

Supplemental Digital Content is available in the text
Keywords: aging, Alzheimer disease, gene expression, NRXN3
Abstract
Background:
Alzheimer disease (AD) is a common neurodegenerative disorder with distinct pathological features, with aging considered the greatest risk factor. We explored how aging contributes to increased AD risk, and determined concurrent and coordinate changes (including genetic and phenotypic modifications) commonly exhibited in both normal aging and AD.
Methods:
Using the Gene Expression Omnibus (GEO) database, we collected 1 healthy aging-related and 3 AD-related datasets of the hippocampal region. The normal aging dataset was divided into 3 age groups: young (20–40 years old), middle-aged (40–60 years old), and elderly (>60 years old). These datasets were used to analyze the differentially expressed genes (DEGs). The Gene Ontology (GO) terms, pathways, and function network analysis of these DEGs were analyzed.
Results:
One thousand two hundred ninety-one DEGs were found to be shared in the natural aging groups and AD patients. Among the shared DEGs, ATP6V1E1, GNG3, NDUFV2, GOT1, USP14, and NAV2 have been previously found in both normal aging individuals and AD patients. Furthermore, using Java Enrichment of Pathways Extended to Topology (JEPETTO) analysis based on Kyoto Encyclopedia of Genes and Genomes (KEGG) database, we determined that changes in aging-related KEGG annotations may contribute to the aging-dependence of AD risk. Interestingly, NRXN3, the second most commonly deregulated gene identified in the present study, is known to carry a mutation in AD patients. According to functional network analysis, NRXN3 plays a critical role in synaptic functions involved in the cognitive decline associated with normal aging and AD.
Conclusion:
Our results indicate that the low expression of aging-related NRXN3 may increase AD risk, though the potential mechanism requires further clarification.
1. Introduction
Alzheimer disease (AD) is one of the most prevalent neurodegenerative disorders worldwide. It is characterized by the accumulation of amyloid beta (Aβ) in senile plaques and hyper-phosphorylated tau in neurofibrillary tangles, as well as by serious neuronal and synaptic dysfunction.[1] AD leads to the progressive loss of cognitive function and is a common cause of dementia. It is strongly correlated with aging and is thus a growing problem in elderly populations.[2] However, even familial forms of AD rarely show clinical symptoms before the fifth decade of life.[3]
Despite the lack of early clinical indicators, AD begins many years before diagnosis, with the long preclinical phase correlated with alterations in brain aging.[4] Normal aging is accompanied by a progressive decline in biological functions, which can increase susceptibility to endogenous and exogenous triggers, thereby exacerbating pre-existing pathological conditions.[5] Alterations in the brain increase with aging at all levels, from molecules to morphology, and at different degrees due to neurotransmitter impairment. Aging also causes constant changes in brain size, vasculature, and cognition, ultimately leading to brain shrinkage.
The human brain is uniquely powerful with respect to cognition and memory. However, many neuronal networks that mediate complex functions are highly vulnerable to aging.[6] For example, aging-related brain atrophy has been widely documented in the hippocampus, thereby impacting hippocampal circuits.[7] The hippocampus plays an important role in memory and cognitive impairment,[8] which are common complaints in healthy aging [9] and among the earliest signs of AD.[10] Synapses, which are highly dynamic structures within neuronal networks, are also critical to cognition and memory. Therefore, precise control of synaptic development and connectivity are essential processes for maintaining accurate neuronal network activity and normal brain function.[11] Aging and AD-related neuronal dysfunction are accompanied by a host of complex alterations in spine numbers and synaptic densities.[12] Impaired hippocampal synaptic function is an early pathological sign of AD, detectable well before the advanced stages of amyloid plaque accumulation and general cell death.[13]
Thus, exploration of the shared pathological changes between AD and normal aging could provide diagnostic clues for their etiology, as well as facilitate the discovery of novel biomarkers and therapeutic drugs for AD. To the best of our knowledge, integrated pathway and network analyses of high-throughput gene expression data from AD patients and age-matched controls have not yet been explored. Therefore, we investigated common transcriptional changes, biological functions, and protein–protein interaction networks in healthy-aging and AD datasets, and further filtered AD-risk single nucleotide polymorphisms (SNPs). By identifying shared gene expression alterations during natural brain aging and AD, we aimed to assess how aging contributes to the increase in AD risk; determine changes common to both aging and AD; and identify aging-related genes carrying high AD risk.
