

In this study, Krishnan et al. present the characterization of DPM-1001 as a potent and highly selective chelator of copper that is orally bioavailable. Treatment of the toxic milk mouse model of Wilson's disease with DPM-1001 lowered the levels of copper in the liver and brain, removing excess copper by excretion in the feces while ameliorating symptoms associated with the disease, suggesting that DPM-1001 should be investigated further as a new therapeutic agent for the treatment of Wilson's disease.
Keywords: Wilson's, chelator, copper, toxic milk
Abstract
The levels of copper, which is an essential element in living organisms, are under tight homeostatic control. Inactivating mutations in ATP7B, a P-type Cu-ATPase that functions in copper excretion, promote aberrant accumulation of the metal, primarily the in liver and brain. This condition underlies Wilson's disease, a severe autosomal recessive disorder characterized by profound hepatic and neurological deficits. Current treatment regimens rely on the use of broad specificity metal chelators as “decoppering” agents; however, there are side effects that limit their effectiveness. Here, we present the characterization of DPM-1001 {methyl 4-[7-hydroxy-10,13-dimethyl-3-({4-[(pyridin-2-ylmethyl)amino]butyl}amino)hexadecahydro-1H-cyclopenta[a]phenanthren-17-yl] pentanoate} as a potent and highly selective chelator of copper that is orally bioavailable. Treatment of cell models, including fibroblasts derived from Wilson's disease patients, eliminated adverse effects associated with copper accumulation. Furthermore, treatment of the toxic milk mouse model of Wilson's disease with DPM-1001 lowered the levels of copper in the liver and brain, removing excess copper by excretion in the feces while ameliorating symptoms associated with the disease. These data suggest that it may be worthwhile to investigate DPM-1001 further as a new therapeutic agent for the treatment of Wilson's disease, with potential for application in other indications associated with elevated copper, including cancer and neurodegenerative diseases.
In biological systems, metals play critical roles in the control of protein function that introduce novel aspects of chemistry and structure. Copper is an essential element in living organisms and an intriguing example in this regard. Its functional significance is enhanced because of its ability to adopt either reduced (Cu+) or oxidized (Cu2+) states. It binds to proteins primarily through histidine and acidic amino acids (Cu2+) or the sulphur-containing residues Met or Cys (Cu+) (Turski and Thiele 2009). There are a wide variety of copper-dependent enzymes. Examples include cytochrome c oxidase, the terminal protein in the mitochondrial electron transport chain that plays a critical role in ATP synthesis; superoxide dismutase, which is involved in scavenging of free radicals; oxidases such as lysyl oxidase, which cross-links the extracellular matrix; dopamine β-hydoxylase, which controls biosynthesis of a neurotransmitter; and transcriptional regulators (Kim et al. 2008; Turski and Thiele 2009; Festa and Thiele 2011). Furthermore, copper can exchange other metals, such as zinc, from their cognate ligands in metalloproteins (Foster et al. 2014). More recently, it has been implicated in the regulation of signal transduction through control of the activity of kinases such as MEK, linking copper to the control of cell growth and its disruption in tumorigenesis and metastasis (Turski and Thiele 2009; Brady et al. 2014). This highlights the fundamental importance of copper to normal cell function. Nevertheless, it can also be toxic, indicating that copper content must be tightly regulated both in the organism as a whole and at the cellular level.
In mammals, diet serves as the primary source of copper. It is absorbed in the gut and transported to the liver, which is the central organ of copper homeostasis and distributes it to other tissues through the bloodstream. In the liver, copper is incorporated into carrier proteins, primarily ceruloplasmin, for release into the bloodstream; however, if copper is in excess, it is excreted in the bile (Mercer and Llanos 2003; Lutsenko et al. 2007). The levels of copper are under complex homeostatic control, including transporters that control influx and efflux, together with specialized chaperones that deliver the metal to its sites of action (Lutsenko et al. 2007). It is transported into cells primarily through copper transporter 1 (CTR1), although other transporters, such as the divalent metal transporter DMT1, may also play a role (Lee et al. 2001). Within the cell, the levels of free copper are kept to a minimum by chaperones that direct the metal to particular targets. These include COX17, which delivers copper to the mitochondria for incorporation into cytochrome c oxidase; CCS, which transfers it to superoxide dismutase for redox regulation; and ATOX1, which directs copper to the P-type Cu-ATPases ATP7A and ATP7B in the trans-Golgi network (Linz and Lutsenko 2007; Lutsenko et al. 2007; Kim et al. 2008). These Cu-ATPases serve a dual function, either delivering copper to the secretory pathway, where it can be incorporated into copper-dependent enzymes, or, under conditions in which copper is present in excess, exporting it from the cell (Linz and Lutsenko 2007; Lutsenko et al. 2007). Disruption of these homeostatic mechanisms is associated with a variety of disease states. In particular, inactivating mutations in the Cu-ATPases are the direct cause of severe disease. ATP7A plays a major role in copper absorption, and loss-of-function mutations lead to decreased levels of copper that result in a rare but severe genetic disorder, Menkes disease (Kaler 2011). This disease, which affects one in 100,000 individuals worldwide, is associated with disruption of normal developmental and neurological function and is characterized by kinky hair, failure to gain weight, and deterioration of the nervous system. It is fatal in early childhood (Kaler 2011; Bandmann et al. 2015). On the other hand, mutations in ATP7B, which functions in copper excretion, lead to accumulation of the metal, resulting in Wilson's disease (Pfeiffer 2007; Rodriguez-Castro et al. 2015).
