

Abstract
Thrombin participates in procoagulation, anticoagulation, and platelet activation. This enzyme contains anion binding exosites, ABE I and ABE II, which attract regulatory biomolecules. As prothrombin is activated to thrombin, pro-ABE I is converted into mature ABE I. Unexpectedly, certain ligands can bind to pro-ABE I specifically. Moreover, knowledge is lacking on changes in conformation and affinity that occur at the individual residue level as pro-ABE I is converted to ABE I. Such changes are transient and failed to be captured by crystallography. Therefore, we employed NMR titrations to monitor development of ABE I using peptides based on Protease Activated Receptor 3 (PAR3). Proton line broadening NMR revealed that PAR3 (44–56) and weaker binding PAR3G (44–56) could already interact with pro-ABE I on prothrombin. 1H-15N Heteronuclear Single Quantum Coherence NMR titrations were then used to probe binding of individual 15N-labeled PAR3G residues (F47, E48, L52, and D54). PAR3G E48 and D54 could interact electrostatically with prothrombin and tightened upon thrombin maturation. The higher affinity for PAR3G D54 suggests the region surrounding thrombin R77a is better oriented to bind D54 than the interaction between PAR3G E48 and thrombin R75. Aromatic PAR3G F47 and aliphatic L52 both reported on significant changes in chemical environment upon conversion of prothrombin to thrombin. The ABE I region surrounding the 30s loop was more affected than the hydrophobic pocket (F34, L65, and I82). Our NMR titrations demonstrate that PAR3 residues document structural rearrangements occurring during exosite maturation that are missed by reported X-ray crystal structures.
GRAPHIC ABSTRACT
INTRODUCTION
Thrombin (Factor IIa) is a multifunctional enzyme that plays critical roles associated with procoagulation and anticoagulation. Procoagulant functions of thrombin include activating other coagulation proteins (zymogen Factors V, VIII, and XIII), converting fibrinogen into fibrin which then polymerizes into a blood clot, and helping to activate platelets by cleaving the protease-activated receptors (PARs). An important anticoagulant function of thrombin is to activate Protein C and thereby help inhibit a set of coagulant proteins.1
Thrombin substrate specificity is regulated by a serine protease active site region, a series of surface loops, and two distant exosites. Similar to trypsin and chymotrypsin2, the catalytic triad of thrombin is composed of His, Asp, and Ser 3 (Figure 1A). The thrombin 60s (or β-insertion) loop regulates entrance into the active site and the autolysis (or γ) loop helps further control substrate specificity. In addition, thrombin is allosterically controlled by a Na+ binding site. 4, 5 Thrombin also has two anion binding exosites, ABE I and ABE II, located on opposite sides of the serine protease active site (Figure 1A). These exosites have been shown to direct substrates to the active site, contribute to opening of the active site region, and help in attracting other regulatory molecules. Ligands that bind to ABE-I include fibrinogen6, thrombomodulin7, Protease Activated Receptors PAR18 and PAR39, and the leech derived inhibitor Hirudin.10, 11 Ligands that target ABE-II include heparin12, heparin analogs13, Factor VIII14, fibrinogen γ′15 and platelet receptor protein GpIbα16. Direct allosteric linkage may exist between the ABE-I and II to modulate thrombin activity.17–20
Figure 1:
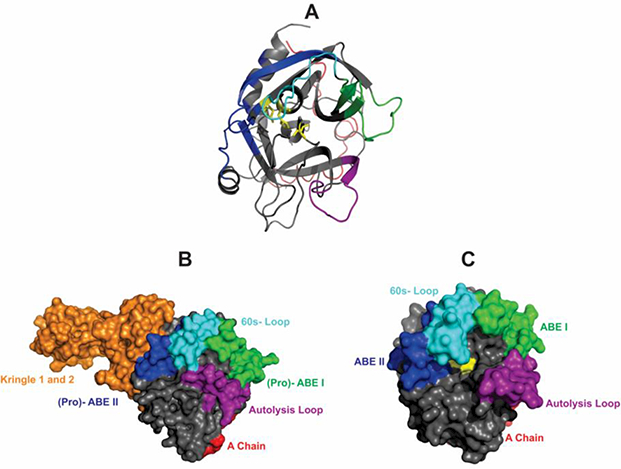
Crystal structures of thrombin (PDB 1PPB) and prothrombin (PDB 4HZH). (A) A ribbon diagram of thrombin in which the catalytic triad (His 57, Asp 102, and Ser 195) is highlighted in yellow, (Pro)- ABE I is shown in green, (Pro)- ABE II is shown in blue, Autolysis Loop is highlighted in purple, 60s- Loop is highlighted in cyan, and the A chain highlighted in red. (B) Space filling model of zymogen Prothrombin missing the Gla Domain. Kringles 1 and 2 are highlighted in orange. The other colored regions match those shown in panel A. (C) Thrombin is generated from Prothrombin following cleavages after Arg 271 and Arg 320. The space filling model highlights the removal of the Kringles (orange), the exposure of the active site (yellow), and full maturation of ABE I (green) and ABE II (blue).
Thrombin is expressed as the zymogen Prothrombin (ProT) (Figure 1B). 21, 22 As ProT is proteolytically converted to thrombin, the immature pro-ABEs develop into the mature, active ABEs I and II (Figure 1C). 21, 23 In ProT, access to pro-ABE II is blocked by the kringle-containing Fragment 1.2 (F1.2). This pro-ABE II region becomes exposed during the cleavage and activation process. 21, 24, 25 By contrast, the pro-ABE I region is not directly affected by a cleavage event. Instead, subsequent conformational changes likely lead to ABE I maturation. 21, 26
Designing new therapeutic anticoagulants that are directed toward individual thrombin exosites is a promising area of investigation. 13, 27, 28 Different segments of the mature exosite surfaces could be targeted without directly occluding the active site with a small molecule. Unexpectedly, there are reports that ABE I - directed ligands based on hirudin 29, 30 and DNA/RNA aptamers31, 32 can already bind to the immature pro-ABE I on ProT.
Similar to hirudin and the aptamers, PAR3 also contain a binding region directed to thrombin ABE I.9 PARs are members of the G-Protein Coupled Receptor family and are involved in platelet activation33, vascular remodeling, and vascular permeability.34 A fragment of PAR3 including amino acid residues 44QNTFEEFPLSDIE56 has been confirmed to bind in a specific manner to thrombin ABE I by X-ray crystallography.9 A review of the crystal structure reveals that thrombin residues located < 4 Å from the PAR3 include F34, L65, R75, Y76, R77a, I82, and K110. Chymotrypsin numbering is used for defining the thrombin sequence. Thrombin R77a is an extra amino acid residue located after E77 which is not found in chymotrypsin.
The PAR3 (44–56) sequence is similar to the ABE I-directed region found within the leech derived inhibitor hirudin.10 PAR3 could thus be a valuable new system to help decipher the specific conformational changes that occur as pro-ABE I on Prothrombin matures to the ABE I on thrombin. An examination of the X-ray crystal structures of Prothrombin24, thrombin-PAR39, and active site inhibited PPACK-thrombin3 reveals no striking differences between their pro-ABE I and ABE I regions (Figure 3).
Figure 3:
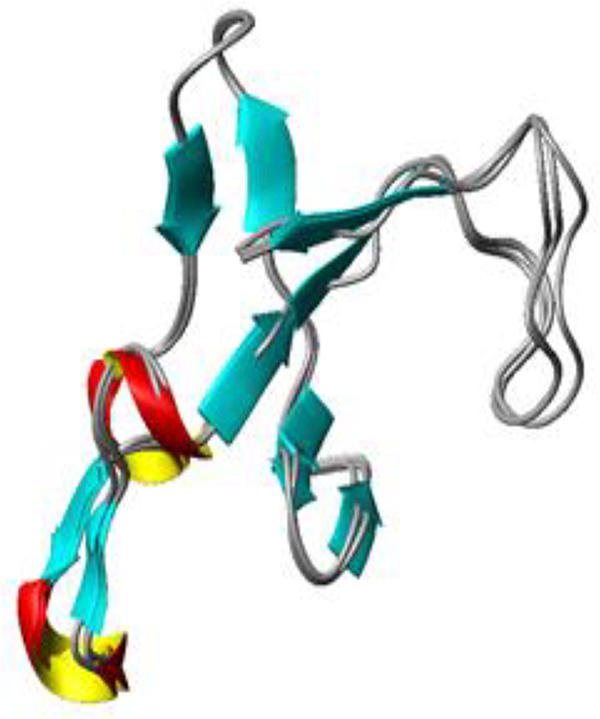
Structural alignment of the ABE I regions of Prothrombin (4HZH), thrombin-PAR3 (2PUX), and PPACK-thrombin (1PPB). Regions encompassing exosite I (residues F34-M84, thrombin straight numbering) were selected and then a backbone alignment was performed using MolMol. The backbone ABE I RMSD values for the different protein-protein comparisons were then determined: ProT and thrombin-PAR3 0.79 A, PPACK-thrombin and thrombin-PAR3 0.47A, ProT and PPACK-thrombin 0.63 Å. This analysis reveals no striking differences for the backbone atoms located in ABE I.