2. Materials and methods
2.1. AD and aging microarray datasets
To explore common features between AD patients and normal aging populations, we downloaded relevant data from the GEO database of the US National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/geo). The data selection criteria were as follows: all datasets should be genome-wide; all tissue should be from brain samples; each AD dataset should include patients and controls; the aging-related dataset should have a broad age distribution, with no individuals suffering from any known neurological or psychiatric disorder; and raw data or gene expression matrices should be available in each dataset. According to the above criteria, we selected 3 AD-related datasets (GSE5281, GSE28146, and GSE48350) and 1 normal brain aging-related dataset (GSE11882) for integrated analysis. All dataset samples were collected from postmortem hippocampus tissue and tested using the Affymetrix Human Genome U133 Plus 2.0 Array (Santa Clara County, CA). Additional information on these datasets is summarized in Table 1. We classified the normal brain dataset into 3 groups: young (20–40 years old), middle-aged (40–60 years old), and elderly (> 60 years old).
Table 1.
Overview of Alzheimer disease- and healthy aging-related datasets.

2.2. Data preprocessing and shared DEGs
The R software package was employed for data preprocessing. All 4 microarray datasets were preprocessed using the robust multichip average (RMA) algorithm in the oligo package.[14] Gene annotation and integration of the AD datasets were performed using a custom written Python code.[15] The 3 AD datasets were further analyzed using metaMA, which applied the empirical Bayes moderated t-statistic and weighted meta-analysis to calculate DEGs in each dataset, and then adjusted the P values with a false discovery rate (FDR).[16] As a result, a Z score was assigned to each DEG, as per previous research.[16] For the aging dataset, we used significance analysis of microarrays (SAM) [17] in the samr R package to identify normal aging-related genes. SAM is a dedicated multiclass-analysis method designed for microarray data, which allocates a score for each gene based on changes in expression relative to the standard deviation of repeated measurements, and generates FDR-adjusted P values.[17] We used this approach to identify increases or decreases in gene expression variance related to aging, as well as positive or negative correlations. Significantly DEGs in the 2 classes of datasets were identified with a FDR value of < 0.05. Genes deregulated in AD and correlated with aging were considered as shared DEGs. The pheatmap package in R was used to show the expression profiles of these shared genes.
2.3. Common topological pathway analysis
To determine the commonly affected cellular pathways in AD patients and normal brain aging groups, we used JEPETTO,[18] which visually analyzes the functional associations of the top-ranked underlying networks between DEGs and known cellular pathways. As a complement to classic pathway enrichment analysis, JEPETTO can identifies novel and significant relationships using protein–protein interaction networks and topological analyses. The KEGG database was searched for the closest topological matches and all pathway visualizations were performed using Cytoscape 3.2.1 software.
2.4. Identification of key aging-related genes in AD
To systematically define key aging-related genes in AD, we downloaded AD-associated SNPs from the Ensemble Variation Database.[19] These genome-wide AD-risk SNPs were divided into 2 groups according to their P values: significant SNPs (P < 10E-08) and suggestive SNPs (P < .001). The shared DEGs with significant SNPs were identified as aging-related genes contributing to AD. The GeneMANIA application in Cytoscape was used to reveal the interactive relationships among these key genes and determine the influenced biological functions.
2.5. Ethical approval
Ethical approval is not necessary because that our study is a systematic review of published literature. We did not make any clinical research and we just collected the data from available publications.
3. Results
3.1. Shared DEGs in AD and aging
We identified 6205 DEGs in the AD patients and healthy controls from the 3 AD-related microarray datasets using the metaMA package in R. The expression profiles of these DEGs were approximately categorized into AD patients and healthy controls (Figure S1); however, the unsupervised clustering method did not distinguish the AD samples into the 2 categories (Figure S2). We also detected 2718 aging-related genes among the 3 different age groups in the normal-aging dataset, and subsequently identified 1291 DEGs in both the AD and normal-aging datasets. As shown in Fig. 1A, the expression profiles of the 1291 shared DEGs were significantly different between the AD patients and healthy controls in the AD-related datasets. Furthermore, as shown in Fig. 1B, the expression profiles of the shared DEGs in the young and middle-aged groups in the normal-aging dataset showed similar expression patterns, whereas the elderly group was markedly different. This difference may be due to the large age gap observed between the first 2 groups and the elderly group, with mean ages of 27, 46, and 82 years, respectively. Therefore, the obtained heatmap of the 1291 shared DEGs showed 2 distinct expression patterns among the 3 age groups.