The incidence of Wilson's disease is estimated to be one in 30,000 worldwide although this may be an underestimate, as its symptoms overlap with other conditions (Bandmann et al. 2015). Wilson's disease is a severe autosomal recessive disorder and, to date, >300 disease-causing mutations have been identified in the ATP7B gene (Lutsenko 2014). The physical burden of the disease is felt in the liver in particular, as this tissue expresses high levels of ATP7B. It begins with a presymptomatic period, during which copper accumulates in the liver. If diagnosis occurs at this stage, the prognosis is good with current therapies (Bandmann et al. 2015); however, without treatment, a variety of hepatic problems are encountered, from enlargement of the liver to hepatitis and cirrhosis and even acute liver failure (Lutsenko 2014; Rodriguez-Castro et al. 2015). As the disease progresses further, it results in the development of speech and cognitive impairment, particular tremors, and dystonia as well as ataxia and Parkinsonism (Bandmann et al. 2015; Rodriguez-Castro et al. 2015). In addition, psychiatric problems, including personality changes, antisocial behavior, anxiety, and depression, appear in Wilson's patients at some time during the course of the disease. Most patients with neurological symptoms also develop Kayser-Fleischer rings, which are formed by copper deposits in the cornea, leading to a brown discoloration that can be diagnostic for the disease (Bandmann et al. 2015). Overall, Wilson's disease can be fatal if not diagnosed and treated early; however, the fact that its symptoms are often indistinguishable from a variety of other diseases makes this challenging. The condition cannot be controlled by switching to a low-copper diet and current treatment strategies depend on “decoppering” agents, the goal of which is to decrease the level of the metal and to try to re-establish normal homeostasis. Unfortunately, the pharmacological agents that are used most frequently are associated with adverse effects and, furthermore, these agents have recently become extremely expensive (Schilsky et al. 2015). Consequently, new, potent, and specific copper chelators are needed for the treatment of Wilson's disease.
In this study, we report the biochemical characterization of a new small molecule chelator and define the mechanism underlying its specificity for copper. Furthermore, we demonstrated that the compound attenuated adverse effects associated with accumulation of copper in cells. Finally, we showed that, when administered orally or intraperitoneally, the compound was effective in decreasing the copper burden in multiple tissues in a mouse model of Wilson's disease while ameliorating symptoms associated with the disease. This compound may represent the basis for an improved approach to the treatment of Wilson's disease.
Results
DPM-1001 {methyl 4-[7-hydroxy-10,13-dimethyl-3-({4-[(pyridin-2-ylmethyl)amino]butyl}amino)hexadecahydro-1H-cyclopenta[a]phenanthren-17-yl] pentanoate} is a specific copper chelator
Previously, we identified DPM-1001 (Fig. 1A) as an inhibitor of PTP1B with the unusual property of also binding copper (Krishnan et al. 2018). In order to examine this chelation property further, we tested whether DPM-1001 displayed specificity for copper by incubating the compound with a series of metal ions and then subjecting the complexes to electrospray ionization mass spectrometry (ESI-MS) analysis. The ESI-MS spectra of the compound in the presence of CuSO4 revealed three peaks, at 568.6, 620, and 727.5 m/z. The peak at 568.6 m/z corresponded to free compound, whereas the peaks at 620 and 727.5 m/z corresponded to the Cu- and CuSO4-bound forms, respectively (Fig. 1B). When we incubated DPM-1001 with a variety of other metals, we found that none formed a complex with the compound (Fig. 1B). This includes silver, which is isoelectronic and similar in size to Cu+, highlighting the exquisite specificity of DPM-1001 for copper.
Figure 1.

DPM-1001 was a specific chelator of copper. (A) Chemical structure of DPM-1001. (B) ESI-MS spectra to examine complex formation between DPM-1001 (1 mM) and the indicated metals (8 mM). (C, top left panel) Chemical structure of DPM-1003. (Bottom left panel) Titration of radiolabeled copper (64Cu) against DPM-1001 and DPM-1003. (Right panel) ESI-MS spectra to examine complex formation between DPM-1003 (1 mM) and the indicated metals (8 mM). (D) Chemical structure of analog 2. (E) ESI-MS spectra to examine complex formation between analog 2 (1 mM) and the indicated metals (8 mM).
Using radiolabeled copper (64Cu) and titrating DPM-1001 against increasing concentrations of 64Cu in a binding assay, we measured a Kd of 75 nM. To investigate the mechanism by which DPM-1001 chelated copper, we generated a series of analogs. Removal of the N1-(pyridin-2-ylmethyl)butane-1,4-diamine tail from the 3 position of the A ring in the cholesterol group was sufficient to abrogate copper binding (Supplemental Fig. 1A), focusing attention on the importance of this tail. Substitution of the pyridyl group with either piperidine or benzene or generating a four-amino substituent with an ether, to produce 4-(pyridin-2-ylmethoxy)butan-1-amine, also impaired binding (Supplemental Fig. 1B). Within the pyridyl group, we moved the N atom from the 2 position in DPM-1001 to the 3 position to generate compound DPM-1003. Interestingly, despite their identical chemical composition, DPM-1003, in contrast to DPM-1001, displayed limited binding to radiolabeled 64Cu and any of the metals tested (Fig. 1C).
In addition, we generated N1,N4-bis(pyridin-2-ylmethyl) butane-1,4-diamine (analog 2), which is a symmetrical compound in which the steroid moiety seen in DPM-1001 was replaced by a second pyridine ring (Fig. 1D). When incubated in the presence of CuSO4, analog 2 yielded a distinct peak at 332 on ESI-MS analysis that corresponded to the Cu(II)-bound form of the compound (Fig. 1E). When titrated against 64Cu, a Kd of 57 nM was obtained. In addition, when we incubated analog 2 against the panel of metals, we observed that, in contrast to DPM-1001, this analog bound to all of the metals tested (Fig. 1E). Overall, these data illustrate that within DPM-1001, the N1-(pyridin-2-ylmethyl)butane-1,4-diamine tail was responsible for copper chelation, whereas the steroid moiety conferred selectivity in copper binding.
