

Abstract
Ciliopathies are pleiotropic human diseases resulting from defects of the primary cilium, and these patients often have cleft lip and palate. IFT88 is required for the assembly and function of the primary cilia, which mediate the activity of key developmental signaling pathways. Through whole exome sequencing of a family of three affected siblings with isolated cleft lip and palate, we discovered that they share a novel missense mutation in IFT88 (c.915G > C, p.E305D), suggesting this gene should be considered a candidate for isolated orofacial clefting. In order to evaluate the function of IFT88 in regulating craniofacial development, we generated Wnt1-Cre;Ift88fl/fl mice to eliminate Ift88 specifically in cranial neural crest (CNC) cells. Wnt1-Cre;Ift88fl/flpups died at birth due to severe craniofacial defects including bilateral cleft lip and palate and tongue agenesis, following the loss of the primary cilia in the CNC-derived palatal mesenchyme. Loss of Ift88 also resulted in a decrease in neural crest cell proliferation during early stages of palatogenesis as well as a downregulation of the Shh signaling pathway in the palatal mesenchyme. Importantly, Osr2KI-Cre;Ift88fl/flmice, in which Ift88 is lost specifically in the palatal mesenchyme, exhibit isolated cleft palate. Taken together, our results demonstrate that IFT88 has a highly conserved function within the primary cilia of the CNC-derived mesenchyme in the lip and palate region in mice and is a strong candidate as an orofacial clefting gene in humans.
Introduction
Orofacial clefting is one of the most common human birth defects occurring at rates of 1/500–1/2500 live births (1). The underlying etiology is complex and multifactorial with a wide range of influences including genetic, environmental, geographic, racial and ethnic, as well as socioeconomic status (2,3). The palate separates the nasal and oral cavities, allowing for the development of speech and efficient swallowing. The patterning and growth of the palatal shelves are mediated by continuous reciprocal epithelial–mesenchymal interactions regulated by multiple signaling pathways and transcriptional factors (4). Cell migration, proliferation and apoptosis in both palate epithelium and cranial neural crest (CNC)-derived ecto-mesenchyme are all involved in mechanisms that contribute to palatogenesis (5–11). A cleft palate may result from intrinsic defects in palatal shelf growth, elevation, midline fusion, or disappearance of the midline epithelium (12). Extensive human genetic studies have attempted to identify the mutations responsible for cleft lip and palate, although the majority of genetic causes remain elusive (13,14). A growing number of genetic and developmental animal models, especially mouse models, have been created to study the mechanisms of craniofacial development because of their remarkable similarities with human defects in palatal growth and morphogenetic processes.
Recent studies have demonstrated that cilia play critical tissue-specific roles in craniofacial development (15). The primary cilium is a microtubule-based organelle that extends from the surface of differentiated cells and functions to mediate intercellular signals and other cues received from its environment (16–18). The primary cilium is composed of functional domains including the basal bodies, transition fibers, transition zone, intraflagellar transport (IFT) machinery, axoneme and ciliary membrane. IFT particles are large complexes of more than 20 proteins organized into two subcomplexes, complex A and B, which mediate bidirectional movement of protein cargo along axonemal microtubules (19). Mutations in proteins of the cilia result in a group of human inherited diseases referred to as ciliopathies (20). Ciliopathies typically comprise a heterogeneous group of congenital diseases with a wide range of phenotypes including polycystic disease, hepatic fibrosis, retinal degeneration, hearing defects, skeletal dysplasia, polydactyly, brain malformations and orofacial clefting (21–23). Examples of human craniofacial ciliopathies include Meckel-Gruber Syndrome [Online Mendelian Inheritance in Man (OMIM):249000], Oro-facial-digital syndrome (OMIM:311200), Joubert Syndrome (OMIM:213300) and Bardet–Biedl Syndrome (OMIM:209900), with orofacial clefting and hypertelorism as their common phenotypes (21,22,24–28). Intraflagellar transport (IFT) 88 (IFT88) is a core component of IFT retrograde complex B, and its role in human disease has yet to be determined. Currently, this gene is considered a gene of unclear clinical significance and is not typically included in clinical exome analyses and reports. Mice with mutation in Ift88 exhibit defects in neural tube patterning, craniofacial abnormalities, polydactyly and left–right axis determination defects (29–32). Although mice with a hypomorphic allele of Ift88 (Tg737orpk) exhibit craniofacial abnormalities including cleft palate and supernumerary teeth, null mutation of Ift88 is embryonic lethal due to severe left–right symmetry defects. To date, the role of IFT88 during craniofacial development has yet to be characterized fully.
Primary cilia regulate signaling cascades and the cell cycle via trafficking of essential ciliary components. Ift88 is localized to the basal body and axoneme of cilia, and loss of IFT88 disrupts the transport of cargo from the tip to the basal bodies (31,33). Previous studies suggest that the primary cilium is involved in the regulation of multiple developmental signaling pathways, including Hedgehog (Hh), canonical Wnt, fibroblast growth factor, platelet-derived growth factor (PDGF) and Notch signaling pathways (16,34–39). Components of the Hh pathway, including the Patched1 (Ptch1), Smoothened (Smo) and Gli transcription factors, are localized in the primary cilia. Ptch1 inhibits Smo by interfering with its localization within the cilia (40). IFT proteins play a role in Shh signaling downstream of Smo and Ptch1, but upstream of Gli1 (41–43). Mutation of IFT genes leads to impaired Hh signaling, resulting in perturbation of neural tube patterning and limb, eye and bone formation (44–46). Moreover, IFT80, IFT122, IFT144 and IFT140 mutations result in a group of human ciliopathies that exhibit craniofacial skeletal and ectodermal abnormalities (47–50). Studies of animal models demonstrate that loss of IFT function leads to disruption of the Shh pathway and defects in the proliferation and differentiation of chondrogenic and osteogenic cells, resulting in chondrodysplasia (47,49,51).
In this study, we performed whole exome sequencing on a family with three affected members who presented with isolated cleft lip and palate (52). We identified a shared missense mutation in exon 14 of the IFT88 gene, consistent with IFT88 as a candidate gene contributing to the phenotype within this family. Despite extensive studies of craniofacial ciliopathies highlighting the importance of primary cilia in CNC development, little is known about IFT88 function in palatogenesis. We disrupted the Ift88 gene in the CNC-derived mesenchyme to investigate the functional requirement for primary cilia in mesenchymal cell fate during palatogenesis and found that it plays a crucial role in craniofacial morphogenesis.
