

Abstract
The aim of this work is to identify the molecular cause of autosomal recessive early onset retinal degeneration in a consanguineous pedigree. Seventeen members of a four-generation Pakistani family were recruited and underwent a detailed ophthalmic examination. Exomes of four affected and two unaffected individuals were sequenced. Variants were filtered using exomeSuite to identify rare potentially pathogenic variants in genes expressed in the retina and/or brain and consistent with the pattern of inheritance. Effect of the variant observed in the gene Intraflagellar Transport Protein 43 (IFT43) was studied by heterologous expression in mIMCD3 and MDCK cells. Expression and sub-cellular localization of IFT43 in the retina and transiently transfected cells was examined by RT-PCR, western blot analysis, and immunohistochemistry. Affected members were diagnosed with early onset non-syndromic progressive retinal degeneration and the presence of bone spicules distributed throughout the retina at younger ages while the older affected members showed severe central choroidal atrophy. Whole-exome sequencing analysis identified a novel homozygous c.100 G > A change in IFT43 segregating with retinal degeneration and not present in ethnicity-matched controls. Immunostaining showed IFT43 localized in the photoreceptors, and to the tip of the cilia in transfected mIMCD3 and MDCK cells. The cilia in mIMCD3 and MDCK cells expressing mutant IFT43 were found to be significantly shorter (P < 0.001) than cells expressing wild-type IFT43. Our studies identified a novel homozygous mutation in the ciliary protein IFT43 as the underlying cause of recessive inherited retinal degeneration. This is the first report demonstrating the involvement of IFT43 in retinal degeneration.
Introduction
Retinal degeneration is a group of inherited conditions that cause irreversible vision loss (1,2). These segregate in autosomal dominant, recessive, X-linked, and mitochondrial patterns (3–6). Retinal degeneration has also been observed to be a component of a large number of inherited syndromic diseases. More than 250 genes have been implicated in retinal degeneration. The majority of these are associated with non-syndromic inherited retinal degeneration (IRD), and some are associated with both syndromic and non-syndromic forms of retinal degeneration (4,7,8). An example of a gene that is implicated in both isolated and syndromic retinal degeneration is BBS5 (9,10). The syndrome that is caused by mutations in this gene is known as Bardet-Biedl syndrome, which is part of the ciliopathy spectrum of disorders. Ciliopathies are a group of genetically and phenotypically heterogeneous conditions caused by dysfunction of primary cilia present in a majority of vertebrate cell types (11–13). The phenotype of these diseases spans a wide spectrum of clinical entities involving nearly all organ systems. Bardet-Biedl syndrome, Joubert syndrome and Sensenbrenner syndrome are all examples of ciliary disorders in which retinal dystrophy may occur. That said, while retinal degeneration is a common feature in Bardet-Biedl and Joubert syndrome, it is much less common in Sensenbrenner syndrome as only a few individuals with retinal degeneration have been reported to date (11,14–18).
Sensenbrenner syndrome, also known as cranio ectodermal dysplasia (CED) and short rib polydactyly syndrome (SRPS) belong to a group of autosomal recessive skeletal dysplasias. These disorders are primarily characterized by skeletal and ectodermal abnormalities, chronic renal failure, heart defects and hepatic fibrosis (19,20). The skeletal dysplasias are genetically heterogeneous and in some cases caused by disruption of intraflagellar transport (IFT) (19,20). One of the genes associated with CED and SRPS phenotypes is IFT43. A homozygous initiation codon mutation (c.1 A > G) in IFT43 was previously reported in two affected members of a consanguineous family of Moroccan descent with CED (21). The same mutation has also been observed to segregate with SRPS in the homozygous state in an additional family (20). This mutation is known to disrupt IFT-A complex regulated retrograde transport (11,21). In addition, a homozygous missense mutation, p.Trp179Arg has been reported in a European pedigree with SRPS (20). The cases with SRPS phenotype and IFT43 mutations were evaluated at 18 weeks gestational age and hence the impact of these mutations on the retina is not known. However, it is remarkable that retinal degeneration was not found in either of the affected children in the Moroccan family as we report here, where non-syndromic early-onset recessive retinal degeneration in a large consanguineous Pakistani pedigree is associated with homozygosity for the c.100 G > A (p.Glu34Lys) IFT43 mutation. These findings along with functional validation demonstrate the involvement of IFT43 in non-syndromic recessive early onset retinal degeneration.
Results
Clinical evaluation of patients
Clinical analysis on four affected members (III: 1, III: 2, III: 7 and III: 8) and two unaffected members (III: 4 and III: 5) of this pedigree (Fig. 1) showed normal physical development, body mass index and had no symptoms of Sensenbrenner syndrome such as polydactyly, short-rid or micromelia. All four affected members were reported to have noticeable night vision abnormalities under the age of 5 years. Fundoscopy of affected individuals (III: 1 and III: 8) (Fig. 2) showed optic nerve pallor, retinal vessel attenuation, and bone spicule pigmentary change anterior to the arcades and in the nasal retina at the age of 30 and 23 years, respectively (Fig. 2). III: 1 also had extensive RPE and choroidal atrophy in each macula; III: 8 had a smaller area of RPE and choroidal atrophy in the macula whereas fundoscopy of a control individual was normal at the age of 32 years (Fig. 2). Full field ERG responses were undetectable to all stimulus conditions in all affected individuals when measured at age 46 (III: 1), 42 (III: 2), 46 (III: 7) and 28 (III: 8) years of age, while an unaffected individual exhibited rod and cone responses within normal ranges at age 30 (III: 4) (Fig. 3).
Figure 1.

Segregation analysis of IFT43 c.100 G > A (p.Glu34Lys) variant in PKRD272 pedigree. Segregation of IFT43 c.100 G > A mutation in pedigree PKRD272 is shown by presenting the genotypes at this locus. Individuals selected for exome sequencing are shown with red outline.
Figure 2.