DPM-1001 improved the viability of cells exposed to high copper
As our data demonstrated that DPM-1001 was a potent copper chelator in vitro, we investigated its ability to bind copper in a cellular context. We used HepG2 cells as a model, including cells in which ATP7B was suppressed by RNAi (Fig. 2A). ATP7B knockdown (ATP7B-KD1) cells displayed greater sensitivity to exposure to copper than wild-type cells. At copper concentrations >0.5 mM, the survival of ATP7B-KD1 cells was <20% (Fig. 2B), highlighting the importance of ATP7B in protecting cells from the effects of excess copper. When viability was tested in the presence of 2 µM DPM-1001, we found that the compound was able to rescue cells from copper-induced cell death (Fig. 2B).
Figure 2.

Treatment with DPM-1001 improved cell viability in the presence of high levels of copper. (A) Representative immunoblot to demonstrate siRNA-induced suppression of ATP7B in HepG2 cells. (B) Survival of control cells with (▴) or without (•) DPM-1001 and ATP7B-KD1 cells with (gray; ▾) and without (▪) DPM-1001 was measured at increasing concentrations of copper, from 0 to 1.5 mM. Cells were preincubated with DPM-1001 at 2 µM for 1 h prior to exposing cells to copper. (C) Survival of control fibroblasts with (▴) and without (▾) DPM-1001 was compared with fibroblasts derived from Wilson's disease patients (GM00032 [left panel], GM00033 [middle panel], and GM05257 [right panel]) with (gray, •) and without (▪) DPM-1001, measured at increasing concentrations of copper, from 0 to 1.5 mM.
The significance of this observation was explored further by testing a panel of six different skin fibroblasts derived from Wilson's disease patients. These cells have been reported to express different mutant forms of ATP7B and display elevated levels of free copper relative to normal fibroblasts (Chan et al. 1980). We measured the survival of these cells in the presence of increasing concentrations of copper and compared the response with that observed in normal skin fibroblasts. Although we observed a varying response to copper in the mutant cells, all were found to be more sensitive to copper-induced cell death than the wild-type cells (Fig. 2C). Interestingly, DPM-1001 also inhibited copper-induced cell death in the fibroblasts derived from Wilson's disease patients (Fig. 2C; Supplemental Fig. 2).
DPM-1001 lowered copper levels in an animal model of Wilson's disease
To investigate further the extent to which DPM-1001 may lower copper levels and ameliorate symptoms associated with Wilson's disease, we used the toxic milk mouse model (Theophilos et al. 1996). The toxic milk mouse is a naturally occurring genetic and phenotypic model of Wilson's disease. A Gly-to-Asp substitution (G775D) renders the ATP7B protein dysfunctional and results in copper accumulation. This has been used widely as a model to understand the human disease (Theophilos et al. 1996; Coronado et al. 2001; Czachor et al. 2002; Przybylkowski et al. 2013).
Tissue copper levels were assessed by two separate methods. Liver tissue was excised from wild-type and toxic milk mice that had been treated with saline or DPM-1001, then fixed and stained with rhodanine, a dye that stains for copper-binding proteins (Thornburg et al. 1985). No signal was detected in liver samples obtained from saline- or DPM-1001-treated wild-type mice. In saline-treated toxic milk mice, we observed bright staining with the dye, indicative of elevated copper levels. In contrast, no obvious staining for rhodanine was observed in liver samples obtained from DPM-1001-treated mice (Fig. 3A). In order to obtain a quantitative measure of copper levels, we used inductively coupled plasma MS (ICP-MS). We detected elevated levels of copper in the livers (Fig. 3B) and brains (Fig. 3C) of saline-treated toxic milk mice compared with control animals, and treatment with DPM-1001 lowered the levels of copper in both tissues. Interestingly, the 3-pyridyl analog DPM-1003, which has the same chemical composition as DPM-1001 but does not chelate copper, did not suppress the levels of the metal in the livers of toxic milk mice (Supplemental Fig. 3), suggesting that the effects of DPM-1001 on tissue copper levels were direct. When we compared the effect of DPM-1001 with the drug candidate chelator tetrathiomolybdate (Brewer 2009; Brewer et al. 2009; Askari et al. 2010), we found that DPM-1001 was as effective as tetrathiomolybdate in removing copper from the liver and brain (Fig. 3C,D) but at both a lower dose and lower frequency of administration.
Figure 3.