The backbone RMSD35 values for these three anion binding exosite regions were less than 0.8 Å. These values indicate that conformational changes may have been missed by crystallographic studies and are transient in nature. NMR could thus provide a critical alternative strategy for directly monitoring exosite maturation. To achieve this goal, we have carried out NMR titration projects comparing the binding of PAR3 amino acid residues (44–56) to the original zymogen state ProT versus the final active protease thrombin. For the first time, the ability of PAR3 fragment (44–56) to target the immature (pro)-ABE I site on ProT could be explored.
Previous hydrogen-deuterium exchange studies coupled with mass spectrometry (HDX-MS) demonstrated that regions of pro-ABE I experience increases in solvent exposure as ProT is converted to thrombin.23 These exposures are hypothesized to be part of the exosite maturation process. NMR removes the issue of only probing segments of a protein and would allow for single amino acid residue analysis. Moreover, NMR has the unique advantage of allowing KD measurements in solution at this individual residue level.
Thrombin is highly dynamic and adopts distinct states in the presence of ligands which enable this enzyme to fulfill its array of protease activities.36–40 Previous NMR studies on thrombin revealed that there are key residues within the 30s and 70s loop regions of ABE I that cannot be monitored by NMR.36–40 Some of these thrombin residues become NMR-visible following introduction of ABE I ligands whereas others do not. Intriguingly, surface loops and the ABE I region can remain flexible even when thrombin is active site inhibited. 37, 38, 40 Because of the flexible nature of thrombin, development of the mature 30s and 70s loop regions can be challenging to probe. To overcome this issue, a novel NMR titration approach was needed. For the current project, the PAR3 (44–56) peptides were 15N-labeled at the amide nitrogen of specific amino acid residues, and the ProT and thrombin remained unlabeled. 41
Critical NMR information could now be collected on how the PAR3 peptide (44–56) responds to the changing environment that occurs upon exosite maturation. NMR titration studies first revealed that the native PAR3 (44–56) sequence bound too tightly to be monitored by NMR. A P51G substitution was successful in lowering the PAR3 affinity. 1D 1H line broadening NMR studies revealed that PAR3G (44–56) could bind to the immature pro-ABE I on ProT and also the mature ABE I on thrombin. 1H-15N-heteronuclear single quantum coherence (HSQC) NMR titrations were then carried out with selectively labeled PAR3G peptides. Acidic PAR3G 15N-E48 and 15N-D54 both entered into higher affinity, intermediate exchange regimes as ProT was converted to thrombin. Strong binding events involving PAR3G D54 could be weakened with the thrombin mutant R77aA thus supporting the presence of a salt bridge. By contrast, PAR3G 15N-F47 and 15N-L52 both bound weaker to ProT/thrombin than the acidic PAR3G residues. Furthermore, 15N-F47 and 15N-L52 were able to document important changes to the hydrophobic exosite environment as pro-ABE I matures to ABE I.
MATERIALS AND METHODS
Materials.
Human plasma Prothrombin (ProT) and thrombin were purchased from Haematologic Technologies, Inc (Essex Junction, VT, USA). Recombinant thrombin mutant R77aA (chymotrypsin numbering) was a kind gift from Dr. Enrico Di Cera and Ms. Leslie Pelc (Saint Louis University School of Medicine, St. Louis, MO, USA).42 R77a is an extra thrombin amino acid residue located after E77 which is not found in chymotrypsin. For the R77aA project, the basic R77a was replaced with a small, neutral A. The active sites of the thrombin species were protected from autolysis by blocking with PPACK (D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone). This serine protease active site inhibitor was purchased from Calbiochem (San Diego, CA, USA). D2O 99.96% was from Cambridge Isotope Laboratories (Andover, MA, USA).
Synthetic Peptides.
A series of PAR3 based peptides were custom synthesized by New England Peptide (Gardner, MA, USA). PAR3 (44Q N T 15F E E F P L S 15D I E56), abbreviated as PAR3 (44–56), is the original PAR3 sequence and amino acid residues F47 and D54 were 15N-labeled at their amide nitrogens. PAR3 (44–56, P51G, 15F47, 15D54) (44Q N T 15F E E F G L S 15D I E56), abbreviated as PAR3GFD (44–56), contains a Pro51 to Gly51 substitution “G” and furthermore the F47 and D54 were 15N-labeled. PAR3 (44–56, P51G, 15E48, 15L52) (44Q N T F 15E E F G 15L S D I E56), abbreviated as PAR3GEL (44–56), also contains the P51G substitution “G” but now E48 and L52 were 15N-labeled. The purity of each synthesized peptide was verified by HPLC and MALDI-TOF mass spectrometry. Initial stock solutions of peptide were solubilized in deionized water and concentration determined by amino acid analysis (AAA Service Laboratory, Inc., Damascus, OR, USA). Peptides were later diluted into 25 mM H3PO4, 150 mM NaCl, 0.2 mM EDTA, pH 6.5 for NMR studies.
The proton chemical shift values for all the PAR3 peptide amino acid residues were determined using a combination of 2D-TOCSY and 2D-transferred NOESY 43, 44 experiments on a Varian Inova 700 MHz NMR. Standard TOCSY and NOESY pulse sequences were employed. As is typical for trNOESY experiments, the ligand-protein complex contained a 10-fold excess of PAR3 (44–56) peptide fragment, and the chemical shifts report on the solution environment encountered by the peptide in the presence of target protein.44 The amide proton 1H chemical shifts assignments derived from the TOCSY and trNOESY experiments were later matched with their respective peaks found in 1H-15N HSQC NMR experiments. Both 1H and 15N values could be followed in the HSQC titrations.
Theoretical Basis for 1D Proton Line Broadening NMR and 1H-15N HSQC NMR.
1D proton line broadening studies were performed to monitor binding of PAR3/PAR3G (44–56) peptide fragment to prothrombin versus thrombin.43, 45, 46 Protein-peptide complexes were prepared with a 10-fold excess of peptide. Peptide protons that undergo interactions experience transient on/off events that cause an increase in the proton transverse relaxation rate and/or cause changes in the observed chemical shift position depending on the timescale of the interaction. The resultant alterations in peptide proton line width/shape reflect the weighted contributions of bound and free populations. This NMR line broadening approach can help map the peptide residue protons that come in direct contact with the protein surface.
1H-15N HSQC NMR titrations were used to assess whether specific 15N-labeled residues located within a peptide ligand exhibited fast, intermediate, or slow exchange on/off a protein surface.47 In these titrations, changes in chemical shift are monitored as a function of protein-ligand ratios. For weak binding, the interaction is under the fast exchange regime (kex >> |Δω|) where kex is the exchange rate of the interaction and Δω is the resonance frequency difference between the bound and free states. Only one signal per 15N-labeled residue is observed reflecting the population-weighted average of bound and free states in terms of chemical shift, intensity, and linewidth. Furthermore, NMR chemical shifts can be used to quantitatively evaluate affinities for individual 15N labeled residues.48, 49 For the moderate affinity, intermediate exchange regime (kex ≈ |Δω|), the 15N peak may disappear during the titration due to extensive line broadening and upon increasing the bound population will later reappear. Finally, for tight binding, in which the interaction is described by the slow exchange regime (kex << |Δω|), two sets of resonances are observed: one corresponding to the free and the other to the bound state. Under this situation, the change in the intensity of the peaks will correspond to the relative populations of each state during the course of the titration.
Sample Preparation and Analysis for 1D Proton Line Broadening Experiments.
All NMR experiments were performed at pH 6.5 and at a temperature of 25 °C. Under these conditions, the highly exchangeable 15N-amide proton could still be readily observed by NMR and the pH was not far from physiological pH 7.4. To prevent autolysis, the serine protease inhibitor PPACK was added to thrombin at a ratio of 4:1, and the mixture was incubated at 37 °C for 30 mins. Previous NMR relaxation studies have confirmed that the ABE I region remains flexible in PPACK-inhibited IIa and can engage in long-range communication across the enzyme surface.40 Plasma versions of ProT and PPACK-thrombin were buffer exchanged into NMR buffer (25 mM H3PO4, 150 mM NaCl, 0.2 mM EDTA, pH 6.5) using a Vivaspin 2 ultrafiltration unit with a 5000 Da molecular weight cutoff (Sartorius, Gottingen, Germany). Protein concentrations were determined using an extinction coefficient E1%280 nm of 18.3 for thrombin and 13.8 for ProT.
Ligand-Protein complexes with ratios of at least 10:1 were then prepared. The complexes included 1 mM PAR3 (44–56) with 74 μM ProT, 1 mM PAR3 (44–56) with 77 μM PPACK-thrombin, 960 μM PAR3G (44–56) with 76 μM ProT, and 1 mM PAR3G (44–56) with 76 μM PPACK-thrombin. Peptide at a minimum concentration of 1mM was used as a free ligand control. The resultant NMR experiments and all others in this project were carried out on a Varian Inova 700 MHz spectrometer with a triple resonance cold probe and pulsed-field Z-axis gradients run at 25 °C. The 1h NMR spectra were processed using Mnova NMR (Mestrelab Research software).
2D 13C-1H HSQC Natural Abundance Experiments.