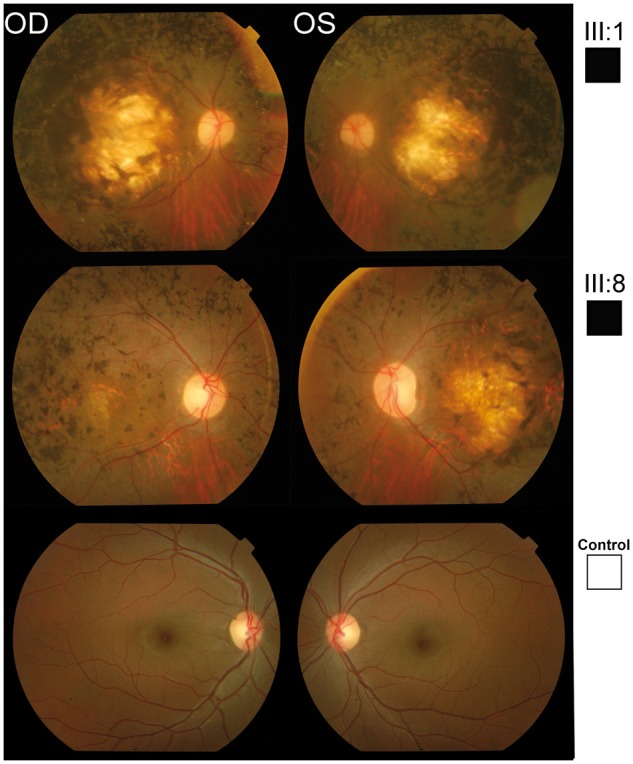
Fundus images from pedigree PKRD272. Fundus images of two affected and one unaffected member. Fundus images of affected individuals (III: 1 and III: 8) showed optic nerve pallor, retinal vessel attenuation, and bone spicule pigmentary change anterior to the arcades and in the nasal retina at the age of 23 and 30 years, respectively. III: 1 had extensive RPE and choroidal atrophy in each macula; III: 8 had a smaller area of RPE and choroidal atrophy in the macula. Color fundus photos of the normal control are without disc pallor, vascular attenuation, pigmentary change or macular RPE or choroidal atrophy.
Figure 3.

ERG response of members of pedigree PKRD272. Electroretinograms of four affected and one unaffected individuals of pedigree PKRD272 are presented: ERG responses of affected members (III: 1, III: 2, III: 7 and III: 8) were undetectable where as the unaffected individual III: 4 showed normal rod and cone responses.
Exome sequencing and analysis of variants
Exomes of four affected (II: 11, II: 12, III: 2 and III: 7) and two unaffected (II: 10 and II: 13) members of the PKRD272 pedigree were captured and sequenced using Nimblegen V2 or V3 sequencing kits. The average base read depth was 99 and the coverage of target sequence ranged from 94 to 99% at 20X. Sequence analysis identified on average 61, 000 variants in each exome. Homozygosity mapping revealed a 18 Mb region on chromosome 14 (chr14: 59000001-77000000) shared by four affected individuals in the homozygous state (Fig. 4A). This region contains 138 genes including RDH11, RDH12 and TTLL5 genes associated with IRD. However, no rare potentially pathogenic variants segregating with IRD in PKRD272 were observed in these three genes. Further analysis of single nucleotide variants (SNVs) and insertion-deletion (INDELs) variants from six members of this family using in-house exomeSuite software (22) identified 180 homozygous variants that were shared by the four affected individuals and heterozygous or absent in the two unaffected members. Further filtering of these for rare (<0.005 frequency) and potentially pathogenic variants in genes expressed in the retina or brain tissue identified two rare candidate variants for the retinal degeneration in this pedigree (23). One candidate variant was c.100 G > A (p.Glu34Lys) in IFT43 and the second candidate variant was c.1256 G > C (p.Ser419Thr) in the SLC38A6 gene. Both changes were located on chromosome 14 about 15 Mb base pairs apart and within the 18 Mb shared homozygous region identified by homozygosity mapping. Both variants are rare, with a frequency of 0.0001 for the IFT43 variant (rs140366557) and 0.000008 for the SLC38A6 variant (rs762713377). Among these, only the c.100 G > A (p.Glu34Lys) change in IFT43 segregated with the retinal degeneration phenotype after analysis of additional members of the PKRD272 pedigree (Fig. 1 and 4B). The glutamic acid residue of p.Glu34Lys is located in a domain that is highly conserved in mammals (Fig. 5A). However, the Chlamydomonas and Drosophila orthologs of IFT43 showed 39% and 22% homology respectively with the human IFT43 (Fig. 5A). The p.Glu34Lys change in IFT43 is predicted to be damaging by PolyPhen2, SIFT and MutationTaster (24,25). These data indicate that this potentially pathogenic variant is the likely cause of retinal dystrophy in the Pakistani family PKRD272. Analysis of 150 ethnicity-matched controls by Sanger sequencing and screening the whole genome data of 800 individuals with no history of IRD did not detect the p.Glu34Lys variant in IFT43. In addition, screening the whole exome variant data of 1771 individuals from IRD pedigrees with Pakistani, Indian, Middle Eastern, Japanese and Caucasian ethnicity did not detect any pathogenic variants in IFT43 segregating with disease. Furthermore, screening the whole genome sequence data of 460 individuals from 120 IRD pedigrees from the above populations also did not identify potential candidate variants in IFT43, suggesting the uncommon nature of the involvement of IFT43 mutations in causing IRD.
Figure 4.

Homozygosity Mapping and Electropherograms of the region containing the c.100 G > A mutation. (A) Homozygosity mapping identified one 18 Mb homozygous region on chromosome 14 (chr14: 59000001-77000000), shared by four affected individuals in PKRD272. The alleles of variants in the region depicted in red are homozygous, the region in blue are heterozygous alleles, while the region in white contains homozygous and heterozygous alleles in the same frequency. (B) Electropherograms showing the sequence of IFT43 at the site of c.100 G > A mutation observed in members of PKRD272. The sequence of the region encompassing the IFT43 c.100 G > A mutation in affected (III: 1), carrier (II: 13) and unaffected (III: 4) individuals from the pedigree PKRD272.
Figure 5.

Conservation of glutamic acid (p.Glu34) and expression profile of IFT43 in ocular tissues. (A) Conserved region surrounding the glutamic acid (p.Glu34) in different species. The glutamic acid residue of p.Glu34Lys is located in a conserved region across different species. (B) The IFT43 transcript showed a high level of expression in retinal tissue as compared with other ocular tissue in 2-month-old mice.