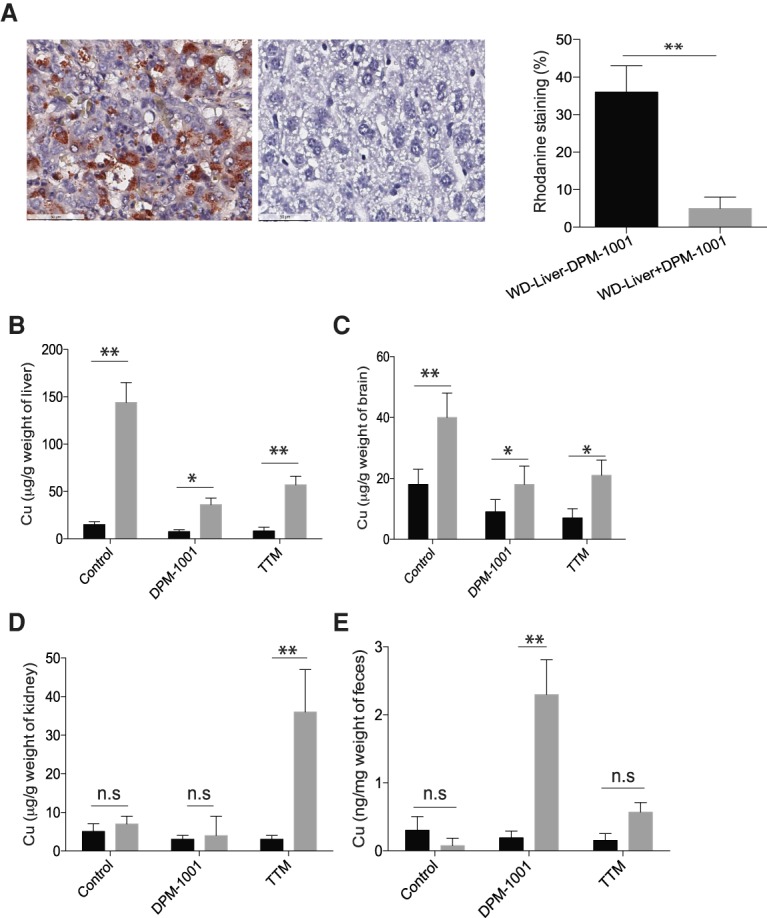
Suppression of copper levels in the toxic milk mouse model of Wilson's disease. (A) Representative sections of liver tissue from the toxic milk mouse model stained with rhodanine following treatment of animals for 2 wk with saline (left panel) or DPM-1001 (right panel). Quantitation is shown at the right. (B–D) Copper levels in the livers (B), brains (C), and kidneys (D) from wild-type (black bars) or toxic milk (gray bars) mice were measured using ICP-MS following treatment with saline, 5 mg/kg DPM-1001 orally every third day, or 5 mg/kg tetrathiomolybdate (TTM) intraperitoneally daily for 2 wk. (E) The levels of copper in the feces of these animals. Data are presented as mean ± SEM. Statistical analysis was performed using ANOVA. (**) P < 0.01; (*) P < 0.1; (ns) not significant.
We investigated the mechanism by which DPM-1001 cleared tissue copper levels. It has been reported that the use of penicillamine as a “decoppering” agent promotes excretion of the metal in the urine (Humann-Ziehank and Bickhardt 2001; Walshe 2011). Therefore, we measured copper levels in kidney samples obtained from saline-treated or DPM-1001-treated wild-type and toxic milk mice and observed no significant difference. In contrast, treatment with tetrathiomolybdate resulted in dramatically elevated copper levels in the kidney (Fig. 3D). We investigated this further by collecting fecal material from the mice and analyzing copper levels. In contrast to toxic milk mice treated with saline, the levels of copper in the feces of DPM-1001-treated toxic milk mice were dramatically elevated. Unlike DPM-1001 treatment, treatment with tetrathiomolybdate resulted in only a small increase in the levels of fecal copper (Fig. 3E).
DPM-1001 ameliorated hepatic complications associated with Wilson's disease
In light of the fact that liver damage and hepatic complications are among the hallmarks of Wilson's disease (Pfeiffer 2007; Lutsenko 2014; Lin et al. 2015), we conducted morphological analyses of the livers obtained from wild-type and toxic milk mice following treatment with saline or DPM-1001. In wild-type livers treated with saline or DPM-1001 and then sectioned and stained for morphological analysis, no obvious abnormalities were detected; normal hepatocyte size, shape, and arrangement were observed. In contrast, liver sections obtained from toxic milk mice treated with saline revealed enlarged hepatocytes with irregular shape and arrangement as well as large cytoplasmic lipid droplets (Fig. 4A). Interestingly, liver sections obtained from toxic milk mice treated with DPM-1001 appeared similar to those from wild-type mice, with normal hepatocyte size, shape, and arrangement. Furthermore, no large lipid droplets were observed, in contrast to liver sections from saline-treated toxic milk mice (Fig 4A).
Figure 4.
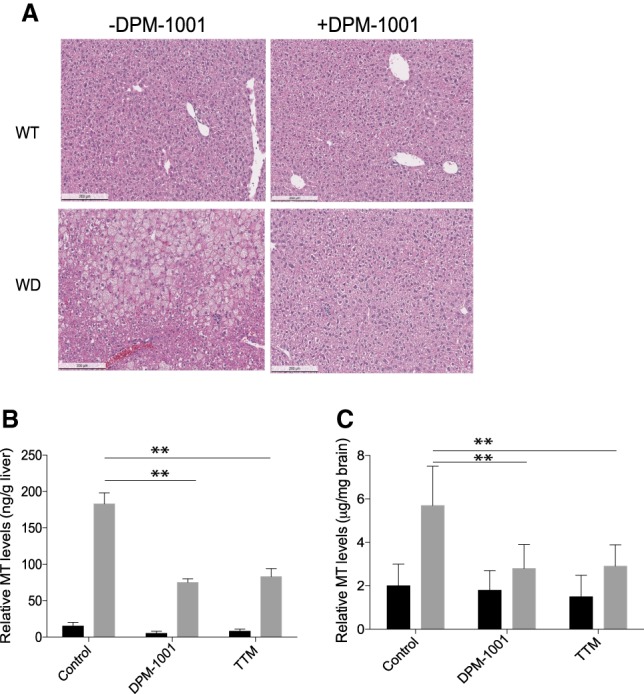
Treatment with DPM-1001 ameliorated symptoms in the toxic milk mouse model of Wilson's disease. (A) H&E staining of representative sections of liver tissue from wild-type (top) and toxic milk (bottom; labeled WD for Wilson's disease) mice that were either untreated (left) or treated with DPM-1001 (right). (B,C). Levels of metallothionein in the livers (B) or brains (C) from wild-type (black bars) and toxic milk (gray bars) mice treated with saline, DPM-1001, or tetrathiomolybdate (TTM) were measured. Data are presented as mean ± SEM. Statistical analysis was performed using ANOVA. (**) P < 0.01.
