

Abstract
The oxygen (O2) concentration is a vital parameter for controlling the survival, proliferation, and differentiation of neural stem cells. A prenatal reduction of O2 levels (hypoxia) often leads to cognitive and behavioral defects, attributable to altered neural development. In this study, we analyzed the effects of O2 levels on human cortical progenitors, the radial glia cells (RGCs), during active neurogenesis, corresponding to the second trimester of gestation. Small changes in O2 levels profoundly affected RGC survival, proliferation, and differentiation. Physiological hypoxia (3% O2) promoted neurogenesis, whereas anoxia (<1% O2) and severe hypoxia (1% O2) arrested the differentiation of human RGCs, mainly by altering the generation of glutamatergic neurons. The in vitro activation of Wnt-β-catenin signaling rescued the proliferation and neuronal differentiation of RGCs subjected to anoxia. Pathologic hypoxia (≤1% O2) also exerted negative effects on gliogenesis, by decreasing the number of O4+ preoligodendrocytes and increasing the number of reactive astrocytes derived from cortical RGCs. O2-dependent alterations in glutamatergic neurogenesis and oligodendrogenesis can lead to significant changes in cortical circuitry formation. A better understanding of the cellular effects caused by changes in O2 levels during human cortical development is essential to elucidating the etiology of numerous neurodevelopmental disorders.
Keywords: cerebral cortex, cortical neurogenesis, hypoxia, neural stem cells, Wnt-β-catenin
Introduction
That changes in O2 levels play an important role during cerebral cortex development is well documented. Reduced O2 levels in the developing brain, whether due to umbilical cord occlusion, obstetric complications, impaired placental function, or premature birth, are often accompanied by hypoxic-ischemic pathologies that severely compromise cortical development (Woodward et al. 2005; Luu et al. 2009; Basovich 2010; Hutter et al. 2010; Volpe 2011; Li et al. 2012). A decrease in O2 levels during the fetal period can impact the development of cortical progenitor cells and result in complex brain anomalies, including a reduction in total brain size and/or cerebral cortex thickness, progressive cerebral ventriculomegaly, and decreases in the amount of subcortical white matter and/or the size of the corpus callosum (Ajayi-Obe et al. 2000; Cannon et al. 2002; Martinussen et al. 2005; Volpe 2011). These hypoxia-related anatomical alterations and fetal hypoxia itself have been broadly associated with both cognitive and behavioral deficits later in life (Weinberger 1987; Akbarian et al. 1993; van der Reijden-Lakeman et al. 1997; Raine et al. 2000; Rees and Harding 2004; Li et al. 2012; Selemon and Zecevic 2015).
O2 concentrations in the germinal zones of the developing forebrain are much lower (1–6% O2) than atmospheric O2 levels (PO2 = 21%, 150 mmHg) (Table 1). This physiological hypoxia is critical in promoting the proliferation and pluripotency of neural stem cells (NSCs) (Simon and Keith 2008; Mohyeldin et al. 2010) and in the proper transition of radial glia cells (RGCs) into glutamatergic neurons in the developing cerebral cortex (Malik et al. 2013). Severe or pathological hypoxia (PO2 ≤ 1%; ≤7 mmHg), however, induces NSC quiescence and apoptosis (Ezashi et al. 2005; Santilli et al. 2010). The effect of hypoxia on neural progenitors from early postnatal human brains, that is, when neurogenesis has ended but gliogenesis is ongoing, has been analyzed (Pistollato et al. 2007), but similar studies of human cortical progenitors during active neurogenesis are lacking.
Table 1.
Comparison of the O2 levels defining physiological and pathological hypoxia in mammals
| Condition | PO2 (mmHg) | PO2 (%) | Reference |
|---|---|---|---|
| Atmosphere | 150 | 21.1 | Reviewed by Carreau et al. (2011) |
| Lungs (tissue) | 42 | 5.6 | |
| Arterial blood | 100 | 13.2 | |
| Venous blood | 40 | 5.3 | |
| Physiological hypoxia (mammalian brain): | |||
| Fetus | <7.6 | ≤1 | Lee et al. (2001), Zhang et al. (2011) |
| Fetal subventricular zone | 18–24 | 2.5–3 | Santilli et al. (2010) |
| Adult subventricular zone | 8–48 | 1–6 | Panchision (2009), Mohyeldin et al. (2010), Zhang et al. (2011) |
| Human adult brain | 6–33 | 0.5–8 | Erecinska and Silver (2001), Ivanovic (2009), Carreau et al. (2011) |
| Pathological hypoxia: | |||
| Stroke (penumbral region in rats) | 30–1.2 | 3.5–0 | Liu et al. (2004, 2006), Zhang et al. (2011) |
| Brain trauma (risk of death in humans) | 15 | 2 | van den Brink et al. (2000) |
| Atmospheric hypoxia | <160 | <21.1 | |
| Mild hypoxia “in vitro” | 19–40 | 2.5–5 | Pistollato et al. (2007), Santilli et al. (2010), De Filippis and Delia (2011) |
| Severe hypoxia/anoxia | <8 | 0–1 | Ivanovic (2009), De Filippis and Delia (2011) |
In this in vitro study, we examined the cellular response to reduced O2 levels of human cortical progenitors (RGCs) during the second trimester of gestation (14–24 gestational weeks, gw). Specifically, we investigated the role of O2 on RGC survival and proliferation as well as the differentiation of these cells into excitatory (glutamatergic) and inhibitory (GABAergic) neurons, astrocytes, and oligodendrocytes, which occurs during the second trimester of gestation (Jakovcevski and Zecevic 2005; Howard et al. 2006; Mo et al. 2007; Mo and Zecevic 2009; Ortega et al. 2013). Our results demonstrate that pathological hypoxia (PO2 ≤ 1% or ≤7 mmHg) reduces the survival, proliferation, and neurogenesis of human cortical RGCs, whereas physiological O2 levels (PO2 3% or ∼21 mm Hg) have the opposite effect. The defects observed in cortical RGC cultures under pathological O2 conditions can, at least in part, be explained by the reduced activation of the Wnt signaling pathway.
Materials and Methods
Human Fetal Cell Cultures
Tissues for cell cultures were dissected from human fetal forebrains (Table 2) ranging in age from 14 to 19 gw and obtained from Advanced Bioscience Resources (ABR) and Human Developmental Biology Resource. Parental consent and the approval of the Ethics Committees of the respective institutions were obtained. None of the fetal brains showed evidence of disease or abnormalities upon ultrasound and neuropathological examinations. The developmental stage was estimated based on the gestational weeks after conception and on the ultrasound findings. Brain tissue was collected in oxygenized Hank's balanced salt solution (Invitrogen) and transported on ice to the laboratory, where it was first dissected and then mechanically and enzymatically dissociated (0.025% trypsin-EDTA, Invitrogen; DNAse I, 2 mg/mL, Sigma-Aldrich). The resulting cells were cultured in poly-d-lysine (Sigma-Aldrich) coated flasks containing proliferation medium (PM), consisting of DMEM/F12 (Invitrogen), B27 supplement (Invitrogen), basic fibroblast growth factor (bFGF; 10 ng/mL, Peprotech), epidermal growth factor (EGF; 10 ng/mL, Millipore), and penicillin/streptomycin (Invitrogen).
Table 2.
Fetal human brain tissues analyzed in this study
| Case no. | Gestational week (gw) | Sex | Application | Technique |
|---|---|---|---|---|
| 1 | 14 | NP | Cultures (in vitro) | WB, ICC |
| 2 | 15 | ♂ | Cultures (in vitro)/tissue imaging | IHC,WB, ICC |
| 3 | 16 | ♂ | Tissue RNA | qPCR |
| 4 | 16 | NP | Cultures (in vitro) | WB, ICC |
| 5 | 17 | NP | Cultures (in vitro) | WB, ICC |
| 6 | 17 | NP | Cultures (in vitro) | WB, ICC |
| 7 | 17 | ♂ | Tissue imaging and RNA | IHC, qPCR |
| 8 | 17 | ♂ | Cultures (in vitro) | WB, ICC, qPCR |
| 9 | 17 | NP | Cultures (in vitro) | qPCR |
| 10 | 17 | ♂ | Tissue RNA | qPCR |
| 11 | 18 | NP | Cultures (in vitro) | WB, ICC |
| 12 | 18 | NP | Cultures (in vitro) | WB, ICC, qPCR |
| 13 | 18 | NP | Tissue imaging | ISH |
| 14 | 18 | ♀ | Tissue imaging | IHC |
| 15 | 19 | NP | Tissue imaging | IHC |
| 16 | 19 | ♀ | Cultures (in vitro) | WB, ICC, qPCR |
| 17 | 22 | ♂ | Tissue imaging | IHC |
| 18 | 22 | NP | Tissue imaging | IHC, ISH |
| 19 | 22 | ♂ | Tissue imaging | IHC |
| 20 | 24 | ♂ | Tissue imaging | IHC, ISH |
Symbols/abbreviations: ♀, female; ♂, male; ICC, immunocytochemistry; IHC, immunohistochemistry; ISH, in situ hybridization; NP, not provided; qPCR, quantitative polymerase chain reaction; WB, western blot.
Enrichment of Human RGCs and Hypoxic Treatments
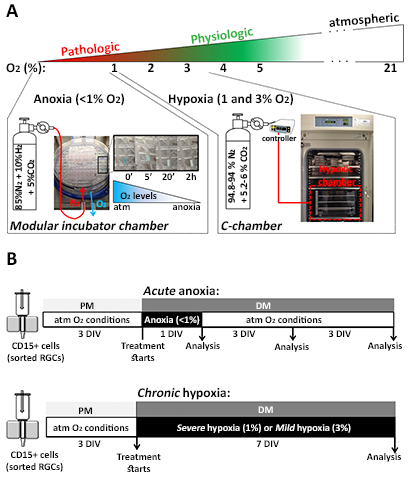
RGCs were isolated from mixed cell cultures derived from different regions of the human forebrain. Since the availability of ganglionic eminences (GEs) was limited, progenitors from the lateral, medial, and caudal GE were pooled to allow comparisons between pallial (cortical) and subpallial (GE) regions. RGCs were enriched from the mixed cell populations obtained from both forebrain areas of second trimester fetuses using MACS® Technology, based on a protocol of immunomagnetic cell sorting using anti-CD15-antibody-coated microbeads (Miltenyi Biotech) as described previously (Ortega et al. 2013; Radonjic et al. 2014b). Human RGCs were cultured on poly-d-lysine-coated 6-well plates and on 12-mm coverslips (Carolina Biologicals) in PM for 1 to 3 days in vitro (DIV) before they were subjected to the hypoxic treatments. For O2 deprivation, cells in PM were shifted to differentiation medium (DM, without bFGF and EGF) and placed into a basic modular incubator chamber (Billups-Rothenberg), in which the influx of nitrogen and CO2 results in the almost complete removal of O2 from the chamber, thus creating anoxic conditions (<1% O2) (Burgers et al. 2008). O2 levels within the chamber were monitored using anoxic strips (Becton-Dickinson) that change from blue to white when the O2 level is <1% (Supplementary Figure 1). To test the effect of longer periods of hypoxia, a C-chamber (BioSpherix) was used, as it provides a more precise control of O2 levels for hypoxic conditions of 1% and 3% O2 (Supplementary Figure 1). Control (atmospheric) conditions were defined as 21% O2. For the pharmacological treatments, the Wnt-β-catenin agonist CHIR99021 (3 μm, Stemgent) and the antagonist XAV939 (5 μm, Stemgent) were added in DM at the beginning of the anoxic stimulus. For the 7-day analysis, the medium was replaced with fresh DM 2 days after a 24-h anoxic stimulus.