Results
Identification of an IFT88 mutation in human patients with cleft lip and cleft palate
We identified a family with recurrent cleft lip and palate of unknown etiology (see Materials and Methods). After Whole Exome Sequencing of DNA from three affected siblings and their unaffected mother, we processed the raw data for variant annotation and filtering, followed by genetic analyses for significance related to the phenotype. Based on the family pedigree information, an autosomal dominant Mendelian inheritance model with incomplete penetrance was considered the best-fit model, although all potentially de novo, homozygous, or compound heterozygous variants were examined and variants shared among affected individuals were assessed with greater scrutiny. Variants with a sequencing depth of coverage <10 or genotype quality <20 were excluded from analysis. Only rare variants with minor allele frequency <1% in the 1000 Genomes Project (www.1000genomes.org; date last accessed August 22, 2015) or Exome Sequencing Project (ESP; esp.gs.washington.edu/drupal/; date last accessed August 22, 2015) reference populations were included for analysis. There were 32 061 unique variants within all four sequenced samples. After removing poor quality variants (Q < 20), poor sequencing depth variants (<10x), and the frequent variants found (>1%) in the 1000 Genomes Project, ExAC database or the ESP (accessed May 2016), there remained 3261 variants. Narrowing the candidates to only variants present in all three samples from the affected children and not in the mother (assuming either a dominant paternal variant with incomplete penetrance or possibly a de novo variant), there were only 46 variants in 34 genes identified. Table 1 summarizes the results of the general variant annotation, functional prediction and population frequencies for the IFT88 mutation identified. The IFT88 mutation resulted in the substitution of the amino acid 305 glutamate with aspartate, which was confirmed via Sanger sequencing (Supplementary Material, Fig. S1). Using numerous databases and in silico tools (53–55), this variant was found to be rare in the population (<0.0001%), and the position was conserved across species, suggesting that a genetic variant at this position would likely be deleterious. We reviewed all candidates individually and none of the genes were associated with already defined human clefting or craniofacial disease. IFT88 was the only gene flagged as a candidate gene because of its association of orofacial clefting in an animal model. It remains a possibility that there is another cause of clefting not detected by sequencing or due to an alternative mechanism (e.g. structural variations such as deletions, duplications, translocations or environmental/teratogenic exposures) (56).
Table 1.
Annotation, functional prediction and population frequencies of the IFT88 single nucleotide variant shared by individuals affected with cleft lip and palate
| General variant information and annotation | |||||||
|---|---|---|---|---|---|---|---|
| Gene | Chr | Position (HG19) | Ref | Alt | Transcript | cDNA | Protein |
| IFT88 | 13 | 21175919 | G | C | NM_175605.3 | c.915G>C | p.E305D |
| Conservation and in silico predictions | |||||||
| GERP ++ | phastCons7 way vertebrate | CADD phred | SIFT | Polyphen2 | Mutation Taster score | FATHMM score | LRT rank score |
| 5.51 | 0.998 | 22.4 | 0.061;0.058 | 0.693;0.992 | 0.999999 | -3.62 | 0.84324 |
| Allele frequencies in various populations and databases | |||||||
| Allele Freq 1000 genomes African populations | Allele Freq 1000 genomes American populations | Allele Freq 1000 genomes Asian populations | Allele Freq 1000 genomes European populations | Allele Freq UK 10K project | Allele Freq TWINS UK project | Allele Freq Exome Variant Server | Allele Freq ExAC |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.0001105 |
Loss of Ift88 in the facial mesenchyme of mice leads to severe craniofacial defects
In order to investigate the molecular and cellular mechanisms underlying Ift88 associated cleft palate, we generated mice with conditional loss of function of Ift88 in the epithelium or mesenchyme by crossing Ift88fl/flmice with K14-Cre and Wnt1-Cre mice, respectively. K14-Cre;Ift88fl/flmice survived and showed no evidence of craniofacial defects, as previously reported (data not shown) (57). In contrast, Wnt1-Cre;Ift88fl/fl mice died at birth and exhibited multiple craniofacial malformations including cleft lip and palate and tongue agenesis (Fig. 1A–E). We examined different stages of development to determine the onset of these defects. At E10.5, after neural crest migration into the facial prominences, Wnt1-Cre;Ift88fl/flembryos appeared indistinguishable from control embryos (Fig. 1F and G). By E12.5, Wnt1-Cre;Ift88fl/flembryos exhibited phenotypes such as cleft lip (Fig. 1H and I). Widening of the frontonasal prominence was detectable in newborn Ift88 fl/flCKO mice (Fig. 1J and K). In newborn mice, the medial edge epithelium had fused and the palatine bones had almost reached the midline of control mice, whereas in Wnt1-Cre;Ift88fl/flmice, the palatine bones were dysmorphic and did not extend toward the midline (Fig. 1L–O).
Figure 1.

Loss of Ift88 in the facial mesenchyme leads to severe craniofacial defects. (A) Newborn (NB) Ift88fl/fl (control) and Wnt1-Cre;Ift88fl/flmice. Arrow indicates milk in the stomach of control mice, whereas arrowhead indicates lack of milk in the stomach of Wnt1-Cre;Ift88fl/flmice. (B–E) Intraoral views of the palates (B, C) and tongues (D, E) of newborn Ift88fl/fl (control) and Wnt1-Cre;Ift88fl/flmice. Arrow indicates cleft palate in Wnt1-Cre;Ift88fl/flmice. (F–K) Frontal view of whole mount E10.5, E12.5 and newborn Ift88fl/fl (control, n = 6 for each stage) and Wnt1-Cre;Ift88fl/flmice (n = 6 for each stage). Arrow indicates cleft lip. Dashed lines delineate the frontonasal prominences. (L–O) H&E staining of sections of newborn Ift88fl/fl (control) and Wnt1-Cre;Ift88fl/flmice. Boxes in L and M are shown magnified in N and O, respectively. Dotted lines in N and O show the fused palatine bone in control mice and dysmorphic palatine bone in Wnt1-Cre;Ift88fl/flmice. Scale bars, L and M, 500 µm; N and O, 200 µm.
Next, we examined the defects in CNC-derived craniofacial bones, such as the premaxilla, maxilla, mandible, and palatine and frontal bones in newborn Wnt1-Cre;Ift88fl/flmice using reconstructed 3D images of micro computed tomography (microCT) scans. The volume of the frontal bone appeared increased in Wnt1-Cre;Ift88fl/flmice (Fig. 2A, B and S). We also found that the shape of the premaxilla was affected, likely resulting from the absence of the anterior portion of the maxilla including the incisors. The maxilla was severely affected in Wnt1-Cre;Ift88fl/flmice. Moreover, the processes of the palatine bone were undetectable in Wnt1-Cre;Ift88fl/flmice (Fig. 2C and D). We found that the length of the mandible had decreased in Wnt1-Cre;Ift88fl/flmice (Fig. 2E and F). Soft tissue microCT scans confirmed the defects in palatal shelf and tongue formation in Wnt1-Cre;Ift88fl/flmice (Fig. 2G–J).
Figure 2.

Skeletal analysis of Wnt1-Cre;Ift88fl/fl mice. (A–F) 3D reconstruction of skulls (A, B), maxilla (C, D), and mandibles (E, F) from microCT scans of Ift88fl/fl (control, n = 3) and Wnt1-Cre;Ift88fl/flmice (n = 3). (G–J) Sections of soft tissue microCT scans from Ift88fl/fl (control) and Wnt1-Cre;Ift88fl/flmice. Asterisk in H indicates the absence of the tongue in Wnt1-Cre;Ift88fl/flmice. Arrow in I indicates the palatal shelf in control mice. (K–R) Alcian Blue (cartilage) and Alizarin Red (bone) staining of Ift88fl/fl (control) and Wnt1Cre;Ift88fl/flmice. Asterisk indicates the absence of the palatal process of the palatine bone. Dashed lines delineate borders of the frontal and parietal bones. Arrow in R indicates the absence of the condylar and coronoid processes. (S) Quantification of the volume of the frontal bone from microCT scans. *P < 0.05. bs, basisphenoid; c, condylar and coronoid process; fb, frontal bone; ib, interparietal bone; md, mandible; mx, maxilla; nb, nasal bone; pb, parietal bone; pmx, premaxilla; ppp, palatal process of palatine. Scale bars, 1 mm.
