

Abstract
Mutations in peripherin 2 (PRPH2), also known as retinal degeneration slow/RDS, lead to various retinal degenerations including retinitis pigmentosa (RP) and macular/pattern dystrophy (MD/PD). PRPH2-associated disease is often characterized by a phenotypic variability even within families carrying the same mutation, raising interest in potential modifiers. PRPH2 oligomerizes with its homologue rod outer segment (OS) membrane protein 1 (ROM1), and non-pathogenic PRPH2/ROM1 mutations, when present together, lead to digenic RP. We asked whether ROM1 could modify the phenotype of a PRPH2 mutation associated with a high degree of intrafamilial phenotypic heterogeneity: Y141C. In vitro, Y141C-Prph2 showed signs of retention in the endoplasmic reticulum (ER), however co-expression with Rom1 rescued this phenotype. In the heterozygous Y141C knockin mouse model (Prph2Y/+), Y141C-Prph2 and Rom1 formed abnormal complexes but were present at normal levels. Abnormal complexes were eliminated in the absence of Rom1 (Prph2Y/+/Rom1-/-) and total Prph2 levels were reduced to those found in the haploinsufficient Prph2+/- RP model. The biochemical changes had functional and structural consequences; while Prph2Y/+ animals exhibited a cone-rod electroretinogram defect, Prph2Y/+/Rom1-/- animals displayed a rod-dominant phenotype and OSs similar to those seen in the Prph2+/-. These data show that ablation of Rom1 results in the conversion of an MD/PD phenotype characterized by cone functional defects and the formation of abnormal Prph2/Rom1 complexes to an RP phenotype characterized by rod-dominant functional defects and reductions in total Prph2 protein. Thus one method by which ROM1 may act as a disease modifier is by contributing to the large variability in PRPH2-associated disease phenotypes.
Introduction
Peripherin 2 (PRPH2), also known as retinal degeneration slow (RDS), is a photoreceptor-specific tetraspanin protein necessary for the formation and function of both rod and cone outer segments (OSs) (1,2). Over 151 mutations in the PRPH2 gene have been identified (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=Prph2; date last accessed December 01, 2016) an extensive number of which have been confirmed to be pathogenic (see (2)). Mutations in PRPH2 are largely autosomal dominant and lead to severe retinal degenerations that range from retinitis pigmentosa (RP) to a variety of macular degenerations (MD), including pattern dystrophy (PD), and can be associated with secondary defects that appear in neighbouring tissues such as the choroid and retinal pigment epithelium (2). In keeping with the severity of diseases associated with PRPH2 mutations, mice lacking Prph2 (Prph2-/-) completely fail to form OSs (1,3), and Prph2 haploinsufficiency (Prph2+/-) also leads to severe structural and functional abnormalities (4).
PRPH2 and its homologue rod outer segment membrane protein 1 (ROM1) form non-covalently linked homo- and hetero-tetramers. These complexes further assemble into covalently linked larger complexes held together by C150 in Prph2 and C153 in Rom1 (5–7). In contrast to PRPH2, no pathogenic mutations in ROM1 have been confirmed in patients. Early reports suggested that a 1-base pair insertion in ROM1 at leucine 114 which creates a frameshift and a putative null allele might be pathogenic (8,9), but it was not found in families large enough to permit co-segregation analyses (10). Combined with data showing that elimination of Rom1 (Rom1-/-) in mice leads to only minor structural and functional defects in photoreceptors (11,12), this has led to the hypothesis that Rom1 plays an ancillary role in the function of Prph2. However, digenic RP has been reported in which double heterozygous patients carrying the L185P mutation in PRPH2 and one of three different mutations in ROM1 (L114 insertion, a one bp insertion at G80, or a G113E substitution) present with disease (10,13,14). This digenic RP phenotype was confirmed in L185P-Prph2 transgenic mice on the Prph2+/-/Rom1+/- genetic background (15). Though the mechanisms of PRPH2/ROM1-associated digenic RP are not completely understood, studies from animals suggest that the mechanism is likely due to abnormalities in Prph2/Rom1 subunit assembly (16) and/or reductions in the total amount of Prph2/Rom1 (15).
PRPH2-associated disease displays an extremely high degree of phenotypic variability both among distinct mutations and among patients carrying the same mutation (2). Research has focused on understanding how different mutations lead to different disease phenotypes (17–19), but as of yet, little is known about what contributes to intrafamilial variability among patients carrying the same mutation. Though many retinal disease genes could hypothetically act as modifiers for PRPH2-associated phenotypes, ROM1 is the most widely explored thus far due both to the known existence of PRPH2/ROM1 digenic RP and to the role of ROM1 as a PRPH2 interacting partner. ROM1 and ABCA4 have been implicated as modifiers of the PRPH2-associated MD phenotype; for example, disease pathology in patients carrying the R172W MD mutation in PRPH2 was significantly worse in the presence of the R229H mutation in ROM1 (20).
Our goal was to verify whether variations in ROM1 could contribute to the phenotypic variability seen in patients carrying the Y141C mutation in PRPH2. Individuals within the same family exhibit retinal phenotypes ranging from pure RP to MD/PD (21). The Y141C mutation occurs in the large second intradiscal loop (D2) of Prph2, a region known to be required for the interaction with Rom1 (22). Proper folding of the D2 loop and assembly of Prph2 complexes is essential for the formation of three intramolecular disulfide bonds and a single intermolecular disulfide bond which are mediated by seven D2 loop cysteines. Cysteine dynamics in the D2 loop are critical for Prph2 function; for example, the C214S mutation creates an unstable, misfolded Prph2 which cannot exit the ER or interact with Rom1 (23,24). The C214S mutation results in a clear haploinsufficiency RP phenotype in patients and mice (23–25). This RP phenotype is similar to that seen in the well-characterized Prph2+/- model of RP (4,26), in which haploinsufficiency results in decreased Prph2 protein levels and a rod-dominant phenotype, but proper assembly of Prph2/Rom1 complexes (4,26). In contrast, studies have suggested that PRPH2-associated MD mutations largely lead to stable mutant proteins, abnormalities in complex formation, and defects in either cone or cone-rod function (19,27,28). We have recently reported that the Y141C mutation causes a cone-rod phenotype, formation of a stable protein product, and abnormalities in Prph2/Rom1 complex assembly. This finding is consistent with previous MD/PD models (19), yet Y141C patients exhibit phenotypes ranging from MD/PD to RP. Here we find that eliminating Rom1 converts the Y141C phenotype from a cone-rod dystrophy to a clear haploinsufficiency RP. These striking results support the hypothesis that ROM1 could be a contributor to intrafamilial phenotypic variability seen in patients with PRPH2 mutations.