Cell Viability Staining and BrdU Labeling
RGC viability was assessed using the Live/Dead viability/cytotoxicity kit (Molecular Probes) according to the manufacturer's instructions. The esterase activity of live cells converts non-fluorescent cell-permeable calcein to green fluorescent calcein, whereas dead cells, because of their damaged membranes, incorporate ethidium homodimer (EthD-1) that binds to nucleic acids, producing red fluorescence.
The thymidine analog 5-bromo-2-deoxyuridine (BrdU, 20 µM; Sigma-Aldrich) was prepared in DM and added to the cultures as distinct pulses at different time points to assess changes in RGC proliferation and the exit of these cells from the cell cycle. The BrdU pulses were stopped by replacing the cell medium. Incorporated BrdU was detected immunohistochemically in fixed cells using a BrdU-specific antibody (Becton-Dickinson).
Reactive O2 Species (ROS) in Live RGCs
ROS levels in RGC cultures were analyzed using the Image-iT TM LIVE green reactive oxygen species detection kit (Molecular Probes), based on 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy- H2DCFDA), a reliable fluorogenic marker of ROS in live cells. As a positive control, tert-butyl hydroperoxide was added to the RGC cultures to induce ROS production. The cells were counterstained by nuclear labeling with DAPI.
Immunocytochemistry and Immunohistochemistry
Cells fixed with 4% paraformaldehyde and blocked for 1 h in phosphate-buffered saline (PBS) containing 0.1% normal goat serum (Gibco) and 0.1% Triton (Sigma-Aldrich) were incubated first with primary antibodies (Table 3) overnight at 4 °C and then for 1 h with the appropriate secondary antibodies conjugated with Alexa488 or Alexa555 fluorophores (1:500, Molecular Probes). Cell nuclei were stained using bisbenzimide (Sigma-Aldrich).
Table 3.
Antibodies used in this study
| Antigen | Host | Clone | Manufacturer | Catalog no. | Method |
|---|---|---|---|---|---|
| ASCL1 | Mouse IgG1 | 24B72D11.1 | BD Pharmingen | 556 604 | WB |
| β-III-tubulin | Mouse polyclonal | Dako | PRB-435P | ICC | |
| β-Actin | Mouse IgG | Thermo Scientific | MA5-15739 | WB | |
| β-Catenin | Mouse IgG1 | 14/β-Catenin | Becton-Dickinson | 610 153 | IHC, WB |
| BLBP | Rabbit IgG | Abcam | ab27171 | ICC | |
| BrdU | Mouse IgG1 | B44 | Becton-Dickinson | 347 580 | ICC |
| DCX (H-280) | Rabbit IgG | Santa Cruz | Sc-28939 | WB, ICC | |
| GABA | Rabbit IgG | Sigma | A2052 | ICC | |
| GFAP | Rabbit IgG | 6F2 | DAKO | M0761 | WB, ICC, IHC |
| GSX2 | Rabbit IgG | Abcam | ab26255 | WB | |
| HIF1α | Mouse IgG | 54/HIF-1α | Becton-Dickinson | 610 958 | WB, ICC, IHC |
| Ki67 | Mouse | MIB1 | DAKO | M7240 | ICC |
| Ki67 | Rabbit IgG | Abcam | ab15580 | ICC | |
| LEF-1 (C12A5) | Rabbit IgG | Cell Signalling | 2230 | WB | |
| LHX6 | Mouse IgG | 3D9 | Sigma | WH0026468M1 | WB |
| NG2 | Rabbit IgG | Millipore | AB5320 | ICC | |
| NKX2.1 | Rabbit IgG | EP1584Y | Abcam | ab76013 | WB |
| O4 | Mouse IgM | Gift of R. Bansal | ICC | ||
| OLIG2 | Rabbit IgG | Chemicon | ab9610 | WB, ICC | |
| S100β | Rabbit IgG | EP1576Y | Abcam | Ab52642 | WB |
| SOX2 (Y-17) | Goat IgG | Santa Cruz | Sc-17320 | WB, ICC | |
| SP8 (C-18) | Goat IgG | Santa Cruz | Sc-104661 | WB | |
| TBR1 | Rabbit IgG | Proteintech | 20932-1-AP | WB | |
| TBR2 | Rabbit IgG | Abcam | ab23345 | WB | |
| Vimentin | Mouse IgG1 | V9 | Sigma | V66630 | WB, ICC |
Abbreviations: WB, western blot; ICC, immunocytochemistry; IHC, immunohistochemistry.
For immunohistochemistry, fetal forebrain blocks were fixed overnight in 4% PFA, cryoprotected in 30% sucrose/PBS, frozen in TissueTek OCT, and sectioned on a cryostat (15-µm sections). Coronal cryosections were incubated for 15 min in 10 mM sodium citrate (Sigma-Aldrich), pH 9.0, at 85 °C for antigen retrieval and then in 3% H2O2 to inhibit endogenous peroxidases. The sections were then placed in blocking solution, consisting of 0.5% blocking reagent (Roche) in Tris-NaCl-Tween buffer (TNT) [0.1 M, pH7.5 Tris-HCl (Thermo Fisher Scientific); 0.15 M NaCl (Thermo Fisher Scientific); and 0.05% Tween], and incubated overnight at 4 °C with primary antibodies to hypoxia-inducible factor (HIF-1α) and glial fibrillary acidic protein (GFAP) (Table 3), followed by antimouse-horseradish-peroxidase (HRP) conjugated secondary antibody (Jackson Immuno-Research Lab) targeting HIF-1α. The samples were developed with the TSA Plus fluorescein system (Perkin Elmer), washed with TNT solution, and incubated first with antirabbit-Alexa555 (Molecular Probes) for GFAP detection and then briefly with bisbenzimide (Sigma-Aldrich) for nuclear staining.
Image Analysis and Statistical Tests
Immunolabeled samples were visualized using an Axioskop microscope (Zeiss) together with Axiovision software and photographed using a digital camera. The images were assembled in Adobe Photoshop (v. 7.0), with consistent quality adjustments for contrast, brightness and color balance. Immunolabeled cells from 9 to 12 predesignated adjacent optical fields and from a minimum of three different human samples per experiment were analyzed using Image J software (National Institutes of Health). Because of the variations among the human samples and the experimental replicates, paired Student's t-tests were used to compare two experimental conditions, and a one-way ANOVA followed by a Bonferroni post hoc test for comparisons of three or more experimental groups.
Western Blot Analysis
The cells were homogenized in lysis buffer [50 mM Tris-HCl pH7.4, 150 mM NaCl, 1% NP-40 (Sigma-Aldrich), 1 mM phenylmethylsulphonyl fluoride (Thermo Fisher Scientific), and protease inhibitor cocktail (Thermo Fisher Scientific)] on ice for 30 min. The lysates were then sonicated to completely disrupt the cell membranes and DNA. Protein extracts were separated by SDS-PAGE and electro-transferred to a nitrocellulose membrane (BioRad). The blocked membranes were incubated first with primary antibodies (Table 3) overnight at 4 °C and then with their corresponding secondary HRP-conjugated antibodies (1:15 000, Thermo Fisher Scientific). Protein signals were detected using the SuperSignal West Pico chemilluminescent system (Thermo Fisher Scientific).
In Situ Hybridization
A plasmid containing the full coding sequence of human HIF-1α was purchased from Addgene (plasmid #21101, Connie Cepko Lab). A PCR from the plasmid sequence was used to add T3 and SP6 DNA-dependent RNA polymerase sequences to the edges of the HIF-1 α sequence to produce the antisense and sense probes, respectively. Digoxigenin-UTP (Roche) labeled riboprobes were generated from the purified PCR product obtained by in vitro transcription. In situ hybridization was performed on cryosections (15 µm) using a protocol described previously (Radonjic et al. 2014b). The specificity of the procedure was assessed using a probe corresponding to the sense strand of HIF-1α.
Quantitative PCR
The expression of genes related to hypoxia and the Wnt-β-catenin signaling pathway was evaluated using real-time PCR. Total RNA was extracted from the cells using a RNA purification kit (Macherey-Nagel) according to the manufacturer's instructions. Reverse transcription was carried using 1 μg of RNA, M-MuLV reverse transcriptase (New England BioLabs), and oligo (dT) primers (Invitrogen) according to the manufacturer's instructions. Quantitative PCR (qPCR) was performed in 96-well reaction plates (Eppendorf) in a CFX96 Connect thermocycler (BioRad). The reactions were prepared according to the standard protocol for SYBR Green qPCR with SsoFast Evagreen Supermix (BioRad). The 5′→3′ sequences of the forward (F) and reverse (R) primers were:
GAPDH: (F) ACCACCATGGAGAAGGC / (R) GGCATGGACTGTGGTCATGA
HIF-1α: (F) TATGAGCCAGAAGAACTTTTAGGC/ (R) CACCTCTTTTGGCAAGCATCCTG
WNT7A: (F) CTGTGGCTGCGACAAAGAGAA / (R) GCCGTGGCACTTACATTCC
AXIN2: (F) CAACACCAGGCGGAACGAA / (R) GCCCAATAAGGAGTGTAAGGACT
LEF-1: (F) TGCCAAATATGAATAACGACCCA / (R) GAGAAAAGTGCTCGTCACTGT
The thermal cycle conditions were 95 °C for 2 min followed by 40 cycles of 15 s at 95 °C, 15 s at 55 °C, and 20 s at 68 °C. All assays were performed in triplicate. The averaged cycle of threshold (Ct) values of the GAPDH triplicates were subtracted from the Ct values of the target genes to obtain the ΔCt. Relative gene expression was determined as 2− ΔCt (ΔΔCt) and expressed relative to the control value.
