

Trafficking kinesin proteins (TRAKs) support mitochondrial trafficking in neurons. Barel, Malicdan, Ben-zeev et al. report the first cases of fatal encephalopathy caused by pathogenic TRAK1 variants, in which aberrant mitochondrial movement and distribution led to abnormal uncoupled respiration. This study provides new understanding of how mitochondrial dynamics relate to disease.
Keywords: TRAK1, mitochondria transport, rare diseases, early-onset epilepsy, neurodegeneration
Abstract
Cellular distribution and dynamics of mitochondria are regulated by several motor proteins and a microtubule network. In neurons, mitochondrial trafficking is crucial because of high energy needs and calcium ion buffering along axons to synapses during neurotransmission. The trafficking kinesin proteins (TRAKs) are well characterized for their role in lysosomal and mitochondrial trafficking in cells, especially neurons. Using whole exome sequencing, we identified homozygous truncating variants in TRAK1 (NM_001042646:c.287-2A > C), in six lethal encephalopathic patients from three unrelated families. The pathogenic variant results in aberrant splicing and significantly reduced gene expression at the RNA and protein levels. In comparison with normal cells, TRAK1-deficient fibroblasts showed irregular mitochondrial distribution, altered mitochondrial motility, reduced mitochondrial membrane potential, and diminished mitochondrial respiration. This study confirms the role of TRAK1 in mitochondrial dynamics and constitutes the first report of this gene in association with a severe neurodevelopmental disorder.
Introduction
The ability to move within cells is crucial for mitochondria to execute many cellular functions, including buffering of calcium ions, regulation of apoptosis and, most importantly, energy generation in the form of ATP. Of all cell types, neurons are particularly sensitive to any impairment in active mitochondrial transport along their axons and dendrites, as they must reach the distant synapses in response to changing requirements for energy and calcium buffering (Cai et al., 2011; Schwarz, 2013). In the past two decades, numerous studies have demonstrated mitochondrial movement in neurons (Strom et al., 2008; Barnhart, 2016), both in kinesin-mediated anterograde (Wang and Schwarz, 2009) and dynein-mediated retrograde directions (Pilling et al., 2006), using the microtubule network and protein complexes that enable their trafficking. Drosophila models of synaptic insufficiency identified the GTPase Mitochondrial Rho (Miro) and trafficking protein kinesin binding 1 (TRAK1)/milton (milt) as important for mitochondrial axonal transport (Stowers et al., 2002; Guo et al., 2005). Other elements of the transport machinery have been demonstrated to interact with TRAK1 and Miro1, including the mitochondrial calcium uniporter (MCU) complex, which regulates the influx of calcium into the mitochondrial matrix (Niescier et al., 2013), and disrupted in schizophrenia 1 (DISC1), hypothesized to contribute to increased risk of psychiatric illness (Ogawa et al., 2014).
Of the numerous proteins involved, the endogenous mammalian TRAK proteins, TRAK1 and TRAK2, were found to function as kinesin adaptors linking kinesin heavy chain (KHC) to mitochondria. Using both gene silencing (shRNAi) and dominant negative methodologies, Brickley and Stephenson (2011) showed that inhibiting the formation or the availability of the TRAK kinesin adaptor in axons of hippocampal pyramidal neurons caused a decrease in mitochondrial mobility. Specific knockdown of TRAK1, and not TRAK2, impaired mitochondrial mobility in neurons, whereas reconstitution of either one rescued TRAK1 shRNAi-induced arrest of mobility.
Further studies elucidating the specific roles of TRAK1 and TRAK2 in mitochondrial trafficking showed that TRAK1 binds to both kinesin-1 and dynein/dynactin, is needed for normal axon outgrowth, and is primarily localized in axons. TRAK2, on the other hand, predominantly interacts with dynein/dynactin, serves a role in dendritic development, and is preferentially localized in dendrites (van Spronsen et al., 2013).
Impairment of mitochondrial transport or dynamics as a whole has been previously linked to several neurodegenerative disorders, including Parkinson’s disease, Alzheimer’s disease and Huntington’s disease (Waterham et al., 2007; Mattson et al., 2008; Schon and Przedborski, 2011). However, thus far, disruption of the TRAK1-mediated mitochondrial trafficking has yet to be associated with a specific human disease. We now report six such cases from three unrelated families of similar origin.
Materials and methods
Patients
Patients were managed at the Metabolic Disease Unit and the Pediatric Neurology Unit at the Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel-Hashomer. Patients were enrolled in the clinical protocol 76-HG-0238, ‘Diagnosis and treatment of patients with inborn errors of metabolism and other genetic disorders’ (identifier: NCT00369421), approved by the NHGRI IRB. All parents of the patients gave written, informed consent.
Whole exome sequencing
Whole exome sequencing of affected Patients A.IV.3 and A.IV.8 (Family A) and one parent (Subject III.2) was performed using an Illumina TruSeq Exome capture kit and the HiSeq 2500 sequencing platform (Illumina). Reads were aligned with human reference genome (hg19; NCBI build 37; Feb. 2009) using Burrows-Wheeler transform (Li and Durbin, 2009). Variant calling was performed with GATK (McKenna et al., 2010) and functionally annotated using KGG-seq (Li et al., 2012). Given the consanguinity in the pedigree, homozygous variants were filtered based on allele frequency less than 0.01 with no reported healthy homozygotes in online databases, dbSNP, 1000G, ESP6500 and ExAC. Likely pathogenicity was assessed if the variant was truncating (splicing or non-sense) or missense; in-frame indels were considered if they were predicted to be pathogenic by online prediction tools, PolyPhen-2, SIFT, CADD and MutationTaster. Confirmation and family screening of identified candidates were performed using direct Sanger sequencing (Applied Biosystems).
Cell culture, RT-PCR, quantitative PCR and western blotting
Dermal fibroblasts derived from affected patients were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 15% foetal bovine serum (FBS), non-essential amino acid medium and pencillin-streptomycin as described (Malicdan et al., 2015). Normal adult and neonatal dermal fibroblasts (ATCC PCS-201-012 and PCS-201-010) were purchased for control experiments. Total RNA was isolated from control and patients using RNeasy® Mini Kit (Qiagen) and treated with DNase (Ambion, Life Technologies) and transcribed to cDNA as described (Malicdan et al., 2015) using high capacity RNA to cDNA kit (Applied Biosystems). To check the splice defect, primers binding to flanking exons (primer sequences are available upon request) were used to amplify the region of interest; polymerase chain reaction (PCR) products were loaded onto a 3% agarose gel and the excised bands were sequenced. Quantitative real-time PCR was performed using specific primers (primer sequences available upon request) with Power SYBR® Green master mix (Applied Biosystems), and ran through a Bio-Rad qPCR machine (CFX96 Touch Real Time PCR detection system) with standard qPCR parameters to analyse the expression of TRAK1 compared with the control gene POLR2A. We used different sets of primers to check the effect of the pathogenic variant on the expression of two predominant transcripts expressed in fibroblasts (NM_001042646.2 or Transcript 1 and NM_014965.4 or Transcript 2). Ct values were analysed with the comparative CT method (Livak and Schmittgen, 2001). The PCR products of the real-time reactions were loaded into 2% agarose gel to compare the band intensity with quantitative PCR results. For western blotting, total cell lysates were prepared from fully confluent T-75 culture flasks. Cells were lysed using RIPA buffer (50 mM Tris, pH 7; 150 mM NaCl; 0.1% SDS; 0.5% sodium deoxycholate; 1% Triton™ X-100; and 1 mM EDTA) supplemented with protease inhibitors (complete, Mini, EDTA-free, Roche). Samples were quantified, electrophoresed on 4–12% Tris-glycine gel and blotted onto nitrocellulose membrane through dry transfer (iBlot®, Invitrogen). Membranes were blocked with Li-Cor blocking buffer (Li-Cor Biosciences) for 1 h, and then incubated with rabbit anti-TRAK1 antibody (HPA005853, Sigma) and mouse anti-β-actin (Sigma). After several washes in phosphate-buffered saline (PBS) with 0.1% Tween, membranes were incubated with the appropriate secondary antibodies (Li-Cor Biosciences) and imaged under the Li-Cor imaging system (Li-Cor Biosciences).