Expression profile of IFT43
Currently, no evidence has been reported for a role of IFT43 in the retina. Analysis of IFT43 expression in the retina was studied by RT-PCR and immunohistochemistry (IHC). qRT-PCR analysis of mouse ocular tissues revealed the expression profile for IFT43 in a 2-month-old mouse with a high level of expression in the retina and minimal expression in the RPE (Fig. 5B). IHC of IFT43 in mouse and human retinal tissue showed that IFT43 is localized predominantly to the photoreceptor outer segment region (Fig. 6). Significant expression was not observed in other layers of the retina. The expression of IFT43 in the retina supports the involvement of this gene in retinal dystrophy.
Figure 6.

Expression profile of IFT43 in mouse and human retina. Immunostaining of retinal sections of a 6-month-old mouse and a 57-year-old human donor revealed IFT43 localized predominantly to the photoreceptor region of the retina (A and C). IFT43 was stained and observed in the red channel and indicated by the red arrow. Rhodopsin (green) and S-Opsin (white) are stained with specific antibodies and nuclei are stained with DAPI (blue). Immunostaining of mouse and human retinal sections with secondary antibodies alone did not reveal positive staining (B and D). RPE, Retinal pigment epithelium; OS, Outer Segments; ONL, Outer Nuclear Layer; OPL, Outer Plexiform Layer; INL, Inner nuclear layer; IPL, Inner Plexiform Layer; GC, Ganglion Cell Layer.
Expression and localization of wild type and mutant IFT43 in mammalian cells
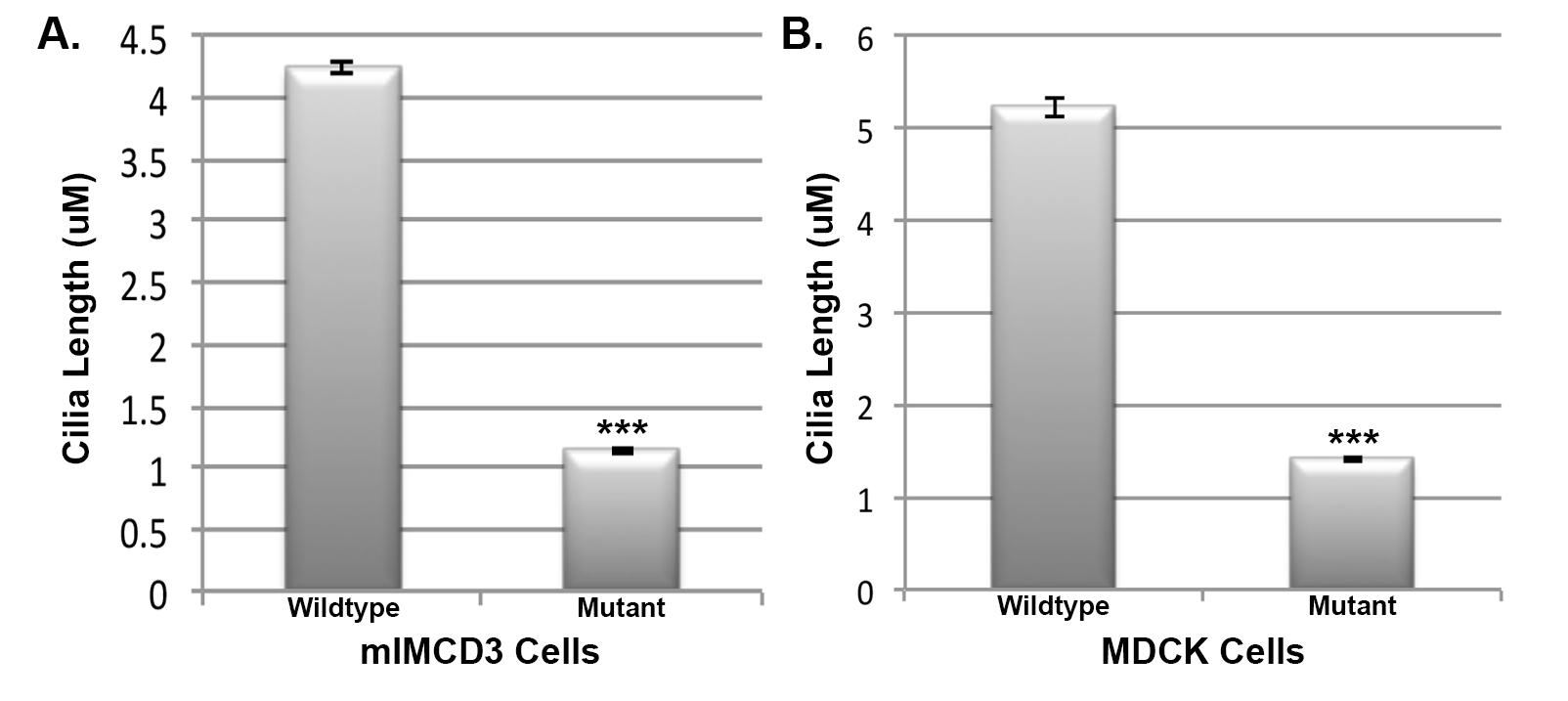
mIMCD3 cells were transfected with mammalian constructs containing wt-IFT43 (Fig. 7A–C) or the Glu34Lys-IFT43 variant (Fig. 7D–F), both tagged with 6X-His. Cells transfected with wt-IFT43 showed cilia of normal length and the localization of IFT43 at the distal tip of cilia as reported previously (26). Localization of some intraflagellar transport proteins (IFTs) also varies at different stages of the cell division cycle (27). Cells transfected with Glu34Lys-IFT43 had cilia that were significantly shorter in length (P < 0.0001) (Supplementary Material, Fig. S1). mIMCD3 cells transfected with wild type and mutant IFT43 constructs were stained with IFT88 and acetylated alpha-tubulin antibodies. Acetylated alpha-tubulin staining revealed normal ciliary structures in cells transfected with the wild type construct, and IFT88 localized to the basal bodies and the distal tip of the cilia as previously reported (Fig. 8A–C) (27). In contrast, acetylated tubulin and IFT88 staining showed shorter to no signal of ciliary structures and colocalization of IFT88 with acetylated tubulin signals in cells transfected with Glu34Lys-IFT43 (Fig. 8D–F). Similar findings were observed when MDCK cells were transfected with wild-type and mutant IFT43 constructs (data not shown). Mock-transfected cells did not show immunostaining with 6x-His antibody.