Results
HIF-1α Expression in Human Fetal RGCs
To estimate the O2-dependent responses of the human developing brain, HIF-1α expression was determined on coronal cryosections of fetal forebrains (15–24 gw, Table 2). HIF-1α is the O2-sensitive subunit of the heterodimeric transcription factor HIF-1. In normoxic conditions (10–20% O2; 70–150 mmHg) HIF-1α is targeted to the proteasome and degraded, but under low O2 conditions it is translocated to the nucleus, where it promotes the expression of multiple genes (Stroka et al. 2001; Carroll and Ashcroft 2005). Thus, HIF-1α expression is a sensitive indicator of physiologically low O2 conditions in tissues. In situ hybridization of cryosections of human fetal brains at 18, 22, and 24 gw revealed HIF-1α mRNA expression in the dorsal telencephalon, mainly in the cortical ventricular zone (VZ) populated by RGCs, and in the cortical plate (CP) populated by young neurons (Fig. 1A–C). HIF-1α was also expressed ventrally, in the VZ of the GE (Fig. 1A,D). A qPCR analysis of tissue samples confirmed HIF-1α expression in both brain areas and showed slightly higher expression in enriched RGC cultures (see below) obtained from the GE than in cortical RGCs (Fig. 1E). Since HIF-1α is regulated at the protein level, human fetal sections (15, 17, 18, 22, and 24 gw) were evaluated histochemically. HIF-1α+ nuclei were observed in the CP and VZ in both the cerebral cortex and the GE, where GFAP+ RGCs were located (Fig. 1F–I). This widespread presence of HIF-1α in the human forebrain during the second trimester is in agreement with the microarray data presented in the Allen Brain Atlas and suggests that hypoxic signaling is required for cortical neurogenesis during human fetal development.
Figure 1.

HIF-1α expression in the human fetal forebrain at 18–22 gw. (A) Partial view of a coronal section through the telencephalon at 18 gw, after HIF-1α in situ hybridization. (B–D) HIF-1α mRNA expression in the CP (B, B′), cortical ventricular zone (CX-VZ; C, C′), and GE VZ (D, D′). Arrowheads indicate HIF-1α expression in RGCs. (E) HIF-1α mRNA expression levels in tissue and sorted RGCs from the cortex and GE. (F–H) Immunohistochemistry for HIF-1α (green) and GFAP (red) in the cortex and GE (F–H, F′–H′). Insets show higher magnifications of the boxed areas. White arrowheads indicate HIF-1α expression in GFAP+ RGCs in the VZ of both the GE and CX. (I) Z-projection image showing a GFAP+ RGC expressing HIF-1α in the cortical VZ. Abbreviations: CP, cortical plate; CX, cortex; GE, ganglionic eminence; IZ, intermediate zone; LV, lateral ventricle; VZ, ventricular zone; SP, subplate; SVZ, subventricular zone. Scale bars: 20 µm. Significant difference: *P < 0.05 in a t-test. Error bars indicate the standard error of the mean (SEM).
The Effect of Hypoxic Stimuli on the Survival of Human Cortical RGCs
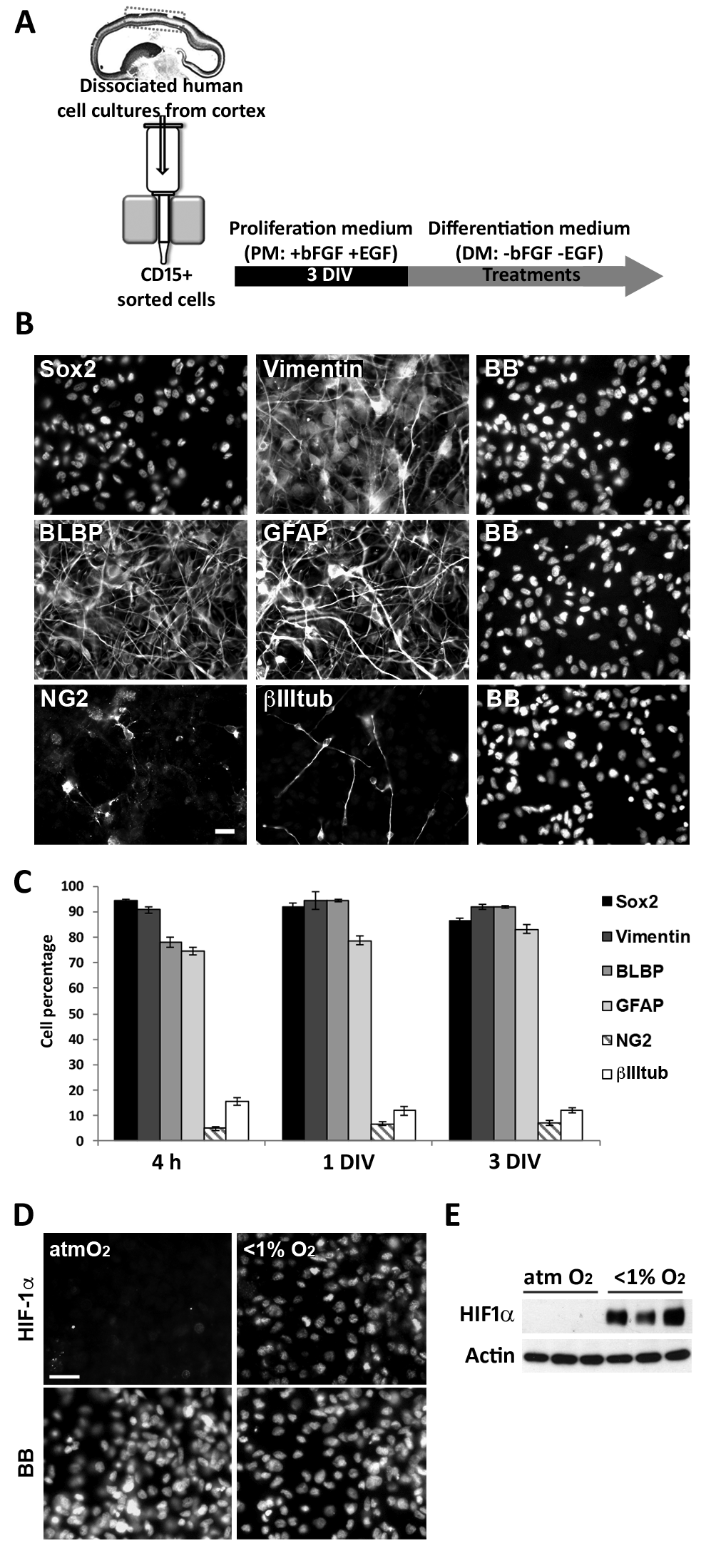
Because species-specific differences in cortical development have been reported (e.g., Letinic et al. 2002; Rakic and Zecevic 2003; Zecevic et al. 2005; Yu and Zecevic 2011; Betizeau et al. 2013; Geschwind and Rakic 2013; Lui et al. 2014; Radonjic et al. 2014a; Pollen et al. 2015) but functional studies in humans are not possible, we used in vitro experiments aimed at providing a better understanding of the factors controlling human cerebral cortex development and, by extension, the etiology of multiple neurodevelopmental disorders. The effects of pathological versus non-pathological O2 levels during fetal development were assessed by examining the in vitro cellular response of human RGCs to distinct hypoxic conditions. RGCs were enriched from the cortical region of human fetal brains ranging in age from 14 to 19 gw (Table 2). This is a critical period for cortical neurogenesis in humans as it is the stage when upper cortical layers are formed (Zecevic et al. 2005; Malik et al. 2013). Magnetic immunosorting of RGCs from dissociated cortical cultures using an anti-CD15 antibody showed that the majority of cells expressed RGC-specific markers 4 h after sorting: 90% of the cells were immunolabeled for SOX2 and Vimentin, and 80% for BLBP and GFAP (Fig. 2A–C). Around 10% of the cells were labeled with the neuronal marker βIII-tubulin, and only 5% with the oligodendrocyte progenitor cell marker NG2. During the next 3 DIV, the cells were cultured in PM, previously shown to maintains the percentages of the distinct cell types (Fig. 2C; Ortega et al. 2013; Radonjic et al. 2014a).
Figure 2.

Characterization of human cortical RGC cultures. (A) Enriched cortical RGC cultures using CD15+ magnetic immunosorting. Sorted cells were grown for 3 DIV in PM and then exposed to anoxia/hypoxia in DM. (B) Immunolabeling of cultures grown 3 DIV after immunosorting using specific RGC (SOX2, BLBP, GFAP, Vimentin), oligodendrocyte progenitor (NG2) and neuronal (BIIItub) markers. (C) Cell percentages from the total population of cells labeled with the different cell-type-specific markers in enriched RGC cultures; >90% of the cells are labeled with RGC markers. Error bars indicate the SEM. (D) Under atmospheric control conditions (21% O2), RGC cultures do not express HIF-1α but after 6 h of anoxia HIF-1α is expressed by the majority of cells. Nuclei were labeled with bisbenzimide (BB). Scale bars: 20 μm. (E) Western blot confirms HIF-1α expression in cultures exposed to anoxia. Actin served as the loading control.
The hypoxic conditions in utero were reproduced by placing RGC cultures in two different hypoxic chambers: one allowing for the nearly absolute absence of O2 (<1%) and the other maintaining O2 levels at 1% or 3% (Supplementary Figure 1A). After 6 h of an anoxic stimulus (<1% O2), corresponding to the peak of HIF-1α expression in the brain of hypoxia-exposed rodents (Stroka et al. 2001), almost 100% of the RGCs were immunolabeled for HIF-1α whereas labeling was not detected in cultures maintained under atmospheric O2 conditions (Fig. 2D). This result, confirmed by western blotting, showed that low O2 levels in vitro efficiently activated hypoxic intracellular signaling in cortical RGCs (Fig. 2E).