To analyze the craniofacial skeleton of newborn Wnt1-Cre;Ift88fl/flmice, we performed Alcian Blue and Alizarin Red staining. We found that the bones of the palate, maxilla, trabecular basal plate, palatine and basisphenoid were either laterally displaced or absent in Wnt1-Cre;Ift88fl/flmice (Fig. 2K–N). The cranium was also severely dysmorphic, with laterally displaced, underdeveloped frontal bones, resulting in an abnormal opening of the skull (Fig. 2O and P). The proximal region of the mandible was strongly affected, including an absence of the condylar and coronoid processes (Fig. 2Q and R). Thus, loss of IFT88 in the CNC-derived mesenchyme results in severe defects in midline fusion of the face and formation of the palatal shelf.
Analysis of putative cellular mechanisms of cleft palate in Wnt1-Cre;Ift88fl/flmice
To investigate the mechanism potentially causing the severe craniofacial defect in Wnt1-Cre;Ift88fl/flmice, we analyzed cell migration, proliferation and apoptosis. First, to assess the migration of mesenchymal progenitors, we generated Wnt1-Cre;Ift88;ZsGreen mice. Control and Wnt1-Cre;Ift88fl/fl;ZsGreen mice appeared indistinguishable at E10.5 (Fig. 3A and B), indicating that the availability of mesenchymal progenitors was unaffected in the absence of Ift88. Next, we examined mesenchymal cell proliferation and survival. We evaluated cell proliferation using phosphohistone H3(PH3), a marker of proliferation. At E13.5, the number of proliferating cells in the palatal shelf was comparable in Wnt1-Cre;Ift88fl/fl and control mice (data not shown). At E14.0, when the palatal shelf was elevated in control embryos, a decrease in proliferation was detectable in the presumptive palatal shelf of Wnt1-Cre;Ift88fl/flmice (Fig. 3C, D and G). In contrast, we found no significant difference in apoptosis in control and Wnt1-Cre;Ift88fl/flmice (Fig. 3E and F). Thus, loss of Ift88 in the CNC-derived mesenchyme resulted in decreased cell proliferation in the palatal shelf during palatogenesis, but migration and apoptosis were unaffected.
Figure 3.
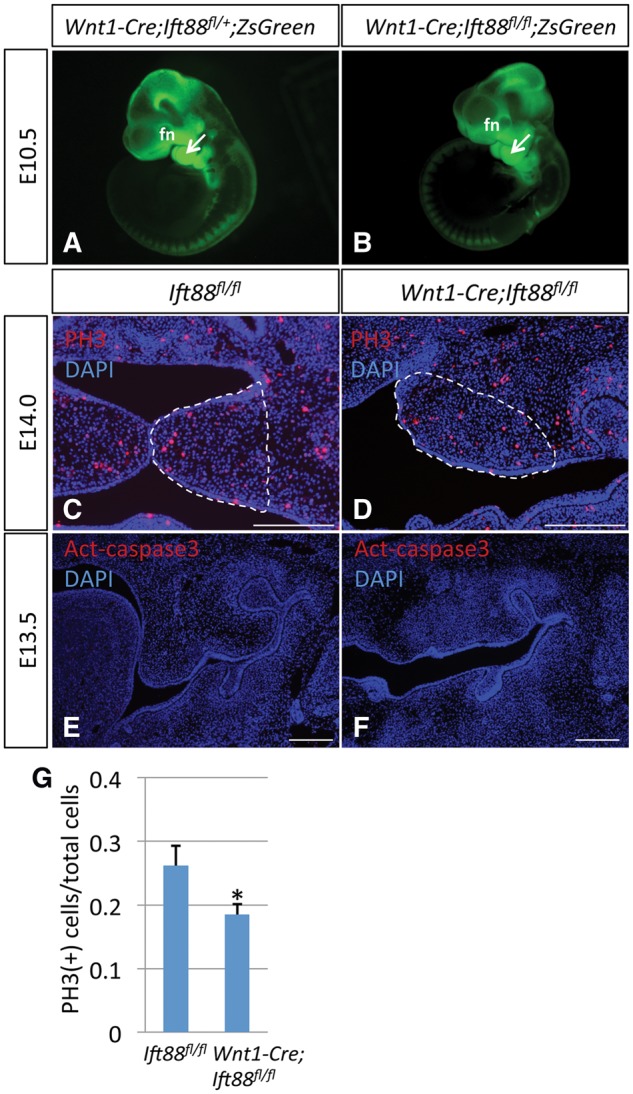
Proliferation, but not CNC migration or apoptosis, is affected in Wnt1-Cre;Ift88fl/fl mice. (A, B) Visualization of E10.5 Wnt1-Cre;Ift88fl/+;ZsGreen (control) and Wnt1-Cre;Ift88fl/fl;ZsGreen mice. fn, frontonasal process. Arrows indicate first branchial arch. (C–F) PH3 staining of E14.0 and Active Caspase 3 staining of E13.5 Ift88fl/fl (control) and Wnt1-Cre;Ift88fl/flmice. Dashed lines delineate areas for counting PH3 positive cells. (G) Quantitation of PH3 staining from (C) and (D). *P < 0.05, n = 3. Scale bars, 100 µm.
Ciliary defects in the palatal mesenchyme of Wnt1-cre;Ift88fl/flmice
We investigated the effect of loss of IFT88 on cilia during palate formation using the cilia markers acetylated α-tubulin and ϒ-tubulin. Acetylated α-tubulin and Ift88 are localized in the axonemes of primary cilia, whereas ϒ-tubulin is expressed in the basal bodies. Acetylated α-tubulin, Ift88 and ϒ-tubulin were all detectable in the palatal epithelium and mesenchyme of E14.5 control mice (Fig. 4A, B, E and F). Although ϒ-tubulin was still detectable in both the epithelium and mesenchyme of Wnt1-Cre;Ift88fl/flmice, expression of acetylated α-tubulin was dramatically reduced and showed a punctuated pattern (Fig. 4G and H). As expected, Ift88 signal was undetectable in the palatal mesenchyme of Wnt1-Cre;Ift88fl/flmice (Fig. 4C and D). Cilia lengths were measured with the National Institutes of Health (NIH) ImageJ software. Statistical analysis confirmed that both the number of ciliated cells and the length of the axonemes had decreased following loss of Ift88 (Fig. 4I and J). These data suggest that primary cilia were absent or severely altered in neural crest cells of Wnt1-Cre;Ift88fl/flmice.
Figure 4.

Ciliary defects in Wnt1-Cre;Ift88fl/flmice. (A–H) Ift88 (green) and acetylated α-tubulin (Ac-tub; red) double immunostaining (A–D) and acetylated α-tubulin (Ac-tub; red) and ϒ-tubulin (ϒ-tub; green) double immunostaining (E–H) of sections of palates from E14.5 Ift88fl/fl (control) and Wnt1-Cre;Ift88fl/flCKO mice. Boxes in A, C, E, G are shown magnified in B, D, F, H, respectively. Inserts in B and F show magnified images of cells indicated by arrows. Dotted lines indicate the boundary between the epithelium (epi) and mesenchyme (mes). (I, J) Quantification of the proportion of ciliated cells (I) and cilia length (J) in the palates of E14.5 Ift88fl/fl (control) and Wnt1-Cre;Ift88fl/flCKO mice. *P < 0.05, n = 3. Scale bars, A, C, E and G, 50 µm; B, D, F and H, 10 µm.