Figure 1.
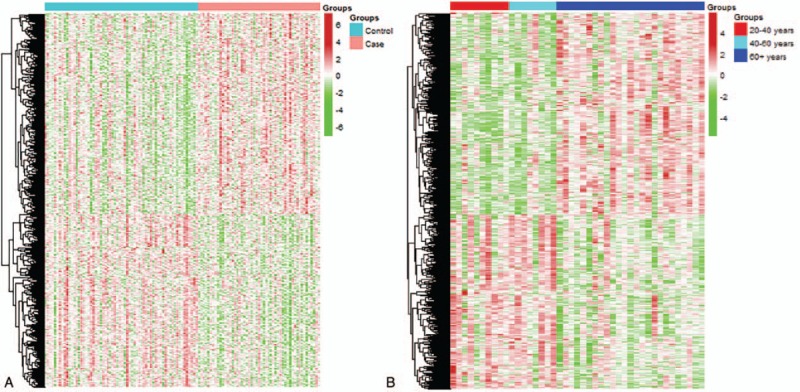
Expression profiles of the 1291 DEGs common to both the AD- and aging-related datasets. Expression values are converted Z-scores.
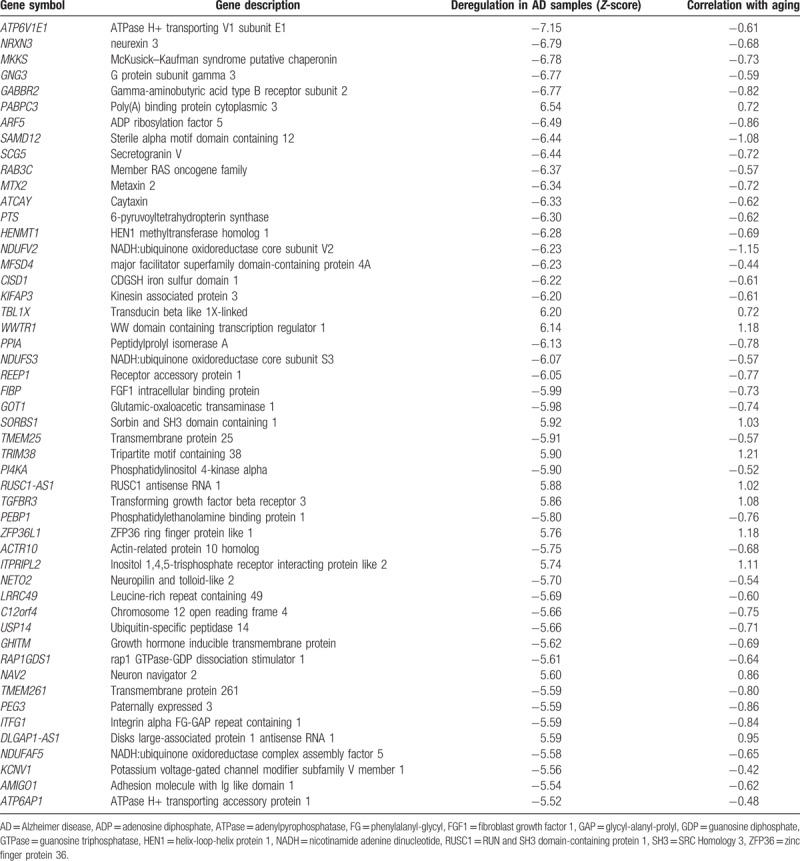
The top 50 shared DEGs sorted by absolute Z-score are listed in Table 2. General comparison of the gene expression alterations in the AD and normal-aging datasets showed that the upregulated genes in AD patients were positively correlated with aging, and most downregulated genes were negatively correlated with aging. Among the top 50 shared genes, ATP6V1E1, GNG3, NDUFV2, GOT1, USP14, and NAV2 have been previously investigated in AD and aging studies. The evidence related to normal brain aging and AD pathogenesis of these 6 genes, along with relevant reference, is summarized in Table S1. Considering the shared gene expression profiles in AD and natural aging, we hypothesized that aging-related hippocampal transcription changes in healthy elderly individuals may increase the risk of AD.
Table 2.
Top 50 differentially expressed genes common to both AD- and aging-related datasets.
