

Abstract
Albumin is a promising surface modifier of nanoparticulate drug delivery systems. Serving as a dysopsonin, albumin can protect circulating nanoparticles (NPs) from the recognition and clearance by the mononuclear phagocytic system (MPS). Albumin may also help transport the NPs to solid tumors based on the increased consumption by cancer cells and interactions with the tumor microenvironment. Several studies have explored the benefits of surface-bound albumin to enhance NP delivery to tumors. However, it remains unknown how the surface modification process affects the conformation of albumin and the performance of the albumin-modified NPs. We use three different surface modification methods including two prevalent approaches (physisorption and interfacial embedding) and a new method based on dopamine polymerization to modify the surface of poly(lactic-co-glycolic acid) NPs with albumin and compare the extent of albumin binding, conformation of the surface-bound albumin, and biological performances of the albumin-coated NPs. We find that the dopamine polymerization method preserves the albumin structure, forming a surface layer that facilitates NP transport and drug delivery into tumors via the interaction with albumin-binding proteins. In contrast, the interfacial embedding method creates NPs with denatured albumin that offers no particular benefit to the interaction with cancer cells but rather promotes the MPS uptake via direct and indirect interactions with scavenger receptor A. This study demonstrates that the surface-bound albumin can bring distinct effects according to the way they interact with NP surface and thus needs to be controlled in order to achieve favorable therapeutic outcomes.
Keywords: Drug delivery, chemotherapy, nanoparticles, albumin, surface modification
Abstract graphic
1. Introduction
Polymeric nanoparticles (NPs) have been considered a promising carrier of an anti-cancer drug due to the potential to alter the pharmacokinetics and toxicity profiles of the drug. A challenge in systemic delivery of NPs is however that they are subject to opsonin binding and removal by the mononuclear phagocytic system (MPS) [1, 2]. A prevalent way of avoiding the MPS removal of NPs is to coat the surface with a hydrophilic layer that reduces the protein adsorption to NPs. For example, synthetic polymers such as polyethylene glycol (PEG) are widely used to prevent the opsonization of NPs and subsequent clearance by the MPS. However, it is suggested that PEG surface can reduce the interactions between NPs and cancer cells [3, 4] and interfere with the endosomal escape of NPs in the cells [5]. For efficient drug delivery with NPs, it will be beneficial to have alternative strategies to modify the NP surface and control their interactions with blood proteins. In this regard, we consider albumin to be a promising surface modifier of NP drug delivery systems.
Albumin is the most abundant protein in human body, responsible for the transport of nutrients and hydrophobic drugs in circulation [6]. Several features make albumin a promising candidate for the modification of NP surfaces. First, albumin serves as a dysopsonin for foreign particles and reduces the binding of other proteins that lead to the MPS uptake [7–10], thereby extending the circulation time of NPs [7, 8]. Second, many cancer cells use albumin as a source of energy and nutrients [11] and tend to show an enhanced uptake of albumin via macropinocytosis [12]. The high demands for albumin may facilitate the transport of albumin-bound compounds to tumors. Third, albumin can reach tumors via not only the paracellular pathway but also the transcellular pathway that involves glycoproteins (gp60) expressed on peritumoral endothelium [6, 13–15]. Albumin can also bind to the secreted protein acidic and rich in cysteine (SPARC), which is overexpressed in head and neck cancers, melanomas, and non-small cell lung cancers [16], increasing the tissue retention of the drug it carries. The effectiveness of Abraxane®, an albumin-bound paclitaxel (PTX) complex, is partly attributed to the ability of albumin to interact with gp60 and/or SPARC [16–18]. Therefore, one may expect that albumin-coated NPs will exploit these physiological features of albumin to achieve favorable biodistribution profiles.
Polymeric NPs can be coated with albumin by various methods [19]. The most common approach is to incubate NPs with albumin and let the protein physically adsorb to the NP surface (physisorption) [10, 20]. Various non-covalent interactions mediate albumin binding to the NPs. Alternatively, albumin can be added during the formation of NPs as an emulsifier (interfacial embedding) [21]. Albumin is added to the continuous phase during emulsification so that it can be embedded on the NP surface. Another approach is to chemically conjugate albumin to reactive functional groups on NP surface, although not always available, via the corresponding side chains of the protein. For example, amine groups of albumin can be conjugated to NPs via carbodiimide chemistry if carboxylic groups are present on the NP surface [22, 23]. However, these methods involve the conditions that may induce structural changes in albumin, such as hydrophobic interactions, interfacial activities, and the exposure to harsh reactants. Albumin undergoing such interactions may expose cryptic epitopes subject to the MPS recognition [24], counteracting the potential benefits of albumin coating. Although several studies report the conformational perturbation of albumin bound to NPs [25–28], the biological consequences of such changes are not entirely clear.
We hypothesize that the method of albumin coating can critically affect the albumin binding efficiency, the conformation of surface-bound albumin, and ultimately the performance of albumin-coated NPs in vivo. To test this hypothesis, we modified the surface of poly(lactic-co-glycolic acid) (PLGA) NPs, a widely used NP platform for drug delivery, with human serum albumin (HSA) by different methods (Fig. 1a). In addition to the physisorption and interfacial embedding approaches, we employed a new surface modification method utilizing dopamine polymerization [29]. This method involves a brief incubation of PLGA NPs in aqueous solution of dopamine, which forms a thin layer of catechol- and amine-rich polymerized dopamine (pD) on NP surface in a mildly oxidative condition [30]. The pD layer accommodates various functional ligands with nucleophilic functional groups such as amines or thiols via Schiff base reaction and Michael addition [31, 32]. An advantage of this method over other chemical conjugation approaches is that it does not require specific functional groups to be present on the NP platform. Also, the reaction condition is mild and does not compromise the activity of conjugated biomolecules [33] or the underlying platform [34]. Therefore, there is a good chance that albumin may maintain the conformational integrity during the immobilization to the PLGA NPs.
Fig. 1.
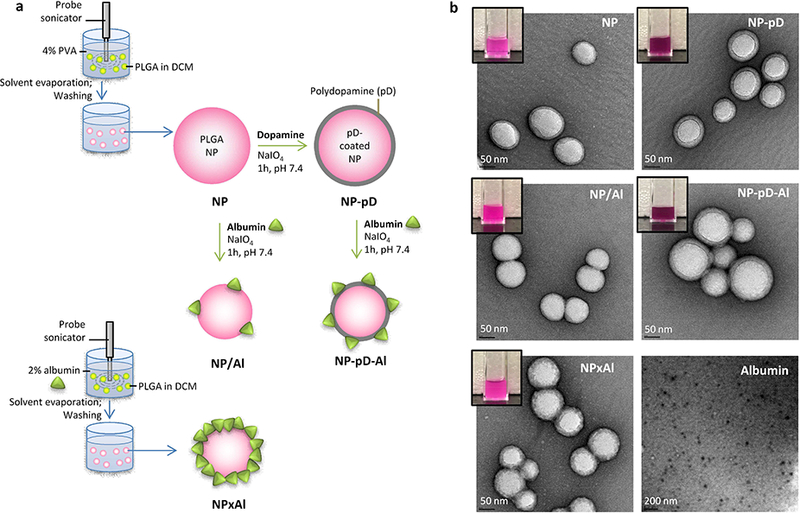
(a) Different methods to conjugate albumin to PLGA NPs. (b) Transmission electron microscopy (TEM) images of rhodamine-labeled NP, NP-pD, NP/Al, NPxAl, NP-pD-Al and albumin, negatively stained with 2% uranyl acetate. Dark purple color of NP-pD and NP-pD-Al suspensions (inset) indicate the presence of polymerized dopamine (pD).
With the three types of albumin-coated PLGA NPs, we compared the extent of albumin binding and the structural integrity of surface-bound albumin. We then examined how these properties affected the binding of serum proteins to NP surface and the interactions of NPs with different types of cells directly relevant to their biodistribution. We find that the albumin conformation plays a critical role in NP-cell interactions and leads to a significant difference in tissue distribution of NPs and drug delivery to tumors. These results suggest that the surface-bound proteins, pre-coated or formed in situ during circulation, can do either good or harm depending on how they interact with the NP surface and thus needs to be controlled in order to achieve favorable therapeutic outcomes.
2. Materials and Methods
2.1. Materials
PLGA (ester endcap, 25–35 kDa, LA:GA= 85:15) and PLGA-Rhodamine B (10–30 kDa, LA:GA= 50:50) were purchased from Akina Inc. (West Lafayette, IN). Dopamine hydrochloride was purchased from Alfa Aesar (Ward Hill, MA). PTX was a gift of Samyang Biopharm (Seoul, Korea). Coomassie Brilliant blue G-250 protein stain and reagents for sodium dodecyl sulfate-acrylamide gel electrophoresis (SDS-PAGE) were purchased from Bio-Rad (Hercules, CA). Human serum albumin (HSA, ≥96% agarose gel electrophoresis), 4-nitrophenyl acetate (p-nitrophenyl acetate, pNPA), polyinosinic acid (poly(I)), and fluorescein-labeled-bovine serum albumin (FITC-BSA) were purchased from Sigma-Aldrich (St. Louis, MO). Collagen-I (Rat Protein, Tail), Hoechst 33342 and Opti-MEM™ I Reduc ed Serum Medium were purchased from Life Technologies (Carlsbad, CA). Transwell polycarbonate insert plates (1 cm2, 3 μm pore size) were purchased from Corning (Pittsburgh, PA). Luciferase Cell Culture Lysis 5× Reagent and Terminal deoxynucleotidyl transferase dUTP nick end labeling kit (DeadEnd Fluorometric TUNEL System) were purchased from Promega (Madison, WI). Mouse SPARC polyclonal antibody was purchased from R&D Systems Inc. (Minneapolis, MN). (3-(4,5-Dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide) (MTT) was purchased from Invitrogen (Eugene, OR). Mouse SPARC or scrambled negative control siRNA were purchased from OriGene (Rockville, MD). Lipofectamine™ RNAiMAX Transfection Reagent was purchased from Invitrogen (Carlsbad, CA). Components of THP1-XBlue-MD2-CD14 cell culture medium and secreted embryonic alkaline phosphatase (SEAP) reporter assay were purchased from InvivoGen (San Diego, CA). Iron oxide (IO) particles (5–10 nm) were purchased from Ocean NanoTech (San Diego, CA). BD cytometric bead array (CBA) with mouse soluble protein flex sets including cytokines, TNF-α, IL-6 and IL-1β were purchased from BD Biosciences (San Jose, CA). Fluorescein labeled Lycopersicon Esculentum (Tomato) Lectin (FITC-lectin) was purchased from Vector Laboratories (Burlingame, CA).
2.2. NP preparation
Albumin-coated PLGA NPs were prepared by different methods (Fig. 1a). First, PLGA (25–30 kDa, 85:15) or rhodamine-labeled PLGA (10–30 kDa, 50:50) NPs were prepared by the single emulsion-solvent evaporation method. Briefly, 50 mg of PLGA was dissolved in 4 mL of dichloromethane (DCM; organic phase) and emulsified in 12 mL of 4% polyvinyl alcohol solution (PVA; aqueous phase) by 2 min probe sonication at 40% amplitude on a 4-s on and 2-s off pulse mode. The emulsion was dispersed in 20 mL of deionized (DI) water, and DCM was evaporated by a rotary evaporator. NPs were collected via centrifugation at 13,600 rcf for 30 min and washed three times using DI water. For surface modification via dopamine polymerization, NPs were incubated in dopamine HCl solution in sodium periodate solution (190 mM, 0.1 M phosphate buffer with pH 7.4) for 1 h at a dopamine HCl-to-NP weight ratio of 0.5/1. When the NPs manifested dark color of polymerized dopamine, they were collected by centrifugation and washed twice with water to remove excess dopamine and pD. The pD-coated NPs (NP-pD) were subsequently incubated with albumin at an albumin-to-NP weight ratio of 4/1 for 1 h in sodium periodate solution (190 mM, 0.1 M phosphate buffer with pH 7.4) to form albumin-coated NPs (NP-pD-Al). The NPs were collected by centrifugation at 13,600 rcf for 20 min at 4 °C and washed twice with DI water. For preparation of albumin-coated NPs by physisorption (NP/Al), plain NPs (instead of NP-pD) were incubated with albumin. The produced NPs were collected via centrifugation at 13,600 rcf for 30 min and washed two times with DI water. For preparation of the NPs with surface-embedded albumin (NPxAl), the organic phase containing PLGA or rhodamine-labeled PLGA was emulsified in 2% albumin solution instead of the PVA solution. When PTX was loaded, PTX was incorporated in the organic phase along with PLGA at the theoretical loading of 16.7 wt%. Briefly, 4 mg of PTX was dissolved with 20 mg of PLGA 35 kDa (AP31, ester endcap, LA:GA=85:15) in 0.8 mL of DCM, emulsified in an aqueous phase of PVA (4%, 4.8 mL), and dispersed in 8 mL of DI water. Iron oxide (IO) loaded NPs (IO@NPs) were prepared by adding IO particles to the PLGA solution at an IO/PLGA weight ratio of 5%. The PTX-loaded (PTX@NPs) or IO@NPs were purified and surface-modified in the same manner as blank NPs.