In addition, we measured metallothionein levels in the liver and brain. We found that metallothionein levels were significantly elevated in both tissues in toxic milk mice compared with wild type. Furthermore, DPM-1001 treatment lowered metallothionein levels in the liver and the brain (Fig. 4B,C). Taken together, the data demonstrated that DPM-1001 lowered tissue copper levels and ameliorated symptoms associated with Wilson's disease.
Discussion
If Wilson's disease is diagnosed in the early, presymptomatic phase, the prognosis is good with current therapeutics. Nevertheless, early diagnosis is challenging, and treatment becomes more difficult as the course of the disease progresses; ultimately, patients may develop fulminant hepatic failure that requires liver transplantation for resolution (Ahmad et al. 2017; Boga et al. 2017). The primary therapeutic strategy is to try to reduce the levels of copper in the patient and re-establish homeostasis. These approaches do not address the underlying mutations in ATP7B that cause the disease and, therefore, such treatments would be expected to be continued throughout the life of the patient, placing an emphasis on avoidance of toxic side effects. There are two approaches to reducing copper levels. Zinc salts, such as zinc acetate, are a first line of therapy, particularly for asymptomatic patients, and are also important as a maintenance therapy for long-term management of Wilson's disease after treatment with chelators (Ranucci et al. 2017). Zinc decreases absorption of copper from the gut by inducing expression of metallothionein in intestinal cells, which then traps both metals, leading to excretion as the mucosal cells are sloughed off and removed in the feces (Brewer 2001). Although this is less toxic than the existing chelators, it does not have the same ability to remove copper from tissues that have become overloaded. Consequently, chelating agents that bind copper directly in the tissues or blood and facilitate its excretion have been developed for use as an initial “decoppering” step. Currently, the standard of care for Wilson's disease includes treatment with the chelating agents D-penicillamine or trientine, with tetrathiomolybdate progressing as an experimental therapy in clinical trials (Rodriguez-Castro et al. 2015).
D-penicillamine, also known as Cuprimine, promotes urinary excretion of copper; however, it is not specific for copper and is a general chelator that has been used to treat heavy metal poisoning (Lyle 1981). This broad specificity metal chelation may contribute to the side effects. Neurological problems associated with D-penicillamine are a particular concern and it has been reported that the deterioration encountered may be irreversible (Brewer et al. 1987). Other problems include swelling and inflammation of the kidneys, hepatotoxicity, and hematological abnormalities, conditions that may be exacerbated by chronic administration regimens. In fact, in one study, ∼30% of patients discontinued treatment due to the severity of the side effects (Bandmann et al. 2015), and it has even been suggested that the drug should not be used as an initial therapy (Brewer 1999).
In light of the problems with D-penicillamine, trientine has been used more extensively. Trientine, also known as Syprine, has a polyamine-like structure that is distinct from D-penicillamine. Although it forms a complex with copper, it has also been reported to bind iron and zinc in vivo (Rodriguez-Castro et al. 2015). It facilitates copper excretion in the urine and is also suggested to impair intestinal absorption and increase excretion in feces. A problem with trientine is that it has a short half-life in humans. It is poorly absorbed from the gastrointestinal tract and what is absorbed is rapidly metabolized, via acetylation, and inactivated (Lu 2010). Although its side effects are less marked than those of D-penicillamine, toxicity problems have been reported (Dahlman et al. 1995; Chang et al. 2013). More recently, tetrathiomolybdate has progressed in clinical trials. It forms a complex with copper that restricts absorption from the gut and limits cellular uptake, eliminating copper in urine and feces. When used initially as an ammonium salt, problems were encountered with the stability of the compound. Subsequently, a bis-choline salt of tetrathiomolybdate, also known as Decuprate/WTX101, has been tested in phase 2 clinical trials (Weiss et al. 2017). Although results were encouraging, some serious adverse events were noted, including 40% of patients who showed elevation of liver enzymes. Further trials will be necessary to establish whether this will be an effective therapeutic agent (Hedera 2017).
Clearly, new approaches are required to treat Wilson's disease. Strategies involving cell and gene therapy to target nonfunctional mutant forms of ATP7B are being considered (Gupta 2014; Jaber et al. 2017; Ranucci et al. 2017) and may bear fruit in the longer term. In this study, we took the approach of characterizing copper binding by a small molecule that is distinct from the known chelators; it may offer an improved strategy for decreasing copper levels that is more immediate. The compound DPM-1001 has several advantageous properties, including the fact that it is highly specific for copper, is orally bioavailable (Krishnan et al. 2018), crosses the blood–brain barrier, and promotes fecal excretion of the metal. The specificity of this compound for copper was striking. Among 14 metals tested, it recognized only copper and did not even chelate silver, which is isoelectronic for Cu+. Structure–activity relationship data established the critical importance of the N1-(pyridin-2-ylmethyl)butane-1,4-diamine tail in metal binding, but the steroid component of the compound confers specificity for copper. To the best of our knowledge, this remarkable specificity is a unique feature of DPM-1001.