Figure 7.

Expression and localization of wt and Glu34Lys-IFT43 in mIMCD3 cells. (A) mIMCD3 cells transfected with wt-IFT43 showed normal appearing cilia when stained with acetylated α-tubulin antibodies (red). (B) The wt-IFT43-6X-His fusion protein is localized at the tip of the cilia (green). (C) Merged image of A and B. (D) Cells transfected with Glu34Lys-IFT43 showed shortened cilia (red). (E) Co-localization of acetylated α-tubulin (red) and mut-IFT43-6X-His (green) in cells transfected with Glu34Lys-IFT43. (F) Merged image of D and E.
Figure 8.

Shortened cilia in mIMCD3 cells transfected with the Glu34Lys-IFT43. (A) mIMCD3 cells transfected with wt IFT43 were stained with the ciliary marker protein acetylated α-tubulin (red). (B) Intraflagellar transport protein IFT88 (green) is localized to the base and the tip of cilia. (C) Merged image of A and B. (D) Cells transfected with Glu34Lys-IFT43 showed shortened cilia (red). (E) Co-Staining of acetylated α-tubulin (red) and IFT88 (green) in cells transfected with Glu34Lys-IFT43 showed shortened cilia. (F) Merged image of (D) and (E).
A transfection efficiency of ∼70% in cells transfected with both wild type and mutant IFT43 was observed. Western blot analysis showed the expression of wild type and mutant IFT43 in transiently transfected cells (Supplementary Material, Fig. S2). The presence of an increased amount of protein was observed in the lane loaded with the Glu34Lys-IFT43 compared with the wild type. This finding suggests the possible formation of higher molecular weight aggregates and accumulation of mutant protein in cells expressing mutant IFT43 (28–30). While a detailed analysis is needed to understand the fate and influence of the mutant protein in cells, the findings suggest that the p.Glu34Lys mutation disrupts the intraflagellar transport machinery causing abnormal cilium structure and/or affects ciliogenesis.
Discussion
Homozygosity mapping and whole exome sequencing in a four-generation pedigree PKRD272 identified a novel, potentially pathogenic homozygous change (p.Glu34Lys) in IFT43 located on chromosome 14. This variant segregated with the early onset retinal degeneration phenotype (Figs 1–3), likely consistent with severe early childhood onset retinal dystrophy (SECORD, ICD-10 H35.5) (31–33) or early onset retinitis pigmentosa (RP) (34). The history of night blindness before age 5 suggests rod cone degeneration, but as adults’ full-field ERG responses were not measurable to either scotopic or photopic stimuli and macular atrophy was present. Although the full-field ERGs were not recorded according to International Society for Clinical Electrophysiology of Vision (ISCEV) standards (35), responses were severely reduced to bright and dim stimuli in both dark and light adapted conditions, consistent with the clinical diagnosis of severe early childhood onset retinal dystrophy. The current report adds IFT43 mutations to the genes that have been associated with early onset RP and macular atrophy, including NMNAT1, AIPL1, and LCA5 (36–39).
Previously, a homozygous missense mutation in the IFT43/C14orf179 was observed in a consanguineous family of Moroccan descent with two siblings diagnosed with Sensenbrenner syndrome (21,40). Remarkably, clinical retinal abnormalities have not been observed in the Moroccan family to date (age 13yrs) (personal communication, Ernie Bongers) (21). Sensenbrenner syndrome is a rare genetically heterogeneous inherited disorder with the involvement of multiple organ systems (19,20). While retinal involvement has not been reported as a common occurrence in Sensenbrenner syndrome, it has been described in a few unrelated families (11,17,18). Although the underlying molecular cause of the disease is unknown in most of these Sensenbrenner patients, Bredrup et al, reported one family with compound heterozygous mutations in WDR19 encoding IFT144 (11). One homozygous start loss mutation c.2 T > A (p.Met1Lys) and another homozygous missense c.535 T > C (p.Trp179Arg) mutation in IFT43 were reported with Short rib polydactyly syndrome in two different families of European descent. The clinical studies on affected cases in these families were performed on fetuses of 18 weeks gestational age and they were reported with hypertelorism. The influence of IFT43 mutations on their retina is not known (20). The retinal degeneration phenotype of PKRD272 and the absence of non-ocular abnormalities indicate that the phenotype associated with the IFT43 p.Glu34Lys mutation is distinctly different from the syndromic phenotypes reported in patients with other IFT43 gene alterations (21,40). Furthermore, these findings also suggest that individuals with Sensenbrenner syndrome and IFT43 mutations may be at risk for developing blindness although the affected patients in the current study were affected with night vision loss before age 5 and had severe vision loss in adulthood.