Electron microscopy
For electron microscopy, the specimens were fixed in 2.5% glutaraldehyde in 0.1 M buffered cacodylate, post-fixed in 1% osmium tetroxide for 1 h, dehydrated in a series of increasing ethanol concentrations, and finally embedded in epoxy resin–Agar mix (Agar Scientific) for 2 days at 60°C. Semi-thin sections were prepared from the blocks, and relevant areas were selected for ultra-thin sections, stained with uranyl acetate and lead citrate. Examination was performed in a Jeol – 1200 EX transmission electron microscope (Jeol).
JC-1 metabolic staining and fluorescence microscopy
Analysis of the metabolic state of the cells was based on the specific characteristics of JC-1 dye, that exhibit a potential-dependent mitochondrial accumulation, indicated by a fluorescent emission shift from green (∼525 nm) to red (∼595 nm). Cells were grown to 70% confluence, incubated for 15 min in growing medium containing 10 µg/ml JC-1. Cells were then washed to remove excess JC-1 and imaged with a 488 nm laser for excitation using a LSM510 Zeiss confocal microscope. Red/green ratios were calculated from the relative red and green intensities. For dual visualization of mitochondria and microtubules, cells were first incubated in 20 nM tetramethylrhodamine methyl ester (TMRM; Life Technologies) for 60 min at room temperature. After rinsing, cells were incubated in 250 nM TubulinTracker Green (Life Technologies) for 30 min at 37°C, then rinsed and placed in recording Hank’s balanced salt solution (HBSS) for fluorescence imaging with standard tetramethylrhodamine (TRITC) and fluorescein isothiocyanate (FITC) filters.
Mitochondrial motility
Cell imaging
For live imaging of the mitochondria, cells were plated on MatTek dishes (MatTek) at a concentration of 25 000 cells/plate and transfected with CellLight® Mitochondria-GFP, BacMam 2.0 (Life Technologies) at a concentration of 40 particles per cell (Kandel et al., 2015). Cells were imaged the following day in recording HBSS (HBSS pH 7.4 with 1.3 mmol/l CaCl2, 0.9 mmol/l MgCl2, 2 mmol/l glutamine, 0.1 g/l heparin, 5.6 mmol/l glucose, and 1% FBS). Cells were imaged using a QImaging QIClick™ camera (QImaging,) (1 × 1 binning, 1392 × 1040 pixels) attached to Olympus IX70 microscope with an Olympus 40 × oil immersion objective lens and PhotoFluor® light source (89 North) (Bonekamp et al., 2013). Computer control of the microscope was facilitated by LUDL programmable filter wheels, shutters, and focus control (LUDL Electronic Products), and images were collected using IPL 3.7 software (BD). For each experiment, cells were visualized using a standard FITC filter, and isolated cells with well-resolved mitochondria were selected. Cells were imaged for 5 min every 3 s.
Mitochondrial motility measurements
We preprocessed raw image files in ImageJ as previously described (Kandel et al., 2015). Supplementary Video 1 shows a raw image sequence of a control cell and Supplementary Video 2 shows the same video after preprocessing in ImageJ. Videos were analysed by a custom Matlab script previously described (Kandel et al., 2015), which was designed to track the movement of individual mitochondria. Net distances travelled by mitochondria were calculated using stored centroid locations from each frame. The scripts and instructions of our algorithm are included online on Github (www.github.com/kandelj/MitoSPT), a freely available software-sharing repository, and are intended for public use with proper citation.
We subjected our analyses to the same restrictions as in our previous work in order to maximize our signal-to-noise ratio (SNR). Only objects measuring between 20 and 500 pixels (∼0.5 to ∼13 μm2 with our microscope’s resolution) and lasting four or more consecutive frames (≥9 s) were considered. Sampling frequency was 3 s, because more frequent sampling increases noise, but less frequent sampling increases the probability of missing true signal.
Statistics
The Matlab function ‘normal parameter estimates’ (normfit) gave the mean and standard deviation of the normal distribution best fitting the distribution of logs of net distances. This information is used together with the ‘normal probability density function’ from Matlab (normpdf) to plot the probability density functions for each mitochondrial population. P-values comparing log distributions were computed using the rank-sum test (ranksum function in Matlab). As acceptance or rejection of the null hypothesis based on P-value is by definition affected by dataset sizes, we simply report the P-values instead of establishing a threshold for significance when comparing our large datasets.
Oxygen consumption measurement in human fibroblasts
The mitochondrial oxygen consumption rate was measured in human fibroblast cells using the Seahorse XF24 Instrument (Seahorse Bioscience). Forty-eight hours prior to the analysis, both wild-type control and patient fibroblast cells were plated on XF-24 plates at 20 000 cells per well and grown in DMEM, which contained 25 mM glucose, 10% FBS and 1% minimum essential medium non-essential amino acid at 37°C with 5% CO2. Immediately before the analysis, the culture medium was replaced by unbuffered DMEM XF Base medium (Seahorse Bioscience) at pH 7.4 supplemented with 5 mM glucose, 2 mM GlutaMAX™ and 1 mM sodium pyruvate, and the plates were incubated at 37°C in a CO2-free incubator for 15 min. The mitochondrial respiration was measured under four conditions: (i) basal conditions; and after the addition of (ii) 1.25 µM oligomycin; (iii) 0.2 µM of the uncoupler, carbonyl cyanide (trifluoromethoxy) phenylhydrazone (FCCP); and (iv) 1 µM of rotenone together with 1.8 µM antimycin A. The drugs were added in the order described above. Each condition involved three cycles of mixing (150 s), waiting (120 s), and measuring (210 s). This series was repeated following each injection.
Results
Clinical features
Family A
Patient A-IV.3 (Fig. 1A) was the first child to first cousin parents of Arab descent. He was born after an uneventful pregnancy via normal delivery. He showed moderate delay in motor and speech development from early on. At 19 months, 1 week after a vesicular rash consistent with varicella infection, he developed acute perioral twitching resembling myokymia, followed by involuntary myoclonic jerks of the right hand, which did not correspond to ictal or interictal EEG changes, and were unresponsive to valproic acid, clonazepam and primidone. At Sheba Medical Center, he was alert although hypotonic with constant perioral and right hand, asynchronous, continuous myoclonic jerks. Awake EEG showed generalized slowing with intermittent spikes asynchronous to the jerks. Further evaluation included elevated inflammatory indices (CRP 112 mg/l; normal reference, <3.0), but all other vasculitic indices were normal. Blood was positive for a shell vial cytomegalovirus (CMV) assay. CSF was acellular with normal glucose and protein levels. Brain MRI showed numerous bilateral subcortical hyperintense white matter foci on fluid-attenuated inversion recovery (FLAIR) sequences (Fig. 2A). A preliminary diagnosis of acute disseminated encephalomyelitis (ADEM) was made. After showing no response to a course of intravenous immunoglobulin (IVIg, 2 g/kg), pulse steroid treatment (30 mg/kg/day for 3 days) was given, which resulted in a transient remission of myoclonus that reappeared a week later, becoming more aggressive and evolving into tonic-clonic status epilepticus resistant to a variety of anticonvulsant drugs, which partially remitted only after high dose midazolam treatment under anaesthesia.