Ift88 is specifically required in the palatal mesenchyme
To focus more precisely on the role of Ift88 in palatogenesis, we generated Osr2KI-Cre;Ift88fl/flmice. Osr2 is specifically expressed in the mesenchyme of the palatal shelves and tooth germ from E12.5 to newborn stage (58). Newborn Osr2KI-Cre;Ift88fl/flmice exhibited cleft palate, but their tongues and mandibles were unaffected (Fig. 5A–F). Histological analysis indicated that palatal shelves in Osr2KI-Cre;Ift88fl/flmice were able to reorient from a vertical to a horizontal position but could not establish contact in the midline (Fig. 5G–L). The cleft palate in these mice suggests that Ift88 is important for CNC derived mesenchyme proliferation and differentiation during palatogenesis.
Figure 5.

Ift88 is specifically required in the palatal mesenchyme. (A, B) Newborn (NB) Ift88fl/fl (control) and Osr2KI-Cre;Ift88fl/flmice. Arrow indicates the partially open eye in Osr2KI-Cre;Ift88fl/flmice. (C–F) Intraoral views of the palates (C) and (D) and tongues (E, F) of newborn Ift88fl/fl (control) and Osr2KI-Cre;Ift88fl/flmice. Dotted line delineates the cleft palate in Osr2KI-Cre;Ift88fl/flmice. (G–L) H&E staining of sections of newborn Ift88fl/fl (control) and Osr2KI-Cre;Ift88fl/flmice. Asterisks indicate cleft palate. ps, palate shelf. Scale bars, 500 µm.
Ciliary defects result in loss of function of Shh signaling in the CNC derived mesenchyme
Based on previous studies reporting that defects in the primary cilia affect Shh signaling, we examined Hh activity in the palate by analyzing Ptch1 and Gli1 activity. Gli transcription factors are direct targets of Hh signaling. We found that Gli1 expression was downregulated in the palatal mesenchyme of E13.0 Wnt1-Cre;Ift88fl/flmice (Fig. 6A and B). We also examined the expression pattern of receptors for the Shh pathway. Ptch1 expression was significantly downregulated in the palatal mesenchyme but was unaffected in the palatal epithelium, consistent with a disruption of Shh signaling (Fig. 6C and D). In parallel, we also found that Axin2 expression level was elevated on the oral side of palatal mesenchyme in Wnt1-Cre;Ift88fl/flmice (Supplementary Material, Fig. S2), suggesting that IFT88-mediated ciliary defects may also affect canonical Wnt signaling pathway during palatogenesis.
Figure 6.
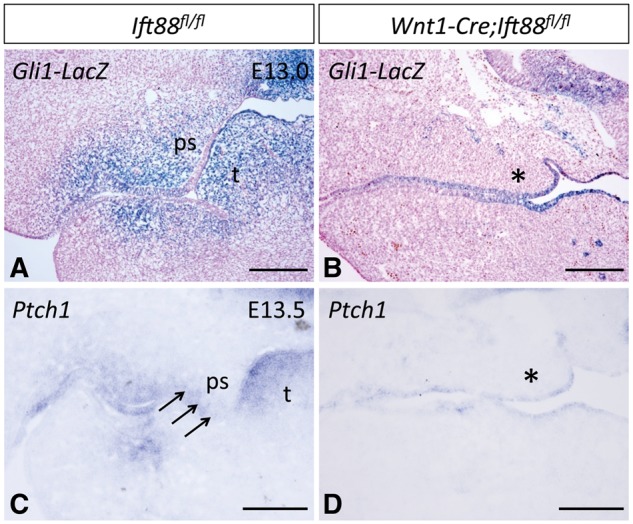
Ciliary defects in Wnt1-Cre;Ift88fl/fl mice result in loss of Shh signaling in the palatal mesenchyme. (A, B) X-gal staining of E13.0 Ift88fl/fl;Gli1-LacZ (control, n = 3) and Wnt1-Cre;Ift88fl/fl;Gli1-LacZ mice (n = 3). Blue color indicates Gli1-LacZ positive cells. (C, D) In situ analysis of Ptch1 (blue staining) in E13.5 Ift88fl/fl(control, n = 6) and Wnt1-Cre;Ift88fl/flmice (n = 6). Arrows indicate the expression of Ptch1 in the mesenchyme. Asterisks indicate the downregulated expression of Gli1 and Ptch1 in the palatal mesenchyme. ps, palatal shelf; t, tongue. Scale bars, 200 µm.
Discussion
In this study, we identified a family with multiple affected members with nonsyndromic cleft lip and palate who share a missense mutation in the IFT88 gene. We have investigated the link between ciliary function and palate development using a conditional knockout of Ift88 in murine CNC cells. These mice exhibited severe craniofacial defects including cleft lip and palate and tongue agenesis. Disruption of primary cilia in CNC cells due to loss of IFT88 also resulted in severe defects in midline fusion of the face. We also generated Osr2KI-Cre;Ift88fl/flmice in which IFT88 is specifically lost from the mesenchyme of the palatal shelves. Osr2KI-Cre;Ift88fl/flmice exhibit complete cleft palate with incomplete penetrance (approximately 30%), recapitulating the phenotype of the human patients with IFT88 mutation. Previous studies have reported multiple organ defects in mice after loss of Ift88, including in the kidney, limb and neural tube. None of the patients we report in this study exhibited any signs nor described symptoms of functional kidney or liver disease. No imaging studies were available and thus we cannot exclude asymptomatic/subclinical renal or hepatic cysts. Similarly, Wnt1-Cre;Ift88fl/fl mice, which specifically target only neural crest cells, did not show evidence of organ abnormalities other than craniofacial defects. Therefore, we propose that IFT88 is a strong candidate for further investigation of its role in human non-syndromic cleft palate.
Role of the cilia in craniofacial development and formation of the palatal shelf
Primary cilia are highly dynamic in their extension and retraction and vary in length, in a manner tightly linked with proliferation and sensitive to molecular and mechanical stimuli. Ift88 is associated with the centrosome throughout the cell cycle and controls cell proliferation by regulating the G1-S transition. Depletion of IFT88 in cultured human or mouse cells induces mitotic defects in vitro (33,59). In our study, the proportion of ciliated cells and cilia length were reduced in the palatal mesenchyme of Wnt1-Cre;Ift88fl/fl mice, and cell proliferation was decreased in the palatal shelf at early stages of palatogenesis. Wnt1-Cre;Ift88fl/fl mice exhibited no defect in CNC migration or apoptosis, suggesting that Ift88 is specifically required in the CNC-derived palatal mesenchymal during palatogenesis. Previous studies have demonstrated that altered cilia function may result in aberrant neural crest cell migration via defects in PDGF-dependent chemotaxis (60). PDGFR-alpha localizes to the axoneme, suggesting that individual ciliary proteins play specific roles in NCC proliferation and/or migration.