3.2. Topological pathway features in AD and aging
We used JEPETTO to assess pathway-level changes in AD patients and normal aging groups. Among the 1291 shared DEGs, 743 were mapped to specific KEGG pathways (Table S2). These topological KEGG pathways included human disease, cellular processes, genetic and environmental information processing, and metabolism processes. Comparative analysis (degree against shortest path length) of the topological properties is shown in Fig. 2. For the topological properties of the shared DEGs in normal aging and AD, matched KEGG annotations were identified for neurodegeneration disease, Huntington disease, apoptosis, AD, ubiquitin-mediated proteolysis, long-term depression, and Parkinson disease. The main KEGG annotations demonstrated the same alterations in cellular processes implicated in normal aging individuals and AD patients. Thus, changes in the aging-related activity of these processes may contribute to the aging-dependent risk of AD.
Figure 2.
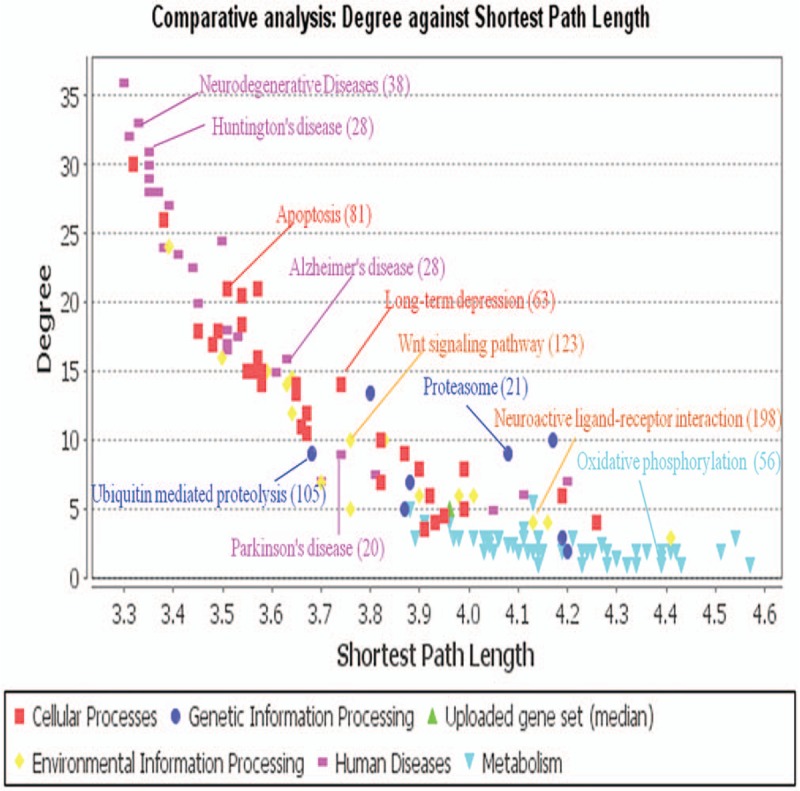
Topological properties of the 1291 DEGs from the AD- and aging-related datasets based on KEGG pathway analysis. Target pathways are labeled with different colors, with some pathway annotations marked with corresponding colors and mapped gene numbers.
3.3. Single nucleotide polymorphisms of shared DEGs
In addition to the contribution of aging and environmental risk elements to the pathogenesis of AD, the disease is also influenced by genetic susceptibility factors. Genome-wide association studies suggest that a significantly large number of genetic variations increase AD risk.[20] We investigated the AD-associated SNPs from previously published studies reported in the Ensemble Variation Database. A total of 548 unique genes with suggestive AD-linked SNPs were obtained, which we mapped to the 1291 shared DEGs. We identified 32 genes carrying mutations and increased AD risk, which were ordered according to their absolute Z-scores (Table S3). The NRXN3 gene, which was the second highest DEG (Z-score: -6.79) and was negatively correlated with the normal aging group (−0.68, see box plot in Fig. 3 and statistics in Table 2), overlapped with the SNP change in AD patients. The expression levels of NRXN3 in both the AD- and aging-related groups exhibited downward trends. We therefore focused on the function of NRXN3 and interacting genes in AD patients and aging populations.
Figure 3.