Results
Rom1 rescues the trafficking defect of Y141C-Prph2 in vitro
We first evaluated the role of Rom1 in modifying Prph2-associated properties in vitro. There are no cultured cells that resemble photoreceptors, but COS-1 and COS-7 cells have been widely used for evaluating various properties of wild-type (WT) and mutant forms of Prph2 (5,6,23). Studies have shown that Prph2 and Rom1 properly interact to form tetramers and larger complexes when expressed in these cells (5,6,23). COS-7 cells were transfected with pcDNA3.1 plasmid carrying WT or mutant forms of Prph2. In addition to Y141C-Prph2, we included two previously characterized mutants (C150S and C214S) known to exhibit defects in complex assembly or trafficking. C150S-Prph2 lacks the cysteine necessary for assembly of covalently linked Prph2/Rom1 higher-order oligomers but retains the ability to form non-covalently linked Prph2 homo- and Prph2/Rom1 hetero-tetramers (5,29). In vivo and in vitro expression of C214S-Prph2 showed that the mutant protein is misfolded; does not bind Rom1; and cannot exit the ER (23,24). Since cysteine dynamics in the D2 loop are critical for Prph2 function, to help determine whether Y141C-associated phenotypes had more to do with the addition of extra cysteine or the loss of a tyrosine, we also included Y141S and Y141F mutants.
To determine whether the mutant proteins could exit the ER, transfected COS cells were co-labelled for Prph2 (red, Fig. 1A) and the ER marker calreticulin (green, Fig. 1A, and an untransfected cell in Supplementary Material, Fig. S1A) or a second ER marker PDI (protein disulfide isomerase, Supplementary Material, Fig. S2A). Y141C-Prph2 largely co-localized with the ER marker (Fig 1A, yellow, arrowheads), as did C214S-Prph2 and Y141S-Prph2 suggesting these three mutations resulted in ER retention. In contrast, WT, C150S-Prph2, and Y141F-Prph2 did not co-localize with the ER marker, suggesting they were able to traffic out of the ER to distinct post ER compartments (Fig. 1A, arrows). Additional examples of these cells can be seen in Supplementary Material, Fig. S1B.
Figure 1.

Y141C-Prph2 shows signs of ER retention. (A,B) COS-7 cells were transiently transfected with pcDNA 3.1 containing WT-, Y141C-, C150S-, C214S-, Y141S-, or Y141F-Prph2. (A) Cells were fixed and labelled using antibodies against Prph2 (red) and calreticulin (green, to visualize the ER), nuclei are counterstained with DAPI (grey). Arrows show Prph2 on post-ER membranes (red), arrowheads indicate some of the regions of co-localization between Prph2 and the ER marker (yellow/orange). Scale bar: 10 µm. (B) Protein extracts were prepared from transiently transfected COS-7 cells and analysed by non-reducing SDS-PAGE/western blot using an antibody specific to Prph2 (RDS-CT). M-monomer, D-dimer, S-streaking consistent with non-specific aggregation.
Prph2 mutants were next evaluated by non-reducing western blot. Non-covalently linked Prph2 complexes run on a non-reducing SDS-PAGE/western blot as monomers at ∼35 kDa while the covalently linked complexes run as dimers at ∼65 kDa. In lysates from transfected COS cells, Y141C-Prph2, Y141S-Prph2 and C214S-Prph2 exhibited both monomer and dimer bands (Fig. 1B, M/D) as well as a consistent streaking pattern (Fig. 1B) which could be due to ER retention. Consistent with this hypothesis, WT, C150S-Prph2, and Y141F-Prph2 did not exhibit this streaking, rather were present as monomers/dimers for WT and Y141F-Prph2 and only monomers for C150S-Prph2.
Rom1 was then co-expressed with Prph2 mutants and cells were triple labelled for Prph2 (red), Rom1 (blue) and calreticulin (green) (Fig. 2A, and additional cells shown in Supplementary Material, Fig. S1C, and with the second ER marker PDI in Supplementary Material, Fig. S2B). As expected based on results from Fig. 1A, in WT, C150S-Prph2, and Y141F-Prph2 transfected cells, Prph2 and Rom1 co-localized on post-ER membranes (Fig. 2A, purple, and arrows), indicating that Prph2 trafficked to the same destination as Rom1 and was not retained in the ER. In contrast, when C214S-Prph2 and Rom1 were co-expressed, Rom1 trafficked out of the ER (Fig 2a, blue) while C214S-Prph2 remained co-localized with the ER marker (Fig. 2A, yellow, and arrowheads). Interestingly, when Y141C-Prph2 and Y141S-Prph2 were co-expressed with Rom1, although some were still co-localized with the ER marker (arrowheads), the majority of Y141C-Prph2 and Y141S-Prph2 adopted a WT distribution in which Prph2 and Rom1 co-localized on post-ER membranes (Fig. 2A, purple, and arrows). This observation suggests that co-expression with Rom1 rescued the trafficking defect associated with the Y141C mutation but not that associated with the C214S mutation.
Figure 2.
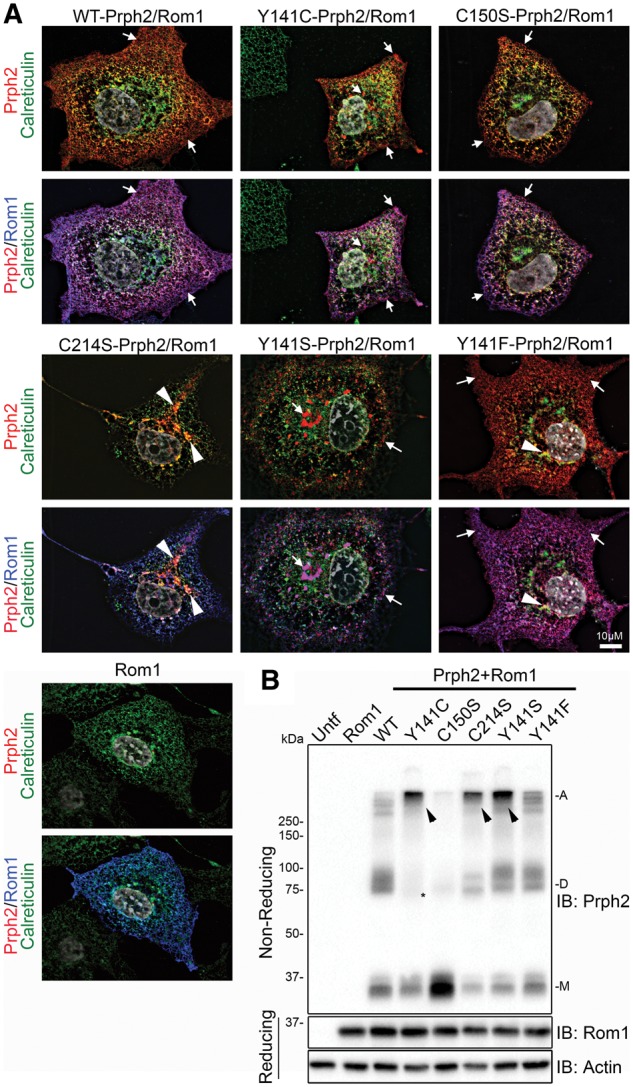
Co-expression of Rom1 with Y141C-Prph2 corrects trafficking defects. (A,B) COS-7 cells were transiently transfected with the indicated WT or mutant Prph2 vectors and Rom1, or Rom-1 alone. (A) Cells were stained for Prph2 (red), Rom1 (blue) and calreticulin (green), with nuclei counterstained with DAPI (grey). Top images show Prph2 and calreticulin only, bottom panels have Rom1 overlaid. Arrows show Prph2 on post-ER membranes (red), arrowheadss indicate some of the regions of co-localization between Prph2 and the ER marker (yellow/orange). Scale bar: 10 µm. (B) Non-reducing SDS-PAGE/western blot analysis of co-transfected cell lysates probed with antibodies to Prph2, Rom1 and actin (as a loading control). Tight, high molecular weight bands (A) are seen in some groups (arrowheads), in addition to the normal monomer (M) and dimer (D) bands. * indicates reduced Prph2 dimers in cultures co-transfected with Rom1 and Y141C-Prph2.