2.3. NP characterization
2.3.1. Particle size, zeta potential, TEM, and PTX content in NPs
NPs were prepared as 1 mg/mL suspension in 10 mM NaCl. The size, polydispersity index (PI) and zeta potential of the produced NPs were measured using a Malvern Zetasizer Nano ZS90 (Worcestershire, UK). NPs were imaged by transmission electron microscopy (TEM) using FEI Tecnai T20 (Hillsboro, OR). Samples were mounted on a 300-mesh Cu grid with Formvar/carbon supporting film and stained with 2% uranyl acetate. pD on the surface of NP-pD was detected and quantified by the Micro Bicinchoninic Acid Assay (Thermo Scientific, Waltham, MA). The PTX loading efficiency, defined as loaded PTX/NP mass, was determined by HPLC. NPs with a known mass were dissolved in 0.5 mL of acetonitrile (ACN). After 1 h, polymers were precipitated with the addition of 0.5 mL of DI water, centrifuged, and the supernatant was filtered with 0.45 μm syringe filter. The samples were analyzed via Agilent 1100 HPLC system (Palo Alto, CA), equipped with Ascentis C18 column (25 cm x 4.6 mm, particle size 5 μm). The mobile phase was a 50:50 mixture of water and ACN, run at a flow rate of 1 mL/min. PTX was detected by a UV detector (227 nm).
2.3.2. PTX release kinetics from NPs
PTX@NPxAl or PTX@NP-pD-Al was suspended in PBS containing 0.2% Tween 80 (PBST) to a concentration equivalent to PTX 4 μg/mL. The NP suspensions were divided into multiple 1 mL aliquots and incubated at 37 °C with constant agitation. At each time point, the aliquots were centrifuged at 13,900 rcf for 10 min. The sampled supernatant was analyzed by HPLC. PTX standards were prepared in 50% ACN.
2.3.3. Albumin content and status
The albumin content in NP was determined by SDS-PAGE. NPs were suspended in phosphate-buffered saline (PBS, pH 7.4), mixed with 4× Laemmli buffer, and boiled at 95 °C for 5 min to separate the surface-bound albumin. The sample buffer-treated slurry was loaded on a 12% SDS-polyacrylamide gel and resolved by electrophoresis at 100 V initially and then at 150 V. The gel was stained with QC Colloidal Coomassie Brilliant Blue and imaged with Azure C300 (Dublin, CA). Albumin standards of known concentrations were treated and analyzed in the same manner. Albumin bands were quantified by the densitometry function of the AzureSpot Analysis Software (Dublin, CA). The albumin content was determined by comparing the band intensities of NP samples and albumin standard solutions.
The status of albumin on NP surface was analyzed by pulse proteolysis [35]. NPxAl and NP-pD-Al (eq. to 0.2 mg/mL albumin) were treated with 0.2 mg/mL of thermolysin in HEPES buffer (pH 7.4, 20 mM) containing 100 mM NaCl and 10 mM CaCl2. After 3 min incubation at room temperature, 10 μL of 50 mM EDTA was added to a 30 μL aliquot to quench proteolysis. As a control, 0.2 mg/mL of native albumin or denatured albumin (boiled at 95 °C for 10 min) were treated in the same manner. The treated samples were analyzed by SDS-PAGE. The intensities of the albumin bands before and after thermolysin treatment were compared to determine the remaining NP surface-bound albumin. The extent of proteolysis was calculated as (1- albumin band intensity after thermolysin treatment/albumin band intensity prior to the treatment) × 100.
The status of albumin on NP surface was also assessed by its esterase-like activity [36, 37]. NP, NP-pD, NP/Al, NPxAl, and NP-pD-Al (20 mg/mL) suspensions in phosphate buffer (pH 7.4, 0.1 M) was incubated with 397 μM of pNPA at room temperature. Immediately and 60 min after the addition of pNPA, supernatants were collected by centrifugation at 13,900 rcf for 8 min at 4 °C. The absorbance of each sample was meas ured at 400 nm to quantify p-nitrophenol, the product of pNPA hydrolysis. The rate of pNPA hydrolylsis was calculated as a measure of esterase-like activity of albumin. The percent active albumin was calculated as the amount of active albumin divided by the amount of total albumin determined by SDS-PAGE.
2.4. Liquid-chromatography mass-spectrometry (LC-MS/MS) analysis of protein corona
Four milligrams of NP, NP-pD, NPxAl and NP-pD-Al were incubated in 1 mL of 90% mouse serum for 2 h at 37 °C with rotation. The NPs were centrifuged at 33,900 rcf for 10 min at 4 °C and washed twice with PBS. The washed NPs were prepared as 4 mg/mL suspension in PBS, mixed with 4× Laemmli buffer, and boiled at 95 °C for 5 min. The sample buffer-treated NP slurry was loaded on a 12% SDS-polyacrylamide gel and resolved with electrophoresis. Protein bands were identified by Coomassie staining and excised for analysis. Gel bands were cut into 1 mm pieces and washed with 50% ACN solution containing 25 mM ammonium bicarbonate (ABC) to remove the stain. After washing, the samples were reduced and alkylated. Sequence grade Lys-C/Trypsin (Promega) was used to enzymatically digest the gel bands. All digestions were carried out in the Barocycler NEP2320 (Pressure Biosciences, Inc.) at 50 °C under 20,000 psi for 1 h. Peptides were recovered from the gel samples using 60% ACN/5% trifluoroacetic acid (TFA)/35% DI water with sonication in an ice bath. The supernatant was removed from the gel and placed in a vacuum centrifuge to dry. The resulting pellets were resuspended in 97% DI water/3% ACN/0.1% formic acid (FA) prior to LC-MS/MS analysis.
Samples were analyzed using the Dionex UltiMate 3000 RSLC Nano System coupled to the Q Exactive™ HF Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific, Waltham, MA). Peptides were loaded onto a C18 PepMap™ 100 trap column (300 μm i.d. × 5 (mm) and washed at a flow rate of 5 μL/min with 98% purified water/2% ACN/0.01% FA. The trap column was switched in-line with the analytical column after 5 min, and peptides were separated by a reverse phase Acclaim™ PepMap™ RSLC C18 (75 μm × 15 cm) analytical column using a 120 min method at a flow rate of 300 nL/min. Mobile phase A was 0.01% FA in water, and mobile phase B was 0.01% FA in 80% ACN. The linear gradient started at 5% B and reached 30% B in 80 min, 45% B in 91 min, and 100% B in 93 min. The column was held at 100% B for the next 5 min and returned to 5% B and held for 20 min. Samples were injected in a random order to the QE HF through the Nanospray Flex™ Ion Source fitted with an emission tip from Thermo Scientific. Data acquisition was performed monitoring the top 20 precursors at 120,000 resolution with an injection time of 100 msec. The peptide masses were searched against a human protein sequence database (Uniprot) using the Mascot Daemon v.2.5.1 (Matrix Science), with peptide mass tolerance of 0.05 Da, fragment mass tolerance of 0.2 Da, 1% false discovery rate (FDR), trypsin digestion, carbamidomethyl cysteine as fixed modification, and oxidized methionine as variable modification. Proteins satisfying the following criteria were selected: (i) protein mass <333 kDa, (ii) exponentially modified protein abundance index (emPAI) >1.0, (iii) spectral count >100, and (iv) peptide count >5. Contaminants (e.g. keratin) were excluded. The percentage of each protein fraction was calculated as the spectral count of individual protein divided by the total protein spectral count.
2.5. Cell culture
Human umbilical vein endothelial cells (HUVEC, ATCC, Manassas, VA) and human brain endothelial cells (hCMEC/D3 cells, donation of Prof. Gregory Knipp) were grown in EGM-2 medium (Lonza, NJ) containing 2% fetal bovine serum (FBS), Normocin (InvivoGen, San Diego, CA), and growth supplements (Lonza, NJ). J774A.1 mouse macrophages (ATCC) were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS, 100 units/mL of penicillin and 100 μg/mL of streptomycin. THP1-XBlue-MD2-CD14 cells (InvivoGen, CA), derived from the human monocytic THP-1 cell line, were maintained in Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% FBS, 2 mM L-glutamine, 1.5 g/L sodium bicarbonate, 10 mM HEPES, 1.0 mM sodium, 100 μg/mL Normocin™ (InvivoGen, San Diego, CA), penicillin (50 IU/mL), streptomycin (50 μg/mL), 200 μg/mL of Zeocin (InvivoGen, San Diego, CA), and 250 μg/mL of G418 (InvivoGen, San Diego, CA). B16F10 mouse melanoma cells (ATCC) were grown in RPMI medium containing 10% FBS, penicillin (100 IU/mL), and streptomycin (100 μg/mL). A549 human non-small cell lung cancer cells (ATCC) were grown in Kaighn’s Modification of Ham’s F-12 Medium (F-12K Medium, ATCC) containing 2 mM L-glutamine and 1500 mg/L sodium bicarbonate, 10% FBS, penicillin (100 IU/mL), and streptomycin (100 μg/mL). All cells were incubated under standard culture conditions (37 °C, 5% CO2, humidified atmosphere).
2.6. NP-cell interactions
2.6.1. Cytocompatibility of NPs
HUVEC and hCMEC/D3 cells were seeded in a 96 well plate precoated with collagen-Ι at a density 10,000 cells per well. B16F10 cells were seeded in a 96 well plate at a density of 3,000 cells per well. THP1-XBlue-MD2-CD14 cells were seeded in a 96 well plate at a density of 100,000 cells per well. After 24 h, the cell culture medium was replaced with fresh serum-supplemented medium containing NPs in different concentrations. After incubation with NPs for 6h (HUVEC and hCMEC/D3 cells) and 24 h (B16F10 and THP1-XBlue-MD2-CD14 cells), cells were collected by centrifugation at 233 rcf for 3 min and washed once with serum-free medium to remove NPs. For THP1-XBlue-MD2-CD14 cells, the supernatants were collected for SEAP assay. A hundred microliters of fresh serum-supplemented medium and 15 μL of MTT solution were added to the cells. After 4 h incubation, 100 μL of stop/solubilization solution was added to the cells and incubated overnight. The absorbance of dissolved formazan was read using a SpectraMax M3 microplate reader (Molecular Device, Sunnyvale, CA) at 565 nm.
2.6.2. NP-cell interactions
HUVEC or hCMEC/D3 cells were seeded in a 12 well plate pre-coated with collagen-I at a density of 50,000 cells per well. J774A.1 macrophages, B16F10, and A549 cells were seeded in a 12 well plate at a density of 50,000 cells per well. After overnight, the medium was replaced with fresh serum-free or serum-supplemented (2% for HUVEC and hCMEC/D3 cells; 10% for J774A.1 macrophages, B16F10, and A549 cells) medium containing 0.1 mg/mL of rhodamine-labeled NPs. To ensure that the NPs are compared at equal fluorescence intensity, the rhodamine-labeled NPs were mixed with non-fluorescent counterparts (made of PLGA, 25–30 kDa, 85:15) (Table S1) as needed. HUVEC, hCMEC/D3, B16F10, and A549 cells were harvested by trypsinization after 1 h incubation with the NPs, and J774A.1 macrophages by scraping after 30 min incubation. Cells were collected by centrifugation at 233 rcf for 3 min, supernatants were discarded, and cell pellets were dispersed in fresh serum-free medium and analyzed with a BD Accuri C6 Flow Cytometer (BD Bioscience, San Jose, CA) with an FL-2 detector (λex/λem = 488 nm/585 nm). For visualization of intracellular NPs, J774A.1 macrophages were plated in a 35 mm glass bottomed dish, incubated with 0.1 mg/mL rhodamine-labeled NPs in serum-supplemented medium, gently rinsed with fresh medium twice, and imaged with a Nikon-A1R confocal microscope (Nikon America Inc., NY). Cells were identified by Hoechst 33342 nuclear staining (5 μg/mL) for 20 min prior to imaging. NPs were detected by λex/λem of 561 nm/ 570–620 nm, and Hoechst-stained nuclei by λex/λem of 407 nm/425–475 nm.