The lack of a syndromic phenotype in members of this Pakistani family with the IFT43 homozygous p.Glu34Lys mutation is intriguing. Similarly, retinal dystrophy has not been reported in patients with Sensenbrenner syndrome due to IFT43 mutations. Genes associated with cranio ectodermal dysplasia include IFT43, IFT52, WDR35/IFT121, WDR19/IFT144 and IFT122 (41–44). All of these are ciliary proteins and members of intraflagellar transport machinery (45). Abnormal retrograde transport of ciliary proteins due to mutations in these genes has been implicated as the underlying cause of the phenotype (41,43,44). IFT43 is a member of the IFT-A complex that is highly conserved through evolution and interacts closely with IFT121 (45,46). The specific function of IFT43 is unknown and no putative structural domains have been identified in this protein, although the proline rich N-terminal half of IFT43 may play an important role in protein-protein interactions (46). The start codon mutation c.1 A > G; p.Met1Val in the IFT43 gene found in the Moroccan patients likely results in the utilization of a downstream initiation codon and generation of a mutant protein shorter by 21 amino acids (21). This mutation causes accumulation of IFT88, suggesting disruption of retrograde transport of ciliary proteins (21). Similarly, fibroblasts of the patient with a 4.5 Mb heterozygous microdeletion on chromosome 14 showed abnormal transport of ciliary proteins; this suggests that haploinsufficiency of IFT43 may impact transport of ciliary proteins as well (40). However, the ciliary structures appear to be normal in the fibroblasts of patients with the start codon (c.1 A > G) mutation and the patient with the heterozygous deletion suggesting normal ciliogenesis (21,40). Contrary to these findings, the fibroblasts from SRPS patients with the homozygous p.Trp179Arg mutation showed abnormal ciliogenesis (20). Consistent with these findings, the cells expressing the p.Glu34Lys mutant IFT43 had significantly shorter ciliary structures indicating abnormal ciliogenesis (Fig. 8 and Supplementary Material, Fig. S1). The missense mutations p.Trp179Arg and p.Glu34Lys mutations may alter the secondary structure of the IFT43 protein leading to abnormal ciliogenesis. Lack of information on the retinal status of the patient with p.Trp179Arg is a limitation, the abnormal ciliogenesis due to the p.Glu34Lys may lead to the retinal degeneration observed in the pedigree PKRK272. Although the mechanism underlying selective retinal degeneration due to the p.Glu34Lys mutation is not known, the absence of pathological changes in non-ocular tissue in affected individuals III: 1, III: 2, III: 7 and III: 8 indicate that photoreceptors may be more sensitive to the p.Glu34Lys mutation than other cells in the body. Photoreceptor cells have a highly evolved ciliary structure that plays a critical role in the development and maintenance of these polarized cells with unique morphology (47–49). The members of IFT machinery participate in ciliogenesis in addition to their role in the bidirectional transport of ciliary proteins in photoreceptors (45,50,51). So far, mutations in four additional members of IFT machinery: IFT27, IFT172, IFT140 and IFT144 have been reported in patients with retinal degenerations (11,52–54). IFT172 and IFT140 are involved in causing both syndromic and non-syndromic RD while mutations in IFT27 and IFT144 are reported only in syndromic RD. The current understanding of IFT function and the impact of its mutations are not sufficient to explain the variation in syndromic and non-syndromic phenotypes. Similar to IFT43, other IRD genes such as USH2A and Bardet Biedl syndrome (BBS) genes are also implicated in both syndromic and non-syndromic IRD and the mechanism underlying this variation in phenotype is not understood. Future studies of the role of IFT43 and the impact of its mutations, particularly in photoreceptors cells and animal models, may facilitate a better understanding of the mechanism underlying syndromic and non-syndromic phenotypes associated with mutations in IFT43 and other IRD genes.
Materials and Methods
Ethics statement
All studies were performed in accordance with the Declaration of Helsinki and the approval of the institutional review boards (IRB) of University of California San Diego, La Jolla, Johns Hopkins University, Baltimore and the National Center of Excellence in Molecular Biology, Lahore, Pakistan. Written informed consent was obtained from all participating subjects. Blood samples were collected from seventeen members of a family (PKRD272) with multiple affected members and multiple consanguineous marriages from Lahore, Pakistan (Fig. 1).
Clinical evaluation
Clinical analysis including electroretinography (ERG), fundus photography, and color vision was performed on four affected members (III: 1, III: 2, III: 7 and III: 8) and two unaffected members of this pedigree (III: 4 and III: 5) (Fig. 1) (55). Patients’ ERG responses were measured at 0 dB while the 30 Hz flicker responses were recorded at 0 dB to a background illumination of 17 to 34 cd/m2 using LKC Technologies, Inc (Gaithersburg, MD).
Genetic analysis
DNA isolation for all available members was performed using Puregene Blood Kit and protocol (Cat No./ID: 158389). Exomes of one affected member (II: 12) were sequenced using Nimblegen V2 kit. Another three affected (II: 11, III: 2 and III: 7) and two unaffected (II: 10 and II: 13) individuals were captured using Nimblegen V3 probe capture kit (Fig. 1). Read mapping and variant calling was performed using BWA and GATK (56). Variants were filtered using exomeSuite as described previously (22). Homozygosity mapping was performed using rare SNPs and INDELs (allele frequency in 1000Genome < 0.001) identified in each 1Mbp window of the genome from four affected and two unaffected individuals (57). Segregation analysis and screening of ethnicity-matched controls were performed by Sanger‘s di-deoxynucleotide sequencing (55).
Expression of IFT43 transcript
Eyes from 2-month-old wild-type C57BL/6 mice were isolated and dissected to collect different ocular tissues. Isolation of RNA was performed by Qiagen RNeasy® Mini Kit (Cat No./ID: 74104) following standard protocol. Reverse transcription and calculation of Ift43 expression relative to the housekeeping genes Gapdh and Actb were performed as described previously (58).
Immunohistochemistry
Cryosections of 6-month-old wild-type C57BL/6 mouse eye and a 57-year-old normal human donor eye were used to perform immunohistochemistry as described previously (59). Rabbit polyclonal anti-IFT43 antibody (1: 100) (Abgent-AP5370c), Anti mouse Rhodopsin (1: 200) (ab3267, Abcam, Cambridge, MA), Anti goat polyclonal OPN1SW (1: 200) (Santa Cruz Biotechnology, Dallas, Texas), AlexaFluor-647-conjugated donkey anti-rabbit secondary antibody (1: 3000) (Invitrogen, Carlsbad, CA, USA), AlexaFluor-488-conjugated donkey anti-mouse secondary antibody (1: 2000) (Invitrogen, Carlsbad, CA, USA) and AlexaFluor-555-conjugated donkey anti-goat secondary antibody (1: 3000) (Invitrogen, Carlsbad, CA, USA) were utilized for staining. Images were captured using Nikon confocal microscope system (A1R+ STORM, Nikon; Melville, NY 11747, USA) and processed using Adobe Photoshop CS6.
Constructs design for mammalian cell transfections
The image clone containing full-length human IFT43 (hIFT43 ENST00000238628.10) cDNA [Clone MGC: 16028 (IMAGE: 3608220)] in the pOTB7 plasmid was purchased from PlasmID Repository at Harvard Medical School (Boston, MA 02115). The sequence of the clone was verified using 5’- GAGATGGAGGATTTGCTCGAC -3’ forward primer and 5’- CAGGTGTGCCTGGCCTGCC -3’ reverse primer. The point mutation (p.Glu34Lys) was introduced by site-directed mutagenesis using primers 5’- GGAGTCAGCGCAGGCCAAGAATCACCTCAATGGCAAGAATTCC -3’ and 5’- GGAATTCTTGCCATTGAGGTGATTCTTGGCCTGCGCTGACTCC -3’. The PCR product was purified using Zymoclean™ Gel DNA Recovery Kit (Zymogen Research, Irvine, CA, USA) and ligated using Gibson cloning (Gibson Assembly Cloning Kit, NEB). Subsequently, the wt-IFT43 and Glu34Lys-IFT43 constructs were generated in pDONRTMP1-P4 and pDONRTMP3-P2 vector using the Multisite Gateway Cloning Kit (Life Technologies, Carlsbad, CA, USA). The wt-IFT43 and Glu34Lys-IFT43 constructs were tagged with polyhistidine at the N-terminus.