Receptors for the Shh pathway are localized to the cilium, and IFT proteins are involved in the trafficking and processing of Gli proteins from full-length isoforms into either activator or repressor forms (41). Mutations in Hh signaling in humans and mice disrupt mediolateral patterning of the neural plate resulting in holoprosencephaly and facial clefting (61–63). Shh signaling plays a crucial role in patterning the palate by stimulating cell proliferation to promote the outgrowth of the palatal shelf. Previous studies have demonstrated that Shh signaling acts downstream of BMP4, Msx1 and Dlx5 signaling and upstream of BMP2, Fgf10 and Foxf signaling during palate formation (10,11,64). In Wnt1-Cre;Ift88fl/fl mice, the Shh pathway was significantly downregulated in the palatal mesenchyme. Interestingly, previous studies have reported that loss of Kif3a, a component of anterograde IFT complex A, leads to an increase in the proliferation of CNC cells due to excessive Hh responsiveness in the facial mesenchyme (65). Loss of anterograde IFT complex A or retrograde IFT complex B results in similar craniofacial phenotypes in mouse models, indicating that bidirectional transport in the primary cilia is required for activation of Shh signaling (16,19,65). The differential effect of mutations in specific IFT proteins on the Hh pathway may be attributable to the specific Gli family member, Gli1, Gli2 or Gli3, functioning in that specific tissue. One mechanism by which ciliary defects are associated with a gain of function of Shh is as a result of loss of Gli3 repression (41,45,66,67). In some tissues, Gli3 expression is directly regulated by Wnt pathway activity and IFT is required for the regulation of the canonical Wnt pathway (68). Our preliminary data suggest that there is elevated Wnt signaling activity in palatal mesenchyme of Wnt1-Cre;Ift88fl/fl mice. Future study will help to address how Ift88 may affect the Wnt signaling in regulating palatogenesis.
Identification of an IFT88 mutation in patients with cleft palate sheds new light on ciliopathies
Our study demonstrates that IFT88 may be a new ciliopathy-related gene involved in cleft palate in humans. We identified a missense mutation in the IFT88 coding sequence of three affected siblings, likely representing a partial loss of IFT88 function. Consistent with this, Wnt1-Cre;Ift88fl/flmice with a total loss of function in IFT88 in CNC cells exhibited a more severe phenotype. In our patients, the mutation occurs in the third of twelve tetratricopeptide (TRP) repeat domains, which are thought to form a scaffold to mediate protein–protein interactions and assembly of multiprotein complexes. Although the mutation we report may be a conservative amino acid substitution, there are multiple examples where a similar substitution of the amino acid glutamate with an aspartate has been found to affect protein function and reported as a disease-causing pathogenic mutation (69–72). IFT88 has been shown previously to interact directly with several genes involved in craniofacial development in humans, two of which (GLI2 and GLI3) have been associated with autosomal dominant human disorders that include cleft lip and/or cleft palate (73,74). Although most ciliopathy genes are inherited in an autosomal recessive fashion, there are several conditions with autosomal dominant inheritance (75). Heterozygous mutations in GLI2 cause Holoprosencephaly 9 (OMIM:610829) and Culler-Jones syndrome (OMIM:615849). Both conditions have multiple congenital anomalies and also include cleft lip and/or cleft palate with variable phenotypes, incomplete penetrance and variable expressivity. Loss of function mutations in GLI3 also cause multiple autosomal dominant conditions including Pallister Hall syndrome (OMIM:146510) and Greig Cephalopolysyndactyly syndrome (OMIM: 175700), which exhibit multiple congenital anomalies including cleft lip and/or cleft palate.
Taken together, our data suggest that IFT88 likely has a highly conserved function within the primary cilia of the CNC-derived mesenchyme in the palate and lip region in both mice and humans. These findings have important implications for clinical studies that aim to identify patients with craniofacial defects and families with high risk of cleft palate.
Materials and Methods
Human subjects
Approval for study on human subjects was obtained from the University of Southern California Institutional Review Board (HS 13-00028). A family with cleft lip and palate of unknown etiology was recruited for participation in the study from Shriner’s Hospital for Children. Informed consent was completed and clinical information including a three-generation pedigree was obtained. The family presented with three teenage children with isolated cleft lip and palate, normal growth and development but severe speech dysfunction related to their clefting. Parents also have two unaffected daughters, one of whom has a daughter with cleft lip and palate (not available for examination). Both parents were nondysmorphic and had normal speech. The father’s skin had diffuse patches of hypopigmentation (likely vitiligo) and his oral exam was significant for a broad uvula and white middle linear groove in his palate (zona pellucida). Oragene saliva kits (DNA Genotek, Inc.) were used to collect saliva samples from the mother and three affected siblings and DNA was extracted using standard protocols (76).
Whole exome sequencing, data analysis and Sanger confirmation
Whole exome sequencing was carried out using the Ion AmpliSeqExomeKit (Life Technologies Inc.) to amplify more than 97% of all consensus coding DNA sequence protein coding exons plus flanking intronic sequences (± 5bp) to create sequencing libraries according to the manufacturer’s instructions. The generated libraries were further amplified on Ion Sphere™ Particles using the Ion OneTouch™ 2 system. Two barcoded libraries were pooled, loaded on Ion PI™ chips and sequenced on an Ion Proton machine. After the sequencing run, the raw data were processed through Torrent Suite™ Software for quality check, sequence alignment and variant calling against the human GRCh37/hg19 reference sequence, from which binary alignment/map and variant call format (VCF) files were generated. Between 35 and 45 million on-target reads were produced for each sample; the mean depth of exome coverage was 85–110× with average uniformity of 90%, and at least 95% of single nucleotide polymorphism (SNP) variants were covered at above 20x, in line with the manufacturer’s technical specifications. The SNP exonic nucleotide transition/transversion rate and SNP quality were checked in the analysis of variance-based online bioinformatics tool Tute Genomics. VCF files were uploaded into Tute Genomics for variant annotation, filtering and candidate variant analyses.
Raw data were processed for variant annotation, which provided information about which gene transcript the variants were located on, nucleotide and protein changes, allele frequency in normal population, zygosity, functional scores, known disease association in ClinVar and ClinVar significance, and Mendelian Inheritance in Man (MIM) number (77). The exonic and splicing variants (relative to hg19) were filtered according to allele frequency and amino acid alteration. Each variant was evaluated for the potential to contribute to orofacial clefting based on a Mendelian (single gene) model. Individual variant analyses and candidate genes were reviewed for potential significance related to the phenotype and possible deleterious effect on craniofacial structures.
The IFT88 mutation identified by whole exome sequencing was confirmed by Sanger sequencing. Forward and reverse PCR primers were designed using the Primer3 online software tool version 0.4.0 (http://bioinfo.ut.ee/primer3-0.4.0/primer3; date last accessed August 22, 2015). The target region was PCR amplified and cycle sequenced using the BigDye Direct Cycle Sequencing kit (Life Technologies), according to the manufacturer’ s instructions. The Sanger sequencing product was separated on an ABI 3730 genetic analyzer and data were analyzed using the commercial software Mutation Surveyor (V4.0.9).
Animal information
We generated mice with conditional loss of function of Ift88 in the epithelium or mesenchyme by crossing Ift88 fl/fl(B6.129P2-Ift88tm1Bky/J, JAX lab) with K14-Cre and Wnt1-Cre mice, respectively. Mating Wnt1-Cre;Ift88fl/+mice with Ift88fl/flmice generated Wnt1-Cre;Ift88fl/flmice. Osr2KI-Cre;Ift88fl/+ mice were crossed with Ift88fl/fl mice to generate Osr2KI-Cre;Ift88fl/flmice. Wnt1-Cre;Ift88fl/fl;ZsGreen mice, Wnt1-Cre;Ift88fl/fl;Gli1-LacZ and Wnt1-Cre;Ift88fl/fl;Axin2-LacZ mice were generated by crossing Wnt1-Cre;Ift88fl/+;ZsGreen,Wnt1-Cre;Ift88fl/+;Gli1-LacZ mice and Wnt1-Cre;Ift88fl/+;Axin2-LacZ mice with Ift88fl/flmice, respectively. All animal studies were performed in accordance with federal regulations and with approval from the Institutional Animal Care and Use Committee (IACUC) at the University of Southern California.