2.6.3. Mechanisms of NP-cell interactions
To determine whether the NPs interact with endothelial cells via the albumin-binding proteins, NP-cell interaction was examined with two endothelial cell lines expressing different levels of albumin-binding proteins. First, differential affinity of the two cell lines for albumin was verified using FITC-BSA. HUVEC or hCMEC/D3 cells were seeded in a 12 well plate pre-coated with collagen-I at a density of 50,000 cells per well. After overnight incubation, the cells were treated with PBS, 0.8 mg/mL of FITC-BSA, or a mixture of FITC-BSA (0.8 mg/mL) and HSA (1.6 mg/mL) in the 10% serum-supplemented medium for 30 min. The cells were then gently rinsed with PBS (pH 7.4) twice and imaged by the Cytation 3 imaging system (Biotek, Winooski, VT) or analyzed by the Accuri C6 flow cytometer (λex/λem = 488 nm/585 nm). After confirming differential endothelial binding of albumin, the cells were incubated with NPs in media containing 2% serum with and without 10 mg/mL HSA (the maximum concentration tolerated by the cells) for 30 min. Cells were gently rinsed twice with serum-free medium to remove free NPs, harvested and dispersed in fresh serum-free medium for flow cytometry analysis (λex/λem = 488 nm/585 nm).
To test whether NP interactions with B16F10, A549 cells, and J774A.1 macrophages were mediated by the scavenger receptors, NP-cell interactions were examined in the presence of poly(I), an inhibitor of class A scavenger receptor. Cells were seeded in a 12 well plate at a density of 50,000 cells per well. After overnight incubation, the culture medium was removed, and the following experiments were performed. J774A.1, B16F10, and A549 cells were pretreated with 0.1 mg/mL of poly(I) in the serum-free medium for 30 min and rinsed once with PBS. The cells were then incubated with 0.1 mg/mL of a rhodamine-labeled NPs in the serum-free or serum-supplemented medium for 30 min (J774A.1) or 1 h (B16F10 and A549). Cells were gently rinsed twice with serum-free medium and analyzed by flow cytometry (λex/λem = 488 nm/585 nm).
To test whether the NP interaction with B16F10 cells was mediated by SPARC, NP-cell interaction was examined in cells with mSparc gene knockdown. B16F10 cells were seeded in a 6 well plate at a density of 200,000 cells per well. After overnight, the cells were transfected with siRNA targeting mSparc or control siRNA using lipofectamine RNAiMAX. Briefly, 0.5 μL SPARC siRNA (10 pmole) was diluted into 125 μL of O pti-MEM medium, followed by the addition of 125 μL of Opti-MEM containing 3 μL of R NAiMAX. The mixture was incubated at room temperature for 5 min prior to the addition to the cells. mSparc knockdown was verified by Western blotting and confirmed to last for 72 h. B16F10 cells were trypsinized at 7 or 18 h post-transfection and plated in a 12 well plate at a density of 50,000 cells per well. After overnight, the cells were incubated with 0.1 mg/mL of rhodamine-labeled NPs in serum-free or serum-supplemented medium for 1 h. Cells were then gently rinsed once with PBS and analyzed by flow cytometry (λex/λem = 488 nm/585 nm).
2.7. NP transport in Transwell system
2.7.1. NP transport across endothelial layer
HUVECs or hCMEC/D3 cells were seeded at a density of 80,000 cells per well in the Transwell insert with a polycarbonate membrane filter (3 μm pore), pre-coated with collagen-I. The apical and basolateral sides were supplied with 0.5 mL and 1.5 mL of complete EGM-2 medium, respectively. Trans-Epithelial Electrical Resistance (TEER) across the Transwell was monitored by EVOM2™ Epithelial Voltohmmeter (World Precision Instruments, FL) daily to confirm the integrity of HUVEC or hCMEC/D3 cell layer [38] throughout the assay. The medium was replaced with fresh complete medium every other day. When the TEER value reached a plateau, 0.1 mg/mL of rhodamine-labeled NPs was added to the apical side of the Transwell with a confluent layer of endothelial cells. After 6 h incubation, the media in apical and basolateral chambers were collected, and the Transwell insert was treated with lysis buffer to lyse the cell layer. The fluorescence intensity of collected media and cell lysate was measured by Synergy™ 4 Multi-Mode Microplate Reader (BioTek, Wi nooski, VT) at λex/λem of 540 nm/580 nm and converted to the NP concentration according to the calibration curve created with each type of NPs in medium or lysis buffer. For TEM imaging, the same experiment was repeated in HUVEC layer with iron oxide-loaded NPs (IO@NPs). After 7 h incubation, the cells were fixed with 2.5% glutaraldehyde for 1 h and imaged by TEM.
2.7.2. NP transport across HUVEC layer to B16F10 cells in a co-culture system
For evaluation of NP transport across the endothelial layer into the underlying cancer cells, NPs were incubated in a co-culture system in which the two cell types were separated by a Transwell insert. HUVECs were grown in the Transwell insert in the same manner as above with daily monitoring of TEER. When the TEER value reached a plateau, the insert (along with complete EGM-2) was transferred to a 12 well plated with B16F10 cells in complete RPMI medium (50,000 cells per well). B16F10 cells were observed with a Cytation 3 imaging system at 6 and 30 h after the transfer to confirm that HUVEC and its medium did not negatively affect the cell status. Rhodamine-labeled NPs (0.1 mg/mL) were added to the apical side of the Transwell insert in the co-culture system. After 6 h incubation, the Transwell insert was removed, and B16F10 cells were incubated for additional 24 h. The B16F10 cells were then lysed with lysis buffer, of which fluorescence intensity was measured by Synergy™ 4 Multi-Mode Microplate Reader with λex/λem of 540 nm/580 nm. An individual calibration curve was created with each type of NPs in lysis buffer to quantify the NPs in the lysate.
2.8. Secreted Embryonic Alkaline Phosphatase reporter assay
THP1-XBlue-MD2-CD14 cells were seeded in a flat-bottom 96-well plate at a density of 100,000 cells per well along with 0.1 mg/mL of rhodamine-labeled NPs in the serum-supplemented medium. After 24 h incubation, the supernatant was treated with the Quanti-Blue reagent (InvivoGen, San Diego, CA) according to the manufacturer’s protocol to quantify the secreted embryonic alkaline phosphatase (SEAP) produced by the THP1-XBlue-MD2-CD14 cells. The absorbance of the Quanti-Blue reagent was monitored over 2 h at 620 nm using a UV-vis spectrophotometer.
2.9. In vivo activity of PTX@NPxAl and PTX@NP-pD-Al
2.9.1. Preparation of B16F10 tumor-bearing mice
All animal procedures were approved by Purdue Animal Care and Use Committee, in conformity with the NIH guidelines for the care and use of laboratory animals. Five to six week old male C57BL/6 mice were obtained from Envigo (Indianapolis, IN) and acclimatized for one week prior to the procedure. A million B16F10 melanoma cells were subcutaneously inoculated in the upper flank of the right hind leg. The animals were used when tumors were palpable (10 days after tumor cell inoculation).
2.9.2. Visualization of NPs in tumor vessels
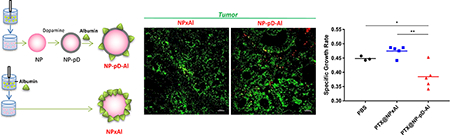
B16F10 tumor-bearing mice received a single intravenous (IV) injection of rhodamine-labeled NPxAl or NP-pD-Al (300 mg NP/kg). After 24 h, the mice were injected with 100 μL of FITC-lectin (1 mg/mL in sterile saline) IV and anesthetized with 3 – 5% isoflurane at a flow rate of 1 L/min, 5 min after the injection. The chest was opened, and the vasculature was perfused with saline through the heart. The tumor, spleen, and liver were harvested, fixed in 10% neutral buffered formalin solution, rinsed twice with PBS, infiltrated with 30% sucrose/PBS solution at 4 °C until the tissues sank, and embedded in optima l cutting temperature (OCT) compound (Fisher Scientific, Pittsburgh, PA). Cryostat sections of each tissue were obtained at a thickness of 16 μm, mounted on a glass slide, and covered wit h a cover glass (0.13 – 0.17 mm, Corning, Pittsburgh, PA). Images were taken with a Nikon A1R confocal microscope. FITC-Lectin-stained blood vessels were detected by λex/λem of 488 nm/500–550 nm. A series of Z stack images (in 5 μm steps) were obtained by a Nikon A1R confocal microscope to visualize intratumoral distribution and colocalization of NPs. The fluorescence distribution in tissues was quantified by the Fiji image analysis software (National Institute of Health, Bethesda, MD) in two- (tumor) or four random fields (liver and spleen) per tissue.
2.9.3. Anti-tumor activity
B16F10 tumor-bearing mice were randomized into 3 groups (n=5 per group for PTX@NPxAl and PTX@NP-pD-Al; n=3 per group for PBS control) and treated with four IV injections of PBS, PTX@NPxAl, and PTX@NP-pD-Al (equivalent to 15 mg/kg of PTX in 200 μL per injection) with a 3-day interval. PTX@NPxAl and PTX@NP-pD-Al were prepared in sterile PBS on the day of treatment. Tumor growth was monitored daily until the end point was reached. The length (L) and width (W) of each tumor were measured by a digital caliper, and the volume (V) was calculated by the modified ellipsoid formula: V= (L × W 2)/2 [39]. Specific growth rate of a tumor was calculated as ΔlogV/Δt (t: time in days) [40].
2.9.4. Tissue and blood analysis
B16F10 tumor-bearing mice received two IV injections of PBS, PTX@NPxAl, and PTX@NP-pD-Al (equivalent to 15 mg/kg of PTX in 200 μL per injection) with a 3-day interval. Animals were monitored in the same way as above. One day after the second injection, mice were sacrificed for the tissue and blood analysis. Blood was collected via cardiac puncture, allowed to clot in a silica-coated tube, and submitted to Purdue Clinical Pathology Laboratory for blood chemistry analysis or stored at –80 °C fo r cytokine analysis. The excised tissues were fixed in 10% neutral buffered formalin solution or frozen at –80 °C for the following analyses.
For evaluation of PTX delivery by NPs, the PTX contents in major organs were analyzed. The frozen tissues were homogenized in PBS using an Omni Tissue Master 125 homogenizer (Kennesaw, GA). One milliliter of the homogenate was mixed with 3 mL of methyl tert-butyl ether (MTBE) and 35 μg of carbamazepine as an internal standard and vortex-mixed for 5 sec for PTX extraction. After centrifugation at 4950 rcf for 5 min, 2.5 mL of MTBE phase was separated, evaporated, and redissolved in a 1:1 mixture of ACN/water for HPLC analysis.
The fixed tissues were embedded in paraffin, sectioned, and stained with hematoxylin and eosin for histological evaluation. For evaluation of pro-apoptotic effect of NPs in tumor, the fixed tumors were analyzed by the TUNEL assay (DeadEnd Fluorometric TUNEL system; Promega, Madison, WI). The TUNEL-stained slides were imaged with a Nikon A1R confocal microscope. Two randomly selected fields per tissue were analyzed with Fiji image analysis software to count apoptotic cells and nuclei. Percent apoptotic cells were calculated as the ratio of the number of apoptotic cells to the number of nuclei.
For evaluation of systemic toxicity and pro-inflammatory effect of NPs, serum chemistry and serum cytokine levels were examined. The concentrations of TNF-α, IL-6, and IL-1β were determined by BD™ CBA Flex set and FCAP array softw are (Franklin, NJ) according to the manufacturer’s instruction.
2.10. Statistical analysis
All statistical analyses were performed with GraphPad Prism 7 (La Jolla, CA). Unless specified otherwise, in vitro data were analyzed by one- or two-way ANOVA test to determine the difference among groups, followed by the Dunnett’s multiple comparisons test or Sidak’s multiple comparisons test. In vivo data were analyzed by one- or two-way ANOVA, followed by the Tukey’s or Sidak’s multiple comparisons test. A value of p < 0.05 was considered statistically significant.
3. Results
3.1. Characterization of albumin-coated NPs
The particle size, PI, and zeta potential of NPs are summarized in Table 1 along with the description. The PI values of NPs ranged from 0.08 and 0.22, which indicates homogeneous NPs. The NPs had average diameters of 160 – 200 nm and n egative zeta potentials, as measured by dynamic light scattering (DLS). In general, the particle sizes measured by DLS were larger than those estimated by TEM (range 80 – 120 nm) (Fig. 1b). This difference is far greater than the thickness of the typically-attributed hydration layer, which is no more than a nanometer [41, 42], suggesting that the NPs might have undergone a mild degree of aggregation in the medium in which the NP size was measured (10 mM NaCl). The aggregation resolved in 50% serum to individual NPs (Table S2); thus, it is unlikely to affect the in vivo performance of the NPs.
Table 1.
