

Abstract
Motivation
Computer simulations are excellent tools for understanding the evolutionary and genetic consequences of complex processes that cannot be analytically predicted and for creating realistic genetic data. There are many software packages that simulate genetic data, but they are typically not fast or memory efficient enough to simulate realistic, individual-level genome-wide SNP/sequence data.
Results
GeneEvolve is a user-friendly and efficient population genetics simulator that handles complex evolutionary and life history scenarios and generates individual-level phenotypes and realistic whole-genome sequence or SNP data. GeneEvolve runs forward-in-time, which allows it to provide a wide range of scenarios for mating systems, selection, population size and structure, migration, recombination and environmental effects. The software is designed to use as input data from real or previously simulated phased haplotypes, allowing it to mimic very closely the properties of real genomic data.
Availability and Implementation
GeneEvolve is freely available at https://github.com/rtahmasbi/GeneEvolve.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
GeneEvolve is C ++ code for simulating individual-level genome-wide data using an object-oriented approach. Unlike coalescent (Kingman, 1982) based simulators, GeneEvolve runs forward-in-time, which allows it to provide a wide range of scenarios for selection, population size and structure, migration, recombination and familial effects. Coalescent approaches are fast but they can have serious limitations when there is a large recombination rate over the simulated genomic region, and they have difficulty modeling many complex scenarios of interest (Davies et al., 2007). On the other hand, leading forward-in-time simulators such as simuPOP (Peng and Kimmel, 2005), Fregene (Chadeau-Hyam et al., 2008), ForSim (Lambert et al., 2008), quantiNemo (Neuenschwander et al., 2008) and SLiM (Messer, 2013) limit the size of genome and/or population for computational efficiency. In general, the forward-in-time simulators are slow and are not memory efficient because they store and work with all the genotypic data in each generation; the computational complexity of these benchmark forward-in-time simulators is O(loci*individuals*populations*generations). In Section 2, we show how genomes and the evolutionary process can be accurately and efficiently simulated by representing chromosomes solely as the identity and termini of haplotypes from a base population, which the user inputs. As a result, GeneEvolve runs in O(individuals*populations*generations), allowing it to easily simulate whole-genome SNP or sequence data. Moreover, this strategy allows GeneEvolve to simulate data that closely mimics all the properties of the (potentially real) data it uses as input for the base population. Finally, GeneEvolve can simulate multiple causes of environmental and genetic influences across a wide range of mating, selection and life history scenarios, and it can track and extract the true identity by descent information between all pairs of individuals in the simulated population.
2 Description
2.1 Features
Full explanation for all features and details are available in the online documentation. Here, we provide a brief overview of the main features of GeneEvolve.
Simulating genotypes: Given a reference panel of phased chromosomes as input, where each chromosome is typed at L bi-allelic sites, the program chooses mates, and each offspring chromosome is a recombination of one of the parent’s two chromosomes, where the recombination rate is defined in a recombination map inputted by the user. The reference chromosomes and recombination rates can be from previously simulated genotype data or, for more realistic simulations, from real, phased SNP/sequence datasets and published recombination maps.
The position of each recombination is saved and we represent each chromosome as a continuous sequences of half-open intervals that denote the termini and identity of haplotypes from the founder population. Clearly, working with a continuous sequence of intervals is much faster and more memory efficient than working with real genotypes (see online documentation, Chapter 3). Therefore, GeneEvolve can simulate very large sequence or SNP datasets. The only difference variant density makes to computational efficiency is in reading and writing the simulated genotypes.
Simulating phenotypes: For each individual i in biparental family f and population p, the phenotype is simulated through
| (1) |
where Ai is the additive genetic (or breeding) value which depends on user inputted additive effects, aj, and the MAFs of the m causal variants (CVs) and Di is the dominance genetic value which depends on the dominance effects, dj, and the MAFs of the CVs. The terms Ei, Fi, Cf and γp are unique, familial, shared sibling (common), and population specific environmental values, respectively (for more information, see the online documentation). Users can also directly specify the variances of each effect (A, D, E, etc.) in the first generation. In this case, GeneEvolve will linearly transform the variance components such that they equal the user-inputted values in the first generation; the variances in subsequent generations may then evolve away from these values as a consequence of assortative mating, selection, drift and so forth. Finally, GeneEvolve can simulate multiple genetically and environmentally correlated phenotypes simultaneously.