Subcellular localization and expression
mIMCD3 cells and MDCK cells were transfected with recombinant expression vectors containing wild type or mutant inserts using Neon® Transfection System (Life Technologies, Carlsbad, CA, USA) as described earlier (30). After 24 h of transfection, cells were collected for further analysis. Immunofluorescence analysis of transfected cells was performed as described earlier (30). Anti-6X His tag® antibody - ChIP Grade (ab9108) antibody (1: 200) (Cambridge, MA 02139-1517), rabbit polyclonal anti-IFT88 antibodies (13967-1-AP) (1: 200) (Proteintech Group, Inc, Rosemont, IL 60018, USA), mouse monoclonal acetylated α-tubulin antibody (sc-23950) (1: 200) (Santa Cruz Biotechnologies, Dallas, TX 75220, USA), donkey-anti-mouse Alexa-Fluor-555 (1: 3000) (Invitrogen, Carlsbad, CA, USA) and donkey anti-rabbit secondary antibody Alexa-Fluor-488 (1: 2000) (Invitrogen, Carlsbad, CA, USA) were used for immunostaining. Images were captured using Nikon confocal microscope system (A1R STORM, Melville, NY, USA). The intracellular localization of wt-IFT43 and Glu34Lys-IFT43 proteins were compared relative to the ciliary marker acetylated-α-tubulin. Image J64 software was used for measuring the length of the cilia in at least 50 transfected cells of each type.
Analysis of wild type and mutant IFT43 protein in transfected cells
Expression of wild type and mutant IFT43 in transiently transfected mIMCD3 cells was evaluated by western blot analysis using anti-6X His tag® antibody - ChIP Grade (ab9108) antibody (1: 2000) (Cambridge, MA 02139-1517) and chicken anti-rabbit IgG-HRP (1: 3000) (sc-2963). After stripping, the membrane was reprobed with monoclonal anti-b-Actin, Clone AC-74 primary antibody (1: 2000) (Sigma Aldrich) and bovine anti-mouse IgG-HRP (1: 3000) (sc-2963). The images of the immunoblots were captured using MyECL gel Imager (Thermo Fisher Scientific) and processed using Adobe Photoshop CS6.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
We would like to thank all the individuals for participating in this research study. We are also grateful to Carlo Rivolta, Department of Medical Genetics, University of Lausanne, Lausanne, Switzerland and Fowzan Alkuray, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia for screening the database of variants observed in their study cohorts for sequence alterations in IFT43.
Conflict of Interest statement. None declared.
Funding
The Foundation Fighting Blindness, Research to Prevent Blindness (unrestricted grant to UCSD, UCSF and the Nelson Trust Award for Retinitis Pigmentosa), Bright Focus Foundation, National Eye Institute Grants (NIH-EY21237, P30-EY22589, P30 2P30CA023100).
Web References
1. Ensemble Genome Browser: http://uswest.ensembl.org/index.html
2. RetNet: http://www.sph.uth.tmc.edu/Retnet/
3. Gene Card: http://www.genecards.org
4. UCSC Genome Browser: http://genome.ucsc.edu/cgi-bin/hgGateway
5. UniGene: https://www.ncbi.nlm.nih.gov/unigene
References
- 1. Heckenlively J.R. (1988) Retinitis Pigmentosa. J.B. Lippincott Company, Philadelphia. [Google Scholar]
- 2. Hartong D.T., Berson E.L., Dryja T.P. (2006) Retinitis pigmentosa. Lancet, 368, 1795–1809. [DOI] [PubMed] [Google Scholar]
- 3. Da Pozzo P., Cardaioli E., Malfatti E., Gallus G.N., Malandrini A., Gaudiano C., Berti G., Invernizzi F., Zeviani M., Federico A. (2009) A novel mutation in the mitochondrial tRNA(Pro) gene associated with late-onset ataxia, retinitis pigmentosa, deafness, leukoencephalopathy and complex I deficiency. Eur. J. Hum. Genet., 17, 1092–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fahim A.T., Daiger S.P., Weleber R.G. (1993) Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Fong C.T., Mefford H.C., Smith R.J.H., Stephens K. (eds.), Nonsyndromic Retinitis Pigmentosa Overview. GeneReviews(R), University of Washington, Seattle (WA), PMID: 20301590, in press. [PubMed] [Google Scholar]
- 5. Avila-Fernandez A., Cantalapiedra D., Aller E., Vallespin E., Aguirre-Lamban J., Blanco-Kelly F., Corton M., Riveiro-Alvarez R., Allikmets R., Trujillo-Tiebas M.J.. et al. (2010) Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Mol. Vis., 16, 2550–2558. [PMC free article] [PubMed] [Google Scholar]
- 6. Farber D.B., Heckenlively J.R., Sparkes R.S., Bateman J.B. (1991) Molecular genetics of retinitis pigmentosa. West. J. Med., 155, 388–399. [PMC free article] [PubMed] [Google Scholar]
- 7. Espinos C., Perez-Garrigues H., Beneyto M., Vilela C., Rodrigo O., Najera C. (1999) [Syndromic hereditary deafness. Usher's syndrome. Oto-neurologic and genetic factors]. An. Otorrinolaringol. Ibero. Am., 26, 83–95. [PubMed] [Google Scholar]
- 8. Blaydon D.C., Mueller R.F., Hutchin T.P., Leroy B.P., Bhattacharya S.S., Bird A.C., Malcolm S., Bitner-Glindzicz M. (2003) The contribution of USH1C mutations to syndromic and non-syndromic deafness in the UK. Clin. Genet., 63, 303–307. [DOI] [PubMed] [Google Scholar]
- 9. Li J.B., Gerdes J.M., Haycraft C.J., Fan Y., Teslovich T.M., May-Simera H., Li H., Blacque O.E., Li L., Leitch C.C.. et al. (2004) Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell, 117, 541–552. [DOI] [PubMed] [Google Scholar]
- 10. Young T.L., Penney L., Woods M.O., Parfrey P.S., Green J.S., Hefferton D., Davidson W.S. (1999) A fifth locus for Bardet-Biedl syndrome maps to chromosome 2q31. Am. J. Hum. Genet., 64, 900–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbo M., Filhol E., Bole-Feysot C.. et al. (2011) Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet., 89, 634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chaki M., Airik R., Ghosh A.K., Giles R.H., Chen R., Slaats G.G., Wang H., Hurd T.W., Zhou W., Cluckey A.. et al. (2012) Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell, 150, 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adams N.A., Awadein A., Toma H.S. (2007) The retinal ciliopathies. Ophthalmic Genet., 28, 113–125. [DOI] [PubMed] [Google Scholar]