Size and Zeta Potential of NPs
| NP type | Description | z-average (d.nm) |
Polydispersity index (PI)a |
Zeta potential (mV)b |
|---|---|---|---|---|
| NP | PLGA NPc | 185 ± 11 | 0.14 ± 0.03 | −1.7 ± 0.5 |
| NP-pD | PLGA NP coated with pD | 188 ± 13 | 0.12 ± 0.02 | −2.2 ± 0.6 |
| NP/Al | PLGA NP with physisorbed | 196 ± 21 | 0.19 ± 0.03 | −2.1 ± 0.3 |
| NPxAl | PLGA NP with albumin | 161 ± 8 | 0.12 ± 0.03 | −12 ± 1 |
| NP-pD-Al | PLGA NP with albumin | 185 ± 16 | 0.08 ± 0.03 | −3.9 ± 0.3 |
| HSA | Free human serum albumin | 8.2 ± 0.2 | 0.22 ± 0.01 | −11 ± 1 |
Polydispersity index (PI), an estimate of the width of the particle size distribution, obtained from the cumulant analysis as described in the International Standard on DLS ISO 13321:1996 and ISO 22412:2008 (Malvern DLS technical note MRK1764−01). PI < 0.1 is considered monodisperse, and > 0.7 is very broad.
Zeta potential measured in 10 mM NaCl.
PLGA NPs made of rhodamine-labeled PLGA.
NPs: n = 7 identically and independently prepared samples (mean ± s.d.) HSA: n = 3 identically and independently prepared samples (mean ± s.d.)
The pD-coated NPs (NP-pD and NP-pD-Al) showed a dark color (Fig. 1b insets) due to the polymerized dopamine (pD) layer [30]. The pD layer on NP-pD was detected and quantified by the Micro BCA assay based on its reducing activity. NP-pD showed greater signal than that of NP (Fig. S1a), and the difference (i.e., pD signal) increased with the NP-pD concentration (Fig. S1b). The pD content in NP-pD was estimated to be 1.5 wt%. NPs coated with albumin via pD layer (NP-pD-Al) had 0.2 ± 0.1 albumin per 100 nm2 (Fig. 2a), whereas physisorption (NP/Al) resulted in inconsistent albumin contents, indicating the essential role of pD in albumin binding (Fig. S2). NPxAl, where albumin was embedded on the NP surface during the emulsification step [21], showed the highest albumin content (4 ± 2 albumin per 100 nm2) (Fig. 2a, Fig. S2). The high albumin density on NP surface is also consistent with the relatively high negative surface charge of NPxAl (Table 1). Given that the average hydrodynamic diameter of albumin is 8.2 nm (Table 1), the number of albumin that can bind in a single layer is estimated to be no more than two per 100 nm2. This suggests that albumin may have adsorbed on NPxAl in multiple layers, which is supported by the relatively rough surface of NPxAl shown in TEM image (Fig 1b).
Fig. 2.
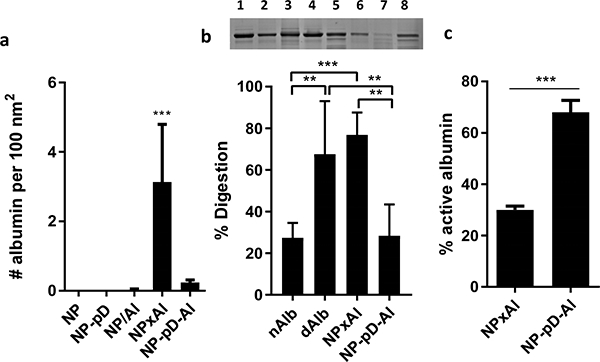
(a) The number of albumin per 100 nm2 of NP surface area. n = 4 identically and independently prepared samples (mean ± s.d.). ***: p < 0.001 vs. NP by Dunnett’s multiple comparisons test following one-way ANOVA. (b) Top: Representative SDS-PAGE gel image of albumin after pulse proteolysis. Native albumin (nAlb), denatured albumin (dAlb), NPxAl, and NP-pD-Al were treated with thermolysin for 3 min. Lane 1: nAlb; Lane 2: dAlb; Lane 3: NPxAl; Lane 4: NP-pD-Al; Lane 5: nAlb + thermolysin; Lane 6: dAlb + thermolysin; Lane 7: NPxAl + thermolysin; and Lane 8: NP-pD-Al + thermolysin. Bottom: % digestion albumin was defined as (1-albumin band intensity after proteolysis / albumin band intensity prior to proteolysis) × 100. N = 5 independently and identically performed experiments (mean ± s.d.). **: p < 0.01 and ***: p < 0.001 by Tukey’s multiple comparisons test following one-way ANOVA. (c) Active albumin content determined by esterase assay. % active albumin = esterase active albumin / the total amount of albumin determined by SDS-PAGE. n = 3 independently and identically performed experiments (mean ± s.d.). ***: p < 0.001 by unpaired t-test.
3.2. Status of surface-bound albumin
To examine how the modification methods affected the status of surface-bound albumin, we analyzed two NPs with consistent albumin contents (NPxAl and NP-pD-Al) with respect to the conformation and functionality. Traditional spectroscopic approaches used in protein conformation studies, such as circular dichroism and synchronous fluorescence, were unsuitable for evaluating the status of surface-bound albumin due to optical interference of NPs. Alternatively, we evaluated the conformation of the surface-bound albumin by pulse proteolysis [35, 43]. The NPs with surface-bound albumin was exposed to thermolysin treatment for a short period, which specifically digests the unfolded proteins [35]. Considering that native albumin (nAlb) is more resistant to pulse proteolysis than denatured albumin (dAlb) (Fig. 2b), the extent of protein unfolding/denaturation can be expressed as the fraction of digested albumin by the thermolysin treatment. The degree of digestion was greater with NPxAl (79 ± 11%) (Fig. 2b) than with NP-pD-Al (31 ± 16%), which are comparable to those of dAlb (74 ± 25%) and nAlb (29 ± 7%), respectively. Similar results were obtained with the esterase-like activity assay [36, 37] (Fig. 2c). Albumin has an esterase-like activity that helps hydrolyze pNPA to p-nitrophenol, which can be quantified spectrophotometrically. The amount of native (active) albumin can be assessed based on the rate of pNPA hydrolysis. The fraction of active albumin per the total surface-bound albumin was 68 ± 5% for NP-pD-Al and 30 ± 2% for NPxAl. The pulse proteolysis and esterase-like activity measurements consistently support that the albumin conformation was better preserved in NP-pD-Al than in NPxAl. We suspect that the high density of albumin on NP surface and the exposure to the organic solvent-water interface with high shear stress during the NP production were detrimental to the albumin stability.
3.3. Effect of albumin-coated NP surface on protein corona formation
As a dysopsonin, albumin is expected to prevent serum protein binding onto the NP surface, thereby reducing the NP recognition by the MPS [44]. We examined how the conformation of surface-bound albumin affected the binding of serum proteins on NP surface (corona formation). NP-pD-Al and NPxAl, as well as their respective albumin-deficient counterparts (NP-pD and NP), were incubated in mouse serum for 2 h. The NPs were washed twice, and the proteins tightly adsorbed to the NPs (i.e., hard corona [45]) were analyzed by SDS-PAGE (Fig. 3a) and LC-MS/MS (Fig. 3b). Although the significance of the loosely-bound proteins (i.e., soft corona) cannot be overlooked, we focused on analyzing hard corona due to the difficulty in reproducible isolation of soft corona. We observed differential patterns of hard corona binding according to the NPs. In all NPs, the most prominent protein band was detected between 50 – 75 kDa, corresponding to mouse serum albumin (for NP and NP-pD) or a mixture of mouse albumin and pre-existing HSA (for NPxAl and NP-pD-Al). It appeared that less protein adsorbed to NP-pD and NP-pD-Al than NP and NPxAl, which is attributable to the hydrophilic nature of the pD-covered surfaces. Noticeably, NPxAl showed an intense band at 25 – 37 kDa. Following the LC-MS/MS analysis, the top 30 most abundant proteins adsorbed onto NP surface were visualized on a heat map (Fig. 3b). As expected from SDS-PAGE, mouse serum albumin (69 kDa) was the major protein (5.5 – 6.8% of total proteins) in all NPs. NPxAl had an additional strong signal corresponding to apolipoprotein E (ApoE, 36 kDa) (4.3% of total proteins), as noticed in SDS-PAGE (Fig 3a). The relative abundance of ApoE was lower in NP, NP-pD, and NP-pD-Al (NP: 1.7%; NP-pD: 1.6%; NP-pD-Al 1.3%). Since ApoE constitutes a minor fraction of serum proteins (60 – 80 μg/mL in mice [46] and 3 0 – 70 μg/mL in humans [47]), we interpret the strong ApoE band intensity to indicate the denatured albumin on NPxAl attracting and enriching ApoE on the NP surface. These results indicate that the status of surface-bound albumin may affect corona formation on circulating NPs.
Fig. 3.
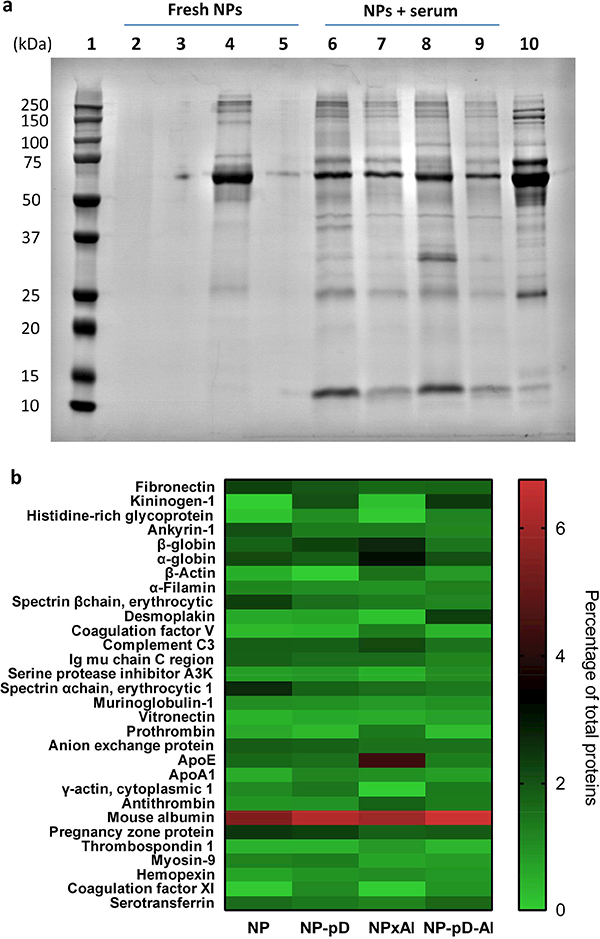
(a) SDS-PAGE of protein corona composition formed on NP, NP-pD, NPxAl, and NP-pD-Al. NPs (4 mg/mL) were incubated in PBS or 90% mouse serum for 2 h and rinsed with PBS twice. Lane 1: size marker; Lane 2: NP; Lane 3: NP-pD; Lane 4: NPxAl; Lane 5: NP-pD-Al; Lane 6–9: NP, NP-pD, NPxAl, and NP-pD-Al incubated in 90% mouse serum; Lane 10: 1% mouse serum. (b) Top 30 most-abundant corona proteins detectable after 2 h mouse serum exposure based on spectral counts in LC-MS/MS analysis.
3.4. Interactions of albumin-coated NPs with different types of cells
We examined the effect of surface-bound albumin on NP-cell interactions using different cell-based models relevant to the delivery of circulating NPs to cancer cells: HUVEC and hCMEC/D3 as endothelial cell models; B16F10 and A549 cells as cancer cell models; and J774A.1 and THP1-XBlue-MD2-CD14 cells as phagocytes in the MPS. To estimate the effect of serum protein binding to NPs, cells were incubated with NPs in both serum-free (serum−) and serum-supplemented (serum+: 2% for endothelial cells and 10% for B16F10, A549, J774A.1, and THP1-XBlue-MD2-CD14 cells) media. Cytocompatibility of NPs was tested in each cell line for the duration of experiment (6 – 24 h). NPs used in a concentration (0.1 mg/mL unless specified otherwise) did not cause more than 20% decrease in mitochondrial activity (Fig. S3). The cells were incubated with fluorescently-labeled NPs for 1 h to establish initial NP-cell interactions and analyzed by flow cytometry.
3.4.1. NP interactions with endothelial cells
We hypothesized that surface-bound albumin would mediate the interaction with endothelial cells via albumin-binding proteins such as gp60. To test this hypothesis, we chose endothelial cells with different gp60 expression levels: HUVEC as a gp60-positive (gp60+) [48, 49] and hCMEC/D3 as a gp60-negative (gp60−) cell line [50–52]. FITC-BSA with 76% sequence homology to HSA [53] bound to HUVEC more than hCMEC/D3 cells, and its binding to HUVEC was competitively inhibited by HSA (Fig. S4). This verifies that HUVEC expressed more albumin-binding proteins than hCMEC/D3 cells.