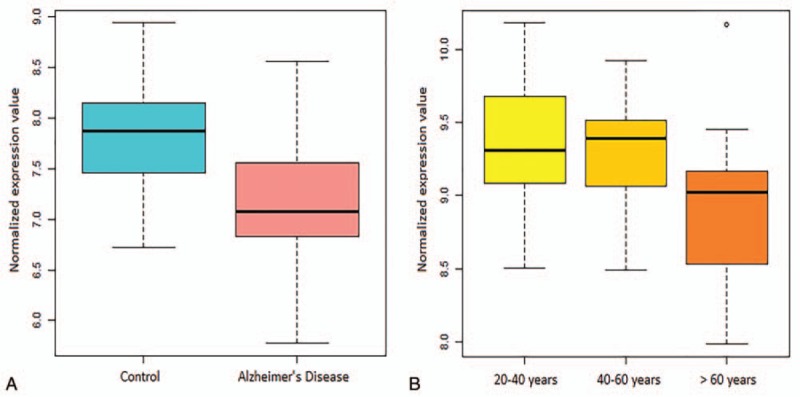
NRXN3 expression levels in AD patients and healthy controls (A) and the 3 age groups (B) were significantly different (FDR-value < 0.05).
3.4. Functional networks of NRXN3 and NRXN3-correlated genes
The functional networks and correlated genes of NRXN3 are shown in Fig. 4. As seen in Fig. 4A, many protein–protein interactions were associated with NRXN3. We used a graph-theoretic clustering algorithm called Molecular Complex Detection (MCODE) [21] to detect the dense functionally connected subnetworks within the large NRXN3 protein–protein interaction network. The first subcluster assigned by MCODE within the NRXN3 network was displayed with 6 yellow nodes (NRXN3, NRXN2, NLGN1, NLGN2, NLGN3, and PTPRT) and red connection lines. The top 5 enriched GO biological processes were neuron cell–cell adhesion, cell adhesion molecule binding, cell–cell adhesion, learning, and positive regulation of excitatory postsynaptic membrane potential (Fig. 4B). Thus, the network and enriched GO annotation results suggested that NRXN3 plays an important role in synapse function and nerve cell activity associated with the other 5 proteins (NRXN2, NLGN1, NLGN2, NLGN3, and PTPRT).
Figure 4.
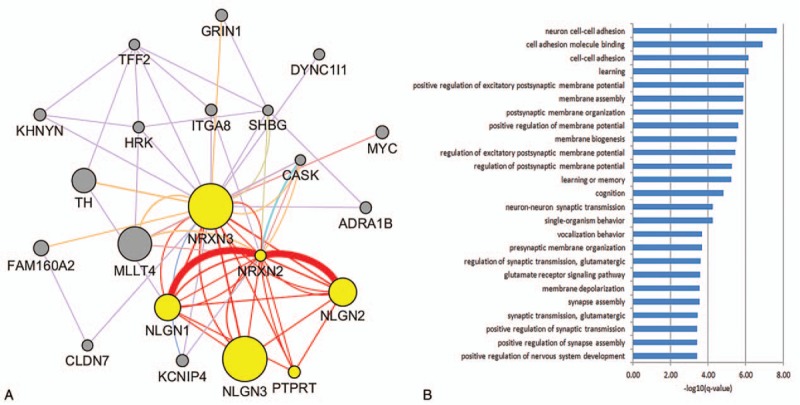
Interaction relationships among NRXN3 and its correlated genes. (A) GeneMANIA results of the NRXN3-enriched network relationships and MCODE plugin assigning the first subcluster with red connected lines. (B) Enriched GO biological processes of the first subcluster genes in the NRXN3 network.
4. Discussion
Hippocampal transcriptional changes in AD patients and normal aging-related groups were investigated, and shared DEGs, pathways, and biological functions were explored. We identified 1291 DEGs in AD patients that were correlated with normal aging. Comparing the genetic susceptibility of these genes, we identified NRXN3 as the most important risk factor contributing to AD, with low expression found in AD patients and the elderly group (Fig. 3). Previous meta-analysis of 5 genome-wide association studies comprised of 1256 SNPs in NRXN1, NRXN2, NRXN3, and NLGN1 identified a NRXN3 gene marker (rs17757879) that showed consistent protective effects, thus concluding that NRXN3 might play a role in susceptibility to AD in males.[22] In the present study, functional enrichment analysis revealed that NRXN3 and its correlated genes affected learning and memory, synaptic plasticity, cell–cell adhesion, and other AD-related pathological processes (Fig. 4).