Before proceeding to 1H-15NHSQC NMR titrations, we performed 1H-13C HSQC natural abundance experiments on both PAR3 (44–56) and PAR3G (44–56) to assess whether the proline to glycine substitution would change the PAR3 peptide conformations. For this project, 1mM PAR3 and 1mM PAR3G were prepared in NMR buffer (25 mM H3PO4, 150 mM NaCl, 0.2 mM EDTA, pH 6.5). Parameters for the 2D 1H-13C HSQC project included 128 transients with 64 complex points in the indirect dimension, sweep width of 8064.5 Hz and 24635.3 Hz for the direct and indirect dimensions, respectively, and 3000 complex points acquired in the direct dimension. 2D 1H-13C HSQC data were processed using NMRPipe and nmrDraw and then further visualized using Sparky. The ability to superimpose the spectra of PAR3 (red) and PAR3G (black) confirmed that replacement of proline with flexible glycine did not change the overall peptide structure (Figure S1)
1D and 2D 1H-15N HSQC NMR Titration.
ProT and active site inhibited PPACK-thrombin were exchanged into NMR buffer using Vivaspin 2 ultrafiltration units with a 5000Da molecular weight cutoff. A series of protein-ligand complexes were then prepared involving the PAR fragments PAR3 (44–56), PAR3GFD (44–56), and PAR3GEL (44–56). ProT and PPACK-thrombin exhibit limited solubility at the high concentrations typically used for NMR titrations. As a result, an alternative titration strategy was employed for the current NMR project. The protein concentration typically started in the 100–200 μM range and was serially diluted while maintaining a constant concentration of 15N-labeled ligand. These conditions were achieved by removing a certain volume of protein-ligand solution and replacing it with the same volume of ligand solution. The titrations were thus initiated with a high protein-ligand ratio and ended with low protein-ligand ratios all while maintaining a constant peptide ligand concentration. The NMR titrations were therefore monitoring the ability of ProT or thrombin to bind to the 15N-labeled PAR3 fragment.
For the first PAR3 binding studies, the starting complexes included 50 μM PAR3 (44–56) in either 137 μM ProT or 210 μM PPACK-thrombin. The serial dilutions resulted in ProT to PAR3 ratios that spanned from 3:1 to 0.1:1. PPACK-thrombin to PAR3 ratios spanned from 4:1 to 0.1:1. For the PAR3Gfd binding studies, starting complexes included 37.5 μΜ PAR3GFD (44–56) in 70 μΜ ProT or in PPACK-thrombin. Ratios of ProT to PAR3GFD during the titrations then spanned from 2:1 to 0.3:1. PPACK-thrombin to PAR3Gfd ratios spanned from 2:1 to 0.05:1.
To evaluate the importance of electrostatic interactions between the 15N-labeled D54 of PAR3Gfd and R77a of wild-type thrombin, a series of 1D and 2D 1H-15N HSQC titrations were carried out involving recombinant thrombin mutant R77aA and PAR3GFD. The stock solution of R77aA thrombin was in 20 mM Tris pH 7.4, 400mM NaCl. After PPACK treatment, active site inhibited R77aA thrombin was buffer exchanged into 25 mM H3PO4, 150 mM NaCl, 0.2 mM EDTA, pH 6.5 using a 3 mL Slide-A-Lyzer Dialysis cassette G2 (Thermo Scientific, Rockford, IL, USA) with a 3500 Da Molecular weight cutoff and then concentrated using a 5000 Da Molecular Weight Cut Off Vivaspin 2 ultrafiltration unit. Starting complexes included 50 μM PAR3Gfd (44–56) with 150 μM PPACK-thrombin (R77aA). PPACK-thrombin (R77aA) to PAR3GFD ratios spanned from 3:1 to 0.1:1.
For the PAR3GEL binding studies, the starting complexes included 50 μM PAR3GEL(44–56) with either 180 μM ProT or 211 μM PPACK-thrombin. Both ProT to PAR3GEL and PPACK-thrombin to PAR3GEL complexes spanned from 4:1 to 0.1:1 protein to ligand ratios.
Parameters for the 1D 1H-15N HSQC titrations included 512 transients, sweep width of 7022.5 Hz, 1242 complex points in the direct dimension with PAR3 and 4096 with the PAR3G complexes. Parameters for the 2D 1H-15N HSQC titrations included 16 transients with 64 complex points in the indirect dimension, sweep width of 7022.5 Hz and 1944.300 Hz for the direct and indirect dimensions, respectively, and 1242 complex points acquired in the direct dimension. 15N labeled free peptide was used as a control for each titration. 1D 1H-15N HSQC data were stacked using Mnova NMR and 2D 1H-15N HSQC data were processed using NMRPipe and nmrDraw and then further visualized using Sparky.
Quantitative estimates of binding interactions between individual 15N-labeled peptide ligand residues and specific proteins were determined using in-house scripts written using Python.50 Information provided to such scripts included the total enzyme [𝑃0 ] and total peptide concentrations [𝐿0 ] employed in the different steps of the HSQC titrations. In addition, the NMR chemical shift difference (Δobs) between each set of free and bound conditions was provided. For the current NMR project, it is important to note that the peptide ligand concentration was kept constant and the protein was serially diluted. As a result, the NMR titrations were measuring the binding of protein to a defined peptide ligand concentration. The equation to determine the binding affinity values (KD) was thus modified so that the denominator now contains [L0] instead of the more typical [Po].47, 48
NMR titrations were performed at least in triplicate to determine KD values involving plasma derived ProT and thrombin. Duplicate titrations were carried out for the thrombin R77aA. An error analysis of the KD values was carried out using a Monte-Carlo approach in which a 10% error was imposed on the serially diluted thrombin concentration. 50
RESULTS
1D Proton Line Broadening NMR Studies with PAR3 (44–56).
One dimensional 1H line broadening NMR experiments were carried out on PAR3 (44–56) in the presence of ProT versus PPACK-thrombin (Figure 4 A-C). The active site inhibitor PPACK (D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone) was used to protect thrombin from autolysis during the NMR experiments. Proton (1H) peak broadening could be detected for aliphatic, amide, and aromatic protons of PAR3 in complex with ProT. Such results indicate that a binding surface for PAR3 (44–56) is already available within the immature pro-ABE I on ProT. Substantial peak line broadening was also observed when the peptide was introduced into a solution of PPACK-thrombin containing the mature exosite ABE I. Such broadening is impressive considering the lower molecular weight of thrombin (37 kDa) versus ProT (72 kDa).45 The overall, extensive thrombin-induced peak broadening provides further justification that mature ABE I is well suited for accommodating PAR3 (44–56). The 1H chemical shift overlaps observed across the different Figure 4 panels make it difficult to quantitatively evaluate individual proton line effects.
Figure 4:

Proton line broadening spectra for PAR3 and PAR3G peptides in the presence of prothrombin and PPACK-thrombin. All NMR samples were in 25mM H3PO4, 150 mM NaCl, 0.2 mM EDTA and 10 % D2O (pH 6.5). (A) 1D 1H-NMR spectrum for 1 mM PAR3 (44– 56) peptide in solution (B) 1D 1H-NMR spectrum for 1mM PAR3 (44–56) peptide in the presence of 74 μM ProT (C) 1D 1H-NMR spectrum for 1 mM PAR3 (44–56) peptide in the presence of 77 μM PPACK-IIa (D) 1D NMR spectmm for 1 mM PAR3G (44– 56) peptide in solution (E) 1D NMR spectrum for 960 μM PAR3G (44–56) peptide in the presence of 76 μM ProT. (F) 1D NMR spectrum for 1000 mM PAR3G (44–56) peptide in the presence of 76 μM PPACK-IIa. Line broadening was observed for residues of PAR3 (44–56) and PAR3G (44–56) when either peptide was bound to prothrombin and PPACK-Ha. The amide protons that were later selected for HSQC titrations are labeled.
15N-HSQC Titration Studies With PAR3 (44–56) Labeled at D54 and F47.
An HSQC titration project was designed to systematically characterize binding of PAR3-based peptides that are selectively labeled at amide nitrogen locations. The X-ray crystal structure of the thrombin - PAR3 (44–56) complex was first consulted to select sets of acidic and hydrophobic amino acid residues of PAR3 that display interactions with the 30s and 70s loop regions on thrombin 9 (Figure 2). 2D total correlation spectroscopy (TOCSY) spectra of the bound PAR3 peptide then provided a valuable guide for choosing amino acid residues with amide chemical shift positions that would be less likely to overlap in the proton dimension during the course of the PAR3-protein 1H-15N HSQC titrations.