Figure 1.

Pedigrees of patients with pathogenic TRAK1 variants. Patients in Families A (A), B (B) and C (C) are depicted. Open symbols represent unaffected individuals, filled symbols designate affected individuals and are homozygous for the splicing variant. N/A = not available. Symbols with small circles within designate carrier status for the splicing variant.
Figure 2.

Neuroimaging and brain biopsy studies. (A) Brain MRI studies of affected cousins from Family A. (i) T2 FLAIR axial image of Patient A-IV.3 at the level of the lateral ventricles demonstrating abnormal signal of the periventricular white matter; (ii) T2 axial image of Patient A-IV.3 at the same level demonstrates grey and white matter atrophy accompanied by signal abnormality of the periventricular white matter; (iii) ADC (apparent diffusion coefficient) calculation of the image of Patient A-IV.10, consistent with diffusion restriction suggesting metabolic compromise; and (iv) Diffusion weighted imaging (DWI) sequence at the same level shows abnormal cortical signal bilaterally in the occipital lobes. (B) Brain biopsy histology of Patients A-IV.3 and A-IV.10. Haematoxylin and eosin staining revealed round-oval shaped structures seen in the neuropil of the grey matter (arrows). Magnification: ×600. (C) Transmission electron micrograph (Patient IV.3) in i shows the distribution of mitochondria at the periphery of the cell; ii is at a higher magnification, demonstrating clusters of the mitochondria (arrows) observed at the cell periphery. Electron micrograph in iii indicates an inclusion body (arrow) in a neuronal process. High magnification of the inclusion body iv indicates that the inclusion consists of aggregates of vesicles.
EEG at that point showed a pattern of multiple independent spike foci (MISF). Single-photon emission computed tomography (SPECT) demonstrated focal increased uptake in the inferior left tempo-parietal area, suspected to be the epileptogenic focus while repeat MRI showed general atrophy more dominant on the right. Brain biopsy from the right occipital region demonstrated mild perivascular lymphocytic infiltrates, fibrillary gliosis involving mainly the grey matter and microglia activation with no evidence for microglial nodules or viral inclusions (Fig. 2B). On electron microscopy inclusion bodies in neuronal processes were detected (Fig. 2B). PCR analysis from tissue biopsy was positive for CMV and negative for all other viruses looked at. Other clinical information is in Table 2. The biopsy was interpreted as consistent with non-specific chronic encephalitis. The diagnostic hypothesis was of bilateral Rasmussen’s encephalitis, possibly related to CMV infection. Despite treatment with five cycles of plasmapheresis and immunoglobulins he continued to deteriorate with loss of all developmental milestones and ongoing multifocal myoclonus, and died at the age of 2.5 years, in another centre.
Table 1.
Demographic, clinical and histopathological characteristics of patients with TRAK1-related encephalopathy
| Patient | 1 (A-IV.3) | 2 (A-IV.10) | 3 (B-V.4) | 4 (B-V.5) | 5 (C-IV.3) | 6 (C-IV.4) |
|---|---|---|---|---|---|---|
| Gender | M | F | M | M | F | F |
| Consanguinity | Yes | Yes | Yes | Yes | Yes | Yes |
| Age of onset (months) | 19 | 14 | 2.5 | 13 | 1.5 | 1 |
| Presenting symptoms | Perioral myoclonus followed by myoclonic hand jerks. | Perioral myoclonus followed by myoclonic unilateral upper limb coarse myoclonus. | Multifocal and generalized myoclonus. | Facial myoclonus followed by bilateral independent upper limb myoclonus. | Exaggerated startle response and recurrent episodes of generalized myoclonic jerks. | Mild myoclonic jerks evolving into episodes of prolonged generalized myoclonus. |
| Developmental delay until rapid regression | + | + | ++ | + | + | Normal |
| Neurological signs | Alert, hypotonia evolving to spasticity, constant perioral and upper limb myoclonus. | Alert, truncal hypotonia and limb spasticity, swallowing difficulties. | Spastic, continuous multifocal myoclonus. | Alert, progressive spasticity, multifocal myoclonus swallowing difficulties. | Multifocal myoclonus, progressive spasticity, poor communication skills. | Alert, multifocal myoclonus, progressive spasticity. |
| Course | Multifocal myoclonus evolved into GTC seizures with recurrent intractable SE resistant to all AEDs, steroids, IVIg. Loss of all developmental milestones at 2 years. | At 17 m episodes of recurrent GTC SE with rapid deterioration and loss of all milestones requiring tube feeding and few weeks later assisted ventilation. | Generalized and partial tonic-clonic seizures from 12 m of age, myoclonic status epilepticus at 22 m of age followed by multifocal EPC. At 3 years intractable SE and loss of all developmental milestones. | At 14 m myoclonic status followed by rapid deterioration and loss of all developmental milestones. | CPR at 4 months after unclear apneic episode. From 7 m recurrent GTCs and episodes of SE. At 28 m rapid deterioration following GTC SE. | At 7 months onset of GTC seizures and episodes of myoclonic status and GTC status. Rapid regression at 20 m after generalized SE. |
| Brain MRI | Numerous bilateral subcortical white matter foci. Second MRI showed generalized atrophy. | Generalized atrophy. | Mild frontal atrophy. | N/A | N/A | Considered normal. |
| Brain biopsy | Mild perivascular lymphocytic infiltrates, fibrillary gliosis, and activation of microglia. EM: inclusion body in a neuronal process. | Increased astrocytosis, activation of microglia, perivascular cuffing by lymphocytes. | N/A | N/A | N/A | N/A |
| Additional laboratory investigation | Blood: elevated CRP, positive CMV shell vial assay. Normal metabolic screen (blood lactate, pyruvate, ammonia, liver enzymes and amino acids profile, urine organic acids profiles). CSF PCR was negative for an array of viruses including CMV. | Blood: positive CMV shell vial assay. Normal metabolic screen. CSF: Negative PCR for viruses. | Blood: Normal metabolic screen (serum lactate, ammonia and blood amino acids and urine organic acids). | Blood: Normal metabolic screen (serum lactate, ammonia and blood amino acids and urine organic acids). | Blood: Normal metabolic screen (plasma, urine and CSF amino acid profiles; urine organic acids profile, sulphite levels, creatinine and guandinoacetate). CSF lactate levels and skin biopsy for NCL were normal. | |
| Age of death (months) | 30 | 18 | 40 | 17 | 60 | Alive |
AED = anti-epileptic drugs; CMV = cytomegalovirus; CRP = C-reactive protein; EM = electron microscopy; EPC = epilepsia partialis continua; GTC = generalized tonic-clonic seizure; IVIg = intravenous immunoglobulins; NCL = neuronal ceroid lipofuscinosis; SE = status epilepticus; N/A = not available or not relevant.
Table 2.