HUVEC interacted with NPxAl to the highest degree, which was reduced in the presence of serum and additional albumin (Fig. 4a). On the other hand, hCMEC/D3 with lower expression of albumin-binding proteins had minimal interactions with all types of NPs (Fig. 4b). These results support that NPxAl interaction with HUVEC was mediated by albumin-binding proteins. Next, we examined if the NP-endothelial cell interaction affects the transendothelial transport of NPs (Fig. 4c). Endothelial cells were cultured on a Transwell insert (pore size 3 μm) until the TEER value reached a plateau (in 8–10 days) indicating the formation of a confluent monolayer (Fig. 4d). Rhodamine-labeled NPs were added to the apical side of the Transwell insert, and their contents in the cell layer and the media of both apical and basolateral sides were quantified after 6 h incubation. NPxAl and NP-pD-Al showed greater transport across the HUVEC layer than NP, NP-pD, and NP/Al (Fig. 4e). With the hCMEC/D3 layer, there was not much difference among the NPs irrespective of the surface modification. This result indicates that albumin-binding proteins were involved in the transport of NPxAl and NP-pD-Al across the HUVEC layer. The amount of NPs crossing the HUVEC layer was similar between NPxAl and NP-pD-Al (NPxAl: 23 ± 2% and NP-pD-Al: 20 ± 2%). Interestingly, more NPxAl were found in the HUVEC layer than NP-pD-Al (16 ± 2% of NPxAl and 10 ± 2% of NP-p D-Al). NPxAl and NP-pD-Al were located in HUVEC layer via TEM using iron oxide (IO) particles as a label (Fig. 4f). The HUVEC layer treated with IO@NPxAl showed dark vesicles with a size ranging from 0.5 to 1.2 μm. In contrast, the HUVEC layer treated with PBS or IO@NP-pD-Al showed much smaller vesicles (0.07 – 0.2 μm) resembling caveolae [54]. Although individual NPs could not be identified, the large vesicles in IO@NPxAl-treated HUVEC are thought to be lysosomes filled with IO@NPxAl, given the morphological similarity to the lysosomes seen in HUVEC internalizing metal and silica particles [55, 56]. The NP content in the HUVEC layer and the TEM image indicate that a greater fraction of NPxAl was stuck in HUVEC during the transport across the cell layer as compared to NP-pD-Al.
Fig. 4.
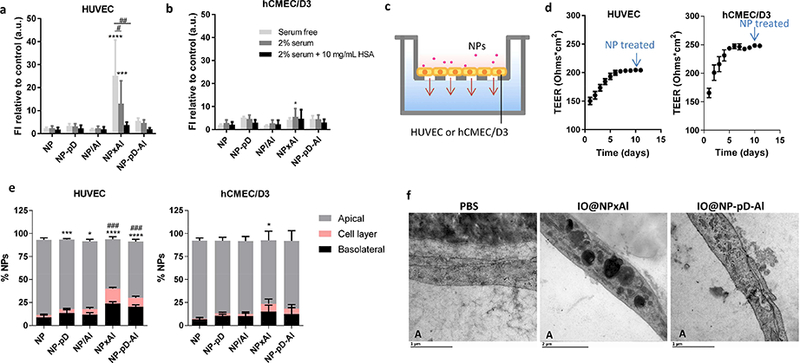
Interactions of NPs with (a) HUVEC (gp60+) and (b) hCMEC/D3 (gp60−) cells in the serum-free, 2% serum-supplemented media (containing 0.7 mg/mL albumin), and 2% serum-supplemented media with additional 10 mg/mL albumin, determined by flow cytometry. Cells were incubated with NPs (0.1 mg/mL) for 1 h. Fluorescence intensity (FI) was normalized by FI of control (untreated cells). n = 3 identically and independently prepared samples (mean ± s.d.). *: p<0.05; ***: p < 0.001; ****: p<0.0001 vs. NP; #: p<0.0005; ##: p < 0.0001 by Dunnett’s multiple comparisons test following two-way ANOVA. (c) Schematic of the Transwell system with endothelial cells seeded on an insert (pore size 3 μm). (d) Trans-Epithelial Electrical Resistance (TEER) of HUVEC and hCMEC/D3 cells layer (n = 36 wells, mean ± s.d.). (e) NP transport across HUVEC and hCMEC/D3 cells layer after 6 h of incubation with rhodamine-labeled NPs (0.1 mg/mL) in 2% serum-supplemented medium. n = 3 identically and independently prepared samples (mean ± s.d.). ###: p<0.0001 vs. NP in cell layer; *: p < 0.05; ***: p < 0.001; ****: p < 0.0001 vs. NP in the basolateral side by Dunnett’s multiple comparisons test following two-way ANOVA. (f) Transmission electron micrographs (TEM) of HUVECs incubated with PBS, iron oxide-loaded NPxAl (IO@NPxAl) or NP-pD-Al (IO@NP-pD-Al) for 6 h. “A” indicates the apical side of Tr answell insert. Scale bars = 1 μm for PBS and IO@NP-pD-Al; 2 μm for IO@NPxAl.
3.4.2. NP interaction with cancer cells
To examine if the albumin-coated NPs interact with cancer cells via albumin-binding proteins such as SPARC, NPs were incubated with A549 [57] and B16F10 cells [58], which are SPARC-positive. With 72% homology between human and murine albumins [59], the albumin-coated NPs were expected to interact with both A549 and B16F10 cells. A549 cells showed the most interaction with NP-pD-Al (Fig. 5a). Although the extent of interaction decreased in serum+ medium, the trend persisted indicating that the albumin/SPARC-mediated uptake mechanism played a dominant role despite the binding of serum proteins to NPs. In contrast, B16F10 cells showed the most interaction with NPxAl in serum− medium, which was significantly reduced in serum+ medium (Fig. 5c). The NP-pD-Al interaction with B16F10 cells was not significantly affected by serum, thereby ranking first among the NPs in serum+ medium. This suggests that NPxAl have another mechanism governing their interactions with B16F10 cells, which was likely disrupted by protein corona components (Fig. 3b).
Fig. 5.
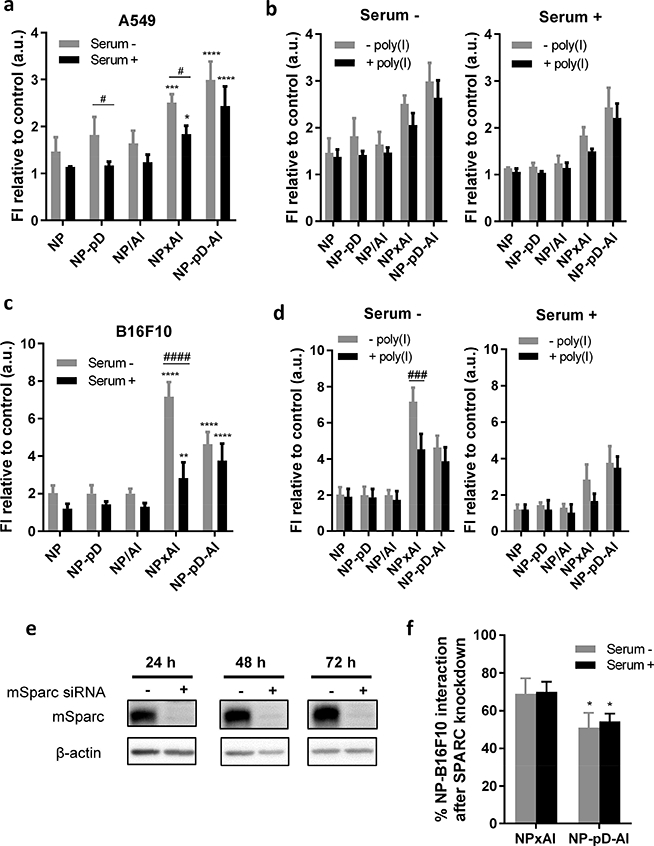
(a) NP interaction with A549 cells after 1 h incubation with NPs (0.1 mg/mL) in the absence and presence of 10% serum. (b) Inhibition of NP-A549 cell interaction by 30 min pre-treatment with polyinosinic acid (poly(I), 0.1 mg/mL) in the absence and presence of 10% serum.(c) NP interaction with B16F10 cells after 1 h incubation with fluorescently labeled NPs in the absence and presence of 10% serum. (d) Inhibition of NP-B16F10 cell interaction by 30 min pre-treatment with poly(I) (0.1 mg/mL) in the absence and presence of 10% serum. n = 3 identically and independently prepared samples (mean ± s.d.). *: p < 0.05; **: p < 0.01; ***: p < 0.001 and ****: p < 0.0001 vs. NP; #: p < 0.05; ###: p < 0.001; ####: p<0.0001 by Sidak’s multiple comparisons test following two-way ANOVA. (e) Validation of SPARC siRNA knockdown in B16F10 cells by western blot. (f) % NP interaction with B16F10 cells after SPARC knockdown relative to wild-type cells. n = 3 identically and independently prepared samples (mean ± s.d.). * p < 0.05 vs. NPxAl by Sidak’s multiple comparisons test following two-way ANOVA.
A notable difference between B16F10 and A549 cells is the expression of scavenger receptors: B16F10 cells express scavenger receptors [58], but A549 cells do not [60]. To test whether the uptake of NPxAl by B16F10 cells is mediated in part by scavenger receptors, we pretreated A549 and B16F10 cells with poly(I), an inhibitor of scavenger receptor A [60]. With A549 cells, neither NPxAl nor NP-pD-Al interaction was affected by the poly(I) pre-treatment (Fig. 5b). However, NPxAl interaction with B16F10 cells in serum− medium was significantly inhibited by poly(I) (Fig. 5d). This indicates that the interaction was mediated partly by the scavenger receptor of B16F10 cells, at least in the absence of serum, likely due to the denatured surface albumin, a substrate of scavenger receptor A [24].
To test if NPxAl and NP-pD-Al interact with cancer cells via SPARC, we examined the NP-cell interactions in B16F10 cells with SPARC silencing. siRNA targeting SPARC almost completely suppressed its expression without affecting cell viability (Fig. 5e). With the SPARC knockdown, NPxAl and NP-pD-Al interactions with B16F10 cells were reduced by 31 ± 8% and 49 ± 8% in serum− medium, respectively (Fig. 5f). The extent of reduction was greater for NP-pD-Al than for NPxAl, which may indicate that SPARC played a more significant role in NP-pD-Al interaction with B16F10 cells as compared to NPxAl’s. This coincides with the previous finding that NPxAl employs an additional mechanism involving scavenger receptors. Serum in the medium had no effect on the extent of reduction (30 ± 5% of NPxAl and 46 ± 4% of NP-pD-Al), consistent with the insensitivity of albumin/SPARC-mediated NP-cell interaction to serum proteins.
3.4.3. NP transport in a HUVEC/B16F10 co-culture system
With the single cell culture systems, we observed that (i) albumin on NP-pD-Al facilitated transendothelial transport and SPARC+ cancer cell interaction of the NPs, whereas (ii) NPxAl faced distinct outcomes, such as endothelial sequestration (Fig. 4e and 4f) and interactions with scavenger receptor A (Fig. 5d) that are easily obscured by serum proteins. We expected that the net result of these interactions would be the enhanced delivery of circulating NP-pD-Al to SPARC+ cancer cells. To assess this in vitro, we designed a Transwell co-culture system with HUVEC (gp60+) on the apical side and B16F10 (SPARC+) cells on the basolateral side that mimic the vessel and underlying tumors, respectively [61]. We first evaluated whether the co-culture condition affected the growth of B16F10 cells by monitoring their morphology and cell density. The results confirmed that neither complete HUVEC culture medium nor HUVECs was detrimental to B16F10 cells (Fig. S5). NPs were provided to the apical side (Fig. 6a) for 6 h when HUVEC was confluent (Fig. 6b), and B16F10 cells in the basolateral side were analyzed after additional 24 h incubation. As expected, NP-pD-Al was delivered best to B16F10 cells (Fig. 6c), confirming that NP-pD-Al was superior to NPxAl in reaching B16F10 cells across the confluent endothelial barrier.
Fig. 6.
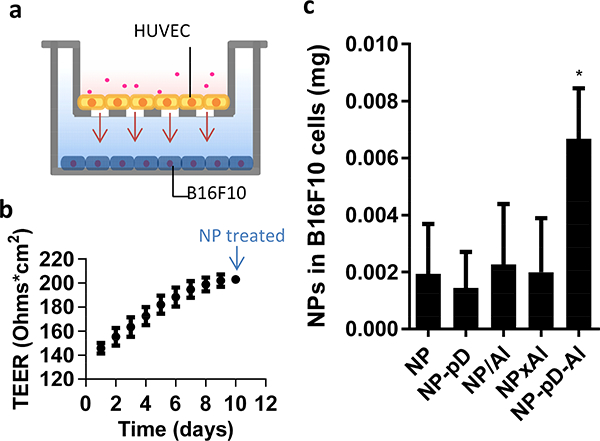
(a) Schematic of a Transwell co-culture system with HUVEC in the insert (pore size 3 μm) and B16F10 cells in the bottom of the basolateral side. (b) TEER of HUVEC layer (n = 24 wells, mean ± s.d.) and (c) NP associated with B16F10 cells, measured at 24 h after 6 h incubation with a Transwell containing 0.1 mg NPs and a HUVEC layer. n = 3 identically and independently performed experiments (mean ± s.d.). *: p < 0.05 vs. NP by Dunnett’s multiple comparisons test following one-way ANOVA.