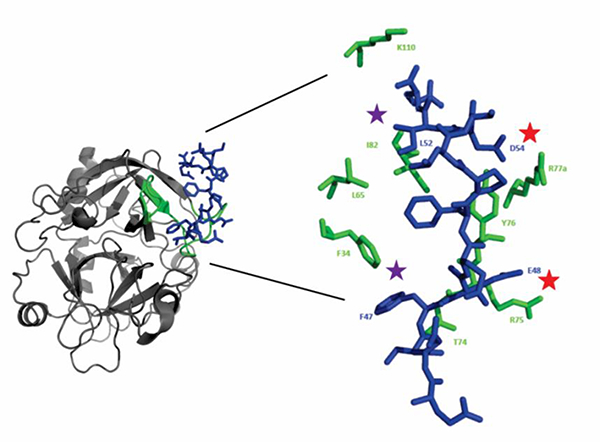
Figure 2:
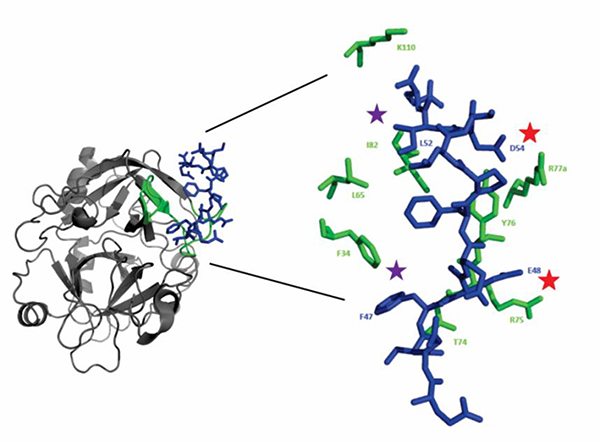
Crystal structure of thrombin in complex with murine PAR3 fragment (44–56). The thrombin surface is rendered in gray ribbons and the PAR3 peptide as blue sticks. Thrombin ABE-I residues that are located < 4 Å from PAR3 (44–56) are displayed as green ribbons or green sticks. The PAR3 residues later chosen for 15N-HSQC titration studies include the acidic residues D54 and E48 (red star) and the hydrophobic residues F47 and L52 (purple star).
The first amino acid residues selected included PAR3 F47 and D54. According to the X-ray crystal structure, acidic PAR3 D54 makes a salt bridge contact with thrombin R77a of the 70s ABE I loop whereas PAR3 F47 exhibits π-π stacking interactions with thrombin F34 in the 30s ABE I loop region 9 (Figure 2). 15N-HSQC NMR titrations were thus carried out with PAR3 (44–56) labeled at 15N-D54 and 15N-F47. These two PAR3 residues both exhibited proton line broadening in the presence of ProT and PPACK-thrombin confirming contact with the target protein in solution (Figure 4A-C).
The HSQC titrations started out with a 3:1 ratio of ProT to PAR3 (containing the 15N-D54 and 15N-F47 residues). Extensive peak broadening was observed for both 15N-D54 and 15N-F47 until the ProT:PAR3 ratios were serially diluted down to 0.3:1 (D45) or 0.6:1 (F47) and lower. With PPACK-thrombin, peaks for both PAR3 residues could not be detected until ratios of 0.3:1 and lower (Figure S2 and S3). These NMR results suggest that PAR3 D54 and F47 can already interact with pro-ABE I on ProT, and tightens in the presence of PPACK-thrombin with a mature ABE I.
Proton Line Broadening NMR and 15N-HSQC NMR Titration Studies With PAR3GFD (44–56) Labeled at D54 and F47.
The extensive line broadening observed for PAR3 (44–56) bound to PPACK-IIa was a major hindrance for 15N-NMR titration studies, and binding affinities could not be determined. To weaken this interaction, a modified sequence was designed in which the P51 was replaced with a flexible glycine (P51G). The resultant PAR3GFD (44–56) showed 1D proton line broadening with both ProT and PPACK-thrombin (Figure 4D-F). Natural abundance 13C HSQC spectra were collected for both PAR3 peptides. As seen in Figure S1, the two spectra show good overlap suggesting that the Pro to Gly substitution has not caused substantial changes to the conformation nor the chemical environments of the two peptides.
1H-15N HSQC titrations were then carried out with PAR3GFD containing 15N-labeled D54 and F47. The ProT to PAR3GFD ratios spanned from 2:1 to 0.3:1 and PPACK-thrombin to PAR3GFD ratios spanned from 2:1 to 0.05:1. Results from 1D displays of the 15N-HSQC studies revealed that the PAR3GFD D54 peak became more broadened than the F47 in the presence of ProT and PPACK-thrombin (Figure S4). Further information about interactions occurring with these two proteins was obtained from examining the 2D HSQC titrations (Figures 5A and 5B). In the presence of ProT, the PAR3GFD residue D54 exhibited changes in chemical shift that resemble a fast exchange scenario corresponding to an interaction with weakened affinity. This interaction creates a new protein environment for the ligand. By contrast, the PAR3GFD residue F47 residue exhibited little changes in chemical shift during the ProT titration suggesting this residue did not experience a substantial change in its chemical environment relative to that of the free peptide (Figure 5A). The project then proceeded to titrations with PPACK-thrombin where additional new effects were observed. The 2D HSQC crosspeak for D54 underwent extensive line broadening and could only be observed for thrombin-PAR3GFD ratios of 0.16:1 to 0.05:1 (Figure 5B). Such a peak broadening reflects improved interactions with the mature ABE I site and is consistent with intermediate exchange conditions. Interestingly, the F47 residue was observed to undergo a wider range of chemical shift changes for the 2:1 to 0.05:1 titration series. The F47 was now experiencing a different chemical environment than what had been observed with ProT containing an immature (pro)-ABE I.
Figure 5:
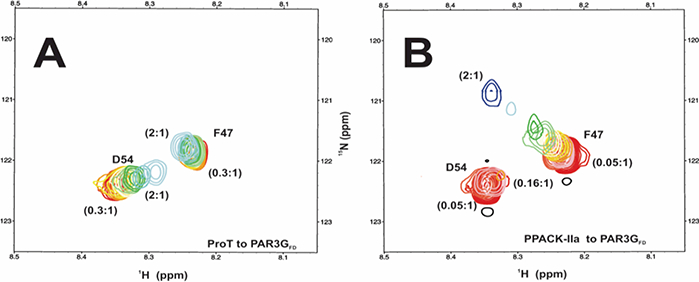
2D 1H-15N HSQC NMR titrations of PAR3GFD (44–56) in the presence of ProT and PPACK-IIa. All NMR samples were in 25mM H3PO4, 150 inM NaCl, 0.2 mM EDTA and 10 % D2O (pH 6.5). (A) For the PAR3GFD binding studies with ProT, starting complexes included 37.5 μM PAR3GFD (44–56, 15N-F47, 15N-D54) in 70 μM ProT. The serial dilutions resulted in ProT to PAR3GFD ratios that spanned from 2:1 to 0.3:1. (B) For PPACK -IIa, starting complexes included 37.5 μM PAR3GFD (44–56, 15N-F47, 15N-D54) in 70 μM PPACK-IIa. The serial dilutions resulted in PPACK-IIa to PAR3GFD ratios that spanned from 2:1 to 0.05:1. Representative data sets are shown. Colors for the HSQC crosspeaks span from blue (highest protein-peptide ratio) to red (free peptide).
The chemical shift changes for the D54 and F47 titrations proceed in the same directions across the NMR panels when probing ProT vs PPACK-IIa (upfield for F47 and downfield for D54). These similarities are consistent with the PAR3 and PAR3G peptides interacting within the same binding region of ProT vs PPACK-IIa. Nonspecific binding to a separate area on ProT is not evident. In further support of this proposal, Andersen PJ et al. confirmed that hirudin (54–65) can bind specifically to the (pro)-exosites of prothrombin and thrombin.30 Moreover, HDX-MS studies demonstrated that hirudin and PAR3 (44–56) both bind to the thrombin ABE I region 65–84.19
NMR HSQC titrations can be used to quantitatively characterize binding interactions for individual 15N-labeled amino acid residues. These calculations work the best when there are distinct changes in chemical shift position when 15N-labeled peptide amino acid residues interact with a target protein. Furthermore, the binding curves should approach saturation. 47, 48 With the current project, peptide concentration remained constant, and the protein solutions were serially diluted. For these protein-peptide ligand systems, affinity estimates could be made for some of the 15N-labeled PAR3G (44–56) amino acid residues. Based on the titration data collected, the D54 had a binding affinity (KD) of 65 ± 12 μM in the presence of ProT and a maximal chemical shift difference (Δωmax) = 0.39± 0.03 ppm. (Table 1, Figure 6B). The interaction became stronger with thrombin, however, the binding of D54 was too tight and the peak broadening was too severe to determine a KD value from the titration curve. The labeled PAR3 F47 residue had an estimated KD value of 64 ± 8 μM and a Δωmax = 0.23± 0.01 ppm for ProT and a KD value of 40 ± 10 μM and a Δωmax = 1.98 ± 0.19 ppm with Thrombin (Table 1, Figures 6A and 6C). Curiously, F47 experienced a marginal increase in affinity upon exosite maturation even though this residue clearly encountered a new environment reflected by its now larger chemical shift spread (Δωmax) (Table 1, Figure 6C).