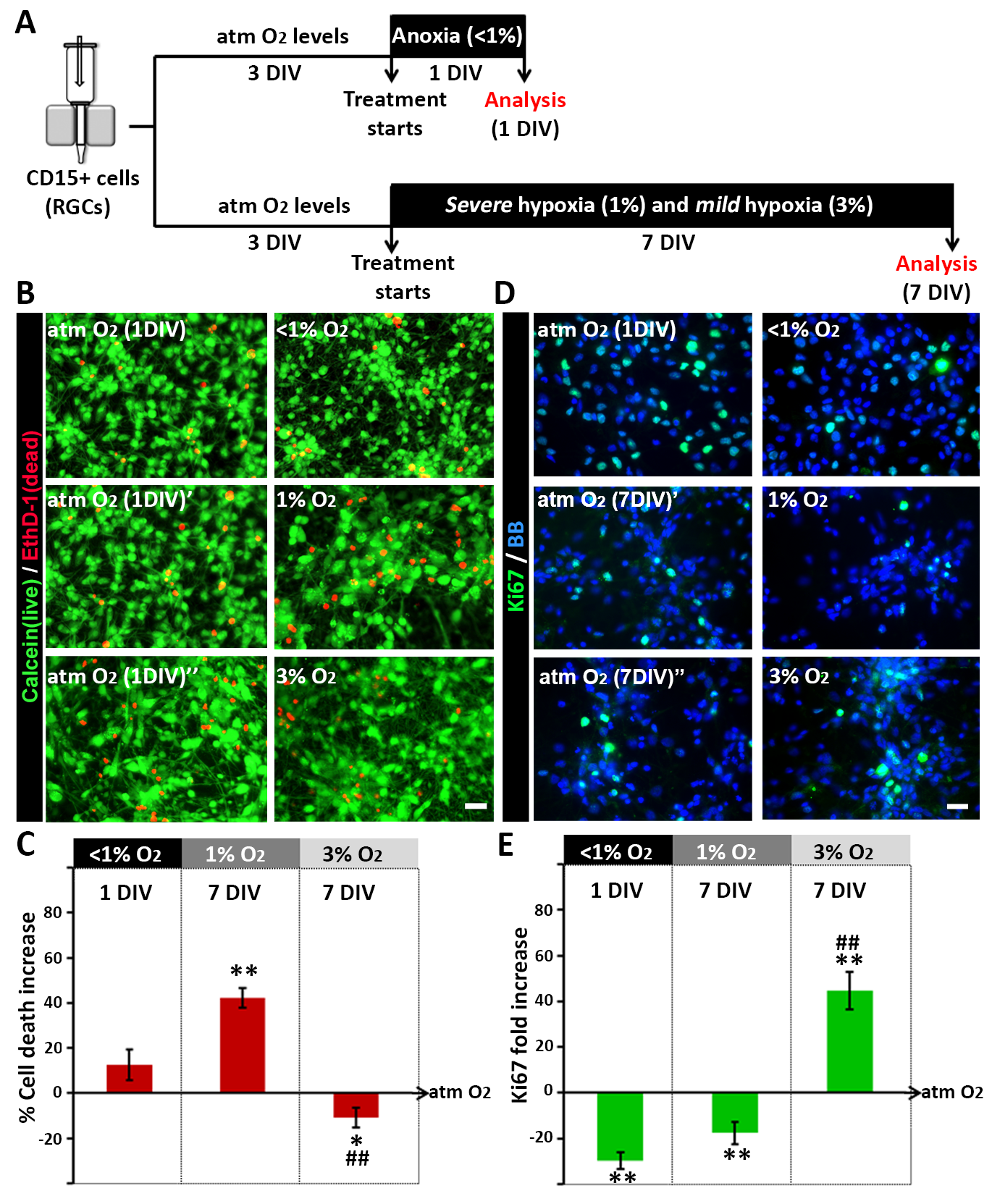
To assess the effects of different levels of hypoxia on RGCs, four different conditions based on data from previously published studies were established (Table 1): atmospheric O2 (21%), as the in vitro “control” condition; two pathologic O2 conditions, “acute anoxia” (24 h at <1% O2) and “chronic severe hypoxia” (7 DIV at 1% O2); and “chronic mild hypoxia” (7 DIV at 3% O2), the physiological condition during development (Supplementary Figure 1B). The survival of RGC cultures (14, 17, and 18 gw) exposed to the various O2 conditions was determined using the Live/Dead assay. The results showed that acute anoxia (<1% O2 for 24 h) did not significantly increase RGC death (Fig. 3A–C), but longer exposures to severe hypoxia (1% O2 for 7 DIV) increased cell death by 40% compared with control cultures (maintained at 21% O2). Notably, the increase in O2 levels from 1% to 3% for 7 DIV significantly reduced RGC death compared with either 1% O2 or atmospheric control conditions (Fig. 3A–C). These observations indicated that both O2 levels and the duration of anoxia/hypoxia affect the survival of human RGCs.
Figure 3.

Effects of different hypoxic conditions on the survival and proliferation of human RGC cultures. (A) Timeline of the experimental procedure. (B) Live/dead assay, based on double-staining for calcein (live cells, green) and EthD-1 (dead cells, red), performed under the different test conditions. (C) Increased cell death in cultures incubated in 1% O2 for 7 DIV versus atmospheric control conditions (atm O2), defined as 0% in the graph. (D) Ki67+ proliferating cells (green) in RGC cultures after 24 h of anoxia or 7 DIV of 1% and 3% O2. Cell nuclei (shown in blue) were labeled with BB. (E) The percentage of proliferating RGCs is significantly reduced in anoxic versus control cultures, and increased in cultures exposed to 3% O2 for 7 DIV compared with either 1% O2 or control cultures (set at 0). Significant differences between the different hypoxic conditions compared with the control (*P < 0.05, **P < 0.01) and between 1% and 3% O2 conditions (##P < 0.01) as demonstrated in a paired t-test or ANOVA. A minimum of three different cases was used per condition. Error bars indicate the SEM.
Effect of Hypoxic Stimuli on the Proliferation of RGCs
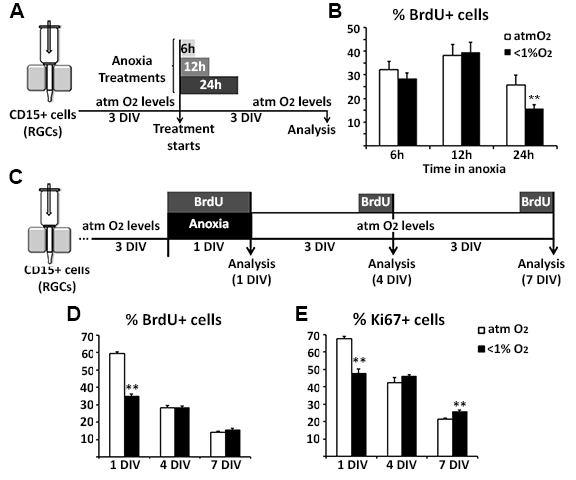
We next analyzed the proliferation of human RGCs under distinct O2 conditions. RGC cultures (14, 17, 18, and 19 gw) treated for 24 h under anoxic conditions showed a 30% reduction of Ki67+ proliferating cells compared with control cultures (Fig. 3D,E). Similar results were obtained by adding the thymidine analog BrdU to the RGC cultures for 16 h after the anoxic stimulus. The number of cells that entered S-phase and therefore took up BrdU was reduced significantly (by 40%) in RGCs cultured for 24 h under anoxic conditions but not for 6 or 12 h Supplementary Figure 2A,B, indicating that RGCs can briefly tolerate extremely low O2 levels. Interestingly, 3 and 6 days after 24-h anoxic stimulus, the number of Ki67+ or BrdU+ cells did not significantly differ between control and anoxic cultures (Supplementary Figure 2C–E), which demonstrated the recovery of proliferative activity in RGC subjected to an acute anoxic stimulus.
In line with the survival data discussed above, an important difference was seen in the proliferative capacity of RGCs maintained in 1% versus 3% O2 for 7 DIV. Thus, compared with controls, the percentage of proliferating (Ki67+) cells was reduced by 17% under 1% O2 but increased by 40% in response to 3% O2 (Fig. 3D,E). These results suggested that the precise regulation of O2 levels from ≤1% to 3% enables the proliferation of multipotent cortical progenitors during human fetal brain development.
O2 Regulation of Cortical Neurogenesis In Vitro
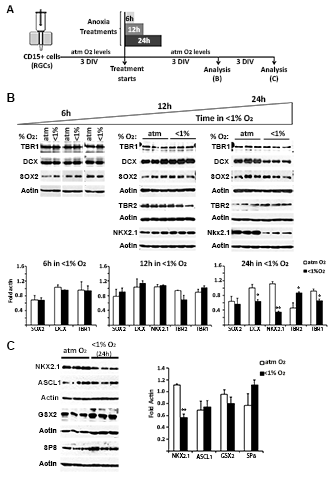
To study the effect of O2 on RGC differentiation into distinct neuronal subtypes, two experimental paradigms were used. In the first, RGC cultures from cortical tissue maintained in DM were subjected to anoxia for 24 h and then analyzed 6 DIV later. In the second, RGC cultures were analyzed after a 7-DIV exposure to chronic severe (1%) or mild (3%) hypoxia (Fig. 4A).
Figure 4.

The effects of distinct hypoxic stimuli on human cortical neurogenesis. (A) Timeline of the experimental procedure. (B) Immunoblots show the effects of anoxia and severe or mild hypoxia on the protein levels of neuronal (DCX), glutamatergic progenitor (TBR2), glutamatergic neuron (TBR1), NKX2.1 progenitor, and GABAergic neuron (LHX6) markers. Histogram of the densitometric values of the fold increase obtained in response to anoxia and from 1% to 3% hypoxia versus the atmospheric control condition (atm O2), set at 100. Actin served as a loading control. (C) Reductions in the number of βIII-tubulin+ neurons, glutamatergic βIII-tubulin+/GABA− cells, and GABA+ cells in cortical RGC cultures exposed to anoxia (<1% O2); severe hypoxia (1% O2) slightly reduces the number of glutamatergic neurons, whereas mild hypoxia (3% O2) increases the percentages of both glutamatergic and GABA+ cells. Cell nuclei are labeled with BB. Scale bar: 20 µm. Significant differences (*P < 0.05, **P < 0.01) compared with the control condition are indicated. A paired t-test was used to compare control versus anoxic conditions and individual human samples. An ANOVA was used to compare responses to control, 1% and 3% O2 conditions. A minimum of three different cases was used per condition. Error bars indicate the SEM.