Mitochondrial motility parameters and statistics in patient and control fibroblasts
| n | Geo-mean of net distances (nm) | P-value versus Control | P-value versus Patient A-1 | |
|---|---|---|---|---|
| Control | 3640 mitochondria (15 cells) | 192.6 | X | X |
| Patient A-IV.3 | 2691 mitochondria (13 cells) | 160.4 | 7.96 × 10−10 | X |
| Patient A-IV.10 | 1505 mitochondria (12 cells) | 142.9 | 2.25 × 10−16 | 0.0012 |
His female cousin (Patient A-IV.10, Fig. 1A) was born at term after an uneventful pregnancy and like Patient A-IV-3 showed mild motor delay before acute onset of disease-related symptoms. At 14 months she presented with peri-oral twitching movements followed by myoclonic jerks of the right arm; EEG was normal. Twitching then evolved within several weeks to generalized tonic-clonic seizures and continuous polymyoclonus which persisted despite treatment with valproic acid and clonazepam. Her condition remained unchanged for the following 3 months, at which point in addition to the myoclonus she developed severe generalized hypotonia and weakness, poor oral intake and decreased alertness. In addition, she developed respiratory distress requiring intubation and mechanical ventilation.
Upon admission at Sheba Medical Center, the infant showed evidence of failure to thrive and had multifocal myoclonus with intermittent generalized tonic seizures. Interictal EEG showed multifocal polyspike wave activity. Blood PCR for CMV showed 200 copies/ml. CSF was acellular with mildly elevated protein concentration (66 mg/dl) and normal glucose and lactate; PCR assays were negative for several viruses including CMV. Brain MRI showed generalized atrophy without focal changes (Fig. 2A). Brain biopsy showed non-specific changes of increased astrocytosis, activation of microglia, and perivascular cuffing by lymphocytes (Fig. 2B). All lymphocytes stained positive for CD3 (T cells) and negative for CD20 (B cells). CMV PCR was negative and no viral inclusions were reported. She continued to deteriorate and died at the age of 18 months.
Family B
Patient B-V.4 (Fig. 1B) was the fourth child to consanguineous (first cousin) parents, born after an uneventful pregnancy via Caesarean section. At 2.5 months of age he began experiencing clonic-like episodes that involved each limb separately intermixed with generalized myoclonic jerks. On examination he was extremely spastic with very brisk tendon joint reflexes up to sustained clonus. EEG was considered normal while brain CT and MRI showed mild frontal atrophy. Myoclonus responded transiently to phenobarbital treatment and a provisional diagnosis of hyperekplexia was made. The clonic/myoclonic episodes relapsed and partially responded to valproic acid; he, however, had myoclonic status epilepticus at 22 months of age. Despite aggressive intravenous treatment only partial remission was achieved and he continued to have right-sided facial and body jerking consistent with epilepsia partialis continua unresponsive to a variety of anticonvulsant drugs. In the meantime, he also showed moderate developmental delay (able to sit at 12 months, able to stand at 18 months; unable to walk; and with significant speech delay). At 3 years, his seizures progressed and was in and out of status epilepticus in a local hospital ICU. He gradually deteriorated, lost independent breathing, and died during a septic episode in the hospital.
His brother (Patient B-V.5, Fig. 1B), the youngest of five siblings, was born after an uneventful Caesarean section. In the first year of life he showed mild developmental delay with mixed tone but no seizures were noticed. At 13 months of age he developed episodes of facial twitching with occasional upper limb myoclonus without loss of consciousness, partially responsive to anticonvulsants; a month later he went into myoclonic status epilepticus accompanied by lethargy. EEG showed normal background activity and occasional bilateral independent spikes. The myoclonic episodes responded to combined anti-epileptic drug (AED) therapy, but he remained lethargic and rapidly lost developmental milestones and became severely spastic, and later required tube feeding due to poor swallowing ability. He died suddenly during night sleep at home at 17 months of age.
Repeat MRIs had shown only mild progressive cortical atrophy. No further workup was performed, but DNA was preserved and analysed after their death given their clinical similarities to the prior patients.
Family C
Patient C-IV.3 was the third child to consanguineous (first cousin) parents of Arab-Muslim descent. She was born following uneventful pregnancy and delivery. At 40 days, an increased startle response was noticed, followed by recurrent episodes of generalized myoclonic jerks of various lengths with normal interictal EEG. At 4 months, she was found cyanotic and apnoeic in bed, but responded to cardiopulmonary resuscitation. From 7 months, she had recurrent generalized tonic-clonic seizures, with occasional episodes of generalized status epilepticus. Seizures were intractable to various AEDs and were intermixed with very frequent polymyoclonus. Despite the intractable seizures, her developmental milestones were only moderately delayed with independent walking at 18 months and ability to say a few words at 2 years. At 28 months, following a prolonged status epilepticus, she abruptly lost all milestones but was able to maintain eye contact. She was still fed by mouth and breathed independently. She died at home at 5 years of age after a pulmonary infection.
Her younger sister (Patient C-IV.4) was also born following normal pregnancy and delivery. Mild myoclonic jerks were noticed by the mother at 1 month of age, however, became continuous and overt at 7 months of age. In addition, she had several prolonged episodes of generalized myoclonus several weeks apart, with normal interictal EEG. At 7 months, she developed generalized tonic-clonic status epilepticus, which subsequently remitted after intravenous medication. Recurrent episodes of AED-unresponsive seizures ensued. Despite the recurrent seizures, she acquired developmental milestones, sitting at 6 months, crawling at 8 months and walking independently at 15 months, and could say a few words and understood simple commands. At 20 months, she had a prolonged status epilepticus requiring hospitalization, and she subsequently lost most of her developmental milestones. Initial evaluation included a brain MRI, which was considered normal, along with other investigations (Table 2). Currently, at 4 years of age, she is alert and maintains good eye contact, however, has no gross motor abilities. She has continuous facial twitching, tongue fasciculations, almost continuous (right > left) arm clonic movements consistent with epilepsia partialis continua, she shows axial hypotonia with severe limb spasticity with evolving joint contractures with +4 deep tendon reflexes and a positive Babinski sign.
Identification of loss of function variant in TRAK1
Exome sequencing performed for Patients A-IV.3 and A-IV.10 and their parent (Patient A-III.2) from Family A (Fig. 1A) identified three rare variants compatible with autosomal recessive inheritance. One was c.287-2A > C, a homozygous splice acceptor site variant in intron 3 of the TRAK1 gene (NM_001042646). The same variant was found in the affected individuals in Families B (Patients B-V.4 and B-V.5) and C (Patients C-IV.3 and C-IV.4). To further confirm the disease segregation of the c.287-2A > C variant, we performed Sanger sequencing of all available DNA. Complete segregation of the variant to disease was found in the three families in this report (Fig. 3A).
Figure 3.

Molecular analyses of TRAK1 variant in patients. (A) Sample chromatogram showing the Sanger confirmation of the variant in the families (wild-type, heterozygous, and homozygous). (B) RT-PCR products obtained with primer pairs (flanking exon 3) for amplification of the mRNA from two affected individuals (Patients A-IV.3 and A-IV.10) and one control. The results indicate two abnormal cDNA fragments in affected individuals compared to control: a 16-bp deleted (mutant isoform A) and 77-bp deleted (mutant isoform B). (C) In comparison to control (top), sequencing of affected individual’s aberrant transcripts shows lack of the first 16 bp of TRAK1 exon 3 (middle) and entire exon 3 (bottom). (D) Schematic diagram of splicing defect, showing the formation of two different mutant transcripts (mutant isoform A and mutant isoform B). (E) Significantly reduced expression of Transcript 1 (NM_001042646) in patients, comparison to control. Results were normalized with the expression of a housekeeping gene, POLR2A. Transcript 2 (NM_014965.4) was undetectable in both Patients A-IV.3 and A-IV.10. (F) Western blot results showing significantly reduced or no expression of TRAK1 in both patients in comparison to control. Beta actin (ACTB) was used as loading control. Lanes 1 to 7 represent: 1, ladder; 2 and 3; control; 4 and 5, Patient A-IV-3; 6 and 7, Patient A-IV-10.