NRXN3 is a synaptic modulator and neurexin (NRXN) family protein that functions in the vertebrate nervous system as a cell adhesion molecule during synaptogenesis and intercellular signaling. Thousands of NRXNs are coded from 3 genes (NRXN1, NRXN2, and NRXN3) by alternative promoter use and extensive alternative splicing [23] in the human genome. NRXN3 is located on chromosome 14q24.3-q31.1 and is the largest (about 1.8 Mb) and most alternatively spliced among the 3 NRXN genes.[24] It was initially implicated in addictions to alcohol,[25] nicotine,[26] and illicit drugs[27]; however, emerging research suggests that NRXN3 also plays a potential role in deregulated synaptic transmission in the Disc1 mouse model of schizophrenia [28] and is involved in body weight gain related to risperidone therapy in the Chinese Han population.[29]
In addition to the production and accumulation of Aβ peptide and hyper-phosphorylated tau protein related to learning and memory in the AD brain, synaptic dysfunction and loss are major pathological correlates of cognitive decline and neuronal damage in elderly individuals and AD patients.[30,31] The first subnetwork obtained from MCODE contained NRXN3 and NRXN2, 2 proteins within the neuroligin (NRXN) family, and NLGN1, NLGN2, and NLGN3, 3 proteins within the NLGN family.[32] These 5 genes are all synaptic cell adhesion molecules found in neurons, with NLGNs localized at the postsynaptic membrane considered to be binding partners of NRXNs located at the pre-synaptic membrane.[33] NRXN-NLGN interactions are important for synaptic maturation and stabilization and contribute to the ratio between excitatory and inhibitory synapses.[34] In addition, through their extracellular domains, NRXNs and NLGNs bind to PTPRT (receptor-type tyrosine-protein phosphatase T), which is exclusively expressed in the central nervous system and regulates synapse formation in hippocampal neurons.[35] Furthermore, NLGN and NRXN functions are potentially regulated by α- and γ-secretases at the synapses or in related signal transduction pathways. This regulation is altered by several mutations in the PSEN1 gene in the substructure of γ-secretases, which can cause early-onset familial AD.[36] Consequently, any alteration in NLGN-NRXN interactions could have considerable influence on synaptic activity and plasticity in aging individuals and AD patients.
Shared mechanisms of synaptic dysfunction in aging and AD have been proposed in recent years. During natural aging, synapse numbers and signal transmissions decrease dramatically in different brain regions of elderly individuals,[37,38] supporting the hypothesis that these synaptic changes are ubiquitous features of the aging brain.[39] In its earliest clinical phase, AD characteristically shows substantial impairment of memory. Mounting evidence suggests that this disease begins with subtle alterations in hippocampal synaptic efficacy before frank neuronal degeneration, and that synaptic dysfunction is caused by diffusible oligomeric assemblies of the amyloid β protein.[30] Studies on synaptic proteins reveal decreased levels of pre-synaptic (synaptophysin) and post-synaptic proteins (synaptopodin and PSD95) [40] in AD patients compared with age-matched controls, suggesting that these proteins are critically involved in AD progression [41] and that synapse and synaptic protein loss are confined to brain regions known to be affected in AD.[42]
On the basis of the NRXN3 functional networks determined here and previous research on the contribution of synaptic dysfunction to cognitive decline in aging individuals and those with AD, we hypothesized that decreasing expression of aging-related NRXN3 in the hippocampus of healthy elderly individuals may increase the risk of AD pathogenesis. Because the transcription data used in this study were not complemented by corresponding single nucleotide variation data for the same samples, the direct relationship between genome and transcription alterations was not assessed. Thus, the shared candidate genes derived from our analysis will require validation using independent complementary genome and transcription data.
In conclusion, we found similar gene expression patterns between AD patients and normal-aging populations. Numerous deregulated aging-related genes were also revealed in the AD patients. Combined with genetic mutation data, downregulation of NRXN3 was identified as the highest risk gene correlated with AD and aging. These results provide a mechanistic explanation for aging-related AD risk. Furthermore, the significantly altered shared genes may have potential application in the identification of novel biomarkers and therapeutic drugs for AD. However, future experiments are required to validate these results and elucidate the potential mechanisms of NRXN3 in AD.
Acknowledgment
The authors thank grants from National Basic Research Program of China (Grant No. 2013CB835100) and National Natural Science Foundation of China (Grant No. 31401142 and No.31401137) for support.
Author contributions
Conceptualization: Junjuan Zheng, Wen-Xing Li, Jia-Qian Liu, Yi-Cheng Guo, Qian Wang, Shao-Xing Dai, Jing-Fei Huang.
Data curation: Junjuan Zheng, Qian Wang, Gong-Hua Li, Shao-Xing Dai, Jing-Fei Huang.
Formal analysis: Junjuan Zheng.