Consistent with its ability to chelate copper in vitro, we observed that DPM-1001 inhibited copper-induced cell death in cell models of Wilson's disease, including fibroblasts derived from patients in which elevated copper had been demonstrated (Chan et al. 1980). Furthermore, when DPM-1001 was administered intraperitoneally or orally to the toxic milk mouse model of Wilson's disease, it lowered copper levels in the liver and brain (Fig. 3). Interestingly, our data indicate that treatment of toxic milk mice with tetrathiomolybdate led to accumulation of copper in the kidney, raising the possibility that such treatment may produce undesirable consequences, including renal damage. Similar increases in copper levels in the kidney have been reported following treatment of either toxic milk mouse (Czachor et al. 2002) or Long-Evans Cinnamon (LEC) rat (Ogra et al. 1995; Komatsu et al. 2000) models of Wilson's disease. Therefore, our data suggest that, in addition to its specificity for copper, DPM-1001 may offer additional advantages as a therapeutic agent to deplete the excess metal in Wilson's disease.
It is now becoming evident that abnormally high levels of copper may have an impact in other indications in addition to Wilson's disease. For example, copper has been implicated in the pathogenesis of neurodegenerative disorders such as Alzheimer's and Parkinson's disease (Cooper 2011; Carboni and Lingor 2015). Copper has been shown to promote amyloid-β production and may also exert effects through increased oxidative stress. Elevated levels of copper have also been noted in the sera of a variety of cancer patients. It has been suggested that copper plays an important role in angiogenesis and tumor invasion. Furthermore, copper has been implicated in the control of proliferation and survival with the identification of MEK in the RAS–MAPK pathway as a copper-dependent enzyme (Turski et al. 2012; Brady et al. 2014). It has been shown that copper chelators, including tetrathiomolybdate, decreased the growth of tumors driven by the BRAF-V600E mutant that is associated with melanoma (Brady et al. 2014). Consequently, such multiple lines of data from several different studies now point to the potential for copper chelation to represent a major step forward in a new approach not only to treat Wilson's disease but also to impact other major indications, including cancer and neurodegenerative diseases. What is needed to bring such an approach to fruition is a potent, selective, and bioavailable copper chelator that is well tolerated. We hope that further studies of DPM-1001 in the future will help to achieve this.
Materials and methods
ESI-MS analysis of the DPM-1001–copper complex
Eight equivalents of M(NO3)2·xH2O or MSO4·xH2O were reacted with 1 eq of DPM-1001, DPM-1003, or analog 2 (1 mM in H2O). The reaction solutions were stirred for 2 h at room temperature, 40°C, and 80°C. Each sample was diluted fivefold in 50% methanol and 0.1% formic acid and loaded into a 500-µL syringe. A Thermo Vantage triple quadrupole mass spectrometer (Thermo Scientific) equipped with an HESI spray source was coupled to an Accela high-performance liquid chromatography system (Thermo Scientific). Mobile phase A consisted of 0.1% formic acid, and mobile phase B consisted of 0.1% formic acid in methanol. Isocratic flow was set to 50% B at 200 µL/min. Samples (200 µL) were injected into the LC flow line via syringe pump at 10 µL/min and electrosprayed into the Vantage triple quadrupole mass spectrometer. A spray voltage of 4.3 kV was used along with a capillary temperature of 350°C. Each full MS scan was 0.5 sec, and data were collected with a scan range of 200–900 m/z over 1 min.
Copper-binding assays
Direct binding assays were performed using radiolabeled copper (64 Cu2+). Varying concentrations of radiolabeled copper were incubated with 100 nM DPM-1001. Excess copper was removed by running the samples through a C18 column. The amount of metal bound to the compound was quantitated directly by scintillation counting.
Cell culture
HepG2 cells purchased from American Type Culture Collection were cultured in RPMI medium containing 10% FBS and 5% CO2 at 37°C. Control (AGO9319) and Wilson's disease patient-derived fibroblasts (GM00032, GM00033, GM05257, GM12158, GM05798, and GM11778) were obtained from Coriell Biorepository and cultured in DMEM, 10% FBS, and 5% CO2 at 37°C.
Cell-based assays
For cell survival assays, 10,000 cells were seeded in a 96-well plate and cultured until fully confluent (90%). These cells were transfected with ATP7B siRNAs or scrambled siRNA (negative control) purchased from Qiagen and Santa Cruz Biotechnology, Inc. Lipofectamine RNAiMAX reagent (Thermo Fisher) and siRNAs were diluted into a final volume of 100 µL in Opti-MEM (Gibco). The mixture was incubated for 30 min at room temperature, following which 800 µL of Opti-MEM was added to the mixture. The above transfection solution was overlaid onto cells at a final concentration of 5 nM siRNA. Transfection of HepG2 cells with scrambled siRNA served as negative control. Twelve hours after the transfection, complete medium was added to each well. Knockdown efficiency was checked 24 h later, following which cells were exposed to varying concentrations of copper (0–1 mM) for 12 h and cell viability was determined using the MTT assay as described previously (Mosmann 1983). To test the ability of DPM-1001 to prevent copper-induced toxicity, cells were preincubated with the compound at 2 µM for 1 h prior to exposing cells to copper.
Animal experiments
C3He-Atp7btx-j mice were obtained from the Jackson Laboratory. All animal experiments were performed according to protocols approved by the Institutional Animal Use and Care Committee of Cold Spring Harbor Laboratory. For short-term treatment, male mice (6–8 wk of age) were administered DPM-1001 intraperitoneally at 5 mg/kg for 2 wk prior to collecting samples for biochemical analysis. At least 10 animals were used in each treatment group.
ICP-MS
Tissue samples from wild-type or toxic milk mice treated with saline, DPM-1001, or tetrathiomolybdate were washed three times with PBS containing 1 mM EDTA (to remove nonspecifically bound copper). Then, 215 µL of concentrated nitric acid (BDH Aristar Ultra) was added to each sample and incubated overnight. Samples were boiled for 1 h at 95°C, and ∼150 µL of each sample was further diluted in 2 mL of 2% nitric acid (freshly made from concentrated nitric acid and Milli-Q water). Samples were analyzed on a Thermo Fisher iCAP Qc ICP mass spectrometer in kinetic energy discrimination (KED) mode against a calibration curve of known copper and phosphorus concentrations, with 20 µg/L Ga (Inorganic Ventures) as an internal standard. Each experiment was carried out twice and each condition was repeated at least three times.