Co-expression with Rom1 also had biochemical consequences for the Y141C and C214S mutations (Fig. 2B). Again, WT Prph2 was present as a primarily monomer and dimer, while C150S was almost exclusively monomer. However, Y141C-Prph2, C214S-Prph2, Y141S-Prph2, and to a lesser extent Y141F-Prph2 exhibited a tight Prph2 band at the very top of the gel instead of the streaking we observed when these mutants were expressed without Rom1 (Fig. 2B, arrowheads). In the case of Y141C, these large abnormal complexes were formed at the expense of the dimer band (Fig. 2B), and replicated previous observations from the Y141C knockin retina (19). Combined these in vitro data suggest that in the absence of Rom1, Y141C protein is not properly processed and trafficked.
Rom1 is essential for Y141C abnormal complex formation in vivo
The in vitro results suggested that the presence/absence of Rom1 affected the behaviour of Prph2 mutants, so to see whether this outcome occurred in in vivo, the Y141C knockin line (19) (Prph2Y/Y and Prph2Y/+) was crossed with the Rom1-/- line (11). Prph2/Rom1 complex assembly was assessed in retinal extracts collected at postnatal day (P) 30 on non-reducing (Fig. 3A, left), and reducing (Fig. 3A, right) blots. Prph2Y/Y and Prph2Y/+ retinas exhibited a tight high molecular weight band at the top of the blot (Fig. 3A, arrows, top left) similar to what was observed in Fig. 2B. These abnormal complexes were likely disulfide-linked since all were reduced to monomers under reducing conditions (Fig. 3A, right), and they contained Rom1 (Fig. 3A, arrows, bottom left). Critically, when Rom1 was removed (Prph2Y/+/Rom1-/- and Prph2Y/Y/Rom1-/-), these high molecular weight complexes were completely abolished (Fig. 3A, arrowheads).
Figure 3.

Elimination of Rom1 prevents formation of abnormal high molecular weight complexes and leads to inner segment accumulation of Y141C-Prph2. (A) Retinal extracts from the indicated genotypes were separated by SDS-PAGE/western blot. Arrows indicate abnormal disulfide linked complexes. Without Rom1 (Rom1-/-) no abnormal complexes are observed (arrowheads). (B) Prph2 levels were measured densitometrically from reducing blots and normalized to actin. To control for cross-blot variability, values for each genotype are plotted as a percent of WT. *P < 0.05, **P < 0.01, ***P < 0.001 by 1-way ANOVA with Bonferroni’s post-hoc comparison. N = 5–6 retinas/genotype.
Prph2 protein levels were quantified densitometrically from western blots (Fig. 3B). Prph2Y/+ retinas had WT levels of Prph2 protein (Fig. 3A and B), suggesting that Y141C-Prph2 protein was stable. Prph2 protein levels in the Rom1-/- were also normal, suggesting that Rom1 was not required for WT Prph2 stability (Fig. 3B). However, the Prph2Y/+/Rom1-/- showed a statistically significant (***P < 0.001 compared to WT, Rom1-/-, or Prph2Y/+) 42% drop in the total amount of Prph2. This reduction brought Prph2 levels in Prph2Y/+/Rom1-/- retinas down to levels indistinguishable from the Prph2+/- retinas (Fig. 3B). Our Prph2 antibodies do not differentiate between WT and Y141C-Prph2, but our results are consistent with the hypothesis that Rom1 positively contributes to the stability of the Y141C protein, and that in the absence of Rom1, Y141C-Prph2 is prone to degradation.
To assess the retinal localization of Y141C-Prph2, we labelled P30 (Fig. 4A) and P120 (Fig. 4B) retinal sections with Prph2 (green). In the WT, Rom1-/-, and Prph2Y/+ retinas, Prph2 was found exclusively in the OS layer. The Prph2Y/Y retina forms only tiny nubs of OS (19) but all remaining Prph2 protein was correctly localized to these OS nubs at P30 (Fig. 4A, arrows). In the P30 Prph2Y/+/Rom1-/- retina, all Prph2 was also restricted to the OS. However, in the Prph2Y/Y/Rom1-/- retina, Prph2 accumulated abnormally in the inner segment (IS) and outer nuclear layer (ONL) (Fig. 4A, arrowheads). At P120, similar to the Prph2Y/Y/Rom1-/- retinas, Prph2Y/Y exhibited some Prph2 mis-localization to the ISs and ONL (Fig. 4B, arrowhead) although some was still detected in the OS (Fig. 4B, arrow). In addition, at P120 a small amount of Prph2 was also detected in the IS in the Prph2Y/+/Rom1-/- (Fig. 4B, arrows), although the majority of Prph2 was still properly localized to the OS. To help visualize the difference between Prph localization in the Prph2Y/Y vs. Prph2Y/Y/Rom1-/-, P120 retinal sections were co-labelled with the IS/axoneme marker acetylated α-tubulin (Fig. 4C, red) and Prph2 (Fig. 4C, green, Prph2Y/+ and Prph2Y/+/Rom1-/-are shown as controls). Even at P120, Y141C-Prph2 in the Prph2Y/Y is clearly detected in small OS structures beyond the IS labeling demarcated in red by acetylated α-tubulin labeling (Fig. 4C, arrows). However, this is not the case in the Prph2Y/Y/Rom1-/- wherein all Prph2 resides in the IS/ONL (Fig. 4C, arrowheads).