- 14. Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C.. et al. (2007) The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet., 39, 875–881. [DOI] [PubMed] [Google Scholar]
- 15. Badano J.L., Ansley S.J., Leitch C.C., Lewis R.A., Lupski J.R., Katsanis N. (2003) Identification of a novel Bardet-Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2. Am. J. Hum. Genet., 72, 650–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Badano J.L., Kim J.C., Hoskins B.E., Lewis R.A., Ansley S.J., Cutler D.J., Castellan C., Beales P.L., Leroux M.R., Katsanis N. (2003) Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet., 12, 1651–1659. [DOI] [PubMed] [Google Scholar]
- 17. Eke T., Woodruff G., Young I.D. (1996) A new oculorenal syndrome: retinal dystrophy and tubulointerstitial nephropathy in cranioectodermal dysplasia. Br. J. Ophthalmol., 80, 490–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Savill G.A., Young I.D., Cunningham R.J., Ansell I.D., Evans J.H. (1997) Chronic tubulo-interstitial nephropathy in children with cranioectodermal dysplasia. Pediatr. Nephrol., 11, 215–217. [DOI] [PubMed] [Google Scholar]
- 19. Young I.D. (1989) Cranioectodermal dysplasia (Sensenbrenner's syndrome). J. Med. Genet., 26, 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duran I., Taylor S.P., Zhang W., Martin J., Qureshi F., Jacques S.M., Wallerstein R., Lachman R.S., Nickerson D.A., Bamshad M.. et al. (2017) Mutations in IFT-A satellite core component genes IFT43 and IFT121 produce short rib polydactyly syndrome with distinctive campomelia. Cilia, 6, 7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arts H.H., Bongers E.M., Mans D.A., van Beersum S.E., Oud M.M., Bolat E., Spruijt L., Cornelissen E.A., Schuurs-Hoeijmakers J.H., de Leeuw N.. et al. (2011) C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J. Med. Genet., 48, 390–395. [DOI] [PubMed] [Google Scholar]
- 22. Maranhao B., Biswas P., Duncan J.L., Branham K.E., Silva G.A., Naeem M.A., Khan S.N., Riazuddin S., Hejtmancik J.F., Heckenlively J.R.. et al. (2014) exomeSuite: Whole exome sequence variant filtering tool for rapid identification of putative disease causing SNVs/indels. Genomics, 103, 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B.. et al. (2016) Analysis of protein-coding genetic variation in 60, 706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adzhubei I., Jordan D.M., Sunyaev S.R. (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet., Chapter 7, Unit7, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ng P.C., Henikoff S. (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res., 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Broekhuis J.R., Verhey K.J., Jansen G., Stieger K. (2014) Regulation of cilium length and intraflagellar transport by the RCK-kinases ICK and MOK in renal epithelial cells. PloS One, 9, e108470.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robert A., Margall-Ducos G., Guidotti J.E., Bregerie O., Celati C., Brechot C., Desdouets C. (2007) The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. J. Cell Sci., 120, 628–637. [DOI] [PubMed] [Google Scholar]
- 28. Johnston J.A., Ward C.L., Kopito R.R. (1998) Aggresomes: a cellular response to misfolded proteins. J. Cell Biol., 143, 1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Markossian K.A., Kurganov B.I. (2004) Protein folding, misfolding, and aggregation. Formation of inclusion bodies and aggresomes. Biochemistry (Mosc), 69, 971–984. [DOI] [PubMed] [Google Scholar]
- 30. Vasireddy V., Vijayasarathy C., Huang J., Wang X.F., Jablonski M.M., Petty H.R., Sieving P.A., Ayyagari R. (2005) Stargardt-like macular dystrophy protein ELOVL4 exerts a dominant negative effect by recruiting wild-type protein into aggresomes. Mol. Vis., 11, 665–676. [PubMed] [Google Scholar]
- 31. Weleber R.G., Michaelides M., Trzupek K.M., Stover N.B., Stone E.M. (2011) The phenotype of Severe Early Childhood Onset Retinal Dystrophy (SECORD) from mutation of RPE65 and differentiation from Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci., 52, 292–302. [DOI] [PubMed] [Google Scholar]
- 32. Mackay D.S., Ocaka L.A., Borman A.D., Sergouniotis P.I., Henderson R.H., Moradi P., Robson A.G., Thompson D.A., Webster A.R., Moore A.T. (2011) Screening of SPATA7 in patients with Leber congenital amaurosis and severe childhood-onset retinal dystrophy reveals disease-causing mutations. Invest. Ophthalmol. Vis. Sci., 52, 3032–3038. [DOI] [PubMed] [Google Scholar]
- 33. Lewis C.A., Batlle I.R., Batlle K.G., Banerjee P., Cideciyan A.V., Huang J., Aleman T.S., Huang Y., Ott J., Gilliam T.C. (1999) Tubby-like protein 1 homozygous splice-site mutation causes early-onset severe retinal degeneration. Invest. Ophthalmol. Vis. Sci., 40, 2106–2114. [PubMed] [Google Scholar]
- 34. Foxman S.G., Heckenlively J.R., Bateman J.B., Wirtschafter J.D. (1985) Classification of congenital and early onset retinitis pigmentosa. Arch. Ophthalmol., 103, 1502–1506. [DOI] [PubMed] [Google Scholar]
- 35. McCulloch D.L., Marmor M.F., Brigell M.G., Hamilton R., Holder G.E., Tzekov R., Bach M. (2015) ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol., 130, 1–12. [DOI] [PubMed] [Google Scholar]