Metallothionein quantitation
Tissue samples (100 mg of liver or 25 mg of brain) were rinsed in PBS to remove excess blood and homogenized using a Dounce homogenizer on ice (PBS, 1% tween). The samples were centrifuged at 5000g for 5 min and the supernatant was collected. Total protein in the supernatant was measured using Bradford reagent, and an equal amount of each sample (based on protein concentration) was used to measure metallothionein levels. Mouse metallothionein ELISA kit was purchased from Lifespan Biosciences, Inc., and the assay was performed according to the user's manual.
Histology
Liver tissues from untreated and DPM-1001-treated wild-type and similarly treated toxic milk mice were sectioned and stained with H&E or rhodanine. Whole-slide digitized images of H&E-stained tissue were captured using the Aperio ScanScope XT automated scanning system. ImageJ software was used to quantitate rhodanine staining.
Competing interest statement
N.K. and N.K.T. serve on the Scientific Advisory Board of DepYmed, Inc., a company that focuses on inhibitors of PTP1B in various disease indications.
Supplementary Material
Acknowledgments
We are very grateful to DepYmed, Inc., for providing the DPM-1001 and DPM-1003 that was used in this study. This research was supported by National Institutes of Health grants CA53840 and GM55989 to N.K.T., Cold Spring Harbor Laboratory Cancer Center Support Grant CA45508, and the Cold Spring Harbor Laboratory–Northwell Alliance. N.K.T. is also grateful for support from the Gladowsky Breast Cancer Foundation, the Don Monti Memorial Research Foundation, the Irving Hansen Foundation, the Estate of Thomas C. Nelson, and the Robertson Research Fund.
Author contributions: N.K. was responsible for all of the data in the manuscript, working in collaboration with K.R. and D.J.P. on mass spectrometry analyses. C.F. assisted in all of the animal studies. N.K. and N.K.T. designed the study and wrote the paper. N.K.T. directed the study.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.314658.118.
References
- Ahmad A, Torrazza-Perez E, Schilsky ML. 2017. Liver transplantation for Wilson disease. Handb Clin Neurol 142: 193–204. [DOI] [PubMed] [Google Scholar]
- Askari F, Innis D, Dick RB, Hou G, Marrero J, Greenson J, Brewer GJ. 2010. Treatment of primary biliary cirrhosis with tetrathiomolybdate: results of a double-blind trial. Transl Res 155: 123–130. [DOI] [PubMed] [Google Scholar]
- Bandmann O, Weiss KH, Kaler SG. 2015. Wilson's disease and other neurological copper disorders. Lancet Neurol 14: 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boga S, Ala A, Schilsky ML. 2017. Hepatic features of Wilson disease. Handb Clin Neurol 142: 91–99. [DOI] [PubMed] [Google Scholar]
- Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, Knapp S, Xiao K, Campbell SL, Thiele DJ, et al. 2014. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 509: 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ. 1999. Penicillamine should not be used as initial therapy in Wilson's disease. Mov Disord 14: 551–554. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. 2001. Zinc acetate for the treatment of Wilson's disease. Expert Opin Pharmacother 2: 1473–1477. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. 2009. The use of copper-lowering therapy with tetrathiomolybdate in medicine. Expert Opin Investig Drugs 18: 89–97. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Terry CA, Aisen AM, Hill GM. 1987. Worsening of neurologic syndrome in patients with Wilson's disease with initial penicillamine therapy. Arch Neurol 44: 490–493. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Askari F, Dick RB, Sitterly J, Fink JK, Carlson M, Kluin KJ, Lorincz MT. 2009. Treatment of Wilson's disease with tetrathiomolybdate: V. Control of free copper by tetrathiomolybdate and a comparison with trientine. Transl Res 154: 70–77. [DOI] [PubMed] [Google Scholar]
- Carboni E, Lingor P. 2015. Insights on the interaction of α-synuclein and metals in the pathophysiology of Parkinson's disease. Metallomics 7: 395–404. [DOI] [PubMed] [Google Scholar]
- Chan WY, Cushing W, Coffman MA, Rennert OM. 1980. Genetic expression of Wilson's disease in cell culture: a diagnostic marker. Science 208: 299–300. [DOI] [PubMed] [Google Scholar]
- Chang H, Xu A, Chen Z, Zhang Y, Tian F, Li T. 2013. Long-term effects of a combination of D-penicillamine and zinc salts in the treatment of Wilson's disease in children. Exp Ther Med 5: 1129–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GJ. 2011. Therapeutic potential of copper chelation with triethylenetetramine in managing diabetes mellitus and Alzheimer's disease. Drugs 71: 1281–1320. [DOI] [PubMed] [Google Scholar]
- Coronado V, Nanji M, Cox DW. 2001. The Jackson toxic milk mouse as a model for copper loading. Mamm Genome 12: 793–795. [DOI] [PubMed] [Google Scholar]
- Czachor JD, Cherian MG, Koropatnick J. 2002. Reduction of copper and metallothionein in toxic milk mice by tetrathiomolybdate, but not deferiprone. J Inorg Biochem 88: 213–222. [DOI] [PubMed] [Google Scholar]
- Dahlman T, Hartvig P, Lofholm M, Nordlinder H, Loof L, Westermark K. 1995. Long-term treatment of Wilson's disease with triethylene tetramine dihydrochloride (trientine). QJM 88: 609–616. [PubMed] [Google Scholar]