The variant was not found in Exome Aggregation Consortium (ExAC) database, Cambridge, MA (http://exac.broadinstitute.org) (accessed 08/2016) nor in ClinVar (Landrum et al., 2016) (http://www.ncbi.nlm.nih.gov/clinvar/) nor in our in-house database (Gahl et al., 2015). The variant is in a highly conserved nucleotide and predicted to affect splicing. To delineate the effect of the variant on TRAK1 mRNA, we generated cDNA from skin fibroblasts from two affected individuals (Patients A-IV.3 and A-IV.10) and an unaffected control. A PCR primer set was designed to amplify flanking exons 1 and 4 with expected product size of 585 base pairs (bp). Electrophoresis in 3% agarose gel of RT-PCR products revealed that the c.287-2A > C variant induces the formation of two mutant isoforms (Fig. 3B) that are less than 585 bp. Direct sequencing of these shortened fragments (Fig. 3C) demonstrated a deletion of the entire exon 3 (NM_001042646.2: c.287_363del) in one fragment and a deletion of the first 16 bp of exon 3 in the other (NM_001042646.2: c.287_302del). This 16 bp deletion is likely created by the activation another potential cryptic acceptor splice site within exon 3, located 16 bp downstream of the original acceptor site. Both exon skipping or the deletion of 16 bp are predicted to shift the frame and create premature termination codons, NM_001042646: p.Leu96Glnfs*46 and p.Leu96Glnfs*5, respectively (Fig. 3D). We checked the effect of splicing on all the predominant isoforms and found similar splicing pattern in all of them (data not shown). As the variant results in the formation of early termination codon, we predicted nonsense mediated decay of the affected transcripts. As expected, analysis of two predominant transcripts of TRAK1 in the fibroblasts from patients compared to control, show very little expression of NM_001042646.2 (0.02% in Patient A-IV.3 and 0.03% in Patient A-IV.10) and no detectable expression of NM_014965.4 in both patients (Fig. 3E). Consistent with this finding, analysis of TRAK1 protein expression using a polyclonal antibody-specific to all the isoforms showed severely reduced expression in both patients (Fig. 3F), indicating that this was a loss-of-function variant.
Apart from the mutation in TRAK1 variant, additional variants in two other genes were identified by whole exome sequencing, namely SHROOM2 [ChrX(GRCh37):g.9863977G > A; NM_001649.3:c.2029G > A; p.Ala677Thr] and ANGPTL3 [Chr1(GRCh37):g.63063479C > T; NM_014495.3(ANGPTL3):c.242C > T; p.Ser81Phe]. Segregation analyses, however, ruled out SHROOM2 and ANGPTL3 as possible causative mutations.
Effects of TRAK1 pathogenic variant in mitochondrial function
Fibroblasts derived from two affected members of Family A (Patients A-IV.hyper3 and A-IV.10) and normal control subjects were stained using JC-1 mitochondrial membrane potential dye. Cells carrying the pathogenic variant in TRAK1 exhibit both irregular patterns of mitochondrial scattering in the cell (Fig. 4A, right panels) compared to normal control cells (Fig. 4A, left panels), as well as a reduced metabolic state, reflected by a lower red/green ratio (Fig. 4B). In addition, mitochondria extended to the cell periphery in control cells, while in patient cells, mitochondria remained clustered around the nucleus (Fig. 5A). We then looked at the status of microtubules, which are involved in proper mitochondrial localization in cells. Microtubules appeared intact in control as well as patient cells, confirming that there was no defect in microtubule structure in any of these cells (Fig. 5A).
Figure 4.
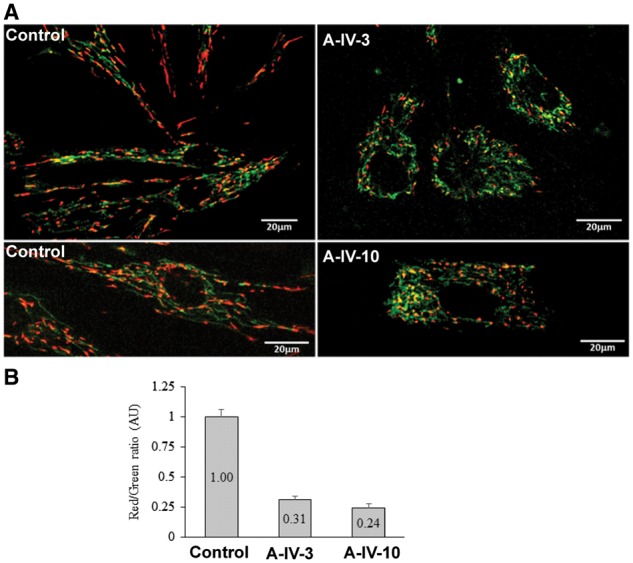
JC-1 metabolic staining. (A) Images from fibroblasts of affected individuals (Patients A-IV-3 and A-IV-10) and unaffected control incubated with JC-1 dye. A fluorescent shift from green to red indicates a potential-dependent mitochondrial accumulation. (B) Red/green ratios were calculated from the relative red and green intensities.
Figure 5.
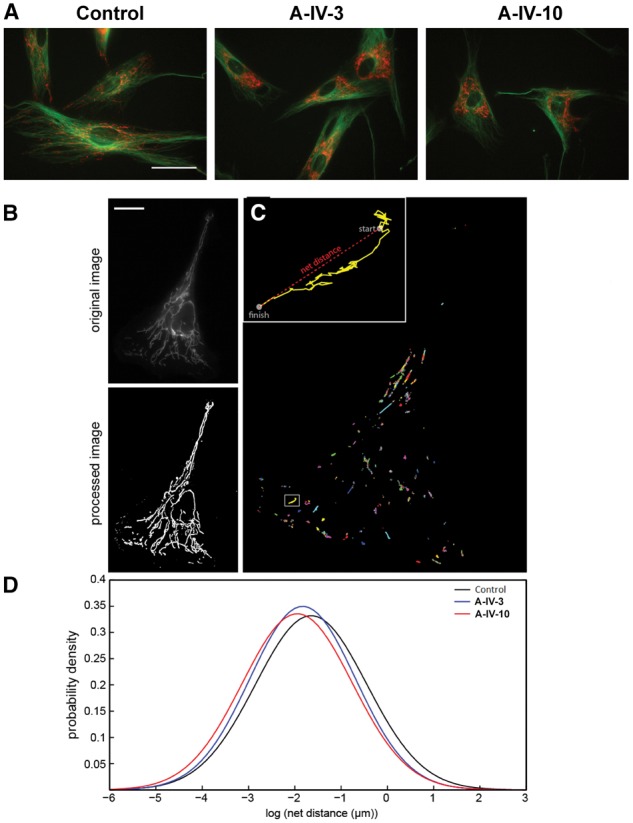
Analysis of mitochondrial movement in fibroblasts. (A) Microtubule staining. Fibroblasts from control and different patients (Patients A-IV.3 and A-IV.10) are stained with TubulinTracker (green) and TMRM (red). Scale bar = 50 µm. (B–D) Images depicting mitochondrial movement. (B) Image of a sample control cell before and after ImageJ processing. Scale bar = 20 µm. (C) Path lengths of all mitochondrial centroids in this cell. Box indicates inset region. Inset: Path length of one particular mitochondrion, with start and finish points indicated. This mitochondrion travelled a total distance of 9.52 µm and a net distance of 2.47 µm. (D) Probability density plots of net distances travelled by control and patient mitochondria.