Table 1:
KDs and |Δωmax| determined from 2D HSQC titrations for 15N labeled PAR3GFD and PAR3GEL bound to human Prothrombin and Thrombin (wildtype and mutant). For these NMR titrations, the peptide ligand concentrations were kept constant and the protein concentrations were serially diluted. Estimated KDs were calculated using in house scripts written in Python. Experimental data employed in the calculations include the individual protein and peptide concentrations and also the 15N-NMR chemical shift differences between each set of free and bound conditions. The plasma derived ProT and IIa titration series were carried out at least in triplicate. The studies with recombinant R77aA-IIa were done in duplicate. Error analysis was carried out using a Monte-Carlo approach assuming a 10% error in the serially diluted protein samples. See Materials and Methods for more details.
| Peptide | Residue | ProT | |Δωmax| (ppm) |
Wild Type PPACK-IIa |
|Δωmax| (ppm) |
R77aA PPACK-IIa |
|---|---|---|---|---|---|---|
| PAR3GFD | F47 | 64 ± 8 μM | 0.23 ±0.01 | 40 ± 10 μM | 1.98± 0.2 | 173 ± 85 μM |
| PAR3GFD | D54 | 65 ± 12 μΜ |
0.39 ±0.03 | Too tight to calculate |
Insufficient data points |
168 ± 88 μM |
| PAR3GEL | L52 | 124 ± 27μM |
0.37 ±0.04 | 47 ± 6 μM | 1.84± 0.08 | salt bridge not involved |
| PAR3GEL | E48 | >200 μM | ΝΑ | Too tight to calculate |
Insufficient data points |
salt bridge not involved |
Figure 6:
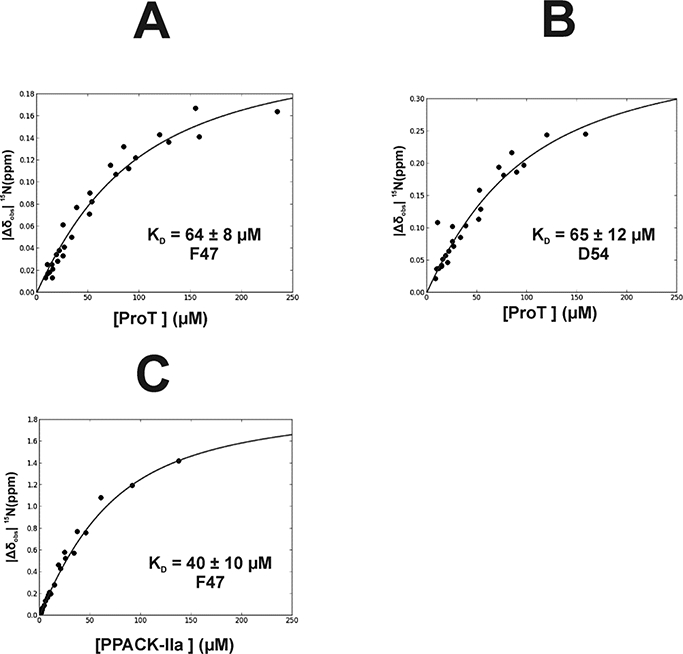
Determination of Binding Affinity (KD) for 15N-labeled F47 and D54 of PAR3GFD interacting with Prothrombin and PPACK-IIa. For this NMR titration series, the peptide ligand concentration was kept constant and the ProT and PPACK-IIa concentrations were serially diluted. As a result, the NMR titrations were measuring the binding of protein to a defined peptide concentration. (A) Interactions between ProT and PAR3G 15N-F47 led to a KD = 64 ± 8 μM, (B) ProT and PAR3G 15N-D54 led to a KD = 65 ± 12 μM, and (C) PPACK-IIa and PAR3G 15N-F47 led to a KD = 40 ± 10 μM. NMR titrations were done in triplicate. The reported KD values were determined using in-house scripts written using Python. The term |Δδobs| 15Nppm = δ15NBound - δ15Ν Free reflects the absolute difference in chemical shift between the bound and free states of the particular 15N-residue. Error analysis was carried out using a Monte-Carlo approach assuming a 10% error in the serially diluted protein samples. See Materials and Methods for more details.
15N-HSQC NMR Titration Studies With PAR3G (44–56) Labeled at D54 and F47 Interacting With the Thrombin Mutant R77aA.
The HSQC titrations described above revealed a significant increase in affinity for PAR3 residue D54 as ProT is converted to thrombin. Acidic PAR3 D54 has been reported to make salt bridge contact with the basic thrombin residue R77a 9 (Figure 2). Interactions between these two residues likely become more effective once the mature ABE I is formed. To probe whether electrostatic interactions are occurring between R77a and D54, 1H-15N HSQC titrations were performed with a thrombin R77aA - PAR3GFD complex (Figure 7). Unlike the wild-type PPACK-thrombin titrations, D54 and F47 chemical shifts could be followed for the full titration series for PPACK-thrombin R77aA: PAR3Gfd ratios of 3:1 to 0.13:1. The D54 crosspeak exhibited a broad range of chemical shifts and even overlapped with the F47 crosspeak for a portion of the titration (Figures 7A and 7B). Note in Figure 7A that at the start of titration (3:1 protein to peptide) the amide proton for PAR3G F47 was at 8.3 ppm and for PAR3G D54 at 8.2 ppm. By a ratio of 0.7 to 1, the two peaks overlapped into a single peak. As the PPACK-R77aA thrombin was further diluted, the F47 and D54 peaks continued to change resonance positions and eventually matched those of the free PAR3GFD peptide. A similar set of trends can be observed in the 2D HSQC crosspeaks shown in Figure 7B. Overall, the 2D crosspeak patterns for both F4 and D54 were consistent with fast exchange conditions. For PAR3G D54, the estimated KD was 168 ± 88 μM and the Δωmax = 1.83 ± 0.62 ppm whereas for PAR3G F47 the values were a KD of 173 ± 85 μM and the Δωmax = 1.79 ± 1.05 ppm (Table 1, Figures S5A and S5B). The removal of the salt bridge between PAR3 D54 and thrombin R77a clearly weakened the interaction of D54 with the ABE I surface, and a KD value could now be estimated. In addition, a roughly 3-fold loss in affinity was observed for PAR3G F47 in the presence of the R77aA thrombin mutant. Even with the weakened affinity, thrombin R77aA still incurred large structural changes as reflected in the >1.8 ppm changes in the maximal chemical shift.
Figure 7:
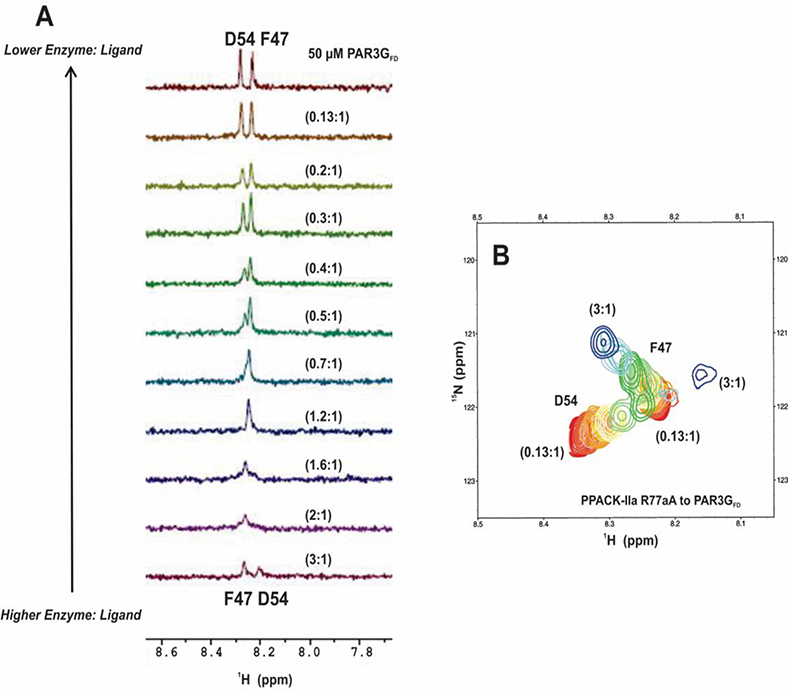
1D and 2D 1H-15N HSQC NMR titrations of PAR3GFD (44–56) in the presence of PPACK-R77aA. All NMR samples were in 25mM H3PO4, 150 mM NaCl, 0.2 mM EDTA and 10 % D2O (pH 6.5). (A) For the 1D HSQC NMR titrations, the starting complexes included 50 μΜ PAR3GFD (44– 56, 15N-F47, 15N-D54) in 150 μM PPACK-R77aA. The serial dilutions resulted in PPACK-R77aA to PAR3GFD ratios tliat spanned from 3:1 to 0.1:1. Note that at the start of titration (3:1 protein to peptide) the amide proton for PAR3G F47 was at 8.3 ppm and for PAR3G D54 at 8.2 ppm. By a ratio of (0.7 to 1), the two peaks overlapped into a single peak. As the PPACK-R77aA thrombin was further diluted, the F47 and D54 peaks contained to change resonance positions and eventually matched those of the free PAR3GFD peptide. Representative data sets are shown. (B) For the 2D 1H-15N HSQC titrations, starting complexes included 50 μM PAR3GFD (44–56,15N-F47,15N-D54) in 150 μΜ PPACK-R77aA. The serial dilutions resulted in PPACK-thrombin to PAR3GFD ratios that spanned from 3:1 to 0.1:1. Representative data sets are shown. Colors for the 2D ‘Η-^Ν HSQC crosspeaks span from blue (highest protein-peptide ratio) to red (free peptide).
15N-HSQC NMR Titration Studies With PAR3G (44–56) Labeled at L52 and E48.