3.4.4. Cellular uptake of albumin-coated NPs by J774A.1 macrophages
Given the tendency to attract serum proteins including ApoE (Fig. 3a) and interact with scavenger receptor A (Fig. 5d), we expected that NPxAl would be more susceptible to phagocytic uptake than other NPs. To test this, we used J774A.1 macrophages as a phagocytic cell model and incubated them with the fluorescently labeled NPs for 30 min. Confocal microscopy showed strong fluorescent signals in the cells treated with NPxAl (Fig. 7a). All other NPs showed minimal signals associated with the macrophages. Consistently, flow cytometry demonstrated the highest fluorescence signal with the macrophages treated with NPxAl, whereas NP-pD-Al, NP, NP-pD, and NP/Al showed much less signals. It is curious that bare NP showed minimal macrophage uptake. We suspect that this may be the function of residual PVA (emulsifier), which is known to remain on the NP surface even after multiple washing and negatively affect the cellular uptake [62]. Macrophage interaction with NPxAl was not reduced in the presence of serum but rather increased (Fig. 7b). In both serum− and serum+ media, NPxAl-J774A.1 interactions were reduced by poly(I) to the same level as other NPs (Fig. 7c), indicating that their interactions were mediated by scavenger receptor A [63]. J774A.1 and B16F10 cells had this pathway in common, but J774A.1 interacted with NPxAl even in the presence of the serum protein binding while B16F10 cells did not. This difference may be attributable to the abundance of scavenger receptors on J774A.1 macrophages as well as the affinity of J774A.1 cells for ApoE in the protein corona of NPxAl [64].
Fig. 7.
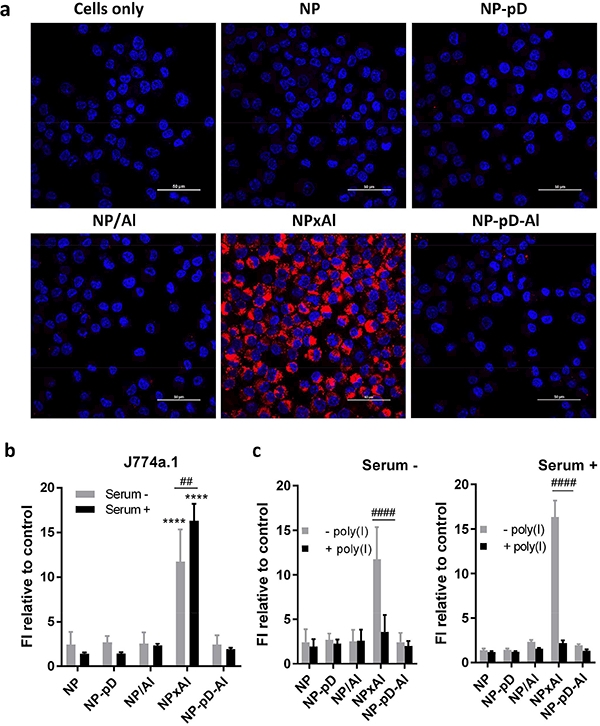
(a) Cellular uptake of NPs by J774A.1 macrophages after 30 min incubation with 0.1 mg/mL NPs in the presence of 10% serum, imaged with confocal microscopy (Red: rhodamine-labeled NPs; Blue: nuclei stained with Hoechst 33342). Scale bars: 50 μm. (b) Cellular uptake of NPs by J774A.1 macrophages, determined by flow cytometry. Macrophages were incubated with fluorescently labeled NPs (0.1 mg/mL) for 30 min in the absence and presence of 10% serum. (c) Inhibition of NP-J774A.1 cell interaction by 30 min pre-treatment with poly(I) in the absence and presence of 10% serum. NPs: 0.1 mg/mL; Poly(I): 0.1 mg/mL. Poly(I) pre-treated cells were incubated with NPs for 30 min. n = 3 identically and independently prepared samples (mean ± s.d.) ****: p < 0.0001 vs. NP by Dunnett’s multiple comparisons test following two-way ANOVA; ##: p < 0.01 and ####: p < 0.0001 by Sidak’s multiple comparisons test following two-way ANOVA.
3.4.5. Evaluation of immunogenicity of albumin-coated NPs
Denatured proteins on NP surface may trigger the activation of immune cells, such as monocytes and neutrophils, to produce pro-inflammatory cytokines [65, 66]. The denatured status of albumin on NPxAl (Fig. 2b, c) may trigger greater pro-inflammatory responses than other NPs. We evaluated the pro-inflammatory potential of NPs using THP1-XBlue-MD2-CD14 cell line, a human monocytic THP-1 cells engineered to express SEAP in response to the activation of NF-κB/activator protein (AP)-1 [66]. THP1-XBlue-MD2-CD14 cells showed most SEAP production (Fig. S6), accompanied by the reduced metabolic activity (Fig. S3d), when incubated with NPxAl. These results indicate that NPxAl serves as a more potent pro-inflammatory stimulus than other NPs. The toxicity is likely a consequence of pro-inflammatory cytokine production, which leads to cell lysis and DNA fragmentation (pyroptosis) [67, 68].
3.4.6. Comparison of NP-pD-Al vs. NP-pD-PEG
To confirm the role of surface albumin of NP-pD-Al, PLGA NPs were modified with amine-terminated methoxyl PEG via the same method (dopamine polymerization) to form NP-pD-PEG and compared with NP-pD-Al in their interactions with J774A.1 macrophages and B16F10 cells. NP-pD-PEG showed minimal signals associated with both types of cells, indicating the lack of cellular interactions (Fig. S7), which is consistent with the known role of PEG [4]. NP-pD-Al was comparable to NP-pD-PEG in the interaction with macrophages but showed greater signals with B16F10 cells than that of NP-pD-PEG. This result further supports that albumin on NP-pD-Al helps reduce phagocytic uptake of the NPs while allowing them to interact with B16F10 cells via albumin-binding proteins.
3.5. Tissue distribution of NP-pD-Al and NPxAl
To examine in vitro-in vivo translatability, we observed the distribution of NPs in tumors, livers, and spleens focusing on their locations relative to blood vessels following IV injection into B16F10 tumor-bearing mice. Immunofluorescence labeling requires a long incubation period and extensive washing after each step [69, 70], which may cause NP loss from the tissues. Instead, we visualized the blood vessels by perfusing FITC-lectin prior to sacrificing the animals. FITC-lectin stains vessels by binding to glycoproteins of endothelial plasmalemma [71] and has been used to locate endothelial cells in studies involving NPs [72, 73]. Due to the leakiness of tumor vasculature, FITC-lectin stain in tumors showed a more diffuse pattern than in liver or spleen, consistent with the literature [74, 75].
Fewer NPxAl were observed in tumors than NP-pD-Al. Most NPxAl appeared to be either in or near the lectin signals, whereas NP-pD-Al rarely overlapped with the lectin signals (Fig. 8a, Fig. S8). By image analysis, NPs in tumors were quantified as NPs (red + yellow) per tumors outlined by lectin staining (green), and the fraction of NPs overlapping with lectin was defined as yellow NPs per total NPs (red + yellow). The analysis found higher accumulation of NP-pD-Al (1.1 ± 0.6% of tissue area in the field of view) in tumors than NPxAl (0.3 ± 0.3%) and much less association of NP-pD-Al (26 ± 14% of total NPs) with blood vessels than NPxAl (83 ±17%) (Fig. 8a). This result indicates that NP-pD-Al reached tumors to a greater extent than NPxAl and translocated across the blood vessels unlike NPxAl that remained in the endothelial layer, consistent with the Transwell studies (Fig. 4e, 4f). More NPxAl (10 ± 4% of tissue area in the field of view) were found in the liver than NP-pD-Al (2 ± 2%) (Fig. 8b, Fig. S9). NPxAl in the liver showed a greater overlap with lectin (23 ± 12% of total NPs) than NP-pD-Al (10 ± 5%). Similarly, the uptake of NPxAl (2.1 ± 0.6% of tissue area in the field of view) in the spleen appeared greater than that of NP-pD-Al (0.6 ± 0.2%), and NPxAl overlap with lectin (33 ± 27% of total NPs) was higher than NP-pD-Al (27 ± 8%) (Fig. 8c, Fig. S10).
Fig. 8.
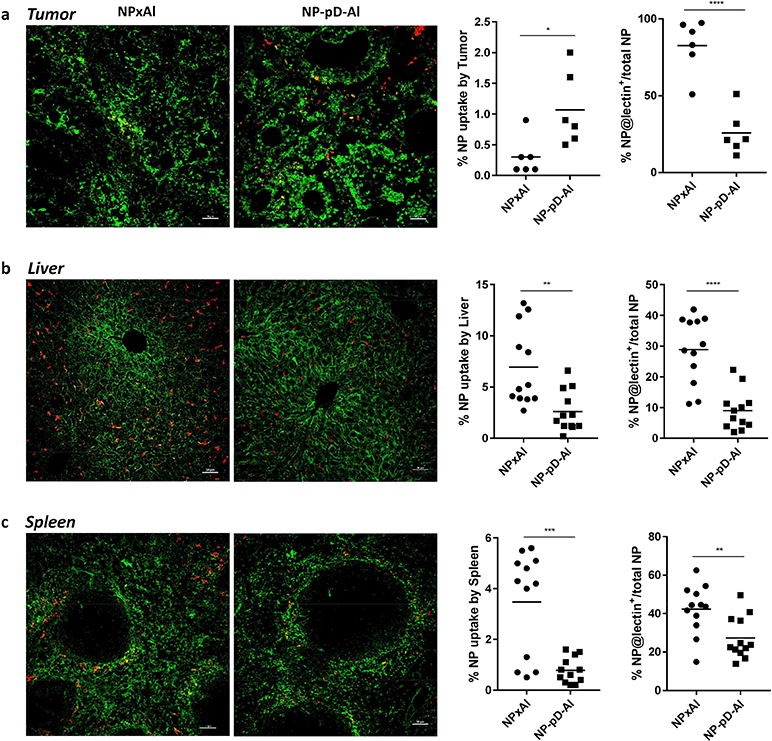
Quantitative analysis of FITC-lectin-stained (a) tumor, (b) liver, and (c) spleen. Mice were administered with a single injection of rhodamine-labeled NPxAl (n=3) or NP-pD-Al (n=3) at 300 mg NPs/kg. After 24 h, FITC-lectin (0.1 mg) was injected 5 min before sacrifice. % NP uptake by tissues was estimated as the area of NP fluorescence (red + yellow) divided by the area of the tissue in the field of view (outlined by green lectin staining). % NP associated with lectin-positive endothelial cells was calculated as the area of NP overlapped with lectin-positive area (yellow) divided by the area of the total NP fluorescence (yellow + red). Randomly selected fields (6 images/group for tumors and 12 images/group for livers and spleens) were analyzed with Fiji image analysis software. Scale bar: 50 μm. *: p < 0.05; **: p < 0.01; ***: p < 0.001 and ****: p < 0.0001 by unpaired t-test.
3.6. In vivo evaluation of PTX-loaded NP-pD-Al and NPxAl
On the basis of reproducible surface modification with albumin and the differential tissue deposition profiles, we chose NP-pD-Al and NPxAl as carriers of PTX and compared their in vivo efficacy in B16F10 tumor-bearing mice. PTX was encapsulated in NP-pD-Al (PTX@NP-pD-Al) and NPxAl (PTX@NPxAl). PTX@NPxAl was smaller and had greater negative charges than PTX@NP-pD-Al (Table S3), similar to blank NPs (Table S1). The drug loading efficiencies of PTX@NPxAl and PTX@NP-pD-Al were 7.9 ± 1.1% and 3.4 ± 0.3%, respectively. Both NPs were washed six times with water to remove loosely bound drugs on NP surface. The difference in drug loading is likely due to the relatively high albumin content on NPxAl, which can serve as a diffusion barrier based on the high affinity for PTX [76] during the purification step. The two NPs showed comparable release kinetics, releasing ~60% PTX in PBS containing Tween 80 in 24 h (Fig. S11). The PTX-loaded NPs were administered to C57BL/6 mice bearing subcutaneous B16F10 tumors when they reached an average size of ~150 mm3. The first group of animals were treated with the NPs four times every three days (q3d × 4) at a dose equivalent to 15 mg/kg PTX per time and monitored for the tumor growth (Fig. 9). The second set of animals were treated with two injections of NPs equivalent to 15 mg/kg with 3 day interval (q3d × 2) and sacrificed one day after the second dose for the analysis of tissues and blood (Fig. 10).
