent intravascular fibrin clot without causing excessive bleeding. Because the intravascular fibrin clot around the platelet thrombus is associated with thrombus expansion and stabilization, this novel effect of outside-in signaling inhibitors is likely to contribute to the potency and anticlotting effect of these new drugs, making them superior to current antiplatelet drugs.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Stefan Offermanns, Max Planck Institute for Heart and Lung Research (Bad Nauheim, Germany), for providing Gα13fl/fl mice; Hartmut Weiler, The Blood Research Institute (Milwaukee, WI), for kindly providing the antifibrin antibody; and Gus Cho at University of Illinois at Chicago for instructing on the laser-induced thrombosis model.
This work is supported in part by National Institutes of Health (NIH), National Heart, Lung and Blood Institute grants HL080264, HL062350, HL125356 (X.D.), and HL132019 (C.Z.), NIH, National Institute of Allergy and Infectious Diseases grant AI044902 (C.Z.) and by NIH, National Heart, Lung and Blood Institute contracts HHSN268201400007C (X.D.) and HHSN268201700002C (M.G.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.P. and Y. Cui equally contributed to performing experiments, analyzing/interpreting data, and preparing the manuscript; M.K.D., N.C., and A.S.-T. performed experiments; M.G. participated in discussions; Y. Chen and C.Z. performed dual biomembrane force probe assay and prepared the manuscript; and X.D. designed research, analyzed/interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: University of Illinois at Chicago (UIC) holds patents relevant to the study. X.D. has ownership interests in DMT, Inc., which licenses the UIC technology.
The current affiliation for Y. Chen is Department of Molecular Medicine, MERU-Roon Research Center on Vascular Biology, The Scripps Research Institute, La Jolla, CA.
Correspondence: Xiaoping Du, 835 S. Wolcott Ave, Room E403, Chicago, IL 60612; e-mail: xdu@uic.edu.
References
- 1.Bevers EM, Comfurius P, van Rijn JL, Hemker HC, Zwaal RF. Generation of prothrombin-converting activity and the exposure of phosphatidylserine at the outer surface of platelets. Eur J Biochem. 1982;122(2):429-436. [DOI] [PubMed] [Google Scholar]
- 2.Niemetz J, Marcus AJ. The stimulatory effect of platelets and platelet membranes on the procoagulant activity of leukocytes. J Clin Invest. 1974;54(6):1437-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vecchione J, Zucker MB. Procoagulant activity of platelets in recalcified plasma. Br J Haematol. 1975;31(4):423-428. [DOI] [PubMed] [Google Scholar]
- 4.Bevers EM, Comfurius P, Zwaal RF. Platelet procoagulant activity: physiological significance and mechanisms of exposure. Blood Rev. 1991;5(3):146-154. [DOI] [PubMed] [Google Scholar]
- 5.Bevers EM, Comfurius P, Zwaal RF. Changes in membrane phospholipid distribution during platelet activation. Biochim Biophys Acta. 1983;736(1):57-66. [DOI] [PubMed] [Google Scholar]
- 6.Geddings JE, Mackman N. New players in haemostasis and thrombosis. Thromb Haemost. 2014;111(4):570-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alberio L, Safa O, Clemetson KJ, Esmon CT, Dale GL. Surface expression and functional characterization of alpha-granule factor V in human platelets: effects of ionophore A23187, thrombin, collagen, and convulxin. Blood. 2000;95(5):1694-1702. [PubMed] [Google Scholar]
- 8.Delaney MK, Liu J, Kim K, et al. Agonist-induced platelet procoagulant activity requires shear and a Rac1-dependent signaling mechanism. Blood. 2014;124(12):1957-1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vandendries ER, Hamilton JR, Coughlin SR, Furie B, Furie BC. Par4 is required for platelet thrombus propagation but not fibrin generation in a mouse model of thrombosis. Proc Natl Acad Sci USA. 2007;104(1):288-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivanciu L, Krishnaswamy S, Camire RM. New insights into the spatiotemporal localization of prothrombinase in vivo. Blood. 2014;124(11):1705-1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673-687. [DOI] [PubMed] [Google Scholar]
- 12.Ginsberg MH, Partridge A, Shattil SJ. Integrin regulation. Curr Opin Cell Biol. 2005;17(5):509-516. [DOI] [PubMed] [Google Scholar]
- 13.Bledzka K, Smyth SS, Plow EF. Integrin αIIbβ3: from discovery to efficacious therapeutic target. Circ Res. 2013;112(8):1189-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong F, García AJ, Mould AP, Humphries MJ, Zhu C. Demonstration of catch bonds between an integrin and its ligand. J Cell Biol. 2009;185(7):1275-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Lee H, Tong H, Schwartz M, Zhu C. Force regulated conformational change of integrin αVβ3. Matrix Biol. 2017;60-61:70-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Estevez B, Du X. New concepts and mechanisms of platelet activation signaling. Physiology (Bethesda). 2017;32(2):162-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tadokoro S, Shattil SJ, Eto K, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302(5642):103-106. [DOI] [PubMed] [Google Scholar]
- 18.Ye F, Kim C, Ginsberg MH. Molecular mechanism of inside-out integrin regulation. J Thromb Haemost. 2011;9(suppl 1):20-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fässler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14(3):325-330. [DOI] [PubMed] [Google Scholar]
- 20.Ma YQ, Qin J, Wu C, Plow EF. Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J Cell Biol. 2008;181(3):439-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr Opin Cell Biol. 2012;24(5):600-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 2001;20(17):4639-4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tzima E, Irani-Tehrani M, Kiosses WB, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437(7057):426-431. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Luo JY, Li B, et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature. 2016;540(7634):579-582. [DOI] [PubMed] [Google Scholar]