Histological analysis
Samples were fixed in 4% paraformaldehyde (PFA) and processed into paraffin-embedded serial sections using routine procedures. Anatomical markers such as eyes and first molars were used to ensure the sections were taken from the same location. For general morphology, deparaffinized sections were stained with hematoxylin and eosin using standard procedures.
MicroCT analysis
Control and Wnt1-Cre;Ift88fl/flnewborn mice were collected, and heads were fixed in 4% PFA at 4 °C overnight. Skull images were acquired using a micro-CT system (Scanco Medical;V1.2a). Visualization and 3D micro-CT reconstruction of the skull were performed using Isosurface parameters in Avizo 7.1 (Visualization Sciences Group). Quantification analyses were carried out on 3D microCT images from control and Wnt1-Cre;Ift88fl/flmice with anatomical landmarks as reported (78).
Alcian blue and alizarin red staining
The three-dimensional architecture of the craniofacial skeleton of Wnt1-Cre;Ift88fl/fland control mice was examined using a modified whole-mount Alcian Blue/Alizarin Red S staining protocol. Newborn mice were fixed in 95% ethanol for 72 h after removal of the skin and internal organs. The skeletons were stained with 0.02% Alcian Blue 8GX for 3 days. The samples were washed with 95% ethanol for 2 h and then treated with 0.5N KOH for 24 h. Once the cartilage was clearly detectable, Alizarin Red staining was performed overnight. Finally, the samples were treated with a series of KOH-glycerol and storedin glycerol with a crystal of thymol.
Apoptosis and proliferation analyses
For proliferation analysis, we performed immunostaining of PH3 (Millipore,06-570;1:200). PH3-positive cells and total number of cells within the palatal mesenchyme were quantitated in three randomly selected sections from each experimental group, with a distance of 30 µm between adjacent sections. Three pairs of samples were analyzed. Apoptosis assays were performed using caspase-3 immunostaining (Abcam, ab2302;1:200) according to the manufacturer’s protocol. Sections were counterstained with DAPI. Images were captured using a fluorescence microscope (Leica DMI 3000B).
Immunostaining
Samples were fixed in 4% PFA in PBS overnight at 4 °C, embedded and sectioned at 8 μm thickness. The sections were first incubated with blocking reagent (PerkinElmer, FP1012) for 2 h at room temperature, then incubated with IFT88 antibody (Proteintech13967-1-AP1:200), acetylated α-tubulin (Sigma,T6793-100UL,1:200) and/or ϒ-tubulin (Sigma,T 5192, 1:200) antibodies at 4 °C overnight, followed by Alexa Fluor 568 and 488 IgG (Invitrogen A11011, 1:200). Sections were counter-stained with DAPI, mounted and imaged with a confocal microscope (Leica Sp5).
X-gal staining
Embryos were collected at E13.0, fixed in 0.2% glutaraldehyde in PBS with 2mM MgCl2 overnight at 4 °C. After dehydration in 15% sucrose at room temperature until the sample sank to the bottom, the samples were soaked in 50% OCT by volume (Sakura Tissue-Tek, 4583) and 30% sucrose at room temperature for 1.5 h before embedding. Samples were sectioned at 8 μm thickness, followed by X-gal staining according to standard protocol, as previously described (79).
In situ hybridization
The expression patterns of Ptch1 were examined by in situ hybridization using digoxigenin-labeled antisense probes following standard procedures (58). Sense probes or PBS solution were used as controls.
Statistical analysis
Two-tailed Student’s t tests were applied for statistical analysis. For all graphs, data are represented as means ±standard deviations (SD). P < 0.05 was considered statistically significant.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We are grateful to Dr. Julie Mayo for critical reading of the manuscript. We thank the patients and their families for allowing us to learn from them and share this information as well as the laboratory staff who made this analysis possible. We thank Operation Smile for the logistical support necessary for the patient study. Dr. Pedro A. Sanchez-Lara coordinated the human data analysis. We thank Dr. Rulang Jiang from Cincinnati Children's Hospital Medical Center for providing Osr2KI-Cre mice and Ptch1 probe.
Conflict of Interest statement. None declared.
Funding
Grants from the National Institute of Dental and Craniofacial Research, National Institutes of Health (R37 DE012711 and U01 DE024421) to Y.C.
References
- 1. Panamonta V., Pradubwong S., Panamonta M., Chowchuen B. (2015) Global birth prevalence of orofacial clefts: a systematic review. J. Med. Assoc. Thai, 98 (Suppl. 7), S11–S21. [PubMed] [Google Scholar]
- 2. Grosen D., Chevrier C., Skytthe A., Bille C., Mølsted K., Sivertsen Å., Murray J.C., Christensen K. (2010) A cohort study of recurrence patterns among more than 54,000 relatives of oral cleft cases in Denmark: support for the multifactorial threshold model of inheritance. J. Med. Genet., 47, 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dixon M.J., Marazita M.L., Beaty T.H., Murray J.C. (2011) Cleft lip and palate: synthesizing genetic and environmental influences. Nat. Rev. Genet., 12, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lan Y., Xu J., Jiang R. (2015) Cellular and molecular mechanisms of palatogenesis. Curr. Top Dev. Biol., 115, 59–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chai Y., Jiang X., Ito Y., Bringas P. Jr, Han J., Rowitch D.H., Soriano P., McMahon A.P., Sucov H.M. (2000) Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development, 127, 1671–1679. [DOI] [PubMed] [Google Scholar]
- 6. Chai Y., Maxson R.E. Jr. (2006) Recent advances in craniofacial morphogenesis. Dev. Dyn., 235, 2353–2375. [DOI] [PubMed] [Google Scholar]
- 7. Gritli-Linde A. (2007) Molecular control of secondary palate development. Dev. Biol., 301, 309–326. [DOI] [PubMed] [Google Scholar]
- 8. Bush J.O., Jiang R. (2012) Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development, 139, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou J., Gao Y., Lan Y., Jia S., Jiang R. (2013) Pax9 regulates a molecular network involving Bmp4, Fgf10, Shh signaling and the Osr2 transcription factor to control palate morphogenesis. Development, 140, 4709–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Han J., Mayo J., Xu X., Li J., Bringas P. Jr., Maas R.L., Rubenstein J.L., Chai Y. (2009) Indirect modulation of Shh signaling by Dlx5 affects the oral-nasal patterning of palate and rescues cleft palate in Msx1-null mice. Development, 136, 4225–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu J., Liu H., Lan Y., Aronow B.J., Kalinichenko V.V., Jiang R. (2016) A Shh-Foxf-Fgf18-Shh molecular circuit regulating palate development. PLoS Genet., 12, e1005769.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grimaldi A., Parada C., Chai Y. (2015) A comprehensive study of soft palate development in mice. PLoS One, 10, e0145018.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bureau A., Parker M.M., Ruczinski I., Taub M.A., Marazita M.L., Murray J.C., Mangold E., Noethen M.M., Ludwig K.U., Hetmanski J.B.. et al. (2014) Whole exome sequencing of distant relatives in multiplex families implicates rare variants in candidate genes for oral clefts. Genetics, 197, 1039–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pengelly R.J., Arias L., Martinez J., Upstill-Goddard R., Seaby E.G., Gibson J., Ennis S., Collins A., Briceno I. (2016) Deleterious coding variants in multi-case families with non-syndromic cleft lip and/or palate phenotypes. Sci. Rep, 6, 30457.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Noda K., Kitami M., Kitami K., Kaku M., Komatsu Y. (2016) Canonical and noncanonical intraflagellar transport regulates craniofacial skeletal development. Proc. Natl Acad. Sci. USA, 113, E2589–E2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang C.F., Schock E.N., Attia A.C., Stottmann R.W., Brugmann S.A. (2015) The ciliary baton: orchestrating neural crest cell development. Curr. Top. Dev. Bio., 111, 97–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bisgrove B.W., Yost H.J. (2006) The roles of cilia in developmental disorders and disease. Development, 133, 4131–4143. [DOI] [PubMed] [Google Scholar]