Mitochondrial motility
Consistent with our previous work (Kandel et al., 2015), net distances travelled by mitochondria in control cells (Fig. 5B) followed a log-normal distribution. We found that mitochondria in patient cells travelled shorter net distances than control mitochondria (Table 2 and Fig. 5C). The geometric mean net distance travelled by control mitochondria was 192.6 nm (n = 3640 mitochondria from 15 cells); Supplementary Video 1 shows representative control mitochondria movement, in which case the geometric mean net distance is 206.8 nm. For Patient A-IV.3, the geometric mean was 160.4 nm (n = 2691 mitochondria from 13 cells), giving a P-value of 7.96 × 10−10 versus control using the rank-sum test. A sample cell from this population is shown in Supplementary Video 3, with the corresponding processed video given as Supplementary Video 4; the mitochondria in this particular cell travelled a geometric mean net distance of 164 nm. Patient A-IV.10 showed an even greater decrease in mitochondrial motility than Patient A-IV.3, with a geometric mean net distance of 142.9 nm (n = 1505 mitochondria from 12 cells). The P-value comparing this population to the control mitochondria was 2.25 × 10−16. Supplementary Video 5 shows a sample cell from Patient A-IV.10, and Supplementary Video 6 shows the corresponding processed video used for analysis; the mitochondria in this cell travelled a geometric mean net distance of 153.8 nm over the course of the 5 min recorded.
Loss of TRAK1 function does not disrupt peroxisomal distribution and motility
Mitochondria and peroxisomes exhibit a close functional interplay and cooperate metabolically (Schrader et al., 2015). Furthermore, Miro1 was also detected on peroxisomes (Costello et al., submitted for publication). We analysed the distribution of peroxisomes in control and patient fibroblasts (Supplementary Fig. 1). In contrast to mitochondria, which cluster around the nucleus in patient cells, peroxisomal distribution was similar to that of control cells (Supplementary Fig. 1A). Peroxisomes were uniformly distributed within the cytoplasm and reached the cell periphery. Like mitochondria, peroxisomes bind to and move along microtubules in mammalian cells (Schrader et al., 2000). To analyse peroxisome motility, control and patient fibroblasts were transfected with a GFP fusion protein bearing a peroxisomal targeting signal (GFP-SKL). GFP-SKL localized exclusively to peroxisomes, and no alterations in peroxisome distribution were observed in live cells, confirming the immunofluorescence results (not shown). Analysis of peroxisome motility revealed no differences in long range, microtubule-dependent motions between controls and patient fibroblasts (Supplementary Fig. 1B). Only a small population of peroxisomes moved in a microtubule-dependent manner at a given time point, which is consistent with published data (Rapp et al., 1996; Wiemer et al., 1997; Schrader et al., 2000). These observations further confirm the integrity of the microtubule network in patient cells and reveal that a loss of TRAK1 function appears to specifically affect mitochondrial distribution and motility in fibroblasts.
Oxygen consumption measurement in human fibroblasts
To understand the metabolic effects in the mitochondria due to the TRAK1 deficiency, we used the Seahorse analyser to measure the cellular oxygen consumption rate. Our results indicated that the maximal respiration capacity in fibroblast cells harbouring the pathogenic variant in TRAK1 are significantly decreased compared to normal fibroblasts shown in Fig. 6.
Figure 6.

Oxygen consumption rates in fibroblasts. Cellular oxygen consumption rates (OCR) for fibroblast cell lines from wild-type and TRAK1 c.287-2 A > C patient (Patient A-IV.3) was measured by XF24 extracellular flux analysis. Oligomycin (1.25 µM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 0.2 µM), and antimycin A (1.8 µM) together with rotenone (1 µM) were injected after 20, 50 and 80 min, respectively. n = 4 plates; *P-value < 0.05.
Discussion
This is the first report of TRAK1 pathogenic variant in patients with neurodevelopmental delay, seizures and fatal encephalopathy. Our patients harbour a biallelic splicing variant in the early coding exon of TRAK1 that results in premature termination during translation. Furthermore, mRNA analysis showed a severely reduced level of normal transcripts; aberrant transcripts produced by the mutant isoforms are likely degraded through nonsense mediated decay. The predicted truncated protein products do not have the C-terminal domain of TRAK1 that is crucial for its interaction with mitochondria, and therefore lose the ability to localize to the mitochondria (Webber et al., 2008). This was supported by our western blot results showing severely reduced expression of TRAK1 using an antibody that recognizes the middle portion of the protein. Given that affected patients from our cohort come from consanguineous background, it is also possible to consider that other deleterious variants may contribute to the disease phenotype. In our analysis, we have identified ANGPTL3, which was associated with familial hypobetalipoproteinemia, and SHROOM2, which has not been associated with human disease. Both candidate genes, however, are unlikely, given the fact that they did not segregate with disease by Sanger sequencing.
Because of a requirement for high energy and calcium buffering during synaptic neurotransmission, the proper distribution of mitochondria in neurons is essential (Brickley et al., 2011). Given the long distances that often separate the neuronal soma from its terminus, and the fact that the majority of mitochondria are generated in the neuronal body, it would be impractical for neurons to rely on the slow diffusion of ATP in order to meet their energy demands. In mammals, the motors that move organelles in an anterograde direction are the kinesins; in particular, the motors of mitochondrial axonal transport belong to the kinesin-1 family, encoded by KIF5A, KIF5B and KIF5C (Sheng and Cai, 2012). The adaptor proteins, TRAK (TRAK1 and TRAK2) and MIRO (Saotome et al., 2008; Macaskill et al., 2009; Wang and Schwarz, 2009), connect the KIF5 motors to the mitochondria. Knockdown of TRAK1 in hippocampal neurons led to significant impairment in mitochondrial motility and affected both anterograde and retrograde transport (Brickley et al., 2011). TRAK1 is also part of the quaternary mitochondrial trafficking complex together with OGT, TRAK2, MIRO, KIF5 (Brickley et al., 2011). Using fibroblasts derived from patients, we showed that TRAK1 deficiency significantly impaired mitochondrial motility; it is possible that the mechanism for mitochondrial movement in fibroblasts is similar to that in neurons.
Cells carrying pathogenic variants in TRAK1 exhibit irregular mitochondrial localization in the cell, which is expected considering the role of TRAK1 in mitochondrial motility, but they also exhibit mitochondrial dysfunction. This was demonstrated by a lower red/green ratio of JC-1 metabolic staining, reflecting an altered mitochondrial membrane potential and a reduction in metabolic state, as well as a reduction in mitochondrial oxygen consumption. Previous studies showed an association between transport defects and altered mitochondrial morphology (Varadi et al., 2004; Fransson et al., 2006), which is tightly related to its function (Anesti and Scorrano, 2006). Another association between motility and function is the increase in mitochondrial calcium uptake following blockage of mitochondrial motility. These clues and our findings imply the possibility of TRAK1 involvement in the regulation of mitochondrial function, in addition to its role in mitochondrial motility.