Anoxia (<1% O2)
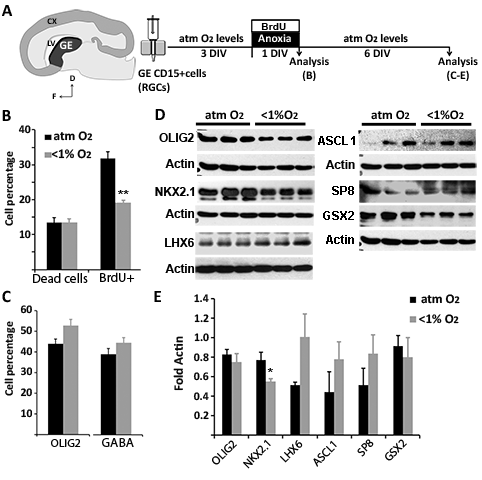
In 14-, 16-, 17-, 18- and 19-gw RGCs cultured for 24 h under anoxic conditions, western blot analysis revealed a significant reduction of the neuronal marker DCX compared with control cultures (Fig. 4B, P < 0.05) and an 18% decrease in the number of βIII-tubulin+ neurons (Fig. 4C). These results suggested that anoxia reduces neurogenesis by cortical RGCs. We then asked whether the generation of excitatory and/or inhibitory neurons was equally affected. In this case, cortical RGCs subjected to anoxia for 24 h but not to shorter anoxic treatments (6 or 12 h) expressed lower levels of the glutamategic marker TBR1 (Fig. 4B and Supplementary Figure 3A, B), which indicated a time-dependent effect of anoxia on the differentiation of cortical RGCs into glutamatergic neurons. However, the simultaneous increase in the glutamatergic progenitor marker TBR2 suggested the arrested differentiation of TBR2+ intermediate progenitors into TBR1+ postmitotic glutamatergic neurons (Fig. 4B and Supplementary Figure 3). That this change was due to specific postmitotic cell death was unlikely, since cell death did not significantly differ in control cultures versus those exposed to 24 h of anoxia either after 3 DIV (atm O2 = 19.64 ± 0.73% vs. <1% O2 = 22.54 ± 1.41%; P = 0.073) or 7 DIV (atm O2 = 21.25 ± 1.63% vs. <1% O2 = 24.11 ± 1.61%; P = 0.215). Moreover, in cultures pulsed with BrdU during the 24-h anoxic stimulus and analyzed after 3 DIV for BrdU and Ki67 double-immunostaining, fewer RGCs were found to have exited the cell cycle (BrdU+Ki67−cells/BrdU+cells × 100: atm O2 = 80 ± 2% vs. <1% O2 = 66 ± 4%; P > 0.02). This result strongly suggested that anoxia arrests cortical progenitor differentiation. GABA−/βIII-tubulin+ cells were used as an indicator of the plethora of possible glutamatergic postmitotic neurons, since individual markers, such as Smi31, Smi32, and TBR1, stain particular subpopulations of glutamatergic cells. In line with the western blot results, there were fewer GABA−/βIII-tubulin+ cells in the anoxic cortical RGC cultures than in control cultures (Fig. 4C). In contrast to the increase in glutamatergic progenitors under anoxic conditions, the level of NKX2.1, a transcription factor expressed by a subset of interneuron progenitors, was reduced (Fig. 4B and Supplementary Figure 3C). At the protein level, there were no changes in LHX6, a downstream effector of NKX2.1, or in other interneuron progenitor markers, such as ASCL1, GSX2, and SP8 (Fig. 4B and Supplementary Figure 3C), and the number of GABA+ cells was reduced only slightly (Fig. 4C). These results demonstrated a specific vulnerability to anoxia of NKX2.1+ progenitors, present in the human cortical subventricular zone during the second trimester of gestation (Radonjic et al. 2014a). In preliminary studies of RGCs isolated from the GE (17 and 19 gw), the reductions in NKX2.1 protein levels in anoxic versus control cells were similar, whereas the levels of the downstream marker LHX6 and of other interneuron markers remained largely unchanged, as did the percentage of GABA+ cells (Supplementary Figure 4).
Chronic Hypoxia (1–3% O2)
When RGC cultures (14, 15, 17, 18, and 19 gw) were exposed for 7 DIV to chronic severe hypoxia (1% O2), although the protein levels of the neuronal marker DCX did not change significantly, there were fewer immunolabeled βIII-tubulin+ cells compared with control cultures (Fig. 4B,C). By contrast, in the 3% O2 cultures, both DCX protein levels and the number of βIII-tubulin+ neurons increased over the control values. Similar changes were observed in the glutamatergic lineage under 1% and 3% O2 conditions, in that TBR1 protein levels and the percentage of glutamatergic neurons (βIII-tubulin+/GABA−) decreased in the 1% O2 and increased in the 3% O2 cultures (Fig. 4B,C). On the other hand, NKX2.1 protein levels in cortical RGC cultures were not changed in 1% and increased in 3% O2 cultures (Fig. 4B). The levels of the postmitotic interneuronal marker LHX6 displayed a similar trend. Accordingly, the percentage of GABA+ cells did not change in response to 1% O2 but it was slightly higher in cells exposed to 3% O2 (Fig. 4C).
Thus, both acute anoxic and chronic severe hypoxia, the two pathological paradigms compared in our study, reduced the generation of glutamatergic neurons but not of GABA+ cells in cortical RGC cultures. Conversely, physiological hypoxic O2 conditions (3% O2) increased both glutamatergic and GABAergic neurogenesis compared with control (atmospheric) O2 levels. Taken together our results show that a change in the O2 level within the narrow range of ≤1% to 3% is sufficient to significantly modify the neuronal differentiation of human cortical RGCs.
Effect of Different O2 Levels on Human Gliogenesis in Vitro
We previously reported that human cortical RGCs produce astrocytes and oligodendrocytes during fetal development (Mo and Zecevic 2009; Ortega et al. 2013). In this study, the possible effects of reduced O2 levels on astrocyte generation were examined. Because of the overlapping expression of several molecular markers in human RGCs and reactive astrocytes, the cells were distinguished using a panel of markers: anti-SOX2 antibody to label RGCs, anti-GFAP and anti-Vimentin antibodies to label both RGCs and mature astrocytes, and anti-S100β antibody to label reactive astrocytes. None of the conditions tested resulted in significant changes in SOX2 and GFAP protein levels (Fig. 5A,B). However, anoxia and severe hypoxia induced increases in the level of S100β and especially in that of Vimentin, in addition to causing more polymorphic Vimentin+ cells (Fig. 5B,C). These observations were indicative of greater astrocytogenesis/astocytic activation under pathologic hypoxia.
Figure 5.

The effect of hypoxic stimuli on glial development. (A) Timeline of the experimental procedure. (B) Western blots show a significant increase in the amount of Vimentin and S100β, but not of other markers of RGCs (SOX2), astroglia (GFAP), or oligodendrocytes (OLIG2) in RGC cultures exposed to anoxia or severe hypoxia versus control cultures (atm O2). Histogram shows the fold increase of the densitometric values obtained in the tested conditions versus atmospheric control set to 100. Actin served as a loading control. (C) Vimentin labeling shows more reactive astroglial morphologies in anoxia and 1% O2 hypoxia than in control conditions (arrowheads; in inset, higher magnification images C′ and C′′ pointed to by arrows). (D) Slight decreases in OLIG2+ cells percentages in RGC cultures exposed to anoxia but not to chronic 1% and 3% O2 hypoxia, compared with control cultures. (E) Reduction of O4+ cells is observed in anoxic versus control cultures. Cell nuclei were labeled with BB. Scale bars: C, F, 50 µm; D, 20 µm. *P < 0.05, **P < 0.01: significant difference between conditions and individual human samples determined by paired t-test and ANOVA. A minimum of three different human cell samples were used per condition. Error bars indicate the SEM.
The effect of different O2 levels on oligodendrogenesis was assessed using the transcription factor OLIG2 as a marker of oligodendroglial differentiation (Jakovcevski and Zecevic 2005; Yuen et al. 2014). Although in a western blot analysis, OLIG2 protein levels did not significantly differ from controls under any of the hypoxic conditions tested (Fig. 5B), the percentage of OLIG2+ cells was slightly reduced in anoxic cultures (Fig. 5D). The 67% reduction in the number of O4+ preoligodendrocytes in anoxic cultures (Fig. 5E) confirmed the selective vulnerability of this cell type to severe hypoxia (Back et al. 2001; Pistollato et al. 2007; De Filippis and Delia 2011). In fact, this scarcity of O4+ preoligodendrocytes, indicative of the very low level of oligodendrocyte maturation in vitro, and the absence of mature MBP+ oligodendrocytes hindered further analysis of the effect of hypoxia on human oligodendrocyte differentiation. Nonetheless, taken together these results showed that pathological hypoxia alters RGC gliogenesis in the second trimester of gestation, by reducing the number of O4+ preoligodendrocytes and increasing the number of reactive astrocytes.