Mating system: Users can specify the correlation (which can change across time) between phenotypes of mates. For multiple phenotypes, the mating phenotype is a user-specified linear combination of each phenotype. User can also choose monogamous or polygamous mating systems and can disallow close inbreeding.
Natural selection: Users can specify the strength and type of natural selection acting on the phenotype. For multiple phenotypes, selection acts upon a user-specified linear combination of each phenotype.
Migration: By defining a migration matrix per generation, users can model Wright’s Island model or arbitrarily more complex models.
Identical by descent segments: GeneEvolve output can be processed by a supplementary program we wrote (available at the main GeneEvolve repository) to identify the lengths and locations of identical by descent segments shared between all pairs of individuals in the final generation.
Output formats: GeneEvolve outputs all effects per individual and per generation (phenotype values, additive and dominant genetic values, etc.) and individual genotypes in ‘.hap’, PLINK and plain text file formats. It also reports basic summary statistics for each generation.
2.2 Performance
As noted above, GeneEvolve’s performance is not a function of number of genetic variants because it tracks haplotype identities and termini rather than all genetic data each generation. It can simulate populations of size 300K with genome-wide SNPs (500K variants) in 18.7 min using 2.5 GB RAM per generation, and does so for genome-wide sequence data (42M variants) in 21.3 min using 2.6 GB RAM per generation (Table 3.1 of supplementary – the time and memory for reading and writing individual-level SNP/sequence data is not considered). To our knowledge, this is not possible with other forward-in-time benchmark softwares. The time and memory usage are also linear functions of sample size. We compare the functionality and runtime of GeneEvolve to several benchmark forward-in-time simulators in Tables 3.4 and 3.5 of supplementary file and in Figure 1, respectively. None of these benchmark software packages could simulate populations of size 100k with more than 400k variants due to the memory or time limitations, but GeneEvolve could run in ∼ 2 h using 6.5 GB RAM.
Fig. 1.
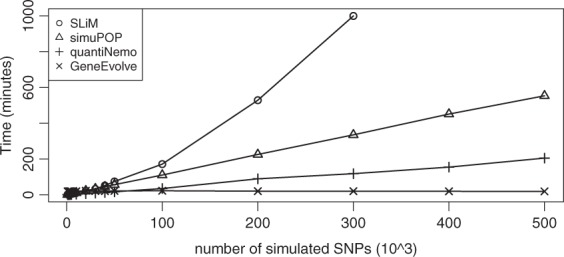
Comparing the simulation time in minutes for populations of size 10k with different numbers of variants
In order to gauge the accuracy and realism of GeneEvolve genetic data, we also checked the MAF over generations, the effects of genetic drift on genetic variation, LD structure and the additive variance under assortative mating. These results are illustrated in the documentation file and show that GeneEvolve accurately models these evolutionary processes and that the genomic properties of the data it simulates are indistinguishable from real data.
3 Discussion
GeneEvolve is a stand-alone and user-friendly genetic simulation program that requires no scripting language. It allows users to simulate complex life events and realistic whole-genome data efficiently for large populations.
Supplementary Material
Acknowledgements
The authors thank Dr. Teresa deCandia, Amanda Wills and Luke Evans for their help in testing and debugging the software.
Funding
This work was supported by the National Institutes of Mental Health grant R01MH100141 to Dr. Keller.
Conflict of Interest: none declared.
References
- Chadeau-Hyam M. et al. (2008) Fregene: simulation of realistic sequence-level data in populations and ascertained samples. BMC Bioinformatics, 9, 364.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J.L. et al. (2007) On recombination-induced multiple and simultaneous coalescent events. Genetics, 177, 2151–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman J.F.C. (1982) The coalescent. Stoch. Process. Appl., 13, 235–248. [Google Scholar]
- Lambert B.W. et al. (2008) ForSim: a tool for exploring the genetic architecture of complex traits with controlled truth. Bioinformatics, 24, 1821–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer P.W. (2013) SLiM: simulating evolution with selection and linkage. Genetics, 194, 1037–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuenschwander S. et al. (2008) quantiNemo: an individual-based program to simulate quantitative traits with explicit genetic architecture in a dynamic metapopulation. Bioinformatics, 24, 1552–1553. [DOI] [PubMed] [Google Scholar]
- Peng B., Kimmel M. (2005) simuPOP: a forward-time population genetics simulation environment. Bioinformatics, 21, 3686–3687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.