- 18. Serra R. (2008) Role of intraflagellar transport and primary cilia in skeletal development. Anat. Rec. (Hoboken), 291, 1049–1061. [DOI] [PubMed] [Google Scholar]
- 19. Taschner M., Bhogaraju S., Lorentzen E. (2012) Architecture and function of IFT complex proteins in ciliogenesis. Differentiation, 83, S12–S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferkol T.W., Leigh M.W. (2012) Ciliopathies: the central role of cilia in a spectrum of pediatric disorders. J. Pediatr, 160, 366–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arts H.H., Doherty D., van Beersum S.E., Parisi M.A., Letteboer S.J., Gorden N.T., Peters T.A., Marker T., Voesenek K., Kartono A.. et al. (2007) Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet., 39, 882–888. [DOI] [PubMed] [Google Scholar]
- 22. Bergmann C., Fliegauf M., Bruchle N.O., Frank V., Olbrich H., Kirschner J., Schermer B., Schmedding I., Kispert A., Kranzlin B.. et al. (2008) Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am. J. Hum. Genet., 82, 959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cortes C.R., Metzis V., Wicking C. (2015) Unmasking the ciliopathies: craniofacial defects and the primary cilium. Wiley Interdiscipl. Rev. Dev. Biol., 4, 637–653. [DOI] [PubMed] [Google Scholar]
- 24. Dowdle W.E., Robinson J.F., Kneist A., Sirerol-Piquer M.S., Frints S.G., Corbit K.C., Zaghloul N.A., van Lijnschoten G., Mulders L., Verver D.E.. et al. (2011) Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am. J. Hum. Genet., 89, 94–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bayram Y., Aydin H., Gambin T., Akdemir Z.C., Atik M.M., Karaca E., Karaman A., Pehlivan D., Jhangiani S.N., Gibbs R.A.. et al. (2015) Exome sequencing identifies a homozygous C5orf42 variant in a Turkish kindred with oral-facial-digital syndrome type VI. Am. J. Hum. Genet. Part a, 167a, 2132–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heon E., Kim G., Qin S., Garrison J.E., Tavares E., Vincent A., Nuangchamnong N., Scott C.A., Slusarski D.C., Sheffield V.C. (2016) Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum. Mol. Genet., 25, 2283–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C.. et al. (2007) The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet., 39, 875–881. [DOI] [PubMed] [Google Scholar]
- 28. Valente E.M., Logan C.V., Mougou-Zerelli S., Lee J.H., Silhavy J.L., Brancati F., Iannicelli M., Travaglini L., Romani S., Illi B.. et al. (2010) Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet., 42, 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murcia N.S., Richards W.G., Yoder B.K., Mucenski M.L., Dunlap J.R., Woychik R.P. (2000) The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development, 127, 2347–2355. [DOI] [PubMed] [Google Scholar]
- 30. Zhang Q., Murcia N.S., Chittenden L.R., Richards W.G., Michaud E.J., Woychik R.P., Yoder B.K. (2003) Loss of the Tg737 protein results in skeletal patterning defects. Dev. Dyn., 227, 78–90. [DOI] [PubMed] [Google Scholar]
- 31. Pazour G.J., Dickert B.L., Vucica Y., Seeley E.S., Rosenbaum J.L., Witman G.B., Cole D.G. (2000) Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J. Cell. Biol., 151, 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ohazama A., Haycraft C.J., Seppala M., Blackburn J., Ghafoor S., Cobourne M., Martinelli D.C., Fan C.M., Peterkova R., Lesot H.. et al. (2009) Primary cilia regulate Shh activity in the control of molar tooth number. Development, 136, 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robert A., Margall-Ducos G., Guidotti J.E., Bregerie O., Celati C., Brechot C., Desdouets C. (2007) The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells (vol 120, pg 628, 2006). J. Cell Sci., 120, 918–918. [DOI] [PubMed] [Google Scholar]
- 34. Chang C.F., Serra R. (2013) Ift88 regulates Hedgehog signaling, Sfrp5 expression, and beta-catenin activity in post-natal growth plate. J. Orthop. Res., 31, 350–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ezratty E.J., Stokes N., Chai S., Shah A.S., Williams S.E., Fuchs E. (2011) A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell, 145, 1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abdelhamed Z.A., Natarajan S., Wheway G., Inglehearn C.F., Toomes C., Johnson C.A., Jagger D.J. (2015) The Meckel-Gruber syndrome protein TMEM67 controls basal body positioning and epithelial branching morphogenesis in mice via the non-canonical Wnt pathway. Dis. Model. Mech, 8, 527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Corbit K.C., Shyer A.E., Dowdle W.E., Gaulden J., Singla V., Chen M.H., Chuang P.T., Reiter J.F. (2008) Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol, 10, 70–76. [DOI] [PubMed] [Google Scholar]
- 38. Chang C.F., Schock E.N., O'Hare E.A., Dodgson J., Cheng H.H., Muir W.M., Edelmann R.E., Delany M.E., Brugmann S.A. (2014) The cellular and molecular etiology of the craniofacial defects in the avian ciliopathic mutant talpid2. Development, 141, 3003–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Koyama E., Young B., Nagayama M., Shibukawa Y., Enomoto-Iwamoto M., Iwamoto M., Maeda Y., Lanske B., Song B., Serra R.. et al. (2007) Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development, 134, 2159–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rohatgi R., Milenkovic L., Scott M.P. (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science (New York, N.Y.), 317, 372–376. [DOI] [PubMed] [Google Scholar]
- 41. Haycraft C.J., Banizs B., Aydin-Son Y., Zhang Q., Michaud E.J., Yoder B.K. (2005) Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet., 1, e53.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huangfu D., Anderson K.V. (2005) Cilia and Hedgehog responsiveness in the mouse. Proc. Natl Acad. Sci. USA, 102, 11325–11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Corbit K.C., Aanstad P., Singla V., Norman A.R., Stainier D.Y., Reiter J.F. (2005) Vertebrate smoothened functions at the primary cilium. Nature, 437, 1018–1021. [DOI] [PubMed] [Google Scholar]
- 44. Huangfu D., Liu A., Rakeman A.S., Murcia N.S., Niswander L., Anderson K.V. (2003) Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature, 426, 83–87. [DOI] [PubMed] [Google Scholar]
- 45. Tran P.V., Haycraft C.J., Besschetnova T.Y., Turbe-Doan A., Stottmann R.W., Herron B.J., Chesebro A.L., Qiu H., Scherz P.J., Shah J.V.. et al. (2008) THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet., 40, 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Caspary T., Larkins C.E., Anderson K.V. (2007) The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell, 12, 767–778. [DOI] [PubMed] [Google Scholar]
- 47. Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J.. et al. (2007) IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet., 39, 727–729. [DOI] [PubMed] [Google Scholar]