Funding acquisition: Shao-Xing Dai, Jing-Fei Huang.
Investigation: Junjuan Zheng.
Methodology: Junjuan Zheng.
Project administration: Shao-Xing Dai, Jing-Fei Huang.
Resources: Junjuan Zheng.
Software: Junjuan Zheng, Qian Wang, Gong-Hua Li, Shao-Xing Dai.
Supervision: Junjuan Zheng, Jing-Fei Huang.
Validation: Junjuan Zheng, Gong-Hua Li.
Visualization: Wen-Xing Li, Jia-Qian Liu, Yi-Cheng Guo, Gong-Hua Li, Jing-Fei Huang.
Writing – original draft: Junjuan Zheng, Wen-Xing Li.
Writing – review & editing: Junjuan Zheng, Wen-Xing Li, Jia-Qian Liu, Yi-Cheng Guo, Qian Wang, Gong-Hua Li, Shao-Xing Dai, Jing-Fei Huang.
Supplementary Material
Footnotes
Abbreviations: Aβ = amyloid beta, AD = Alzheimer disease, DEGs = differentially expressed genes, FDR = false discovery rate, GEO = Gene Expression Omnibus, JEPETTO = Java Enrichment of Pathways Extended to Topology, KEGG = Kyoto Encyclopedia of Genes and Genomes, MCODE = molecular complex detection, NCBI = National Center for Biotechnology Information, NLGN = neuroligin, NRXN = neurexin, PTPRT = receptor-type tyrosine-protein phosphatase T, RMA = robust multichip average, SAM = significance analysis of microarrays, SNPs = single nucleotide polymorphisms.
J-JZ, W-XL, and J-QL contributed equally to this work.
All authors have read and approved the final manuscript.
The funder had no role in study design, data collection and analysis, decision to publish, and preparation of the manuscript.
The authors of this work have nothing to disclose.
The authors declare that they have no competing interests.
Supplemental Digital Content is available for this article.
References
- [1].Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol 2009;118:5–36. [DOI] [PubMed] [Google Scholar]
- [2].Rocca WA, Petersen RC, Knopman DS, et al. Trends in the incidence and prevalence of Alzheimer's disease, dementia, and cognitive impairment in the United States. Alzheimers Dement 2011;7:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ridge PG, Ebbert MT, Kauwe JS. Genetics of Alzheimer's disease. BioMed Res Int 2013;vol. 2013, Article ID 254954, 13 pages, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Reiman EM, Quiroz YT, Fleisher AS, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol 2012;11:1048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Han G, Wang J, Zeng F, et al. Characteristic transformation of blood transcriptome in Alzheimer's disease. J Alzheimers Dis 2013;35:373–86. [DOI] [PubMed] [Google Scholar]
- [6].Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science 1997;278:412–9. [DOI] [PubMed] [Google Scholar]
- [7].Du AT, Schuff N, Chao LL, et al. Age effects on atrophy rates of entorhinal cortex and hippocampus. Neurobiol Aging 2006;27:733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li WX, Dai SX, Liu JQ, et al. Integrated analysis of Alzheimer's disease and schizophrenia dataset revealed different expression pattern in learning and memory. J Alzheimers Dis 2016;51:417–25. [DOI] [PubMed] [Google Scholar]
- [9].Wang M, Gamo NJ, Yang Y, et al. Neuronal basis of age-related working memory decline. Nature 2011;476:210–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Alzheimer's Association. 2016 Alzheimer's disease facts and figures. Alzheimers Dement 2016;12:459–509. [DOI] [PubMed] [Google Scholar]
- [11].Van Spronsen M, Hoogenraad CC. Synapse pathology in psychiatric and neurologic disease. Curr Neurol Neurosci Rep 2010;10:207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci 2012;13:240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Marchetti C, Marie H. Hippocampal synaptic plasticity in Alzheimer's disease: what have we learned so far from transgenic models? Rev Neurosci 2011;22:373–402. [DOI] [PubMed] [Google Scholar]
- [14].Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010;26:2363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li WX, Dai SX, Wang Q, et al. Integrated analysis of ischemic stroke datasets revealed sex and age difference in anti-stroke targets. PeerJ 2016;4:e2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Marot G, Foulley JL, Mayer CD, et al. Moderated effect size and P-value combinations for microarray meta-analyses. Bioinformatics 2009;25:2692–9. [DOI] [PubMed] [Google Scholar]