Fig. 9.
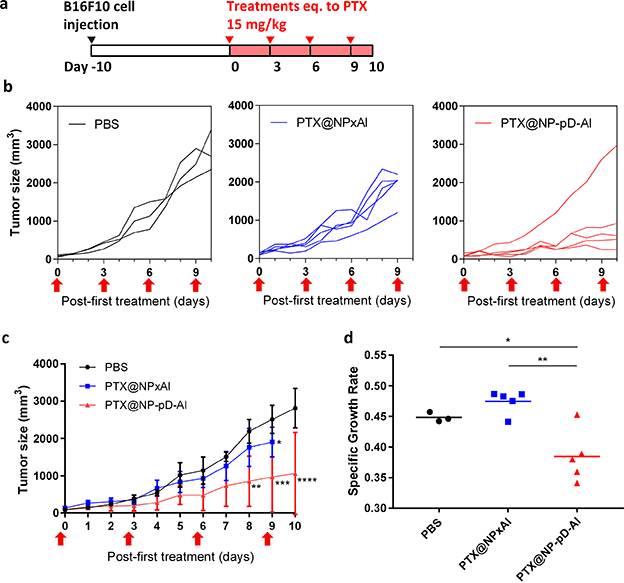
In vivo activity of PTX@NPxAl and PTX@NP-pD-Al in C57BL/6mice bearing B16F10 tumor. (a) Dosing schedule of PTX-loaded NPs. (b) Tumor growth curves of individual animals treated with PBS (n=3), PTX@NPxAl (n=5), or PTX@NP-pD-Al (n=5). Arrows indicate treatment times. (c) Average tumor size (mm3). *: p < 0.05; **: p <0.01; ***: p < 0.001; ****: p <0.0001 vs. PBS at each time point by Sidak’s multiple comparisons test following two-way ANOVA. (d) Specific growth rate of B16F10 tumor ΔlogV/Δt (V: tumor volumes; t: time in days). *: p < 0.05; **: p < 0.01 by Tukey’s multiple comparisons test following one-way ANOVA.
Fig. 10.
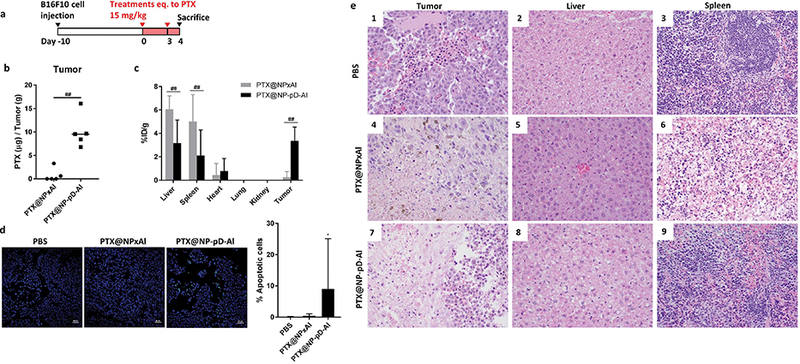
In vivo activity of PTX@NPxAl and PTX@NP-pD-Al in C57BL/6mice bearing B16F10 tumor. (a) Dosing schedule of PTX-loaded NPs, (b) PTX content in B16F10 tumors. PTX@NPxAl (n = 5); PTX@NP-pD-Al (n = 5). ##: p < 0.01 by non-parametric Mann-Whitney test. (c) % injected PTX dose per gram of each tissues (%ID/g) of PTX@NPxAl or PTX@NP-pD-Al in B16F10 tumor bearing mice 24h after i.v. injection. %ID/g is defined as percentage of injected dose per gram of tissue weight. ##: p < 0.01 by Sidak’s multiple comparisons test following two-way ANOVA. (d) Representative photographs of TUNEL-stained B16F10 tumor sections and quantitative analysis of TUNEL-stained sections. % apoptotic cells = number of apoptotic cells / total number of nuclei measured by Fiji image analysis software (6 random fields for PBS and 10 random fields for PTX@NPxAl and PTX@NP-pD-Al groups). Scale bars 50 μm. *: p < 0.05 vs. PBS by Dunn’s multiple comparisons test following Kruskal-Wallis test. (e) Photomicrographs (40× objective) of PBS treated (1–3) animals with rare tumor hemorrhage, rare vacuolar change within the liver, and normal spleen pathology. PTX@NPxAl treated animals (4–6) demonstrating multifocal tumor necrosis, multifocal vacuolar change within the liver, and lymphoid depletion within the spleen. PTX@NP-pD-Al treated animals (7–9) demonstrating marked tumor necrosis, multifocal vacuolar change within the liver, and lymphoid depletion within the spleen. For low magnification images (20× objective), see Supporting Fig. 14.
3.6.1. Anti-tumor activity of PTX@NPs
PTX-loaded NPs were administered to the tumor-bearing mice by tail vein injection (Fig. 9a). The growth of B16F10 tumors was significantly attenuated by PTX@NP-pD-Al as compared to the PBS control but not by PTX@NPxAl (Fig. 9b and c). PTX@NP-pD-Al treatment resulted in a significantly lower tumor growth rate as compared to PTX@NPxAl (p < 0.01) and PBS control (p < 0.05) (Fig. 9d). This result is consistent with the NP distribution in tumor tissues (Fig. 8a). None of the animals experienced significant weight loss throughout the 10-day study period (Fig. S12). However, all the mice treated with PTX@NPxAl were found dead after the fourth injection and showed splenomegaly upon necropsy (data not shown).
3.6.2. Tissue and blood analysis
PTX distribution in organs, the serum chemistry and serum cytokine levels were evaluated after two injections of NPs with the same dose of PTX (15 mg/kg) and dosing interval (q3d × 2) (Fig. 10a). Although the difference in tumor size did not clearly manifest at the point of sacrifice (after two injections), the growth trend (Fig. S13) was consistent with the previous study (Fig. 9). Notably, PTX@NPxAl treatment resulted in substantial elevation in the serum level of alanine aminotransferase (ALT), an indicator of liver damage [77] (Table S4). PTX@NP-pD-Al-treated animals showed normal ALT levels except for one. Serum levels of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) appeared elevated in the PTX@NPxAl group though statistically insignificant but not in the PTX@NP-pD-Al group.
Next, the effect of PTX@NP treatment was evaluated on the tissue level. The PTX content in the tumor was significantly higher in the PTX@NP-pD-Al group (10.1 ± 3.5 μg/g) than in the PTX@NPxAl group (0.8 ± 1.4 μg/g) ( p < 0.05, Mann-Whitney test) (Fig. 10b). The extent of difference was greater than that estimated from the image analysis of NP distribution in tumor section (1.1 ± 0.6% of NP-pD-Al vs. 0.3 ± 0.3% of NPxAl, Fig. 8a). This may be explained by the greater fraction of NPxAl associated with endothelial layer (25.7 ± 14.2% of NP-pD-Al vs. 82.7 ± 17.4% of NPxAl, Fig. 8a), which are prone to partial drug release into circulation. PTX@NPxAl-treated animals showed twice as much PTX in the liver (18.2 ± 3.4 μg/g) as those with PTX@NP-pD-Al (9.5 ± 5.9 μg/g) ( Fig. S14). The spleen showed a similar trend as the liver with 15.1 ± 6.9 μg/g in PTX@NPxA l-treated mice and 6.3 ± 6.6 μg/g in PTX@NP-pD-Al-treated ones. The percent injected PTX dose per gram of each tissue (%ID/g) demonstrated a greater level of PTX in tumor and lower levels in liver and spleen with PTX@NP-pD-Al than with PTX@NPxAl (Fig. 10c), as expected from the images of NPs in those tissues (Fig. 8). It is noteworthy that the PTX@NP-pD-Al group the amount of PTX accumulated in the tumor (3.4 ± 1.2%ID/g) was similar to that in the liver (3.2 ± 2.0%ID/g), because NPs (including PTX@NPxAl) typically exhibit relatively high accumulation in the liver at 24 h post-treatment [78]. The PTX contents in the kidneys and the lungs were below the detection limit, suggesting minimal accumulation of both NPs in these organs. The extent of tumor apoptosis, estimated by the TUNEL assay, reflected the PTX levels in tumors (Fig. 10d, Fig. S15). The PTX@NP-pD-Al-treated animals showed higher numbers of apoptotic cells than those of the PBS control or PTX@NPxAl groups. Histological evaluation showed a consistent trend as tissue levels of PTX (Fig. 10e, Fig. S16). All five animals treated with PTX@NP-pD-Al showed tumors with signs of necrosis characterized by shrunken, hypereosinophilic tumor cells with shrunken or absent nuclei, whereas only 1 out of 5 with PTX@NPxAl showed a similar pattern of necrosis. In the liver, PTX@NP-pD-Al induced a few foci of hepatocellular degeneration associated with scattered clusters of lymphocytes and plasma cells in 3 out of 5 animals and vacuolar change in 2 out of 5. PTX@NPxAl induced hepatocellular degeneration in all five animals and vacuolar change in 4 out of 5. In the spleen, all five animals treated with PTX@NP-pD-Al and 3 out of 5 PTX@NPxAl-treated animals demonstrated mild to moderate lymphoid depletion. Histological findings support the efficacy of treatment with PTX@NP-pD-Al in this model with marginal alterations to major parenchymal tissues.
4. Discussion
Surface chemistry of NPs is critical to the therapeutic outcomes of the NP-mediated drug delivery. Albumin is considered a promising surface modifier of NPs due to the dysopsonin function [7], increased consumption by cancer cells [11], and the physiological transport mechanism favoring deposition in tumors [6]. In this study, we investigated how the method to attach albumin on NP surface affects the status of surface-bound albumin and its functions in transporting NPs to tumors. Albumin-modified NPs were produced in three different methods (Fig. 1) – physisorption, interfacial embedding, an d conjugation via pD adhesive layer – and compared with respect to their physicochemical properties, the status of surface-bound albumin, their interactions with different types of cells and organ distribution following systemic administration. With physisorption (NP/Al), albumin was bound to NPs so weakly that it did not stay on the NP surface consistently, thus making little contribution to the NP performance in the following studies. The interfacial embedding approach (NPxAl) achieved a relatively high extent of albumin coating. However, pulse proteolysis and esterase-like activity assay indicated that the surface-bound albumin did not maintain the native conformation likely due to the prolonged exposure to water/organic solvent interface and the high density of albumin on the NP surface (Fig. 2). In contrast, the albumin conjugated via pD adhesive layer (NP-pD-Al) retained the native conformation relatively well, although the absolute amount of albumin per NP surface was far less than that of NPxAl.
The significance of protein conformation in protein-bound NPs has been recognized with various types of NPs and model proteins [79]. The protein activity may depend on the orientation and number of the surface-bound proteins, as shown with gold NPs and a model enzyme [80]. The NP structure, size, surface chemistry, charge, and surface shape are another factors governing the protein activity [79, 81]. For example, small NPs with high curvature have a greater chance to cause protein denaturation than larger NPs with relatively plain surface [82]. The altered conformation of the surface-bound proteins can dictate cellular interaction profiles of the NPs, making them show the opposite behaviors with similar diameters and surface charges Our study shows that the status of the surface-bound albumin can also vary with the method of surface modification and it impacts the interactions of NPs with biological interfaces relevant to drug delivery significantly.