Activation of Wnt-β-Catenin Signaling in Cortical RGCs Rescues the Negative Effects of Anoxia
Wnts (wingless) are morphogens with important regulatory roles in cortical development, including the proliferation and differentiation of distinct cortical progenitors (Chenn and Walsh 2003; Munji et al. 2011; Gan et al. 2014). O2 levels modulate Wnt expression during perinatal cortical development in rodents (Yuen et al. 2014). Since proliferation and neurogenesis were reduced in human RGC cultures under pathological hypoxia, we asked whether the Wnt signaling pathway mediated these effects. As expected, human fetal RGCs in the cortical VZ expressed β-catenin, the key intracellular element of the canonical Wnt signaling pathway (Wnt-β-catenin; Fig. 6A). We then examined the expression of WNT7A, which occurs in germinal regions of the cortex (Grove et al. 1998; Chenn 2008) and enhances both NSC proliferation and neuronal differentiation (Qu et al. 2013). RGCs cultured in 3% O2 expressed high levels of WNT7A. This result as well as the increased proliferation and neurogenesis of RGCs in 3% O2 (Figs 3D,E, 4B,C, 6B) was consistent with previous studies showing higher Wnt-β-catenin activation in NSCs exposed to similar hypoxic conditions, which led to increases in cell proliferation, neuronal differentiation, and maturation (Mazumdar et al. 2010; Cui et al. 2011; Varela-Nallar et al. 2014). However, acute anoxia and severe hypoxia reduced WNT7A mRNA levels in RGC cultures. Anoxic RGC cultures, characterized by diminished RGC proliferation and neuronal differentiation (Figs 3 and 4), also displayed lower Wnt-β-catenin activation, as shown by the very large reductions in the mRNA levels of AXIN2 and LEF-1, two endogenous Wnt-β-catenin target genes (Jho et al. 2002; Fancy et al. 2011; Bowman et al. 2013), and in β-catenin and LEF-1 protein levels (Fig. 6C–E). To confirm the role of Wnt-β-catenin signaling in RGCs under pathologic O2 levels, the Wnt-β-catenin signaling pathway was activated using the Wnt-β-catenin agonist CHIR99021 and inhibited using the antagonist XAV939 (Fig. 6F and Supplementary Figure 5). As expected, CHIR99021 stimulated the proliferation and neuronal differentiation of cortical RGCs (16, 17, and 18 gw), as shown by the increases in proliferating Ki67+ cells (Fig. 6G), the percentage of βIII-tubulin+ neurons, and DCX protein levels (Fig. 6H–J), respectively. This treatment was sufficient to reverse the decreases in RGC proliferation (Fig. 6G) and neurogenesis (Fig. 6H–J) caused by anoxia. A recovery of neurogenesis was not achieved in anoxic RGC cultures treated with a combination of CHIR99021 and XAV939, demonstrating the specificity and importance of Wnt-β-catenin signaling in the anoxia-induced reduction of neurogenesis. However, the Wnt-β-catenin antagonist did not completely block the action of the agonist on RGC proliferation, probably due to the faster and more powerful short-term effect of the agonist CHIR99021 at the analyzed time point (1 DIV). Taken together, our results demonstrated that O2 levels modulate the proliferation and differentiation of human cortical RGCs, by modifying the Wnt-β-catenin signaling pathway.
Figure 6.

Reactivation of Wnt-β-catenin signaling reverses the effects of anoxia in cortical RGC cultures. (A) Immunolabeling of β-catenin expression in the CP and VZ of the human cerebral cortex at 22 gw. Abbreviations: CP, cortical plate; LV, lateral ventricle; VZ, ventricular zone. Scale bar: 20 μm. (B) qPCR analysis shows changes in WNT7A mRNA levels in cortical RGCs exposed to distinct hypoxic stimuli versus control conditions (atmospheric, atm O2), represented by a dashed line. (C) The decrease in AXIN2 and LEF-1 mRNA levels in anoxic versus control RGC cultures reflects a reduction in Wnt-β-catenin activation in anoxic RGCs. (D, E) Western blot demonstrates the decreased β-catenin and LEF-1 protein levels in anoxic RGC cultures. Significant differences between hypoxic and control conditions (*P < 0.05; **P < 0.01) and between 1% and 3% O2 conditions (##P < 0.01) are indicated. Error bars show the SEM. (F) Timeline of the experimental procedure. (G) Quantification of proliferating Ki67+ cells in control and anoxic cultures in the presence or absence of a Wnt-β-catenin agonist (CHIR999021) or antagonist (XAV939). (H) The percentage of βIII-tubulin+ neurons from all cells in cortical RGC cultures exposed to anoxia and the Wnt agonist/antagonist. The number of neurons recovered in cultures treated with the Wnt-β-catenin agonist. (I, J) Western blot of the neuronal marker DCX confirms the βIII-tubulin immunolabeling results. Actin served as a loading control. *P < 0.05, **P < 0.01, show significant differences compared with the non-treated-control condition (DMSO; atm); #P < 0.05, ##P < 0.01 show differences between the respectives treated-anoxic (CHIR/CHIR+XAV; <1%) and treated-control conditions (CHIR/CHIR+XAV; atm). Paired t-test was used to compare control versus anoxic conditions and multiple experimental conditions were compared using ANOVA. A minimum of three different cases were used per condition. Error bars indicate the SEM.
Discussion
In this study, we first showed that HIF-1α is expressed in human fetal forebrain, and specifically in the VZ and CP of the cerebral cortex, during the second trimester of gestation. Consistent with previous studies on HIF-1α in various animal models (Stroka et al. 2001; Keith et al. 2012; Yuen et al. 2014), our findings revealed the importance of low O2 levels in the maintenance of human cortical progenitors and in neurogenesis. Although HIF-1α expression differed in pallial (cortex) versus subpallial (GE) RGCs, our preliminary results did not show differences in the responses of these two progenitor populations to hypoxia. Additional experiments are, however, needed to better understand the effects of O2 levels on the development of multiple neural cell types in distinct forebrain regions.
Our study also allowed us to define the narrow boundary between physiological (3%) and pathologically low (≤1%) O2 levels that determines the survival, proliferation, and differentiation of human cortical RGCs during fetal development. Three results are worth stressing 1) the improved survival and proliferation of RGCs cultured in physiological versus atmospheric (control) or pathological O2 conditions; 2) glutamatergic neurogenesis by human RGCs is more sensitive to pathological hypoxia than GABAergic neurogenesis; this can interfere with formation of cortical circuitry and impair the balance between cortical excitation and inhibition; and 3) the importance of Wnt-β-catenin signaling in the pathophysiology of fetal hypoxia.
Survival and Proliferation of Human Cortical RGCs in Response to Different O2 Levels
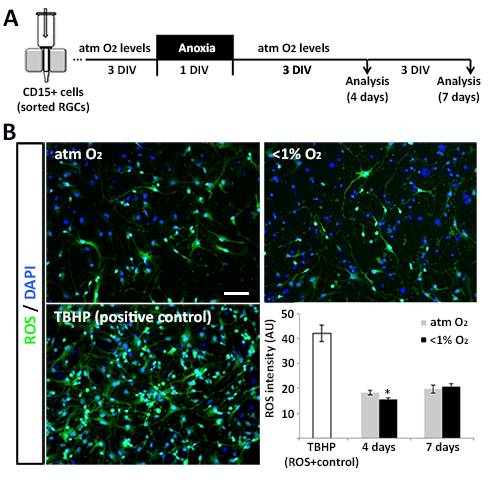
Under the atmospheric O2 levels commonly used for in vitro experiments, cell metabolism generates high levels of ROS, thus increasing the risk of DNA damage and cell senescence (Busuttil et al. 2003). Under physiological O2 conditions (1–6% O2), by contrast, neural progenitors are able to maintain the self-renewal of multiple types of NSCs (Studer et al. 2000; Pistollato et al. 2007; Mohyeldin et al. 2010; Santilli et al. 2010), whereas extremely low (≤1%) O2 levels drive neural progenitors into quiescence and/or apoptosis (De Filippis and Delia 2011). Here we showed that human fetal cortical progenitor cells (RGCs) are resistant to brief episodes of acute anoxia, as there were no significant changes in cell death or ROS levels in cells cultured in <1% O2 for 24 h (Supplementary Figure 6) versus in atmospheric control cultures. A previous study in rodents also showed that, in the presence of sufficient amounts of glucose in vitro, anoxia stimulates NSC survival and proliferation (Burgers et al. 2008). In the glucose-rich cell medium commonly used in vitro, NSCs survived chronic anoxia, whereas in the analogous in vivo condition of perinatal hypoxic-ischemic injury NSCs are highly vulnerable (Romanko et al. 2004). In our model, an extension of the duration of severe hypoxia (1% O2) to 7 DIV resulted in a significant decrease in cell survival, probably because glucose levels were unable to compensate for the effects of prolonged and substantial reduction in O2 levels.
The reduction in RGC proliferation during pathological hypoxia suggested that NSC self-renewal is a highly energy-demanding process and anaerobic glycolysis might not provide enough ATP to RGCs (Erecinska and Silver 2001; De Filippis and Delia, 2011; Wheaton and Chandel 2011). The energetic cellular failure under pathological hypoxia provokes a disregulation of cellular ion metabolism that leads to intracellular calcium accumulation (Vannucci et al. 2001; Kristian 2004). The precise regulation of calcium in cortical RGCs is vital for the proliferation of these cells and their subsequent differentiation as well as for the migration of cortical neurons (Komuro and Rakic 1996; Weissman et al. 2004; Rash et al. 2016). It is therefore likely that O2 levels interact with multiple mechanisms that simultaneously regulate RGC proliferation. Here we showed that the Wnt-β-catenin pathway is downregulated in RGCs under anoxic conditions and its pharmacological reactivation restores the proliferative potential of these cells. This is in agreement with previous studies showing that Wnt-β-catenin signaling is downregulated under anoxic conditions, whereas mild hypoxia induces the activation of this pathway and proliferation of neural progenitors (Mazumdar et al. 2010; Varela-Nallar et al. 2014). The stabilization of HIF-1α in mild hypoxic conditions potentiates other signaling pathways, such as Oct4 and Notch, which also promote stemness in multiple stem cell systems (Gustafsson et al. 2005; Kaidi et al. 2007; Simon and Keith 2008; Yoshida et al. 2009). Under atmospheric conditions, however, decreased HIF-1α levels activate the tumor suppressors p53 and p21, which increase mitotic arrest (Gustafsson et al. 2005; Pistollato et al. 2007). Consistent with these findings, the proliferation of cortical RGCs was higher in 3% O2 conditions than in atmospheric control conditions. Taken together, these results show that the proliferative activity of human RGCs can be modulated by multiple O2-dependent mechanisms acting at different cellular levels.
Differentiation of Human Cortical RGCs at Different O2 Levels
O2 levels control progenitor cell differentiation, as previously shown in studies in which a reduction in O2 levels changed the genetic profile of human embryonic stem cells and promoted the neuronal to astroglial switch (Xie et al. 2014). While severe hypoxia (≤1% O2) supports a slight increase in the astroglial differentiation of NSCs, mild hypoxia (2–5% O2) promotes NSC differentiation into neurons and oligodendrocytes (Ezashi et al. 2005; Pistollato et al. 2007; Santilli et al. 2010). Similarly, in human fetal RGC cultures kept at ≤1% O2, neurogenesis and oligodendrogenesis (OLIG2+/O4+ cells) are reduced and astrocytogenesis is slightly increased. The high energy level needed for O4+ preoligodendrocytes to differentiate into myelin-producing MBP+ oligodendrocytes makes this cell type especially vulnerable to hypoxia (Back et al. 2001; Pistollato et al. 2007; De Filippis and Delia 2011). The small glial changes observed in our study may have been due to the fact that the neural progenitors were derived from fetal brains of ~20 gw, when neurogenesis prevails over gliogenesis. Accordingly, the most robust change observed in our study was the neuronal differentiation of cortical RGCs.