- [17].Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 2001;98:5116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Winterhalter C, Widera P, Krasnogor N. JEPETTO: a Cytoscape plugin for gene set enrichment and topological analysis based on interaction networks. Bioinformatics 2014;30:1029–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen Y, Cunningham F, Rios D, et al. Ensembl variation resources. BMC Genomics 2010;11:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cuyvers E, Sleegers K. Genetic variations underlying Alzheimer's disease: evidence from genome-wide association studies and beyond. Lancet Neurol 2016;15:857–68. [DOI] [PubMed] [Google Scholar]
- [21].Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Martinez-Mir A, Gonzalez-Perez A, Gayan J, et al. Genetic study of neurexin and neuroligin genes in Alzheimer's disease. J Alzheimers Dis 2013;35:403–12. [DOI] [PubMed] [Google Scholar]
- [23].Ichtchenko K, Nguyen, Sudhof TC. Structures, alternative splicing, and neurexin binding of multiple neuroligins. J Biol Chem 1996;271:2676–82. [DOI] [PubMed] [Google Scholar]
- [24].Rowen L, Young J, Birditt B, et al. Analysis of the human neurexin genes: alternative splicing and the generation of protein diversity. Genomics 2002;79:587–97. [DOI] [PubMed] [Google Scholar]
- [25].Bierut LJ, Madden PA, Breslau N, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet 2007;16:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Novak G, Boukhadra J, Shaikh SA, et al. Association of a polymorphism in the NRXN3 gene with the degree of smoking in schizophrenia: a preliminary study. World J Biol Psychiatry 2009;10:929–35. [DOI] [PubMed] [Google Scholar]
- [27].Lachman HM, Fann CS, Bartzis M, et al. Genomewide suggestive linkage of opioid dependence to chromosome 14q. Hum Mol Genet 2007;16:1327–34. [DOI] [PubMed] [Google Scholar]
- [28].Brown SM, Clapcote SJ, Millar JK, et al. Synaptic modulators Nrxn1 and Nrxn3 are disregulated in a Disc1 mouse model of schizophrenia. Molecular Psychiatry 2011;16:585–7. [DOI] [PubMed] [Google Scholar]
- [29].Hu X, Zhang J, Jin C, et al. Association study of NRXN3 polymorphisms with schizophrenia and risperidone-induced bodyweight gain in Chinese Han population. Prog Neuropsychopharmacol Biol Psychiatry 2013;43:197–202. [DOI] [PubMed] [Google Scholar]
- [30].Selkoe DJ. Alzheimer's disease is a synaptic failure. Science 2002;298:789–91. [DOI] [PubMed] [Google Scholar]
- [31].DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration 1996;5:417–21. [DOI] [PubMed] [Google Scholar]
- [32].Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008;455:903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ushkaryov YA, Petrenko AG, Geppert M, et al. Neurexins - synaptic cell-surface proteins related to the alpha-latrotoxin receptor and laminin. Science 1992;257:50–6. [DOI] [PubMed] [Google Scholar]
- [34].Lise MF, El-Husseini A. The neuroligin and neurexin families: from structure to function at the synapse. Cell Mol Life Sci 2006;63:1833–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lim SH, Kwon SK, Lee MK, et al. Synapse formation regulated by protein tyrosine phosphatase receptor T through interaction with cell adhesion molecules and Fyn. EMBO J 2009;28:3564–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bot N, Schweizer C, Ben Halima S, et al. Processing of the synaptic cell adhesion molecule neurexin-3beta by Alzheimer disease alpha- and gamma-secretases. J Biol Chem 2011;286:2762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Manczak M, Kandimalla R, Fry D, et al. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer's disease. Hum Mol Genet 2016;25:5148–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bertoni-Freddari C, Fattoretti P, Paoloni R, et al. Synaptic structural dynamics and aging. Gerontology 1996;42:170–80. [DOI] [PubMed] [Google Scholar]
- [39].Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med 2008;14:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Almeida CG, Tampellini D, Takahashi RH, et al. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis 2005;20:187–98. [DOI] [PubMed] [Google Scholar]
- [41].Reddy PH, Mani G, Park BS, et al. Differential loss of synaptic proteins in Alzheimer's disease: implications for synaptic dysfunction. J Alzheimers Dis 2005;7:103–17. discussion 173-180. [DOI] [PubMed] [Google Scholar]
- [42].Gylys KH, Fein JA, Yang F, et al. Synaptic changes in Alzheimer's disease. Am J Pathol 2004;165:1809–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.