Two additional PAR3 residues were chosen to probe the environments of the 30s and 70s loop regions of (pro)-ABE I. The E48 of PAR3 (44–56) makes a salt bridge with R75 of thrombin and L52 of PAR3 (44–56) is positioned within a hydrophobic pocket containing the thrombin residues F34, L65 and I82 (Figure 2).9 As observed with F47 and D54, the two new PAR3 residues E48 and L52 also exhibited 1D proton line broadening when complexed with ProT and PPACK-IIa (Figure 4). Thus, HSQC NMR titrations could be carried out with PAR3G (44–56) labeled at 15N-E48 and 15N-L52.
The titration ratios for ProT-PAR3GEL and (PPACK-thrombin)-PAR3GEL spanned from 4:1 to 0.1:1. For the ProT bound complex, 1D and 2D displays of HSQC titration peaks could be followed for all the titrations points (Figure S6A and Figure 8). Unlike PAR3Gfd, the binding affinities for L52 and E48 were weaker resulting in KD values of 124 ± 27 μM and > 200 μM, respectively (Table 1, Figure S7A and S7B). Moreover, both E48 and L52 exhibited fewer changes in chemical shift position. These results suggested that E48 and L52 did not encounter much of a change in binding environment when they were tethered weakly to the surface of ProT. Furthermore, we propose that ProT R75 is less available for interacting with E48 of PAR3 than the distinct interactions already observed between ProT R77a and D54 of PAR3. When the peptide was titrated with PPACK-thrombin (Figure S6B and Figure 8B), L52 exhibited a greater change in chemical shift position. By contrast, E48 resembled intermediate exchange on/off the thrombin surface. Extensive peak broadening could already be observed at 1.2:1 protein to ligand ratios. In the presence of PPACK-thrombin, L52 showed a 3-fold improvement in Kd (47 ± 6 μM) and E48 underwent extensive peak broadening preventing a KD value from being calculated. For L52, a Δωmax = 0.37 ± 0.04 ppm obtained for ProT and was increased to Δωmax =1.84 ± 0.08 ppm upon conversion to thrombin (Table 1, Figures S7A and S7C).
Figure 8:
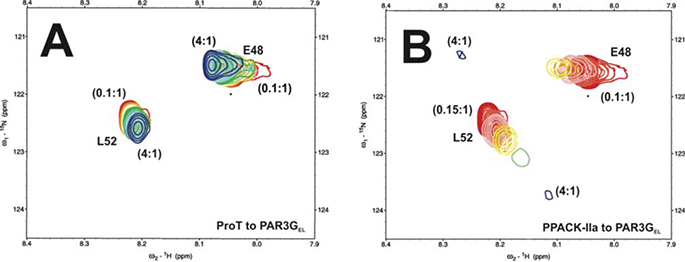
2D 1H-15N HSQC NMR titrations of PAR3GEL (44–56) in the presence of ProT and PPACK-IIa. All NMR samples were in 25mM H3PO4, 150 mM NaCl, 0.2 inM EDTA and 10 % D2O (pH 6.5). (A) For the PAR3GEL binding studies with ProT, starting complexes included 50 μM PAR3GEL (44–56, 15N-E48, 15N-L52) in 180 μM ProT. The serial dilutions resulted in ProT to PAR3G ratios that spanned from 4:1 to 0.1:1. (B) For the PPACK - IIa, starting complexes included 50 μM PAR3GEL (44–56, 15N-E48,15N-L52) in 211 μM PPACK-Ha. The serial dilutions resulted in PPACK-Ha to PAR3GEL ratios that spanned from 4:1 to 0.1:1. Representative data sets are shown. Colors for the 2D 1H-15N HSQC crosspeaks span from blue (highest protein-peptide ratio) to red (free peptide).
DISCUSSION
With thrombosis becoming a global disease burden, the urge to develop better anticoagulants has increased.51 Novel oral anticoagulants 52, 53 that target the active sites of FXa and thrombin 54 have shown much medical promise but reversing their therapeutic activities during heavy bleeding scenarios can be challenging.55, 56 Drug candidates 27, 28, 57 that target thrombin exosite ABE I are an alternative strategy. Thrombin is, however, proteolytically derived from zymogen Prothrombin (Figure 1). Unexpectedly, some ABE I ligands can already bind to the immature pro-ABE I site on this zymogen.29, 30, 32, 58, 59 Future ABE I directed therapeutics might therefore be designed to target or avoid specific regions of pro-ABE I on ProT versus ABE I on thrombin.
With our NMR titration approaches, we characterized, for the first time, the binding of ABE I directed PAR3 peptides to ProT versus PPACK-thrombin. Previous NMR relaxation studies had already revealed that communication from ABE I toward the active site region is still preserved with PPACK-thrombin.37 Our 1D 1H line broadening NMR results demonstrated that PAR3 (44–56) and its weaker binding version PAR3G (44–56) could interact with both ProT and PPACK-thrombin (Figure 4).9 NMR 1H-15N-HSQC titration studies were then carried out with a series of 15N-labeled PAR3 peptides to characterize ability to bind ProT versus PPACK-thrombin (Figure 5–8, Figure S1–6). Such an approach has the distinct advantage of monitoring exosite maturation at the individual amino acid residue level. Isothermal titration calorimetry, fluorescence, and surface plasmon resonance all provide a global KD fit using all peptide amino acid residues. NMR, by contrast, preserves the ability to measure individual binding interactions in solution and thus document their contributions. Moreover, NMR works best with weaker affinity systems.
For the current NMR project, PAR3 peptides were prepared with 15N-amide labeling of F47, E48, L52, and D54. HSQC titrations were performed at the typical NMR pH of 6.5. The acidic and hydrophobic residues 15N-D54 and 15N-F47 on PAR3 (44–56) were examined first. This native PAR3 sequence bound too tightly to thrombin hindering ability to assess KD values by NMR titration methods. Prior fluorescence titrations revealed a KD of 2 μM for a related PAR3 sequence thus confirming our observation of intermediate/slow exchange conditions in the NMR titrations.60 Greater success was achieved with PAR3Gfd (44–56) in which P51 was replaced with G51. With ProT, PAR3G D54 and F47 both had individual KD values in the 65 μM range. The immature pro-ABE I can thus accommodate both residues to a similar extent. Upon maturation to ABE I, PAR3G D54 bound too tightly to PPACK-thrombin for binding affinity calculation. By contrast, the F47 binding affinity increased only modestly as ProT was converted to thrombin. Interestingly, the F47 also documented a substantial change in chemical environment (Δωmax =1.8 ppm) within the 30s loop region following activation to thrombin. Structural rearrangements are proposed to occur in the vicinity of the ProT/thrombin residue F34 but do not result in a significant change in affinity (Figure 2).
The PAR3G D54 residue was hypothesized to be influenced by its electrostatic interaction with thrombin R77a (Figure 2).9 This acidic PAR3 residue may have properties similar to those found in hirudin.61 The hirudin sequence (56FEEI59) has already been proposed to electrostatically steer toward the ABE I surface.61, 62 Ionic tethering between acidic hirudin residues and specific basic residues on ABE I might then be promoted followed by stabilizing hydrophobic interactions.61, 62 These hypotheses could now be tested with the PAR3G sequence and thrombin R77aA. As predicted, the affinity for D54 weakened 3-fold upon loss of its salt bridge partner. In response, the F47 also displayed a much weaker affinity value.
PAR3G residues E48 and L52 provided the opportunity to probe two additional regions of ProT versus PPACK-thrombin. E48 makes a salt bridge with thrombin R75, a neighboring region of the 70s loop. PAR3G L52 interacts with a cluster of hydrophobic residues including thrombin F34, L65, and I82 (Figure 2).9 Curiously, both E48 and L52 exhibited only minor changes in chemical shift upon binding to ProT. Furthermore, the estimated individual KD values for L52 and E48 bound to ProT were two or more-fold weaker than those for D54 and F47. The pro-ABE I surface on ProT may not accommodate these two residues as well as it does PAR3 D54 and F47. Similar to PAR3G D54, there was extensive line broadening for E48 upon conversion to thrombin and KD values could not be determined. By contrast, L52 exhibited a broad series of chemical shift changes during the thrombin titration (Δωmax =1.8 ppm). Interestingly, the estimated KD for L52 binding (KD = 47 ± 6 μM) to PPACK-thrombin was similar to the value determined for F47 (KD = 40 ± 10 μM). Both L52 and F47 interact at the more hydrophobic surface area of ABE I with a common overlapping influence from thrombin F34 (Figure 2).
CONCLUSIONS
Overall, our solution NMR studies have elucidated changes in exosite environment that occur as zymogen ProT is converted to active thrombin. 1D proton line broadening NMR and 1H-15N-HSQC studies demonstrated that PAR3G E48 and D54 could already interact with the ProT and the affinity increased upon maturation to thrombin. In the NMR studies, the 3-fold tighter binding affinity value for D54 versus E48 suggests that the 70s loop region surrounding thrombin R77a may be better oriented to bind PAR3G D54 than the interaction between thrombin R75 and PAR3G E48 (Figure 2). Moreover, a close review of the crystal structure overlays in Fig 3 suggests that the 70s loop region of ProT may assume an orientation that is preconfigured to that of PAR3-thrombin and PPACK-thrombin. As the ionic PAR3G D54 - ProT/IIa R77a and PAR3G E48-ProT/IIa R75 interactions are stabilized, the PAR3G F47 and L52 are proposed to take further advantage of interacting with thrombin F34 (within the 30–40s loop) and the thrombin hydrophobic cluster F34, L65, and I82 (Figure 2). The aromatic PAR3G F47 and the aliphatic PAR3G L52 both reported on significant changes in the chemical environment upon conversion of ProT to thrombin. The region surrounding the ProT/IIa 30s loop appears to be more affected than the hydrophobic pocket containing F34, L65, and I82.