Figure 4.

Elimination of Rom1 leads to accumulation of Y141C-Prph2 in the inner segment. Retinal sections collected at P30 (A,D,E) or P120 (B,C) were labelled for Prph2 (green A-E) and acetylated α-tubulin (red, C) or rhodopsin (red, D,E) with nuclei counterstained with DAPI (blue). Arrowheads indicate localization of Prph2 in the IS or ONL, arrows indicated localization of Y141C-Prph2 in the OS. (D,E) Arrows indicate accumulation of rhodopsin but not Prph2 in the IS tips. Scale bars: 25 µm. OS: outer segment, IS: inner segment, ONL: outer nuclear layer, INL: inner nuclear layer.
Accumulation of OS proteins in the IS/ONL can arise for at least two different reasons: 1) protein mis-folding/ER retention and 2) lack of OSs, i.e. when there is no significant OS structure to reside in, OS proteins accumulate in other cellular compartments (30). Murine ISs are quite small, and thus co-localization studies with an ER marker do not give as clear an outcome as in tissue culture studies. Thus to help differentiate between these two possibilities, we took advantage of the fact that in the absence of OSs in the Prph2-/-, rhodopsin falls in the second category. Rhodopsin is found in the distal tips of the connecting cilia in addition to accumulating in the IS/ONL (Fig. 4D, arrows) suggesting that it can traffic, but due to the absence of OS structures, it also accumulates in the cell body. When we labelled Prph2Y/Y/Rom1-/- retinas for rhodopsin (red) and Prph2 (green), rhodopsin exhibited the same pattern as in the Prph2-/-, namely enrichment at the distal tips of the ISs (Fig. 4E, left, arrows), with additional accumulation in the ONL. In contrast, Y141C-Prph2 in the Prph2Y/Y/Rom1-/- retina did not exhibit this pattern. Rather Y141C-Prph2 was evenly distributed throughout the ISs and did not accumulate in the IS tips, suggesting it did not traffic to the connecting cilium. These data support the hypothesis that in the absence of Rom1 or WT Prph2, Y141C-Prph2 does not traffic correctly.
Elimination of Rom1 shifts the pattern dystrophy phenotype of Y141C towards RP
The effect of these biochemical changes on the Y141C knockin clinical phenotypes was next assessed. Electroretinography (ERG) was performed at P30 and (representative traces are shown in Fig. 5A and quantification in Fig. 5B). Patients with Y141C-associated disease carry only one mutant PRPH2 allele (autosomal dominant disease), so we focused evaluation of clinical phenotypes on the heterozygous animals. The Prph2Y/+ retina exhibited a cone-rod phenotype where rod function was improved while cone function was impaired compared to the Prph2+/- RP model (Fig. 5B). Eliminating Rom1 switched this cone-rod phenotype to a pure RP phenotype wherein rod function was severely compromised but cone function was not. In the Prph2Y/+/Rom1-/- animals, maximum scotopic a-wave responses were significantly reduced compared to Prph2Y/+ (as well as WT and Rom1-/-) and were not significantly different from those in the Prph2+/- (Fig. 5B, left). Consistent with this conversion to an RP phenotype, photopic b-wave responses, a measure of cone function, were significantly improved in the Prph2Y/+/Rom1-/- compared to Prph2Y/+ and were actually recovered to levels seen in the WT and Prph2+/- (which have normal photopic ERG at P30) (Fig. 5B, right). ERG values in the Prph2-/-, Prph2Y/Y and Prph2Y/Y/Rom1-/- were extremely low at P30, as expected based on the low levels of Prph2 in these retinas, (Supplementary Material, Fig. S3).
Figure 5.

Lacking Rom1 in the Prph2Y/+ alters the PD functional phenotype to RP. (A) Shown are representative waveforms from full-field ERGs collected at P30 under either scotopic (left) or photopic (right) conditions. (B) Plotted are mean ±SEM scotopic a-wave (left) or photopic b-wave (right) amplitudes at P30. *P < 0.05, **P < 0.01, ***P < 0.001 by 1-way ANOVA with Bonferroni’s post-hoc comparison. N = 5–15 animals/genotype.
Removing Rom1 also had an effect on the fundus flecking phenotype we had previously reported in the Prph2Y/+ at P180 (19). In the Prph2Y/+, the fundus exhibits characteristic whitish yellow punctate flecks. These are widespread across the whole retina and arrows in Fig. 6A highlight a few of the flecks. In the Prph2Y/+/Rom1-/- this phenotype was ameliorated; many retinas showed no flecks at all (Supplementary Material, Fig. S4A); while others showed only a minimal number of these flecks (Fig. 6A). Instead of discrete flecks, Prph2Y/+/Rom1-/- retinas sometimes exhibited a mottled appearance (Supplementary Material, Fig. S4A, arrowheads). Flecking was also observed at P180 in the Prph2Y/Y and to some degree in the Prph2-/-, but once again was eliminated in the Prph2Y/Y/Rom1-/- (Supplementary Material, Fig. S4B).
Figure 6.

Eliminating Rom1 ameliorates yellow flecking found in the Prph2Y/+ and leads to the formation of OS whorls. (A) Animals of the indicated genotypes underwent fundus imaging at P180. Arrows indicate discrete, punctate flecks apparent in Prph2Y/+ retinas. N = 4–6 animals/genotype. (B,C) Eyes at P180 were analysed by LM/EM, arrowheads show abnormalities in the base of Rom1-/- OSs, arrows show whorl OSs in Prph2Y/+/Rom1-/-. Scale bars: 25 µm (B), 2 µm (C).
To evaluate whether these ERG/fundus phenotypes correlated with retinal photoreceptor degeneration, the number of rows of photoreceptor nuclei was evaluated in light micrographs at P180. At this age, the Rom1-/-, Prph2+/-, and Prph2Y/+ exhibited degeneration of the ONL compared to WT, showing ∼5–6 rows of nuclei in the central retina rather than 9–10 seen in the WT. However, this degeneration was worsened in the Prph2Y/+/Rom1-/-, which only had 3–4 rows of photoreceptor nuclei (Fig. 6B). When retinal thin sections were evaluated by EM, Rom1-/- OS ultrastructure was largely normal apart from abnormalities in disc sizing and alignment at the base of the OS consistent with previous report (11) (Fig. 6C, arrowhead), while the Prph2+/- exhibited the expected OS whorls (Fig. 6C). Disc alignment and OS length were significantly improved in the Prph2Y/+ in comparison to the Prph2+/- retina (Fig. 6C, and low-magnification in Supplementary Material, Fig. S5). However, elimination of Rom1 drastically worsened the OS structure in the Prph2Y/+/Rom1-/- vs. Prph2Y/+ or Prph2+/-. OSs in the Prph2Y/+/Rom1-/- had a similar whorl like morphology to those in the Prph2+/- but were smaller and were not as tightly stacked as those in the Prph2Y/+ (Fig. 6C, arrows). In addition, many photoreceptor cells in the Prph2Y/+/Rom1-/- lacked OSs entirely (Supplementary Material, Fig. S5). At P180, significant degeneration was seen in the Prph2-/- retina with ∼3 rows of nuclei remaining in the ONL. However, the degeneration was worse in the Prph2Y/Y and Prph2Y/Y/Rom1-/- with only 1–2 rows of nuclei (Supplementary Material, Fig. S6A). On the EM level, no OSs were seen in either the Prph2Y/Y or Prph2Y/Y/Rom1-/- (Supplementary Material, Fig. S6B), and most photoreceptors in the Prph2Y/Y/Rom1-/- also lacked ISs, in contrast to the Prph2Y/Y retina.