- 48. Walczak-Sztulpa J., Eggenschwiler J., Osborn D., Brown D.A., Emma F., Klingenberg C., Hennekam R.C., Torre G., Garshasbi M., Tzschach A.. et al. (2010) Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet., 86, 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbo M., Filhol E., Bole-Feysot C.. et al. (2011) Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet., 89, 634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Perrault I., Saunier S., Hanein S., Filhol E., Bizet A.A., Collins F., Salih M.A., Gerber S., Delphin N., Bigot K.. et al. (2012) Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet., 90, 864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ashe A., Butterfield N.C., Town L., Courtney A.D., Cooper A.N., Ferguson C., Barry R., Olsson F., Liem K.F. Jr., Parton R.G.. et al. (2012) Mutations in mouse Ift144 model the craniofacial, limb and rib defects in skeletal ciliopathies. Hum. Mol. Genet., 21, 1808–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brown T.L., Meloche T.M. (2016) Exome sequencing a review of new strategies for rare genomic disease research. Genomics, 108, 109–114. [DOI] [PubMed] [Google Scholar]
- 53. Dong C., Wei P., Jian X., Gibbs R., Boerwinkle E., Wang K., Liu X. (2015) Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet., 24, 2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jian X., Boerwinkle E., Liu X. (2014) In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic. Acids Res., 42, 13534–13544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu X., Wu C., Li C., Boerwinkle E. (2016) dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat., 37, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sanchez-Lara P.A. (2015) Clinical and genomic approaches for the diagnosis of craniofacial disorders. Curr. Top. Dev. Biol., 115, 543–559. [DOI] [PubMed] [Google Scholar]
- 57. Croyle M.J., Lehman J.M., O'Connor A.K., Wong S.Y., Malarkey E.B., Iribarne D., Dowdle W.E., Schoeb T.R., Verney Z.M., Athar M.. et al. (2011) Role of epidermal primary cilia in the homeostasis of skin and hair follicles. Development, 138, 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Parada C., Li J., Iwata J., Suzuki A., Chai Y. (2013) CTGF mediates Smad-dependent transforming growth factor beta signaling to regulate mesenchymal cell proliferation during palate development. Mol. Cell. Biol., 33, 3482–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Delaval B., Bright A., Lawson N.D., Doxsey S. (2011) The cilia protein IFT88 is required for spindle orientation in mitosis. Nat. Cell. Biol., 13, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schneider L., Clement C.A., Teilmann S.C., Pazour G.J., Hoffmann E.K., Satir P., Christensen S.T. (2005) PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol., 15, 1861–1866. [DOI] [PubMed] [Google Scholar]
- 61. Belloni E., Muenke M., Roessler E., Traverso G., Siegel-Bartelt J., Frumkin A., Mitchell H.F., Donis-Keller H., Helms C., Hing A.V.. et al. (1996) Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet., 14, 353–356. [DOI] [PubMed] [Google Scholar]
- 62. Hu D., Helms J.A. (1999) The role of sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development, 126, 4873–4884. [DOI] [PubMed] [Google Scholar]
- 63. Jeong J., Mao J., Tenzen T., Kottmann A.H., McMahon A.P. (2004) Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev., 18, 937–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang Z., Song Y., Zhao X., Zhang X., Fermin C., Chen Y. (2002) Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development, 129, 4135–4146. [DOI] [PubMed] [Google Scholar]
- 65. Brugmann S.A., Allen N.C., James A.W., Mekonnen Z., Madan E., Helms J.A. (2010) A primary cilia-dependent etiology for midline facial disorders. Hum. Mol. Genet., 19, 1577–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. May S.R., Ashique A.M., Karlen M., Wang B., Shen Y., Zarbalis K., Reiter J., Ericson J., Peterson A.S. (2005) Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol., 287, 378–389. [DOI] [PubMed] [Google Scholar]
- 67. Chang C.F., Chang Y.T., Millington G., Brugmann S.A. (2016) Craniofacial ciliopathies reveal specific requirements for GLI proteins during development of the facial midline. PLoS Genet., 12, e1006351.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Alvarez-Medina R., Cayuso J., Okubo T., Takada S., Marti E. (2008) Wnt canonical pathway restricts graded Shh/Gli patterning activity through the regulation of Gli3 expression. Development, 135, 237–247. [DOI] [PubMed] [Google Scholar]
- 69. Bujakowska K.M., Zhang Q., Siemiatkowska A.M., Liu Q., Place E., Falk M.J., Consugar M., Lancelot M.E., Antonio A., Lonjou C.. et al. (2015) Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum. Mol. Genet., 24, 230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zaghloul N.A., Liu Y., Gerdes J.M., Gascue C., Oh E.C., Leitch C.C., Bromberg Y., Binkley J., Leibel R.L., Sidow A.. et al. (2010) Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl Acad. Sci. USA, 107, 10602–10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hoskins B.E., Thorn A., Scambler P.J., Beales P.L. (2003) Evaluation of multiplex capillary heteroduplex analysis: a rapid and sensitive mutation screening technique. Hum. Mutat., 22, 151–157. [DOI] [PubMed] [Google Scholar]
- 72. Ge Z., Bowles K., Goetz K., Scholl H.P., Wang F., Wang X., Xu S., Wang K., Wang H.. et al. (2015) NGS-based molecular diagnosis of 105 eyeGENE((R)) probands with Retinitis Pigmentosa. Sci. Rep., 5, 18287.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Malicki J., Avidor-Reiss T. (2014) From the cytoplasm into the cilium: bon voyage. Organogenesis, 10, 138–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liem K.F. Jr., Ashe A., He M., Satir P., Moran J., Beier D., Wicking C., Anderson K.V. (2012) The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J. Cell. Biol., 197, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lindstrand A., Frangakis S., Carvalho C.M., Richardson E.B., McFadden K.A., Willer J.R., Pehlivan D., Liu P., Pediaditakis I.L., Sabo A.. et al. (2016) Copy-number variation contributes to the mutational load of Bardet-Biedl syndrome. Am. J. Hum. Genet., 99, 318–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nunes A.P., Oliveira I.O., Santos B.R., Millech C., Silva L.P., Gonzalez D.A., Hallal P.C., Menezes A.M., Araujo C.L., Barros F.C. (2012) Quality of DNA extracted from saliva samples collected with the Oragene DNA self-collection kit. BMC Med. Res. Methodol., 12, 65.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shen L., Diroma M.A., Gonzalez M., Navarro-Gomez D., Leipzig J., Lott M.T., van Oven M., Wallace D.C., Muraresku C.C., Zolkipli-Cunningham Z.. et al. (2016) MSeqDR: a centralized knowledge repository and bioinformatics web resource to facilitate genomic investigations in mitochondrial disease. Hum. Mutat., 37, 540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ho T.V., Iwata J., Ho H.A., Grimes W.C., Park S., Sanchez-Lara P.A., Chai Y. (2015) Integration of comprehensive 3D microCT and signaling analysis reveals differential regulatory mechanisms of craniofacial bone development. Dev. Bio.l, 400, 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Li J., Feng J., Liu Y., Ho T.V., Grimes W., Ho H.A., Park S., Wang S., Chai Y. (2015) BMP-SHH signaling network controls epithelial stem cell fate via regulation of its niche in the developing tooth. Dev. Cell, 33, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.