Curiously, no striking differences are observed between the crystal structures of immature pro-ABE I in ProT versus the mature ABE I regions of thrombin-ligand complexes (Figure 3). Moreover, there are no X-ray crystal structures for a ligand bound to the pro-ABE I region of ProT. We can speculate that there are transient structural states within this coagulation protein system that are differentially explored by ProT and thrombin. Some of these states may be shared whereas others may be unique. Interestingly, ProT already possesses a binding competent state. Additional research has implicated transient, or lowly populated, structures as playing an essential role in molecular interactions. 63–65 Thus, we believe that the mature thrombin likely visits a binding competent state more often than ProT thereby promoting the higher affinity of PAR3 for thrombin.
In conclusion, our NMR titration studies have clearly revealed that individual PAR3 amino acid residues are documenting structural rearrangements that occur upon ABE I maturation. Moreover, these amino acid residues make individual contributions to the overall binding affinity. The knowledge gained from this NMR project may be used to help decipher the characteristics features of pro-ABE I versus ABE I that become accessible to physiological ligands or future drug candidates.
Supplementary Material
Figure S1: 2D 1H-13C natural abundance HSQC NMR for PAR3 (44–56) and PAR3G (44–56). Figure S2:1D 15N-HSQC NMR titrations of PAR3FD (44–56) in the presence of ProT and PPACK- IIa. Figure S3: 2D 15N HSQC NMR titrations of PAR3FD (44–56) in the presence of ProT and PPACK- IIa. Figure S4:1D 15N-HSQC NMR titrations of PAR3GFD (44–56) in the presence of ProT and PPACK- IIa. Figure S5: 1D 15N HSQC NMR titrations of PAR3GEL (44–56) in the presence of ProT and PPACK- IIa. Figure S6: Determination of Binding Affinity (KD) for 15N-labeled F47 and D54 of PAR3GFD interacting with PPACK-R77aA IIa. Figure S7: Determination of Binding Affinity (KD) for 15N-labeled L52 and E48 of PAR3GEL interacting with Prothrombin and PPACK-IIa.
ACKNOWLEDGEMENTS
The authors thank N. Stolowich for help with NMR instrumentation. We are grateful for the generous gift of thrombin mutant R77aA provided by Enrico Di Cera and Leslie Pelc from St. Louis University School of Medicine. We thank M.V. Malovichko for initiating HSQC titration projects with thrombin ligands in the lab. Finally, we appreciate critical discussions on this research with K.N. Mouapi, and B.A. Anokhin. This research was supported by grants NIH R01HL68440 (MCM), NIH R15HL120068 (MCM), a University of Louisville Research Initiation Grant (MCM), and by start-up funds provided by the James Graham Brown Foundation (TMS).
Footnotes
The authors declare no competing financial interest
REFERENCES
- (1).Huntington JA (2005) Molecular recognition mechanisms of thrombin. Journal of Thrombosis and Haemostasis 3, 1861–1872. [DOI] [PubMed] [Google Scholar]
- (2).Kraut J (1977) Serine proteases: structure and mechanism of catalysis. Annu Rev Biochem 46, 331–358. [DOI] [PubMed] [Google Scholar]
- (3).Bode W, Turk D, and Karshikov A (1992) The refined 1.9-A X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci 1, 426–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).De Filippis V, De Dea E, Lucatello F, and Frasson R (2005) Effect of Na(+) binding on the conformation, stability and molecular recognition properties of thrombin. Biochem J 390, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Di Cera E, Page MJ, Bah A, Bush-Pelc LA, and Garvey LC (2007) Thrombin allostery. Phys Chem Chem Phys 9, 1291–1306. [DOI] [PubMed] [Google Scholar]
- (6).Pechik I, Madrazo J, Mosesson MW, Hernandez I, Gilliland GL, and Medved L (2004) Crystal structure of the complex between thrombin and the central “E” region of fibrin. Proc Natl Acad Sci U S A 101, 2718–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fuentes-Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, Morser J, Light DR, and Bode W (2000) Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature 404, 518–525. [DOI] [PubMed] [Google Scholar]
- (8).Gandhi PS, Chen Z, and Di Cera E (2010) Crystal structure of thrombin bound to the uncleaved extracellular fragment of PAR1. J Biol Chem 285, 15393–15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bah A, Chen Z, Bush-Pelc LA, Mathews FS, and Di Cera E (2007) Crystal structures of murine thrombin in complex with the extracellular fragments of murine protease-activated receptors PAR3 and PAR4. Proc Natl Acad Sci U S A 104, 11603–11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rydel TJ, Ravichandran KG, Tulinsky A, Bode W, Huber R, Roitsch C, and Fenton JW 2nd. (1990) The structure of a complex of recombinant hirudin and human alpha-thrombin. Science 249, 277–280. [DOI] [PubMed] [Google Scholar]
- (11).Rydel TJ, Tulinsky A, Bode W, and Huber R (1991) Refined structure of the hirudin-thrombin complex. J Mol Biol 221, 583–601. [DOI] [PubMed] [Google Scholar]
- (12).Carter WJ, Cama E, and Huntington JA (2005) Crystal structure of thrombin bound to heparin. J Biol Chem 280, 2745–2749. [DOI] [PubMed] [Google Scholar]
- (13).Sidhu PS, Liang A, Mehta AY, Abdel Aziz MH, Zhou Q, and Desai UR (2011) Rational design of potent, small, synthetic allosteric inhibitors of thrombin. J Med Chem 54, 5522–5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Esmon CT, and Lollar P (1996) Involvement of thrombin anion-binding exosites 1 and 2 in the activation of factor V and factor VIII. J Biol Chem 271, 13882–13887. [DOI] [PubMed] [Google Scholar]
- (15).Sabo TM, Farrell DH, and Maurer MC (2006) Conformational analysis of gamma’ peptide (410–427) interactions with thrombin anion binding exosite II. Biochemistry 45, 7434–7445. [DOI] [PubMed] [Google Scholar]
- (16).Sabo TM, and Maurer MC (2009) Biophysical investigation of GpIbalpha binding to thrombin anion binding exosite II. Biochemistry 48, 7110–7122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Fredenburgh JC, Stafford AR, and Weitz JI (1997) Evidence for allosteric linkage between exosites 1 and 2 of thrombin. J Biol Chem 272, 25493–25499. [DOI] [PubMed] [Google Scholar]
- (18).Petrera NS, Stafford AR, Leslie BA, Kretz CA, Fredenburgh JC, and Weitz JI (2009) Long range communication between exosites 1 and 2 modulates thrombin function. J Biol Chem 284, 25620–25629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Malovichko MV, Sabo TM, and Maurer MC (2013) Ligand binding to anion binding exosites regulates conformational properties of thrombin. J Biol Chem 288, 8667–8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).De Candia E, Hall SW, Rutella S, Landolfi R, Andrews RK, and De Cristofaro R (2001) Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of Par-1 on intact platelets. J Biol Chem 276, 4692–4698. [DOI] [PubMed] [Google Scholar]
- (21).Krishnaswamy S (2013) The transition of prothrombin to thrombin. J Thromb Haemost 11 Suppl 1, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Brufatto N, and Nesheim ME (2003) Analysis of the kinetics of prothrombin activation and evidence that two equilibrating forms of prothrombinase are involved in the process. J Biol Chem 278, 6755–6764. [DOI] [PubMed] [Google Scholar]
- (23).Koeppe JR, and Komives EA (2006) Amide H/2H exchange reveals a mechanism of thrombin activation. Biochemistry 45, 7724–7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pozzi N, Chen Z, Gohara DW, Niu W, Heyduk T, and Di Cera E (2013) Crystal structure of prothrombin reveals conformational flexibility and mechanism of activation. J Biol Chem 288, 22734–22744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Pozzi N, Bystranowska D, Zuo X, and Di Cera E (2016) Structural Architecture of Prothrombin in Solution Revealed by Single Molecule Spectroscopy. J Biol Chem 291, 18107–18116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wu Q, Picard V, Aiach M, and Sadler JE (1994) Activation-induced exposure of the thrombin anion-binding exosite. Interactions of recombinant mutant prothrombins with thrombomodulin and a thrombin exosite-specific antibody. J Biol Chem 269, 3725–3730. [PubMed] [Google Scholar]