Discussion
Here, we show biochemical and cell biological evidence demonstrating that removal of Rom1 significantly alters the disease phenotype in mice carrying the Y141C mutation in Prph2. This manifests as a conversion from a cone-rod PD-like phenotype in the Prph2Y/+ to a rod-dominant RP phenotype in the Prph2Y/+/Rom1-/-. Prph2-associated RP is typically caused by haploinsufficiency, while MD/PD mutations typically arise due to gain-of-function defects. Our data are consistent with this framework; upon eliminating Rom1 (Prph2Y/+/Rom1-/-) Prph2 levels drop to those seen in the haploinsufficient Prph2+/-, and phenotypes convert to those characteristic of RP. These data also strongly suggest that in the absence of Rom1, Y141C-Prph2 protein is unstable and marked for degradation. This hypothesis is supported by our in vitro work showing that Y141C-Prph2 is retained in the ER when expressed alone, but when co-expressed with Rom1 it traffics to post-ER membranes similar to WT Prph2.
The Y141C mutation resides in the Prph2-D2 loop, a critical region for Prph2 function which is known to be the area responsible for Prph2/Rom1 oligomerization (22). Of particular importance for Prph2 folding and oligomerization are the seven D2 loop cysteines, six of which mediate intramolecular disulfide bonds while the seventh mediates intermolecular disulfide bonds. One of the key findings from our prior work was that animals carrying the Y141C mutation exhibited abnormally large disulfide linked oligomers containing Prph2 and Rom1. A question arising from this observation was whether these large complexes arose due to novel disulfide bonds formed by the additional cysteine at position 141 or whether they were formed by cysteines previously involved in intramolecular bonding but now available for intermolecular bonding due to alterations in the structure of the D2 loop. Linked to this is the question of whether the effects of the Y141C mutation are due to the loss of the bulky tyrosine residue or due to the addition of a cysteine. We find that in contrast to the Y141C-Prph2, the Y141F-Prph2 is properly trafficked out of the ER, suggesting that the bulky tyrosine residue (as mimicked by phenylalanine in the case of Y141F) is critical for proper folding of the Prph2 D2 loop. Likewise, the Y141S and Y141C mutations have identical effects on protein localization (i.e. majority retained in the ER unless expressed with Rom1). This suggests that a cysteine specifically is not needed for the Y141C phenotype, but rather that replacement of tyrosine with a smaller and less bulky amino acid such as serine leads to the same phenotypic outcome. Though we do not have direct evidence of it, the logical conclusion from this observation is that the disulfide bonds linking the abnormal complexes seen both in vitro and in vivo in the presence of the Y141C protein are not held together by C141.
Although Y141C resides within the area of the D2 loop known to be required for Prph2/Rom1 binding (22), the Y141C mutation does not eliminate Rom1 binding (19). Prph2 and Rom1 are thought to assemble into tetramers in the IS, likely in the ER (31). Coupled with the fact that in the absence of Rom1, Y141C protein is retained in the ER, this suggests that Rom1 and Y141C-Prph2 form tetramers or other complexes that can exit the ER while Y141C-Prph2 alone cannot. Alternatively, Rom1 may promote improved folding of Prph2 or protect mutant Prph2 from the machinery which degrades misfolded proteins, but our data do not permit these possibilities to be distinguished from one another. It is worth noting, however, that the presence of Rom1 cannot “save” all mutant forms of Prph2. For example, C214S is retained in the ER of transfected cells with or without Rom1 and similar abnormalities have been previously observed in transgenic retinas (24). Since C214S-Prph2 cannot bind Rom1 (24), it is not surprising that co-expression of C214S-Prph2 and Rom1 does not correct the ER retention of C214S-Prph2.
Prior work in this field has shown that mutations which lead to haploinsufficiency (e.g. loss-of-function mutations such as C214S) cause rod-dominant RP (23,24). In contrast, cones are not affected until much later by Prph2 haploinsufficiency; rather cone-dominant MD/PD is associated with the formation of abnormal Prph2/Rom1 complexes (19,27). Our findings on C214S and Y141C suggest that Prph2 proteins in which Rom1 binding is ablated will likely be degraded, and therefore lead to RP/haploinsufficiency, while Prph2 mutants which retain the ability to bind Rom1 will lead to the abnormal complex formation and MD/PD.
Paradoxically, the ability of Rom1 to promote exit of Y141C-Prph2 from the ER and trafficking to the OS likely contributes to the PD/MD phenotype in patients since it leads to the formation of the large abnormal Y141C-Prph2/Rom1 complexes that are found in the OS in Prph2Y/+ and Prph2Y/Y animals. Large abnormal complexes are a notable feature of models of Prph2-associated MD/PD in contrast to the haploinsufficiency phenotype seen in Prph2Y/+/Rom1-/- animals and other Prph2 RP models (4,19,24,27). The differences in the role of Prph2 in rods vs. cones have been well documented (30,32–34); rods appear to be more sensitive to the total amount of Prph2 than cones (thus haploinsufficiency results in RP phenotypes) while cones are more sensitive to the correct assembly of Prph2/Rom1 complexes (thus MD/PD phenotypes when mutant protein is stable but forms abnormal oligomers). The role of Rom1 in these differences is less well understood; in large part because overall changes in the absence of Rom1 are less severe (11) and because fewer studies have evaluated the role of Rom1 in the retina. We have shown that cone cells overexpressing a Rom1 transgene have structural and functional defects, suggesting that excess Rom1 can be toxic in cones (35), but it is not clear whether this would be the case in rods. Eliminating Rom1 leads to alterations in disc sizing (11) and it is possible that the role of Rom1 in disc sizing/alignment is different in rods vs. cones.