Studies in animal models and in human fetal tissue have demonstrated that physiological hypoxia in utero is necessary for the differentiation of RGCs into intermediate progenitors and ultimately into postmitotic neurons, whereas cortical neurogenesis is severely reduced following the premature exposure of preterm animals to atmospheric O2 (Malik et al. 2013). Similarly, in our in vitro system, human cortical RGCs produced fewer neurons under atmospheric O2 conditions than in response to 3% O2, and even fewer neurons when exposed to pathological hypoxia (≤1% O2). These results are in line with the reduction in cortical gray matter in individuals who suffered fetal hypoxia during the last trimester of gestation (Cannon et al. 2002; Martinussen et al. 2005). We found that in cortical RGC cultures exposed to analogous conditions, the anoxia-induced reduction of neurogenesis correlated with reduced WNT7A expression/Wnt-β-catenin activation. Reactivation of the Wnt-β-catenin pathway reversed anoxia-impaired neurogenesis, demonstrating the importance of this signaling pathway in pathological hypoxia.
Effect of Hypoxia on the Generation of Distinct Neuronal Cell Types
Because stress conditions producing hypoxia/ischemia during fetal development can induce various neurodevelopmental disorders (van der Reijden-Lakeman et al. 1997; Rees and Harding 2004; Basovich 2010), we asked whether fetal pathological hypoxia impairs the ratio of glutamatergic to GABAergic neurons during the second trimester of gestation. The balance between excitation and inhibition is crucial for proper cortical function, and its disturbance has been linked to psychiatric and neurological disorders in affected individuals (Rubenstein and Merzenich 2003; Haider et al. 2006; Marín 2012). In immortalized human NSCs maintained at 2.5–5% O2, the generation of GABAergic neurons was increased, whereas glutamatergic neuron production remained unchanged (Santilli et al. 2010). In our study, glutamatergic neurogenesis from human fetal RGCs increased in response to 3% O2 and decreased under pathological hypoxia (≤1% O2). These results are of clinical significance, since a reduction in glutamatergic function has been associated with schizophrenia, which has been reported in individuals who suffered prenatal hypoxia (Cannon et al. 2002; Lewis et al. 2003; Marsman et al. 2013). However, in our in vitro system, GABAergic cell production was not affected by the reduced O2 levels, although cortical NKX2.1+ progenitors were particularly sensitive to anoxia. In previous work we showed that, unlike in rodents, the fetal cortex of humans includes a subset of NKX2.1+ cells that generate both cortical parvalbumin (PV) and somatostatin interneurons (Jakovcevski et al. 2011; Zecevic et al. 2011; Radonjic et al. 2014a). The differentiation of these specific interneuron cell subtypes is impaired in mice subjected to perinatal hypoxia (Komitova et al. 2013). Although at 20 gw the percentage of NKX2.1+ cells among all cortical subventricular zone cells is relatively small (4–5%) (Radonjic et al. 2014a), the clinical relevance of the sensitivity of these primate-specific cortical progenitors to O2 levels is the association between impaired cortical PV-interneuronal circuitry and psychiatric conditions such as schizophrenia and bipolar disorder (Torrey et al. 2005; Volman et al. 2011; Lewis et al. 2012; Powell et al. 2012).
The present study contributes to a better understanding of the effect of fetal hypoxia on cortical progenitors and, consequently, on human cortical development. We propose that, although clinically difficult to detect, hypoxic episodes during fetal development lead to neurodevelopmental changes in the cortex that may give rise to learning and behavioral deficits. The in vitro paradigm proposed herein is, at least for now, the only way to study the behavior of human cortical progenitor cells (RGCs) in response to changing O2 levels. Nonetheless, because of the complexity of the in vivo environment, including the influence of other cellular elements and the complex neurovascular niche that surrounds human radial glia progenitors, a variety of approaches will be necessary to obtain a complete picture of the effect of O2 levels on human cortical development.
Supplementary Material
Supplementary material can be found at: http://www.cercor.oxfordjournals.org/
Funding
This work was support by the National Institute of Health (R01 NSO41489) and subcontract to 5R01DA023999-07).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Notes
We thank Addgene for supplying the pCAG- HIF-1α plasmid (#21101), from the lab of Connie Cepko (Harvard Medical School, Boston, MA), used to produce the probes for the in situ hybridization experiments. Human fetal tissue was procured from Advanced Bioscience Resources (ABR, Alameda, CA), and the Joint Medical Research Council/Welcome Trust (grant no. 099175/Z/12/Z Human Developmental Biology Resource http://hdbr.org), Newcastle upon Tyne, England. Conflict of Interest: None declared.
References
- Ajayi-Obe M, Saeed N, Cowan FM, Rutherford MA, Edwards AD.. 2000. Reduced development of cerebral cortex in extremely preterm infants. Lancet. 356:1162–1163. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Bunney WE Jr, Potkin SG, Wigal SB, Hagman JO, Sandman CA, Jones EG.. 1993. Altered distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Arch Gen Psychiatry. 50:169–177. [DOI] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC.. 2001. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 21:1302–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basovich SN. 2010. The role of hypoxia in mental development and in the treatment of mental disorders: a review. Biosci Trends. 4:288–296. [PubMed] [Google Scholar]
- Betizeau M, Cortay V, Patti D, Pfister S, Gautier E, Bellemin-Menard A, Afanassieff M, Huissoud C, Douglas RJ, Kennedy H, et al. 2013. Precursor diversity and complexity of lineage relationships in the outer subventricular zone of the primate. Neuron. 80:442–457. [DOI] [PubMed] [Google Scholar]
- Bowman AN, van Amerongen R, Palmer TD, Nusse R.. 2013. Lineage tracing with Axin2 reveals distinct developmental and adult populations of Wnt/beta-catenin-responsive neural stem cells. Proc Natl Acad Sci USA. 110:7324–7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers HF, Schelshorn DW, Wagner W, Kuschinsky W, Maurer MH.. 2008. Acute anoxia stimulates proliferation in adult neural stem cells from the rat brain. Exp Brain Res. 188:33–43. [DOI] [PubMed] [Google Scholar]
- Busuttil RA, Rubio M, Dolle ME, Campisi J, Vijg J.. 2003. Oxygen accelerates the accumulation of mutations during the senescence and immortalization of murine cells in culture. Aging Cell. 2:287–294. [DOI] [PubMed] [Google Scholar]
- Cannon TD, van Erp TG, Rosso IM, Huttunen M, Lonnqvist J, Pirkola T, Salonen O, Valanne L, Poutanen VP, Standertskjold-Nordenstam CG.. 2002. Fetal hypoxia and structural brain abnormalities in schizophrenic patients, their siblings, and controls. Arch Gen Psychiatry. 59:35–41. [DOI] [PubMed] [Google Scholar]
- Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C.. 2011. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 15:1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll VA, Ashcroft M.. 2005. Targeting the molecular basis for tumour hypoxia. Expert Rev Mol Med. 7:1–16. [DOI] [PubMed] [Google Scholar]
- Chenn A. 2008. Wnt/beta-catenin signaling in cerebral cortical development. Organogenesis. 4:76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, Walsh CA.. 2003. Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb Cortex. 13:599–606. [DOI] [PubMed] [Google Scholar]
- Cui XP, Xing Y, Chen JM, Dong SW, Ying DJ, Yew DT.. 2011. Wnt/beta-catenin is involved in the proliferation of hippocampal neural stem cells induced by hypoxia. Ir J Med Sci. 180:387–393. [DOI] [PubMed] [Google Scholar]
- De Filippis L, Delia D.. 2011. Hypoxia in the regulation of neural stem cells. Cell Mol Life Sci. 68:2831–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erecinska M, Silver IA.. 2001. Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol. 128:263–276. [DOI] [PubMed] [Google Scholar]
- Ezashi T, Das P, Roberts RM.. 2005. Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci USA. 102:4783–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, et al. 2011. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci. 14:1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Q, Lee A, Suzuki R, Yamagami T, Stokes A, Nguyen BC, Pleasure D, Wang J, Chen HW, Zhou CJ.. 2014. Pax6 mediates ss-catenin signaling for self-renewal and neurogenesis by neocortical radial glial stem cells. Stem Cells. 32:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Rakic P.. 2013. Cortical evolution: judge the brain by its cover. Neuron. 80:633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove EA, Tole S, Limon J, Yip L, Ragsdale CW.. 1998. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development. 125:2315–2325. [DOI] [PubMed] [Google Scholar]
- Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M.. 2005. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 9:617–628. [DOI] [PubMed] [Google Scholar]
- Haider B, Duque A, Hasenstaub AR, McCormick DA.. 2006. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 26:4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard B, Chen Y, Zecevic N.. 2006. Cortical progenitor cells in the developing human telencephalon. Glia. 53:57–66. [DOI] [PubMed] [Google Scholar]
- Hutter D, Kingdom J, Jaeggi E.. 2010. Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: a review. Int J Pediatr. 2010:401323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic Z. 2009. Hypoxia or in situ normoxia: the stem cell paradigm. J Cell Physiol. 219:271–275. [DOI] [PubMed] [Google Scholar]
- Jakovcevski I, Mayer N, Zecevic N.. 2011. Multiple origins of human neocortical interneurons are supported by distinct expression of transcription factors. Cereb Cortex. 21:1771–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski I, Zecevic N.. 2005. Sequence of oligodendrocyte development in the human fetal telencephalon. Glia. 49:480–491. [DOI] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F.. 2002. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 22:1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidi A, Williams AC, Paraskeva C.. 2007. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 9:210–217. [DOI] [PubMed] [Google Scholar]
- Keith B, Johnson RS, Simon MC.. 2012. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 12:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komitova M, Xenos D, Salmaso N, Tran KM, Brand T, Schwartz ML, Ment L, Vaccarino FM.. 2013. Hypoxia-induced developmental delays of inhibitory interneurons are reversed by environmental enrichment in the postnatal mouse forebrain. J Neurosci. 33:13375–13387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro H, Rakic P.. 1996. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron. 17:275–285. [DOI] [PubMed] [Google Scholar]
- Kristian T. 2004. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium. 36:221–233. [DOI] [PubMed] [Google Scholar]