- 25.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104(6):1606-1615. [DOI] [PubMed] [Google Scholar]
- 26.Law DA, Nannizzi-Alaimo L, Phillips DR. Outside-in integrin signal transduction. Alpha IIb beta 3-(GP IIb IIIa) tyrosine phosphorylation induced by platelet aggregation. J Biol Chem. 1996;271(18):10811-10815. [DOI] [PubMed] [Google Scholar]
- 27.Flevaris P, Li Z, Zhang G, Zheng Y, Liu J, Du X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood. 2009;113(4):893-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong H, Shen B, Flevaris P, et al. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin “outside-in” signaling. Science. 2010;327(5963):340-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen B, Zhao X, O’Brien KA, et al. A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature. 2013;503(7474):131-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30(12):2341-2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obergfell A, Eto K, Mocsai A, et al. Coordinate interactions of Csk, Src, and Syk kinases with [alpha]IIb[beta]3 initiate integrin signaling to the cytoskeleton. J Cell Biol. 2002;157(2):265-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS, Ginsberg MH, Shattil SJ. Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc Natl Acad Sci USA. 2003;100(23):13298-13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boylan B, Gao C, Rathore V, Gill JC, Newman DK, Newman PJ. Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood. 2008;112(7):2780-2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moers A, Nieswandt B, Massberg S, et al. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat Med. 2003;9(11):1418-1422. [DOI] [PubMed] [Google Scholar]
- 35.Li Z, Zhang G, Le Breton GC, Gao X, Malik AB, Du X. Two waves of platelet secretion induced by thromboxane A2 receptor and a critical role for phosphoinositide 3-kinases. J Biol Chem. 2003;278(33):30725-30731. [DOI] [PubMed] [Google Scholar]
- 36.Estevez B, Kim K, Delaney MK, et al. Signaling-mediated cooperativity between glycoprotein Ib-IX and protease-activated receptors in thrombin-induced platelet activation. Blood. 2016;127(5):626-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flevaris P, Stojanovic A, Gong H, Chishti A, Welch E, Du X. A molecular switch that controls cell spreading and retraction. J Cell Biol. 2007;179(3):553-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schoenwaelder SM, Yuan Y, Josefsson EC, et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood. 2009;114(3):663-666. [DOI] [PubMed] [Google Scholar]
- 39.Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327(5965):580-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lacroix R, Robert S, Poncelet P, Kasthuri RS, Key NS, Dignat-George F; ISTH SSC Workshop. Standardization of platelet-derived microparticle enumeration by flow cytometry with calibrated beads: results of the International Society on Thrombosis and Haemostasis SSC Collaborative workshop. J Thromb Haemost. 2010;8(11):2571-2574. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, Liu B, Ju L, et al. Fluorescence biomembrane force probe: concurrent quantitation of receptor-ligand kinetics and binding-induced intracellular signaling on a single cell. J Vis Exp. 2015;4(102):e52975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosetti F, Chen Y, Sen M, et al. A lupus-associated mac-1 variant has defects in integrin allostery and interaction with ligands under force [published online ahead of print 11 March 2015]. Cell Reports. doi:10.1016/j.celrep.2015.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ju L, Chen Y, Li K, et al. Dual biomembrane force probe enables single-cell mechanical analysis of signal crosstalk between multiple molecular species. Sci Rep. 2017;7(1):14185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho J, Kennedy DR, Lin L, et al. Protein disulfide isomerase capture during thrombus formation in vivo depends on the presence of β3 integrins. Blood. 2012;120(3):647-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atkinson BT, Jasuja R, Chen VM, Nandivada P, Furie B, Furie BC. Laser-induced endothelial cell activation supports fibrin formation. Blood. 2010;116(22):4675-4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pontiggia L, Steiner B, Ulrichts H, Deckmyn H, Forestier M, Beer JH. Platelet microparticle formation and thrombin generation under high shear are effectively suppressed by a monoclonal antibody against GPIba. Thromb Haemost. 2006;96(6):774-780. [PubMed] [Google Scholar]
- 47.Rao LV, Pendurthi UR. Regulation of tissue factor coagulant activity on cell surfaces. J Thromb Haemost. 2012;10(11):2242-2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Segawa K, Nagata S. An apoptotic ‘eat me’ signal: phosphatidylserine exposure. Trends Cell Biol. 2015;25(11):639-650. [DOI] [PubMed] [Google Scholar]
- 49.Holme PA, Orvim U, Hamers MJ, et al. Shear-induced platelet activation and platelet microparticle formation at blood flow conditions as in arteries with a severe stenosis. Arterioscler Thromb Vasc Biol. 1997;17(4):646-653. [DOI] [PubMed] [Google Scholar]
- 50.Reininger AJ, Heijnen HF, Schumann H, Specht HM, Schramm W, Ruggeri ZM. Mechanism of platelet adhesion to von Willebrand factor and microparticle formation under high shear stress. Blood. 2006;107(9):3537-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H, Kim A, David T, et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell. 2012;151(1):111-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Powling MJ, Hardisty RM. Glycoprotein IIb-IIIa complex and Ca2+ influx into stimulated platelets. Blood. 1985;66(3):731-734. [PubMed] [Google Scholar]
- 53.Yamaguchi A, Yamamoto N, Kitagawa H, Tanoue K, Yamazaki H. Ca2+ influx mediated through the GPIIb/IIIa complex during platelet activation. FEBS Lett. 1987;225(1-2):228-232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.