Our data provide proof-of-principle as well as a mechanism by which Rom1 could act as one of many potential contributors to intrafamilial phenotypic variability in PRPH2-associated disease by converting phenotypes from cone-targeted gain-of-function disease to rod-targeted loss-of-function disease. In the previously reported cases where ROM1 mutations have been shown clinically to act as PRPH2 disease modifiers by worsening phenotypes (for example in patients heterozygous for the R172W MD mutation in PRPH2), patients were heterozygous for the ROM1 variant (20), suggesting one mutant ROM1 allele may be sufficient. The ROM1 mutation found in that study (R229H) resides in the D2 loop, but as ROM1 variants have not been characterized, R229H may be a null allele or a variant with gain-of-function activity. In future clinical studies, it will be interesting to see whether ROM1 variants are observed in patients with the Y141C mutation. The various studies documenting phenotypic heterogeneity in Y141C patients do not specify whether the ROM1 gene was screened for sequence changes (21,36,37), so these patients could carry unidentified ROM1 variants. In general, screening for ROM1 in clinical studies reporting on RP and MD mutations has been infrequent, likely because no known pathogenic mutations in ROM1 have been verified. However, as more patients are screened using next generation sequencing methods, simultaneous analysis of many genes is becoming more common. This ability to more easily screen many genes will facilitate evaluation of potential genetic modifiers or coexisting pathogenic mutations in multiple genes, and may advance our understanding of phenotypic variability in patients with inherited retinal degenerations.
Materials and methods
Transfection and vectors
Constructs carrying WT, C214S Prph2 cDNA or Rom1 cDNA under the control of the CMV promoter in the pcDNA3.1 vector have been described previously (23). Site-directed mutagenesis was used to generate vectors containing the C150S, Y141C, Y141S, and Y141F mutations in the Prph2 cDNA. All vectors were sequenced prior to use. For immunocytochemistry (ICC), COS-7 cells were seeded onto poly-L-lysine (P4832, Sigma) coated 22 mm cover slips in 6 well plates and transfected with 2 µg of each vector (either 2 µg Prph2 alone or 2 µg Prph2 and 2 µg Rom1 for double transfection). For protein chemistry, COS-7 cells were seeded into 10 cm culture dishes and transfected with 5 µg of each vector. Calcium phosphate transfection was used as described previously (38); in brief, 2X BBS buffer (50 mM BES, 280 mM NaCl, 1.4 mM Na2HPO4, pH 6.96) was added to 250 mM CaCl2 containing the appropriate amount of dissolved vector drop by drop then allowed to sit for 30 min prior to being gently added to the culture media (∼70% confluent). This was allowed to sit overnight, the media was refreshed and allowed to sit for an additional 24 h prior to harvesting and downstream processing.
Animals
All animal work was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Oklahoma and the University of Houston, and conformed to the guidelines set by the Association for Research in Vision and Ophthalmology (ARVO). Y141C-Prph2 knock-in mice were previously characterized (19). The Prph2-/- and Rom1-/- mice were bred from founders originally provided by Dr. Neeraj Agarwal (National Eye Institute, Bethesda MD) and Dr. Roderick McInnes (McGill University, Montreal, Canada), respectively. Animals were reared under cyclic lighting conditions (12 h L/D, ∼30 lux). All animals for the current study were P30, P120 or P180 as indicated in the results section.
Immunofluorescence and imaging
For in vitro experiments, 48 h following transfection, the cover slips were washed with PBS and fixed with 4% paraformaldehyde (PF) for 30 min. For in vivo experiments, eyes were enucleated, fixed with 4% paraformaldehyde, dissected, and cryoprotected as described previously (39). 10 µm retinal cryosections were collected. Slides or coverslips were then subjected to immunofluorescence analysis following procedures described previously (40). Briefly, slides/cover slips were washed, blocked with 5% BSA, 2% donkey serum, 1% fish gelatin, 1% triton, and 1X PBS pH7.0, and incubated overnight with the indicated antibodies. Primary antibodies used were calreticulin (chicken, Abcam, ab2908), protein disulfide isomerase (PDI, mouse, Abcam, ab2792) Prph2-2B7 (mouse, in-house (27)), RDS-CT (rabbit, in-house (22)), Rom1 (rabbit, in-house (22)), and rhodopsin mAB 1D4 (mouse, generously provided by Dr. Robert Molday, University of British Columbia, Vancouver, Canada). After washing, cover slips/slides were incubated in appropriate secondary antibodies conjugated to Alexa488, Alexa555, or Alexa647 (Life Technologies, Grand Island, NY) and DAPI to counterstain nuclei. All experiments were repeated at least three times and on three independent eyes.
After mounting, retinal sections or cells on coverslips were imaged using an Olympus BX-62 (Olympus, Tokyo, Japan) microscope equipped with a spinning disc confocal unit and a Hamamatsu C-4742 camera. Images in Figs. 1 and4A,D,E, Supplementary Materials, S1 and S2, and were captured at 100x (oil 1.4 NA) and shown are single planes from a confocal stack. Images in Fig. 4B were collected under epifluorescent conditions at 40x (air, 0.9 NA), while images in Supplementary Material Fig. S2 and Fig. 4C were captured at 100x under epifluorescent conditions. Images were captured using equivalent exposure times and normalized to secondary antibody only controls. In some cases, longer exposure times were needed (for Prph2Y/Y and Prph2Y/Y/Rom1-/-), but images were still normalized to secondary only controls from the same slide. All images were analysed and deconvolved using Slidebook v5 software (Intelligent Imaging Innovations, Denver, CO).
Protein extraction and analysis
For transfected cells, cultures were washed three times with 1x PBS and cells were scraped on ice in the presence of 1X PBS + protease inhibitor. Cells were pelleted gently by centrifugation and re-suspended in lysis buffer (1x PBS pH 7.0 containing 1% triton-X 100, 5 mM EDTA, 5 mg/ml n-ethylmaleimide, and a standard protease inhibitor cocktail). Retinal extracts were prepared using 150 µl of chilled (4ºC) lysis buffer per retina. Samples (cells or retinas) were briefly sonicated and incubated for ∼1 h at 4ºC prior to a 30 min spin at 20,000xg at 4ºC to remove insoluble debris. Protein concentration was assayed by using a colorimetric protein assay (Bradford reagent from Bio-rad, Hercules, CA). All extracts were kept on ice and used immediately to avoid any non-specific post-extraction aggregation. Western blots, SDS-PAGE, and immunoprecipitation (IP) were performed as previously described (19,27,31). Blots were imaged using ChemiDoc™ MP imaging system (Bio-rad) and densitometric analysis was performed using Image Lab software v4.1 (Bio-rad). All experiments were repeated at least three times with extracts from three independent eyes.