Our patients main clinical features were of paroxysmal events of two main types: First, all presented with polymyoclonus with onset from 1–15 months of age. These myoclonic jerks tended to worsen and become more overt with elapsing time and evolved into ‘multifocal’ epilepsia partialis continua involving different body parts. The myoclonic jerks were not always accompanied by ictal EEG changes. Second, a few months after the myocolonic events, all patients developed generalized tonic-clonic seizures with high tendency to evolve into generalized tonic-clonic status epilepticus both resistant to most AEDs; status epilepticus events required sedation by anaesthetic drugs. When generalized tonic-clonic seizures started to appear, epileptiform activity in the form of multifocal and generalized spike wave were identified on the interictal EEG. In addition, all patients that were either hypotonic or initally with normal tone developed progressive spasticity. Their developmental course was unique: patients had mild-to-moderate developmental delay in the first 1–2 years of life, but all patients rapidly regressed and lost global developmental milestones consistently after an episode of generalized status epilepticus. All patients except Patient C-IV-4, who is still alive, although severely handicapped at 5 years of age, died within weeks to months after their abrupt regression.
Dominant mutations in KIF5A, a neuron specific kinesin protein that interacts with TRAK1 as its linker protein to mitochondria (Glater et al., 2006) has been associated with spastic paraplegia type 10 both in its pure and complicated variants with onset from early childhood to early adulthood (Reid et al., 2002; Goizet et al., 2009). The main mechanism considered to cause both progressive spasticity and peripheral neuropathy in KIF5A mutated patients and models is related to anterograde axonal deficient mitochondrial transport caused by dysfunctional KIF5A. It is possible that the spasticity seen in our patients can be explained by secondary dysfunction of KIF5A because of its abnormal linking to mitochondria when TRAK1 is dysfunctional or to the primary role of TRAK1 in mitochondrial transport in axons. Recently, a de novo variant in the cargo binding domain of KIF5A was reported in patients with myoclonic seizures, hypotonia, profound neurodevelopmental delay and progressive leukoencephalopathy (Rydzanicz et al., 2016). The phenotype is consistent with the existing information that more than 75% of conditional Kif5a knockout mice undergo seizures and die within 3 weeks (Xia et al., 2003). One KIF5A patient presented with myoclonus resembling that of our patients and, similar to them, lacked obvious EEG abnormalities, suggesting either subcortical or peripheral myoclonus.
Another explanation to the progressive spasticity in our patients relates to the role of TRAK1 as regulating cargo other than mitochondria. Variants in the mouse orthologue of human TRAK1 (Trak1/hyrt mutant mice) are associated with low levels of γ-aminobutyric acid type A (GABAA) receptors, and hence deficiency of GABA-mediated neural inhibition, in the CNS of hypertonic mice (Gilbert et al., 2006). Histological examination and staining with haematoxylin and eosin from CNS samples of the Trak1/hyrt mutant mice revealed oval-shaped inclusion bodies that appeared vesicular in nature (as opposed to the typical protein aggregates seen in neurodegenerative diseases). They had two layers of membrane and were present in the neuronal processes of the grey matter, rather than in the cell bodies. A role of TRAK1 in the regulation of endocytic trafficking of the GABAA receptors was then postulated, as a mechanistic explanation for motor disinhibition and hypertonicity in the mouse model.
In fact, all our patients showed progressive spasticity. Although pyramidal signs and limb spasticity may develop in mitochondrial disorders usually related to white matter involvement, hypotonia is a more common sign. One may speculate that the severe spasticity in our patients is at least partially related to the effect of TRAK1 variant on the GABAA receptors in the lower motor neurons leading to decreased motor neuron inhibition by GABAA as described in Trak1/hyrt mice (Gilbert et al., 2006). Because of the profound generalized encephalopathy and epilepsy in our patients, the clinical management did not include anti-spastic drugs such as baclofen (a GABAA agonist). Nevertheless, this treatment should be considered when facing a patient with TRAK1 variant(s) in the future.
The presenting paroxysmal symptom of our patients was of multifocal myoclonus evolving in most to epilepsia partialis continua. Epilepsia partialis continua is a condition that may develop in patients with inflammatory brain disorder (e.g. Rasmussen’s encephalitis) (Kravljanac et al., 2013) but would also be consistent with mitochondrial disorders (Riquet et al., 2008; El Sabbagh et al., 2010; Desguerre et al., 2014). Although no good pathophysiological explanation for its development in mitochondrial diseases exists, seizures could be related to patchy cellular mitochondrial dysfunction either because of heteroplasmy in mitochondrially inherited disorders [as in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) or uneven mitochondrial depletion in various neurons (as in POLG1)]. We hypothesize that this type of seizure can result from defective mitochondrial motility in neurons related to their length in TRAK1 deficiency. With progression of the disorder, the epilepsy became more generalized, accompanied by other neurological deficits including developmental delay and after the acute regression to brainstem involvement leading to early death. At no point were mitochondrial-related metabolic derangements noticed; specifically, there were no elevations of lactate in blood or CSF, blood amino acids, or urine organic acids, nor was there evidence for extra CNS systemic involvement. This could be explained by the fact that the respiratory chain pathway inside the mitochondria was only mildly affected by pathogenic TRAK1 variants but the ‘supply’ of these mildly dysfunctional mitochondria to the active axons caused much greater ‘localized’ energy deficits; normal mitochondrial motility may also be more important for neurons than for other cells. The absence of systemic involvement or a biochemical metabolic signature necessitates direct genetic testing in patients with similar neurologic phenotypes.
The direct association between TRAK1 and seizure disorder was raised by Chioza et al. (2009). In a study looking at genome-wide single nucleotide polymorphism-based high density linkage in 41 pedigree families with at least two affected members with childhood absence, TRAK1 was demonstrated as the strongest candidate within a susceptibility locus on chromosome 3p23-p14.
An additional explanation for the role of TRAK1 in epilepsy besides its effect on mitochondrial functions, can perhaps be related to its association with the intracellular loop of GABAA receptor b3 subunits (Stephenson, 2014). Homozygous mutations in the GABAA receptor b3 subunits were found to be related to familial absence epilepsy and de novo mutations to severe early epileptic encephalopathies while this gene knockout mice is used as a model for absence epilepsy; all suggesting that dysfunctional GABAa B3 subunit related to abnormal TRAK1 guidance may lead to an epileptic phenotype (DeLorey et al., 1998; Tanaka et al., 2008; Epi4K Consortium et al., 2013).
In summary, we describe for the first time a devastating human disorder associated with a null allele in the TRAK1 gene and subsequent dysfunction of mitochondrial trafficking. The complex disease manifestations may be explained by two separate, or perhaps combined, mechanisms of mitochondrial and GABAergic receptor dysfunction. While using a fibroblast model we managed to prove the mitochondrial dysfunction, further investigations into the gene’s product and its defects in various models may lead to better understanding of the complicated underlying pathology, and perhaps pave the path for mechanism-directed interventions.
Supplementary Material
Acknowledgements
The authors wish to thank the patients and their families for their kind support and assistance. G.R. is a member of the Sagol Neuroscience Network and holds the Djerassi Chair for Oncology at the Tel-Aviv University, Israel.