- Lee YM, Jeong CH, Koo SY, Son MJ, Song HS, Bae SK, Raleigh JA, Chung HY, Yoo MA, Kim KW.. 2001. Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: a possible signal for vessel development. Dev Dyn. 220:175–186. [DOI] [PubMed] [Google Scholar]
- Letinic K, Zoncu R, Rakic P.. 2002. Origin of GABAergic neurons in the human neocortex. Nature. 417:645–649. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW.. 2012. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 35:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Glantz LA, Pierri JN, Sweet RA.. 2003. Altered cortical glutamate neurotransmission in schizophrenia: evidence from morphological studies of pyramidal neurons. Ann NY Acad Sci. 1003:102–112. [DOI] [PubMed] [Google Scholar]
- Li Y, Gonzalez P, Zhang L.. 2012. Fetal stress and programming of hypoxic/ischemic-sensitive phenotype in the neonatal brain: mechanisms and possible interventions. Prog Neurobiol. 98:145–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu W, Ding W, Miyake M, Rosenberg GA, Liu KJ.. 2006. Electron paramagnetic resonance-guided normobaric hyperoxia treatment protects the brain by maintaining penumbral oxygenation in a rat model of transient focal cerebral ischemia. J Cereb Blood Flow Metab. 26:1274–1284. [DOI] [PubMed] [Google Scholar]
- Liu S, Shi H, Liu W, Furuichi T, Timmins GS, Liu KJ.. 2004. Interstitial pO2 in ischemic penumbra and core are differentially affected following transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 24:343–349. [DOI] [PubMed] [Google Scholar]
- Lui JH, Nowakowski TJ, Pollen AA, Javaherian A, Kriegstein AR, Oldham MC.. 2014. Radial glia require PDGFD-PDGFRbeta signalling in human but not mouse neocortex. Nature. 515:264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu TM, Ment LR, Schneider KC, Katz KH, Allan WC, Vohr BR.. 2009. Lasting effects of preterm birth and neonatal brain hemorrhage at 12 years of age. Pediatrics. 123:1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Vinukonda G, Vose LR, Diamond D, Bhimavarapu BB, Hu F, Zia MT, Hevner R, Zecevic N, Ballabh P.. 2013. Neurogenesis continues in the third trimester of pregnancy and is suppressed by premature birth. J Neurosci. 33:411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín O. 2012. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 13:107–120. [DOI] [PubMed] [Google Scholar]
- Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE.. 2013. Glutamate in schizophrenia: a focused review and meta-analysis of (1)H-MRS studies. Schizophr Bull. 39:120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinussen M, Fischl B, Larsson HB, Skranes J, Kulseng S, Vangberg TR, Vik T, Brubakk AM, Haraldseth O, Dale AM.. 2005. Cerebral cortex thickness in 15-year-old adolescents with low birth weight measured by an automated MRI-based method. Brain. 128:2588–2596. [DOI] [PubMed] [Google Scholar]
- Mazumdar J, O'Brien WT, Johnson RS, LaManna JC, Chavez JC, Klein PS, Simon MC.. 2010. O2 regulates stem cells through Wnt/beta-catenin signalling. Nat Cell Biol. 12:1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Z, Moore AR, Filipovic R, Ogawa Y, Kazuhiro I, Antic SD, Zecevic N.. 2007. Human cortical neurons originate from radial glia and neuron-restricted progenitors. J Neurosci. 27:4132–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Z, Zecevic N.. 2009. Human fetal radial glia cells generate oligodendrocytes in vitro. Glia. 57:490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A.. 2010. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 7:150–161. [DOI] [PubMed] [Google Scholar]
- Munji RN, Choe Y, Li G, Siegenthaler JA, Pleasure SJ.. 2011. Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci. 31:1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega JA, Radonjic NV, Zecevic N.. 2013. Sonic hedgehog promotes generation and maintenance of human forebrain Olig2 progenitors. Front Cell Neurosci. 7:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchision DM. 2009. The role of oxygen in regulating neural stem cells in development and disease. J Cell Physiol. 220:562–568. [DOI] [PubMed] [Google Scholar]
- Pistollato F, Chen HL, Schwartz PH, Basso G, Panchision DM.. 2007. Oxygen tension controls the expansion of human CNS precursors and the generation of astrocytes and oligodendrocytes. MCN. 35:424–435. [DOI] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, Shuga J, Liu SJ, Oldham MC, Diaz A, et al. 2015. Molecular identity of human outer radial glia during cortical development. Cell. 163:55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SB, Sejnowski TJ, Behrens MM.. 2012. Behavioral and neurochemical consequences of cortical oxidative stress on parvalbumin-interneuron maturation in rodent models of schizophrenia. Neuropharmacology. 62:1322–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Q, Sun G, Murai K, Ye P, Li W, Asuelime G, Cheung YT, Shi Y.. 2013. Wnt7a regulates multiple steps of neurogenesis. MCB. 33:2551–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radonjic NV, Ayoub AE, Memi F, Yu X, Maroof A, Jakovcevski I, Anderson SA, Rakic P, Zecevic N.. 2014. a. Diversity of cortical interneurons in primates: the role of the dorsal proliferative niche. Cell Rep. 9:2139–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radonjic NV, Memi F, Ortega JA, Glidden N, Zhan H, Zecevic N.. 2014. b. The role of sonic hedgehog in the specification of human cortical progenitors in vitro. Cereb Cortex. 26:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raine A, Lencz T, Bihrle S, LaCasse L, Colletti P.. 2000. Reduced prefrontal gray matter volume and reduced autonomic activity in antisocial personality disorder. Arch Gen Psychiatry. 57:119–127. [DOI] [PubMed] [Google Scholar]
- Rakic S, Zecevic N.. 2003. Emerging complexity of layer I in human cerebral cortex. Cereb Cortex. 13:1072–1083. [DOI] [PubMed] [Google Scholar]
- Rash BG, Ackman JB, Rakic P.. 2016. Bidirectional radial Ca(2+) activity regulates neurogenesis and migration during early cortical column formation. Sci Adv. 2:e1501733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees S, Harding R.. 2004. Brain development during fetal life: Influences of the intra-uterine environment. Neurosci Lett. 361:111–114. [DOI] [PubMed] [Google Scholar]
- Romanko MJ, Rothstein RP, Levison SW.. 2004. Neural stem cells in the subventricular zone are resilient to hypoxia/ischemia whereas progenitors are vulnerable. J Cereb Blood Flow Metab. 24:814–825. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM.. 2003. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2:255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santilli G, Lamorte G, Carlessi L, Ferrari D, Rota Nodari L, Binda E, Delia D, Vescovi AL, De Filippis L.. 2010. Mild hypoxia enhances proliferation and multipotency of human neural stem cells. PLoS One. 5:e8575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selemon LD, Zecevic N.. 2015. Schizophrenia: a tale of two critical periods for prefrontal cortical development. Transl Psychiatry. 5:e623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MC, Keith B.. 2008. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol. 9:285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroka DM, Burkhardt T, Desbaillets I, Wenger RH, Neil DA, Bauer C, Gassmann M, Candinas D.. 2001. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 15:2445–2453. [DOI] [PubMed] [Google Scholar]
- Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B, McKay R.. 2000. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci. 20:7377–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrey EF, Barci BM, Webster MJ, Bartko JJ, Meador-Woodruff JH, Knable MB.. 2005. Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol Psychiatry. 57:252–260. [DOI] [PubMed] [Google Scholar]
- van den Brink WA, van Santbrink H, Steyerberg EW, Avezaat CJ, Suazo JA, Hogesteeger C, Jansen WJ, Kloos LM, Vermeulen J, Maas AI.. 2000. Brain oxygen tension in severe head injury. Neurosurgery. 46:868–876; discussion 876–868. [DOI] [PubMed] [Google Scholar]
- van der Reijden-Lakeman IE, de Sonneville LM, Swaab-Barneveld HJ, Slijper FM, Verhulst FC.. 1997. Evaluation of attention before and after 2 years of growth hormone treatment in intrauterine growth retarded children. J Clin Exp Neuropsychol. 19:101–118. [DOI] [PubMed] [Google Scholar]
- Vannucci RC, Brucklacher RM, Vannucci SJ.. 2001. Intracellular calcium accumulation during the evolution of hypoxic-ischemic brain damage in the immature rat. Dev Brain Res. 126:117–120. [DOI] [PubMed] [Google Scholar]
- Varela-Nallar L, Rojas-Abalos M, Abbott AC, Moya EA, Iturriaga R, Inestrosa NC.. 2014. Chronic hypoxia induces the activation of the Wnt/beta-catenin signaling pathway and stimulates hippocampal neurogenesis in wild-type and APPswe-PS1DeltaE9 transgenic mice in vivo. Front Cel Neurosci. 8:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volman V, Behrens MM, Sejnowski TJ.. 2011. Downregulation of parvalbumin at cortical GABA synapses reduces network gamma oscillatory activity. J Neurosci. 31:18137–18148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ. 2011. Systemic inflammation, oligodendroglial maturation, and the encephalopathy of prematurity. Ann Neurol. 70:525–529. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. 1987. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 44:660–669. [DOI] [PubMed] [Google Scholar]
- Weissman TA, Riquelme PA, Ivic L, Flint AC, Kriegstein AR.. 2004. Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron. 43:647–661. [DOI] [PubMed] [Google Scholar]
- Wheaton WW, Chandel NS.. 2011. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol. 300:C385–C393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward LJ, Edgin JO, Thompson D, Inder TE.. 2005. Object working memory deficits predicted by early brain injury and development in the preterm infant. Brain. 128:2578–2587. [DOI] [PubMed] [Google Scholar]
- Xie Y, Zhang J, Lin Y, Gaeta X, Meng X, Wisidagama DR, Cinkornpumin J, Koehler CM, Malone CS, Teitell MA, et al. 2014. Defining the role of oxygen tension in human neural progenitor fate. Stem Cell Rep. 3:743–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S.. 2009. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell. 5:237–241. [DOI] [PubMed] [Google Scholar]
- Yu X, Zecevic N.. 2011. Dorsal radial glial cells have the potential to generate cortical interneurons in human but not in mouse brain. J Neurosci. 31:2413–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SP, Zahed H, Maltepe E, Rowitch DH.. 2014. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell. 158:383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecevic N, Chen Y, Filipovic R.. 2005. Contributions of cortical subventricular zone to the development of the human cerebral cortex. J Comp Neurol. 491:109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecevic N, Hu F, Jakovcevski I.. 2011. Cortical interneurons in the developing human neocortex. Dev Neurobiol. 71:18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Zhu L, Fan M.. 2011. Oxygen, a key factor regulating cell behavior during neurogenesis and cerebral diseases. Front Mol Neurosci. 4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.