Electroretinography and fundus imaging
Full-field ERG was performed as described previously (18,29,34). Briefly, mice were dark-adapted overnight, then eyes were dilated using 1% cyclogyl (Pharmaceutical Systems, Inc., Tulsa, OK). Animals were anaesthetized by intramuscular injection of 85 mg/kg ketamine and 14 mg/kg xylazine (Butler Schein Animal Health, Dublin, OH). ERG stimulus was presented using the UTAS system (LKC, Gaithersburg, MD) with a platinum wire loop electrode in contact with the cornea through a layer of methylcellulose (Pharmaceutical Systems Inc.). Rod photoreceptor function (scotopic) was assessed with a single strobe flash stimulus of 157 cd s/m2 presented to the dark adapted mouse. Cone photoreceptor function (photopic) was analysed by averaging responses to 25 flashes at 157 cd s/m2 following a 5 min light adaptation with background light at an intensity of 29.03 cd/m2. Each cohort contained between 5–15 animals, specific numbers are as follows: WT N = 15, Rom1-/- N = 6, Prph2+/- N = 15, Prph2Y/+ N = 14, Prph2Y/+/Rom1-/- N = 13, Prph2-/- N = 9, Prph2Y/Y N = 5, Prph2Y/Y/Rom1-/-N = 7. Fundus imaging was performed as described previously (19) using the Micron IV system (Phoenix Research Laboratories, Pleasanton, CA). Animals were anaesthetized/dilated as for ERG but were not dark-adapted. All images were captured using StreamPix® software (Phoenix Research Labs).
Light and electron microscopy
Eyes were enucleated, fixed, and processed for plastic embedding as described previously (19,29). Thin sections (600–800 Å) collected on copper 75/300 mesh grids were then stained with 2% uranyl acetate and Reynolds’ lead citrate for TEM. Semithin sections were used for LM imaging. EM images were collected using a JEOL 100CX electron microscope at an accelerating voltage of 60kV. Light micrographs were captured at 40x. EM images were collected at 10,000x (Fig. 5, and4A, D, E bottom Supplementary Material, Fig. S6B) and 3,000x (Supplementary Materials, Fig. S5, and top S6B).
Statistical analysis
Where appropriate, between group differences were assessed for statistical significance using 1-way ANOVA coupled with Bonferroni’s post-hoc comparison. Significance was set at P < 0.05. Throughout the manuscript *P < 0.05, **P < 0.01, and ***P < 0.001 for indicated pairwise comparisons.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors thank Dr. Muayyad Al-Ubaidi, Dr. Rahel Zulliger, Dr. David Sherry, Ms. Barb Nagel, and Mr. Marc Banworth for technical support. We thank Dr. Neeraj Agarwal, Dr. Roderick McInnes, and Dr. Robert Molday for the provision of animal lines and reagents as indicated in the text.
Conflict of Interest statement. None declared.
Funding
This work was supported by the National Eye Institute (R01EY010609-MIN), the Foundation Fighting Blindness (MIN), the Oklahoma Center for the Advancement of Science and Technology (SMC, HR14-150), and the Presbyterian Health Foundation (SMC).
References
- 1. Jansen H.G., Sanyal S. (1984) Development and degeneration of retina in rds mutant mice: electron microscopy. J. Comp. Neurol., 224, 71–84. [DOI] [PubMed] [Google Scholar]
- 2. Boon C.J., den Hollander A.I., Hoyng C.B., Cremers F.P., Klevering B.J., Keunen J.E. (2008) The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res., 27, 213–235. [DOI] [PubMed] [Google Scholar]
- 3. Reuter J.H., Sanyal S. (1984) Development and degeneration of retina in rds mutant mice: the electroretinogram. Neurosci. Lett., 48, 231–237. [DOI] [PubMed] [Google Scholar]
- 4. Cheng T., Peachey N.S., Li S., Goto Y., Cao Y., Naash M.I. (1997) The effect of peripherin/rds haploinsufficiency on rod and cone photoreceptors. J. Neurosci., 17, 8118–8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goldberg A.F., Loewen C.J., Molday R.S. (1998) Cysteine residues of photoreceptor peripherin/rds: role in subunit assembly and autosomal dominant retinitis pigmentosa. Biochemistry, 37, 680–685. [DOI] [PubMed] [Google Scholar]
- 6. Loewen C.J., Molday R.S. (2000) Disulfide-mediated oligomerization of Peripherin/Rds and Rom-1 in photoreceptor disk membranes. Implications for photoreceptor outer segment morphogenesis and degeneration. J. Biol. Chem., 275, 5370–5378. [DOI] [PubMed] [Google Scholar]
- 7. Goldberg A.F., Molday R.S. (1996) Subunit composition of the peripherin/rds-rom-1 disk rim complex from rod photoreceptors: hydrodynamic evidence for a tetrameric quaternary structure. Biochemistry, 35, 6144–6149. [DOI] [PubMed] [Google Scholar]
- 8. Sakuma H., Inana G., Murakami A., Yajima T., Weleber R.G., Murphey W.H., Gass J.D., Hotta Y., Hayakawa M., Fujiki K., et al. (1995) A heterozygous putative null mutation in ROM1 without a mutation in peripherin/RDS in a family with retinitis pigmentosa. Genomics, 27, 384–386. [DOI] [PubMed] [Google Scholar]
- 9. Bascom R.A., Liu L., Heckenlively J.R., Stone E.M., McInnes R.R. (1995) Mutation analysis of the ROM1 gene in retinitis pigmentosa. Hum. Mol. Genet., 4, 1895–1902. [DOI] [PubMed] [Google Scholar]
- 10. Dryja T.P., Hahn L.B., Kajiwara K., Berson E.L. (1997) Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci., 38, 1972–1982. [PubMed] [Google Scholar]
- 11. Clarke G., Goldberg A.F., Vidgen D., Collins L., Ploder L., Schwarz L., Molday L.L., Rossant J., Szel A., Molday R.S., et al. (2000) Rom-1 is required for rod photoreceptor viability and the regulation of disk morphogenesis. Nat. Genet., 25, 67–73. [DOI] [PubMed] [Google Scholar]
- 12. Sato H., Suzuki T., Ikeda K., Masuya H., Sezutsu H., Kaneda H., Kobayashi K., Miura I., Kurihara Y., Yokokura S., et al. (2010) A monogenic dominant mutation in Rom1 generated by N-ethyl-N-nitrosourea mutagenesis causes retinal degeneration in mice. Mol. Vis., 16, 378–391. [PMC free article] [PubMed] [Google Scholar]
- 13. Kajiwara K., Berson E.L., Dryja T.P. (1994) Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science, 264, 1604–1608. [DOI] [PubMed] [Google Scholar]