Glossary
Abbreviation
- TRAK
trafficking kinesin protein
Funding
D.M.E. and J.K. are supported by the Office of Naval Research (ONR) Grant N000141410538. M.S. is supported by the BBSRC (BB/K006231/1), a Wellcome Trust Institutional Strategic Support Award (WT097835MF, WT105618MA), and a Marie Curie Initial Training Network (ITN) action PerFuMe (316723). M.C.V.M., J.S., H.P., C.F., T.V. and W.A.G. are supported by the NGHRI Intramural Research Program. G.R. is supported by the Kahn Family Foundation and the Israeli Centers of Excellence (I-CORE) Program (ISF grant no. 41/11).
Supplementary material
Supplementary material is available at Brain online.
References
- Anesti V, Scorrano L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim Biophys Acta 2006; 1757: 692–9. [DOI] [PubMed] [Google Scholar]
- Barnhart EL. Mechanics of mitochondrial motility in neurons. Curr Opin Cell Biol 2016; 38: 90–9. [DOI] [PubMed] [Google Scholar]
- Bonekamp NA, Islinger M, Lazaro MG, Schrader M. Cytochemical detection of peroxisomes and mitochondria. Methods Mol Biol 2013; 931: 467–82. [DOI] [PubMed] [Google Scholar]
- Brickley K, Pozo K, Stephenson FA. N-acetylglucosamine transferase is an integral component of a kinesin-directed mitochondrial trafficking complex. Biochim Biophys Acta 2011; 1813: 269–81. [DOI] [PubMed] [Google Scholar]
- Brickley K, Stephenson FA. Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J Biol Chem 2011; 286: 18079–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Davis ML, Sheng ZH. Regulation of axonal mitochondrial transport and its impact on synaptic transmission. Neurosci Res 2011; 70: 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chioza BA, Aicardi J, Aschauer H, Brouwer O, Callenbach P, Covanis A. et al. Genome wide high density SNP-based linkage analysis of childhood absence epilepsy identifies a susceptibility locus on chromosome 3p23-p14. Epilepsy Res 2009; 87: 247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A. et al. Olsen mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci 1998; 18: 8505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desguerre I, Hully M, Rio M, Nabbout R. Mitochondrial disorders and epilepsy. Rev Neurol 2014; 170: 375–80. [DOI] [PubMed] [Google Scholar]
- El Sabbagh S, Lebre AS, Bahi-Buisson N, Delonlay P, Soufflet C, Boddaert N. et al. Epileptic phenotypes in children with respiratory chain disorders. Epilepsia 2010; 51: 1225–35. [DOI] [PubMed] [Google Scholar]
- Epi4K Consortium; Epilepsy Phenome/Genome Project, Allen AS, Berkovic SF, Cossette P, Delanty Net al. De novo mutations in epileptic encephalopathies. Nature 2013; 501: 217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson A, Ruusala A, Aspenstom P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun 2006; 344: 500–10. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Wise AL, Ashley EA. The undiagnosed diseases network of the national institutes of health: a national extension. JAMA 2015; 314: 1797–8. [DOI] [PubMed] [Google Scholar]
- Gilbert SL, Zhang L, Forster ML, Anderson JR, Iwase T, Soliven B. et al. Trak1 mutation disrupts GABA(A) receptor homeostasis in hypertonic mice. Nat Genet 2006; 38: 245–50. [DOI] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol 2006; 173: 545–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goizet C, Boukhris A, Mundwiller E, Tallaksen C, Forlani S, Toutain A. et al. Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum Mutat 2009; 30: E376–85. [DOI] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M. et al. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 2005; 47: 379–93. [DOI] [PubMed] [Google Scholar]
- Kandel J, Chou P, Eckmann DM. Automated detection of whole-cell mitochondrial motility and its dependence on cytoarchitectural integrity. Biotechnol Bioeng 2015; 112: 1395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravljanac R, Djuric M, Jovic N, Djordjevic M, Zamurovic D, Pekmezovic T. Etiology, clinical features and outcome of epilepsia partialis continua in cohort of 51 children. Epilepsy Res 2013; 104: 112–17. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016; 44: D862–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MX, Gui HS, Kwan JS, Bao SY, Sham PC. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res 2012; 40: e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001; 25: 402–8. [DOI] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A. et al. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 2009; 61: 541–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malicdan MC, Vilboux T, Stephen J, Maglic D, Mian L, Konzman D. et al. Mutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes. J Med Genet 2015; 52: 830–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008; 60: 748–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niescier RF, Chang KT, Min KT. Miro, MCU, and calcium: bridging our understanding of mitochondrial movement in axons. Front Cell Neurosci 2013; 7: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa F, Malavasi EL, Crummie DK, Eykelenboom JE, Soares DC, Mackie S. et al. DISC1 complexes with TRAK1 and Miro1 to modulate anterograde axonal mitochondrial trafficking. Hum Mol Genet 2014; 23: 906–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell 2006; 17: 2057–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp S, Saffrich R, Anton M, Jakle U, Ansorge W, Gorgas K. et al. Microtubule-based peroxisome movement. J Cell Sci 1996; 109( Pt 4): 837–49. [DOI] [PubMed] [Google Scholar]
- Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK. et al. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet 2002; 71: 1189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquet A, Auvin S, Cuisset JM, Lamblin MD, Sablonniere B, Cuvellier JC. et al. Epilepsia partialis continua and defects in the mitochondrial respiratory chain. Epilepsy Res 2008; 78: 1–6. [DOI] [PubMed] [Google Scholar]
- Rydzanicz M, Jagla M, Kosinska J, Tomasik T, Sobczak A, Pollak A. et al. KIF5A de novo mutation associated with myoclonic seizures and neonatal onset progressive leukoencephalopathy. Clin Genet 2016; Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P. et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA 2008; 105: 20728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron 2011; 70: 1033–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader M, Costello J, Godinho LF, Islinger M. Peroxisome-mitochondria interplay and disease. J Inherit Metab Dis 2015; 38: 681–702. [DOI] [PubMed] [Google Scholar]
- Schrader M, King SJ, Stroh TA, Schroer TA. Real time imaging reveals a peroxisomal reticulum in living cells. J Cell Sci 2000; 113( Pt 20): 3663–71. [DOI] [PubMed] [Google Scholar]
- Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 2013; 5(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 2012; 13: 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson FA. Revisiting the TRAK family of proteins as mediators of GABAA receptor trafficking. Neurochem Res 2014; 39: 992–6. [DOI] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 2002; 36: 1063–77. [DOI] [PubMed] [Google Scholar]
- Strom AL, Gal J, Shi P, Kasarskis EJ, Hayward LJ, Zhu H. Retrograde axonal transport and motor neuron disease. J Neurochem 2008; 106: 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Olsen RW, Medina MT, Schwartz E, Alonso ME, Duron RM. et al. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am J Hum Genet 2008; 82: 1249–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M. et al. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 2013; 77: 485–502. [DOI] [PubMed] [Google Scholar]
- Varadi A, Johnson-Cadwell LI, Cirulli V, Yoon Y, Allan VJ, Rutter GA. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J Cell Sci 2004; 117: 4389–400. [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 2009; 136: 163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 2007; 356: 1736–41. [DOI] [PubMed] [Google Scholar]
- Webber E, Li L, Chin LS. Hypertonia-associated protein Trak1 is a novel regulator of endosome-to-lysosome trafficking. J Mol Biol 2008; 382: 638–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemer EA, Wenzel T, Deerinck TJ, Ellisman MH, Subramani S. Visualization of the peroxisomal compartment in living mammalian cells: dynamic behavior and association with microtubules. J Cell Biol 1997; 136: 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia CH, Roberts EA, Her LS, Liu X, Williams DS, Cleveland DW. et al. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol 2003; 161: 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.