- Festa RA, Thiele DJ. 2011. Copper: an essential metal in biology. Curr Biol 21: R877–R883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster AW, Osman D, Robinson NJ. 2014. Metal preferences and metallation. J Biol Chem 289: 28095–28103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S. 2014. Cell therapy to remove excess copper in Wilson's disease. Ann N Y Acad Sci 1315: 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedera P. 2017. Update on the clinical management of Wilson's disease. Appl Clin Genet 10: 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humann-Ziehank E, Bickhardt K. 2001. Effects of D-penicillamine on urinary copper excretion in high-copper supplemented sheep. J Vet Med A Physiol Pathol Clin Med 48: 537–544. [DOI] [PubMed] [Google Scholar]
- Jaber FL, Sharma Y, Gupta S. 2017. Demonstrating potential of cell therapy for Wilson's disease with the long-evans cinnamon rat model. Methods Mol Biol 1506: 161–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaler SG. 2011. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat Rev Neurol 7: 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BE, Nevitt T, Thiele DJ. 2008. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol 4: 176–185. [DOI] [PubMed] [Google Scholar]
- Komatsu Y, Sadakata I, Ogra Y, Suzuki KT. 2000. Excretion of copper complexed with thiomolybdate into the bile and blood in LEC rats. Chem Biol Interact 124: 217–231. [DOI] [PubMed] [Google Scholar]
- Krishnan N, Konidaris KF, Gasser G, Tonks NK. 2018. A potent, selective, and orally bioavailable inhibitor of the protein-tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models. J Biol Chem 293: 1517–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Prohaska JR, Thiele DJ. 2001. Essential role for mammalian copper transporter Ctr1 in copper homeostasis and embryonic development. Proc Natl Acad Sci 98: 6842–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Wang D, Ding N, Zheng C. 2015. Hepatic manifestations in Wilson's disease: report of 110 cases. Hepatogastroenterology 62: 657–660. [PubMed] [Google Scholar]
- Linz R, Lutsenko S. 2007. Copper-transporting ATPases ATP7A and ATP7B: cousins, not twins. J Bioenerg Biomembr 39: 403–407. [DOI] [PubMed] [Google Scholar]
- Lu J. 2010. Triethylenetetramine pharmacology and its clinical applications. Mol Cancer Ther 9: 2458–2467. [DOI] [PubMed] [Google Scholar]
- Lutsenko S. 2014. Modifying factors and phenotypic diversity in Wilson's disease. Ann N Y Acad Sci 1315: 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. 2007. Function and regulation of human copper-transporting ATPases. Physiol Rev 87: 1011–1046. [DOI] [PubMed] [Google Scholar]
- Lyle WH. 1981. Penicillamine in metal poisoning. J Rheumatol Suppl 7: 96–99. [PubMed] [Google Scholar]
- Mercer JF, Llanos RM. 2003. Molecular and cellular aspects of copper transport in developing mammals. J Nutr 133: 1481S–1484S. [DOI] [PubMed] [Google Scholar]
- Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65: 55–63. [DOI] [PubMed] [Google Scholar]
- Ogra Y, Ohmichi M, Suzuki KT. 1995. Systemic dispositions of molybdenum and copper after tetrathiomolybdate injection in LEC rats. J Trace Elem Med Biol 9: 165–169. [DOI] [PubMed] [Google Scholar]
- Pfeiffer RF. 2007. Wilson's disease. Semin Neurol 27: 123–132. [DOI] [PubMed] [Google Scholar]
- Przybylkowski A, Gromadzka G, Wawer A, Bulska E, Jablonka-Salach K, Grygorowicz T, Schnejder-Pacholek A, Czlonkowski A. 2013. Neurochemical and behavioral characteristics of toxic milk mice: an animal model of Wilson's disease. Neurochem Res 38: 2037–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranucci G, Polishchuck R, Iorio R. 2017. Wilson's disease: prospective developments towards new therapies. World J Gastroenterol 23: 5451–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Castro KI, Hevia-Urrutia FJ, Sturniolo GC. 2015. Wilson's disease: a review of what we have learned. World J Hepatol 7: 2859–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilsky ML, Roberts EA, Hahn S, Askari F. 2015. Costly choices for treating Wilson's disease. Hepatology 61: 1106–1108. [DOI] [PubMed] [Google Scholar]
- Theophilos MB, Cox DW, Mercer JF. 1996. The toxic milk mouse is a murine model of Wilson disease. Hum Mol Genet 5: 1619–1624. [DOI] [PubMed] [Google Scholar]
- Thornburg LP, Beissenherz M, Dolan M, Raisbeck MF. 1985. Histochemical demonstration of copper and copper-associated protein in the canine liver. Vet Pathol 22: 327–332. [DOI] [PubMed] [Google Scholar]
- Turski ML, Thiele DJ. 2009. New roles for copper metabolism in cell proliferation, signaling, and disease. J Biol Chem 284: 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turski ML, Brady DC, Kim HJ, Kim BE, Nose Y, Counter CM, Winge DR, Thiele DJ. 2012. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol Cell Biol 32: 1284–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walshe JM. 2011. The pattern of urinary copper excretion and its response to treatment in patients with Wilson's disease. QJM 104: 775–778. [DOI] [PubMed] [Google Scholar]
- Weiss KH, Askari FK, Czlonkowska A, Ferenci P, Bronstein JM, Bega D, Ala A, Nicholl D, Flint S, Olsson L, et al. 2017. Bis-choline tetrathiomolybdate in patients with Wilson's disease: an open-label, multicentre, phase 2 study. Lancet Gastroenterol Hepatol 2: 869–876. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.