- 14. Sullivan L.S., Bowne S.J., Birch D.G., Hughbanks-Wheaton D., Heckenlively J.R., Lewis R.A., Garcia C.A., Ruiz R.S., Blanton S.H., Northrup H., et al. (2006) Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest. Ophthalmol. Vis. Sci., 47, 3052–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kedzierski W., Nusinowitz S., Birch D., Clarke G., McInnes R.R., Bok D., Travis G.H. (2001) Deficiency of rds/peripherin causes photoreceptor death in mouse models of digenic and dominant retinitis pigmentosa. Proc. Natl Acad. Sci. USA, 98, 7718–7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goldberg A.F., Molday R.S. (1996) Defective subunit assembly underlies a digenic form of retinitis pigmentosa linked to mutations in peripherin/rds and rom-1. Proc. Natl Acad. Sci. USA, 93, 13726–13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kedzierski W., Lloyd M., Birch D.G., Bok D., Travis G.H. (1997) Generation and analysis of transgenic mice expressing P216L-substituted rds/peripherin in rod photoreceptors. Invest. Ophthalmol. Vis. Sci., 38, 498–509. [PubMed] [Google Scholar]
- 18. Ding X.Q., Nour M., Ritter L.M., Goldberg A.F., Fliesler S.J., Naash M.I. (2004) The R172W mutation in peripherin/rds causes a cone-rod dystrophy in transgenic mice. Hum. Mol. Genet., 13, 2075–2087. [DOI] [PubMed] [Google Scholar]
- 19. Stuck M.W., Conley S.M., Naash M.I. (2014) The Y141C knockin mutation in RDS leads to complex phenotypes in the mouse. Hum. Mol. Genet., 23, 6260–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poloschek C.M., Bach M., Lagreze W.A., Glaus E., Lemke J.R., Berger W., Neidhardt J. (2010) ABCA4 and ROM1: implications for modification of the PRPH2-associated macular dystrophy phenotype. Invest. Ophthalmol. Vis. Sci., 51, 4253–4265. [DOI] [PubMed] [Google Scholar]
- 21. Vaclavik V., Tran H.V., Gaillard M.C., Schorderet D.F., Munier F.L. (2012) Pattern dystrophy with high intrafamilial variability associated with Y141C mutation in the peripherin/RDS gene and successful treatment of subfoveal CNV related to multifocal pattern type with anti-VEGF (ranibizumab) intravitreal injections. Retina, 32, 1942–1949. [DOI] [PubMed] [Google Scholar]
- 22. Ding X.Q., Stricker H.M., Naash M.I. (2005) Role of the second intradiscal loop of peripherin/rds in homo and hetero associations. Biochemistry, 44, 4897–4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Conley S.M., Stricker H.M., Naash M.I. (2010) Biochemical analysis of phenotypic diversity associated with mutations in codon 244 of the retinal degeneration slow gene. Biochemistry, 49, 905–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stricker H.M., Ding X.Q., Quiambao A., Fliesler S.J., Naash M.I. (2005) The Cys214–>Ser mutation in peripherin/rds causes a loss-of-function phenotype in transgenic mice. Biochem. J., 388, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saga M., Mashima Y., Akeo K., Oguchi Y., Kudoh J., Shimizu N. (1993) A novel Cys-214-Ser mutation in the peripherin/RDS gene in a Japanese family with autosomal dominant retinitis pigmentosa. Hum. Genet., 92, 519–521. [DOI] [PubMed] [Google Scholar]
- 26. Hawkins R.K., Jansen H.G., Sanyal S. (1985) Development and degeneration of retina in rds mutant mice: photoreceptor abnormalities in the heterozygotes. Exp. Eye Res., 41, 701–720. [DOI] [PubMed] [Google Scholar]
- 27. Conley S.M., Stuck M.W., Burnett J.L., Chakraborty D., Azadi S., Fliesler S.J., Naash M.I. (2014) Insights into the mechanisms of macular degeneration associated with the R172W mutation in RDS. Hum. Mol. Genet., 23, 3102–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chakraborty D., Conley S.M., Zulliger R., (2016) The K153Del PRPH2 mutation differentially impacts photoreceptor structure and function. Hum. Mol. Genet, pii: ddw193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chakraborty D., Ding X.Q., Conley S.M., Fliesler S.J., Naash M.I. (2009) Differential requirements for retinal degeneration slow intermolecular disulfide-linked oligomerization in rods versus cones. Hum. Mol. Genet., 18, 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chakraborty D., Conley S.M., Al-Ubaidi M.R., Naash M.I. (2014) Initiation of rod outer segment disc formation requires RDS. PLoS One, 9, e98939.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chakraborty D., Ding X.Q., Fliesler S.J., Naash M.I. (2008) Outer segment oligomerization of Rds: evidence from mouse models and subcellular fractionation. Biochemistry, 47, 1144–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Conley S.M., Al-Ubaidi M.R., Han Z., Naash M.I. (2014) Rim formation is not a prerequisite for distribution of cone photoreceptor outer segment proteins. Faseb J., 28, 3468–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Farjo R., Fliesler S.J., Naash M.I. (2007) Effect of Rds abundance on cone outer segment morphogenesis, photoreceptor gene expression, and outer limiting membrane integrity. J. Comp. Neurol., 504, 619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Farjo R., Skaggs J.S., Nagel B.A., Quiambao A.B., Nash Z.A., Fliesler S.J., Naash M.I. (2006) Retention of function without normal disc morphogenesis occurs in cone but not rod photoreceptors. J. Cell Biol., 173, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chakraborty D., Conley S.M., Nash Z., Ding X.Q., Naash M.I. (2012) Overexpression of ROM-1 in the cone-dominant retina. Adv. Exp. Med. Biol., 723, 633–639. [DOI] [PubMed] [Google Scholar]
- 36. Francis P.J., Schultz D.W., Gregory A.M., Schain M.B., Barra R., Majewski J., Ott J., Acott T., Weleber R.G., Klein M.L. (2005) Genetic and phenotypic heterogeneity in pattern dystrophy. Br. J. Ophthalmol., 89, 1115–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sohocki M.M., Daiger S.P., Bowne S.J., Rodriquez J.A., Northrup H., Heckenlively J.R., Birch D.G., Mintz-Hittner H., Ruiz R.S., Lewis R.A., et al. (2001) Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat., 17, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zulliger R., Conley S.M., Mwoyosvi M.L., Stuck M.W., Azadi S., Naash M.I. (2015) SNAREs Interact with Retinal Degeneration Slow and Rod Outer Segment Membrane Protein-1 during Conventional and Unconventional Outer Segment Targeting. PLoS One, 10, e0138508.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chakraborty D., Conley S.M., Stuck M.W., Naash M.I. (2010) Differences in RDS trafficking, assembly and function in cones versus rods: insights from studies of C150S-RDS. Hum. Mol. Genet., 19, 4799–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stuck M.W., Conley S.M., Naash M.I. (2012) Defects in the outer limiting membrane are associated with rosette development in the nrl retina. PLoS One, 7, e32484.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.