- 36. Falk M.J., Zhang Q., Nakamaru-Ogiso E., Kannabiran C., Fonseca-Kelly Z., Chakarova C., Audo I., Mackay D.S., Zeitz C., Borman A.D.. et al. (2012) NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet., 44, 1040–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perrault I., Hanein S., Zanlonghi X., Serre V., Nicouleau M., Defoort-Delhemmes S., Delphin N., Fares-Taie L., Gerber S., Xerri O.. et al. (2012) Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat. Genet., 44, 975–977. [DOI] [PubMed] [Google Scholar]
- 38. Dharmaraj S., Leroy B.P., Sohocki M.M., Koenekoop R.K., Perrault I., Anwar K., Khaliq S., Devi R.S., Birch D.G., De Pool E.. et al. (2004) The phenotype of Leber congenital amaurosis in patients with AIPL1 mutations. Arch. Ophthalmol., 122, 1029–1037. [DOI] [PubMed] [Google Scholar]
- 39. Mohamed M.D., Topping N.C., Jafri H., Raashed Y., McKibbin M.A., Inglehearn C.F. (2003) Progression of phenotype in Leber's congenital amaurosis with a mutation at the LCA5 locus. Br. J. Ophthalmol., 87, 473–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stokman M.F., Oud M.M., van Binsbergen E., Slaats G.G., Nicolaou N., Renkema K.Y., Nijman I.J., Roepman R., Giles R.H., Arts H.H.. et al. (2016) De novo 14q24.2q24.3 microdeletion including IFT43 is associated with intellectual disability, skeletal anomalies, cardiac anomalies, and myopia. Am. J. Med. Genet. A, 170, 1566–1569. [DOI] [PubMed] [Google Scholar]
- 41. Fu W., Wang L., Kim S., Li J., Dynlacht B.D. (2016) Role for the IFT-A complex in selective transport to the primary cilium. Cell Reports, 17, 1505–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Girisha K.M., Shukla A., Trujillano D., Bhavani G.S., Hebbar M., Kadavigere R., Rolfs A. (2016) A homozygous nonsense variant in IFT52 is associated with a human skeletal ciliopathy. Clin. Genet., 90, 536–539. [DOI] [PubMed] [Google Scholar]
- 43. Liem K.F. Jr., Ashe A., He M., Satir P., Moran J., Beier D., Wicking C., Anderson K.V. (2012) The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J. Cell Biol., 197, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alazami A.M., Seidahmed M.Z., Alzahrani F., Mohammed A.O., Alkuraya F.S. (2014) Novel IFT122 mutation associated with impaired ciliogenesis and cranioectodermal dysplasia. Mol. Genet. Genomic Med., 2, 103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sedmak T., Wolfrum U. (2010) Intraflagellar transport molecules in ciliary and nonciliary cells of the retina. J. Cell Biol., 189, 171–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Behal R.H., Miller M.S., Qin H., Lucker B.F., Jones A., Cole D.G. (2012) Subunit interactions and organization of the Chlamydomonas reinhardtii intraflagellar transport complex A proteins. J. Biol. Chem., 287, 11689–11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roepman R., Wolfrum U. (2007) Protein networks and complexes in photoreceptor cilia. Subcell. Biochem., 43, 209–235. [DOI] [PubMed] [Google Scholar]
- 48. Bhowmick R., Li M., Sun J., Baker S.A., Insinna C., Besharse J.C. (2009) Photoreceptor IFT complexes containing chaperones, guanylyl cyclase 1 and rhodopsin. Traffic, 10, 648–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horst C.J., Johnson L.V., Besharse J.C. (1990) Transmembrane assemblage of the photoreceptor connecting cilium and motile cilium transition zone contain a common immunologic epitope. Cell Motil. Cytoskeleton, 17, 329–344. [DOI] [PubMed] [Google Scholar]
- 50. Wang J., Deretic D. (2014) Molecular complexes that direct rhodopsin transport to primary cilia. Prog. Retin. Eye Res., 38, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu B., Zhu X., Wang L., Liang Y., Feng Q., Pan J. (2017) Functional exploration of the IFT-A complex in intraflagellar transport and ciliogenesis. PLoS Genet., 13, e1006627.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bujakowska K.M., Zhang Q., Siemiatkowska A.M., Liu Q., Place E., Falk M.J., Consugar M., Lancelot M.E., Antonio A., Lonjou C.. et al. (2015) Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum. Mol. Genet., 24, 230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aldahmesh M.A., Li Y., Alhashem A., Anazi S., Alkuraya H., Hashem M., Awaji A.A., Sogaty S., Alkharashi A., Alzahrani S.. et al. (2014) IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Hum. Mol. Genet., 23, 3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Perrault I., Saunier S., Hanein S., Filhol E., Bizet A., Collins F., Salih M., Silva E., Baudouin V., Oud M.. et al. (2012) Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet., 1, O28. 864870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Duncan J.L., Biswas P., Kozak I., Navani M., Syed R., Soudry S., Menghini M., Caruso R.C., Jeffrey B.G., Heckenlively J.R.. et al. (2014) Ocular phenotype of a family with FAM161A-associated retinal degeneration. Ophthalmic Genet., 37, 44–52, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Duncan J.L., Roorda A., Navani M., Vishweswaraiah S., Syed R., Soudry S., Ratnam K., Gudiseva H.V., Lee P., Gaasterland T.. et al. (2012) Identification of a novel mutation in the CDHR1 gene in a family with recessive retinal degeneration. Arch. Ophthalmol., 130, 1301–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M.. et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome. Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mandal M.N., Vasireddy V., Jablonski M.M., Wang X., Heckenlively J.R., Hughes B.A., Reddy G.B., Ayyagari R. (2006) Spatial and temporal expression of MFRP and its interaction with CTRP5. Invest. Ophthalmol. Vis. Sci., 47, 5514–5521. [DOI] [PubMed] [Google Scholar]
- 59. Chavali V.R., Khan N.W., Cukras C.A., Bartsch D.U., Jablonski M.M., Ayyagari R. (2011) A CTRP5 gene S163R mutation knock-in mouse model for late-onset retinal degeneration. Hum. Mol. Genet., 20, 2000–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.