- (27).Mehta AY, Thakkar JN, Mohammed BM, Martin EJ, Brophy DF, Kishimoto T, and Desai UR (2014) Targeting the GPIbalpha binding site of thrombin to simultaneously induce dual anticoagulant and antiplatelet effects. J Med Chem 57, 3030–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mehta AY, Mohammed BM, Martin EJ, Brophy DF, Gailani D, and Desai UR (2016) Allosterism-based simultaneous, dual anticoagulant and antiplatelet action: allosteric inhibitor targeting the glycoprotein Ibalpha-binding and heparin-binding site of thrombin. J Thromb Haemost 14, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ni F, Ning Q, Jackson CM, and Fenton JW 2nd. (1993) Thrombin exosite for fibrinogen recognition is partially accessible in prothrombin. J Biol Chem 268, 16899–16902. [PubMed] [Google Scholar]
- (30).Anderson PJ, Nesset A, Dharmawardana KR, and Bock PE (2000) Characterization of proexosite I on prothrombin. J Biol Chem 275, 16428–16434. [DOI] [PubMed] [Google Scholar]
- (31).Kretz CA, Stafford AR, Fredenburgh JC, and Weitz JI (2006) HD1, a thrombin-directed aptamer, binds exosite 1 on prothrombin with high affinity and inhibits its activation by prothrombinase. J Biol Chem 281, 37477–37485. [DOI] [PubMed] [Google Scholar]
- (32).Bompiani KM, Monroe DM, Church FC, and Sullenger BA (2012) A high affinity, antidote-controllable prothrombin and thrombin-binding RNA aptamer inhibits thrombin generation and thrombin activity. J Thromb Haemost 10, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV Jr., Tam C, and Coughlin SR (1998) A dual thrombin receptor system for platelet activation. Nature 394, 690–694. [DOI] [PubMed] [Google Scholar]
- (34).Steinberg SF (2005) The cardiovascular actions of protease-activated receptors. Mol Pharmacol 67, 2–11. [DOI] [PubMed] [Google Scholar]
- (35).Koradi R, Billeter M, and Wuthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14, 51–55, 29–32. [DOI] [PubMed] [Google Scholar]
- (36).Lechtenberg BC, Johnson DJ, Freund SM, and Huntington JA (2010) NMR resonance assignments of thrombin reveal the conformational and dynamic effects of ligation. Proc Natl Acad Sci U S A 107, 14087–14092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Fuglestad B, Gasper PM, Tonelli M, McCammon JA, Markwick PR, and Komives EA (2012) The dynamic structure of thrombin in solution. Biophys J 103, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Fuglestad B, Gasper PM, McCammon JA, Markwick PR, and Komives EA (2013) Correlated motions and residual frustration in thrombin. J Phys Chem B 117, 12857–12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lechtenberg BC, Freund SM, and Huntington JA (2012) An ensemble view of thrombin allostery. Biol Chem 393, 889–898. [DOI] [PubMed] [Google Scholar]
- (40).Handley LD, Fuglestad B, Stearns K, Tonelli M, Fenwick RB, Markwick PR, and Komives EA (2017) NMR reveals a dynamic allosteric pathway in thrombin. Sci Rep 7, 39575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Markin CJ, and Spyracopoulos L (2012) Increased precision for analysis of protein-ligand dissociation constants determined from chemical shift titrations. J Biomol NMR 53, 125–138. [DOI] [PubMed] [Google Scholar]
- (42).Marino F, Pelc LA, Vogt A, Gandhi PS, and Di Cera E (2010) Engineering thrombin for selective specificity toward protein C and PAR1. J Biol Chem 285, 19145–19152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ni F (1994) Recent developments in transferred NOE methods. Prog Nucl Magn Reson Spectrosc 26, 517–606. [Google Scholar]
- (44).Cleary DB, Trumbo TA, and Maurer MC (2002) Protease-activated receptor 4-like peptides bind to thrombin through an optimized interaction with the enzyme active site surface. Arch Biochem Biophys 403, 179–188. [DOI] [PubMed] [Google Scholar]
- (45).Campbell AP, and Sykes BD (1993) The two-dimensional transferred nuclear Overhauser effect: theory and practice. Annu Rev Biophys Biomol Struct 22, 99–122. [DOI] [PubMed] [Google Scholar]
- (46).Ni F, and Scheraga HA (1994) Use of the transferred nuclear Overhauser effect to determine the conformations of ligands bound to proteins. Accounts of chemical research 27, 257–264. [Google Scholar]
- (47).Teilum K, Kunze MB, Erlendsson S, and Kragelund BB (2017) (S)Pinning down protein interactions by NMR. Protein Sci 26, 436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fielding L (2003) NMR methods for the determination of protein-ligand dissociation constants. Curr Top Med Chem 3, 39–53. [DOI] [PubMed] [Google Scholar]
- (49).Kleckner IR, and Foster MP (2011) An introduction to NMR-based approaches for measuring protein dynamics. Biochim Biophys Acta 1814, 942–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Iconaru LI, Ban D, Bharatham K, Ramanathan A, Zhang W, Shelat AA, Zuo J, and Kriwacki RW (2015) Discovery of Small Molecules that Inhibit the Disordered Protein, p27(Kip1). Sci Rep 5, 15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, Hylek EM, Kakkar TL, Konstantinides SV, McCumber M, Ozaki Y, Wendelboe A, and Weitz JI (2014) Thrombosis: a major contributor to global disease burden. Semin Thromb Hemost 40, 724–735. [DOI] [PubMed] [Google Scholar]
- (52).Weitz JI, Lensing AWA, Prins MH, Bauersachs R, Beyer-Westendorf J, Bounameaux H, Brighton TA, Cohen AT, Davidson BL, Decousus H, Freitas MCS, Holberg G, Kakkar AK, Haskell L, van Bellen B, Pap AF, Berkowitz SD, Verhamme P, Wells PS, and Prandoni P (2017) Rivaroxaban or Aspirin for Extended Treatment of Venous Thromboembolism. N Engl J Med 376, 1211–1222. [DOI] [PubMed] [Google Scholar]
- (53).Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A, Pogue J, Reilly PA, Themeles E, Varrone J, Wang S, Alings M, Xavier D, Zhu J, Diaz R, Lewis BS, Darius H, Diener HC, Joyner CD, and Wallentin L (2009) Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 361, 1139–1151. [DOI] [PubMed] [Google Scholar]
- (54).Salmeron Febres LM, and Cuenca Manteca J (2017) Direct Oral Anticoagulants in the Treatment of Venous Thromboembolic Disease. Ann Vasc Surg. [DOI] [PubMed] [Google Scholar]
- (55).Dager WE, and Banares L (2017) Reversing the anticoagulation effects of dabigatran. Hosp Pract (1995) 45, 29–38. [DOI] [PubMed] [Google Scholar]
- (56).Weitz JI (2017) Reversal of Direct Oral Anticoagulants: Current Status and Future Directions. Semin Respir Crit Care Med 38, 40–50. [DOI] [PubMed] [Google Scholar]
- (57).Bock PE, Panizzi P, and Verhamme IM (2007) Exosites in the substrate specificity of blood coagulation reactions. J Thromb Haemost 5 Suppl 1, 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Kretz CA, Stafford AR, Fredenburgh JC, and Weitz JI (2015) HD1, a thrombin-directed aptamer, binds exosite 1 on prothrombin with high affinity and inhibits its activation by prothrombinase. J Biol Chem 290, 4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Monteiro RQ, Bock PE, Bianconi ML, and Zingali RB (2001) Characterization of bothrojaracin interaction with human prothrombin. Protein Sci 10, 1897–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Krem MM, and Di Cera E (2003) Dissecting substrate recognition by thrombin using the inactive mutant S195A. Biophys Chem 100, 315–323. [DOI] [PubMed] [Google Scholar]
- (61).Myles T, Le Bonniec BF, Betz A, and Stone SR (2001) Electrostatic steering and ionic tethering in the formation of thrombin-hirudin complexes: the role of the thrombin anion-binding exosite-I. Biochemistry 40, 4972–4979. [DOI] [PubMed] [Google Scholar]
- (62).Myles T, Le Bonniec BF, and Stone SR (2001) The dual role of thrombin’s anion-binding exosite-I in the recognition and cleavage of the protease-activated receptor 1. Eur J Biochem 268, 70–77. [DOI] [PubMed] [Google Scholar]
- (63).Boehr DD, Nussinov R, and Wright PE (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol 5, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Tzeng SR, and Kalodimos CG (2012) Protein activity regulation by conformational entropy. Nature 488, 236–240. [DOI] [PubMed] [Google Scholar]
- (65).Wei G, Xi W, Nussinov R, and Ma B (2016) Protein Ensembles: How Does Nature Harness Thermodynamic Fluctuations for Life? The Diverse Functional Roles of Conformational Ensembles in the Cell. Chem Rev 116, 6516–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: 2D 1H-13C natural abundance HSQC NMR for PAR3 (44–56) and PAR3G (44–56). Figure S2:1D 15N-HSQC NMR titrations of PAR3FD (44–56) in the presence of ProT and PPACK- IIa. Figure S3: 2D 15N HSQC NMR titrations of PAR3FD (44–56) in the presence of ProT and PPACK- IIa. Figure S4:1D 15N-HSQC NMR titrations of PAR3GFD (44–56) in the presence of ProT and PPACK- IIa. Figure S5: 1D 15N HSQC NMR titrations of PAR3GEL (44–56) in the presence of ProT and PPACK- IIa. Figure S6: Determination of Binding Affinity (KD) for 15N-labeled F47 and D54 of PAR3GFD interacting with PPACK-R77aA IIa. Figure S7: Determination of Binding Affinity (KD) for 15N-labeled L52 and E48 of PAR3GEL interacting with Prothrombin and PPACK-IIa.