The differential albumin status induced several notable differences in NP behaviors. First, NPxAl bound to a greater amount of proteins than NP-pD-Al when incubated in serum (Fig. 3). Among the bound serum proteins was ApoE, which interacts with scavenger receptor A and mediates macrophage binding to surfaces [83]. Second, NP-pD-Al and NPxAl showed distinct interactions with endothelial cells (Fig. 4), cancer cells (Fig. 5), and phagocytes (Fig. 7). NP-pD- Al was transported across the endothelial layer better than other NPs with no albumin coating. Although NPxAl crossed the endothelial layer to a similar extent, a large part of the NPs was also sequestered in the cell layer during the transport, which may not be a desirable event unless the endothelial cells are the intended target. Moreover, NP-pD-Al had robust interactions with cancer cells expressing SPARC such as A549 and B16F10 cells, irrespective of the presence of serum. On the other hand, NPxAl partly interacted with cancer cells via scavenger receptor A, which was easily obscured in serum-containing medium. Consequently, NP-pD-Al showed greater interactions with A549 and B16F10 cells than NPxAl in serum-containing medium, despite the relatively small albumin content. In the HUVEC/B16F10 co-culture model, NP-pD-Al was superior to NPxAl in reaching B16F10 cells in the basolateral side across the endothelial layer (Fig. 6). NPxAl performed less favorably in the co-culture model than in the B16F10 single culture model. Given that NPxAl and NP-pD-Al showed comparable transendothelial migration in a Transwell single culture, the poor interaction of NPxAl with B16F10 cells may not be explained by the serum-induced fouling of scavenger receptors alone. We suspect that NPxAl may have acquired additional corona based on cellular proteins during transendothelial migration (as it did with serum proteins), further losing the ability to interact with B16F10 cells. Phagocytes such as macrophages showed high affinity for NPxAl, likely due to the denatured surface albumin as well as ApoE of the protein corona. In contrast, other NPs including NP-pD-Al showed minimal interaction with the macrophages, suggesting the lack of such proteins on the surface. Taken together, these results indicate that the NP-pD-Al is preferable to NPxAl for drug delivery to tumors, based on the tendency to translocate across endothelial layer with minimal sequestration in the cells, the ability to interact with SPARC+ cancer cells in a serum-insensitive manner, and the minimal interaction with phagocytes.
To verify this, we evaluated NP distribution and NP-mediated delivery of PTX in C57BL/6 mice bearing B16F10 melanoma. We observed greater accumulation of NP-pD-Al in tumor and less in the liver and spleen compared to those of NPxAl by microscopic evaluation of each tissue (Fig. 8). It also revealed differential association of the NPs with tumor blood vessels (Fig. 8a), consistent with the in vitro observation in the Transwell system (Fig. 4e). More NPxAl was associated with blood vessels in tumors than NP-pD-Al, with few penetrating into tumor tissues. NPxAl was also associated with endothelial cells in the liver to a greater extent than NP-pD-Al, likely due to the scavenger receptor A expressed on the liver sinusoidal endothelial cells [84]. The PTX content in the tumor measured at 24 h after two doses of NPs (15 mg/kg, q3d × 2) showed a consistent trend as the tissue distribution of NPs. NP-pD-Al delivered a greater amount of PTX to tumors and less to the liver and spleen than NPxAl (Fig. 10). These findings were further corroborated by tumor necrosis and vacuolar change of the liver in PTX@NP-pD-Al treated animals compared to those in PTX@NPxAl treated animals. The animals treated with PTX@NPxAl showed signs of greater liver damage and systemic inflammation (serum levels of pro-inflammatory cytokines). With four doses of NPs (q3d × 4), PTX@NP-pD-Al brought better tumor suppression activity than PTX@NPxAl (Fig. 9). Notably, all the animals treated with PTX@NPxAl died after the last treatment. The administered PTX dose (15 mg/kg, q3d × 4) was lower than the reported maximum tolerated doses of Taxol (20 mg/kg/day x 3 [85]) or Abraxane (60 mg/kg, q3d × 5 [86]) and thus considered sub-lethal. Hence, the abrupt death of the PTX@NPxAl-treated animals is more likely linked to the pro-inflammatory effects of NPxAl, which have been consistently manifested in the SEAP assay indicating the activation of NF-κB/AP-1 in the in vitro monocyte model as well as the elevated levels of pro-inflammatory cytokines in blood and splenomegaly observed at necropsy.
5. Conclusion
In summary, we demonstrate that albumin, only with native conformation, can serve as a favorable surface modifier for the systemic delivery of anti-cancer drugs to tumors. In this study, the protein structure depends largely on the method of surface modification, where the pD-mediated conjugation well preserved protein conformation during surface modification as opposed to interfacial embedding. NP-pD-Al with native albumin interacted with albumin-binding proteins such as SPARC, better reaching tumors with less interaction with phagocytes compared to NPxAl. In contrast, the denatured albumin on NPxAl acted as a substrate of scavenger receptor A for macrophages not only in itself but also by promoting the adsorption of protein corona including ApoE and increased the MPS uptake. This finding may be applicable to other proteins considered for the functionalization of NPs or those acquired during circulation; therefore, it is necessary to make a deliberate effort to control the protein binding to the surface in engineering therapeutic NPs.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health NIBIB R01EB017791 and the Lilly Endowment Gift Graduate Research Award to H.H. This work was also supported by the Indiana Clinical and Translational Sciences Institute, funded in part by grant #UL1 TR001108 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. We thank Samyang Biopharm (Seoul, Korea) for the kind donation of paclitaxel for the gift support. We also thank Victoria E. Hedrick at the Purdue Bindley Bioscience Center for the help with LC-MS/MS analysis of protein corona and Laurie Mueller at the Purdue Life Science Microscopy Facility for the help with TEM imaging of endothelial layer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Appendix A. Supporting Data
Supporting data related to this article can be found on the internet.
Data Availability
The raw data required to reproduce these findings and the processed data required to reproduce these findings are available upon request.
References
- [1].Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng. 14 (2012) 1–16. [DOI] [PubMed] [Google Scholar]
- [2].Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 134 (2012) 2139–47. [DOI] [PubMed] [Google Scholar]
- [3].Hatakeyama H, Akita H, Harashima H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: a strategy for overcoming the PEG dilemma. Adv Drug Deliv Rev. 63 (2011) 152–60. [DOI] [PubMed] [Google Scholar]
- [4].Hatakeyama H, Akita H, Harashima H. The polyethyleneglycol dilemma: advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol Pharm Bull. 36 (2013) 892–9. [DOI] [PubMed] [Google Scholar]
- [5].Gref R, Luck M, Quellec P, Marchand M, Dellacherie E, Harnisch S, et al. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces. 18 (2000) 301–13. [DOI] [PubMed] [Google Scholar]
- [6].Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 72 (2010) 463–93. [DOI] [PubMed] [Google Scholar]
- [7].Ogawara K-i, Furumoto K, Nagayama S, Minato K, Higaki K, Kai T, et al. Pre-coating with serum albumin reduces receptor-mediated hepatic disposition of polystyrene nanosphere: implications for rational design of nanoparticles. J Control Release. 100 (2004) 451–5. [DOI] [PubMed] [Google Scholar]
- [8].Furumoto K, Yokoe J-I, Ogawara K-i, Amano S, Takaguchi M, Higaki K, et al. Effect of coupling of albumin onto surface of PEG liposome on its in vivo disposition. Int J Pharm. 329 (2007) 110–6. [DOI] [PubMed] [Google Scholar]
- [9].Beukers H, Deierkauf FA, Blom CP, Deierkauf M, Riemersma JC. Effects of albumin on the phagocytosis of polystyrene spherules by rabbit polymorphonuclear leucocytes. J Cell Physiol. 97 (1978) 29–36. [DOI] [PubMed] [Google Scholar]
- [10].Peng Q, Zhang S, Yang Q, Zhang T, Wei XQ, Jiang L, et al. Preformed albumin corona, a protective coating for nanoparticles based drug delivery system. Biomaterials. 34 (2013) 8521–30. [DOI] [PubMed] [Google Scholar]
- [11].Stehle G, Sinn H, Wunder A, Schrenk HH, Stewart JC, Hartung G, et al. Plasma protein (albumin) catabolism by the tumor itself--implications for tumor metabolism and the genesis of cachexia. Crit Rev Oncol Hematol. 26 (1997) 77–100. [DOI] [PubMed] [Google Scholar]
- [12].Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 497 (2013) 633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vogel SM, Minshall RD, Pilipovic M, Tiruppathi C, Malik AB. Albumin uptake and transcytosis in endothelial cells in vivo induced by albumin-binding protein. Am J Physiol Lung Cell Mol Physiol. 281 (2001) L1512–22. [DOI] [PubMed] [Google Scholar]
- [14].Tiruppathi C, Song W, Bergenfeldt M, Sass P, Malik AB. Gp60 activation mediates albumin transcytosis in endothelial cells by tyrosine kinase-dependent pathway. J Biol Chem. 272 (1997) 25968–75. [DOI] [PubMed] [Google Scholar]
- [15].Schnitzer JE, Oh P. Albondin-mediated capillary permeability to albumin. Differential role of receptors in endothelial transcytosis and endocytosis of native and modified albumins. J Biol Chem. 269 (1994) 6072–82. [PubMed] [Google Scholar]
- [16].Elsadek B, Kratz F. Impact of albumin on drug delivery - New applications on the horizon. J Control Release. 157 (2012) 4–28. [DOI] [PubMed] [Google Scholar]
- [17].Trieu V, Damascelli B, Soon-Shiong P, Desai N. SPARC expression in head and neck cancer correlates with tumor response to nanoparticle albumin-bound paclitaxel (nab-paclitaxel, ABI-007, Abraxane). Cancer Res. 66 (2006) 1050. [Google Scholar]
- [18].Iglesias J nab-Paclitaxel (Abraxane(®)): an a lbumin-bound cytotoxic exploiting natural delivery mechanisms into tumors. Breast Cancer Res. 11 (2009) S21–S.20030873 [Google Scholar]
- [19].Mariam J, Sivakami S, Dongre PM. Albumin corona on nanoparticles - a strategic approach in drug delivery. Drug deliv. 23 (2016) 2668–76. [DOI] [PubMed] [Google Scholar]
- [20].Peng Q, Wei XQ, Yang Q, Zhang S, Zhang T, Shao XR, et al. Enhanced biostability of nanoparticle-based drug delivery systems by albumin corona. Nanomedicine (London, England). 10 (2015) 205–14. [DOI] [PubMed] [Google Scholar]
- [21].Wohlfart S, Khalansky AS, Gelperina S, Maksimenko O, Bernreuther C, Glatzel M, et al. Efficient chemotherapy of rat glioblastoma using doxorubicin-loaded PLGA nanoparticles with different stabilizers. PloS One. 6 (2011) e19121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Manoochehri S, Darvishi B, Kamalinia G, Amini M, Fallah M, Ostad SN, et al. Surface modification of PLGA nanoparticles via human serum albumin conjugation for controlled delivery of docetaxel. Daru. (2013) 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jeong H, Huh M, Lee SJ, Koo H, Kwon IC, Jeong SY, et al. Photosensitizer-conjugated human serum albumin nanoparticles for effective photodynamic therapy. Theranostics. 1 (2011) 230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mortimer GM, Butcher NJ, Musumeci AW, Deng ZJ, Martin DJ, Minchin RF. Cryptic epitopes of albumin determine mononuclear phagocyte system clearance of nanomaterials. ACS Nano. 8 (2014) 3357–66. [DOI] [PubMed] [Google Scholar]
- [25].Fleischer CC, Payne CK. Secondary Structure of Corona Proteins Determines the Cell Surface Receptors Used by Nanoparticles. The Journal of Physical Chemistry B. 118 (2014) 14017–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Asha Jhonsi M, Kathiravan A, Renganathan R. Spectroscopic studies on the interaction of colloidal capped CdS nanoparticles with bovine serum albumin. Colloids Surf B Biointerfaces. 72 (2009) 167–72. [DOI] [PubMed] [Google Scholar]
- [27].Liu Y, Ji F, Liu R. The interaction of bovine serum albumin with doxorubicin-loaded superparamagnetic iron oxide nanoparticles: spectroscope and molecular modelling identification. Nanotoxicology. 7 (2013) 97–104. [DOI] [PubMed] [Google Scholar]
- [28].Kong L, Hu J, Qin D, Yan P. Interaction of Ifosfamide-Loaded Superparamagnetic Iron Oxide Nanoparticles with Human Serum Albumin—A Biop hysical Study. J Pharm Innov. 10 (2015) 13–20. [Google Scholar]
- [29].Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-inspired surface chemistry for multifunctional coatings. Science. 318 (2007) 426–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Park J, Brust TF, Lee HJ, Lee SC, Watts VJ, Yeo Y. Polydopamine-Based Simple and Versatile Surface Modification of Polymeric Nano Drug Carriers. ACS Nano. 8 (2014) 3347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu Y, Ai K, Lu L. Polydopamine and its derivative materials: synthesis and promising applications in energy, environmental, and biomedical fields. Chem Rev. 114 (2014) 5057–115. [DOI] [PubMed] [Google Scholar]
- [32].Hong S, Na YS, Choi S, Song IT, Kim WY, Lee H. Non-Covalent Self-Assembly and Covalent Polymerization Co-Contribute to Polydopamine Formation. Adv Funct Mater. 22 (2012) 4711–7. [Google Scholar]
- [33].Lee H, Rho J, Messersmith PB. Facile Conjugation of Biomolecules onto Surfaces via Mussel Adhesive Protein Inspired Coatings. Advanced materials (Deerfield Beach, Fla). 21 (2009) 431–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yang SH, Kang SM, Lee KB, Chung TD, Lee H, Choi IS. Mussel-inspired encapsulation and functionalization of individual yeast cells. J Am Chem Soc. 133 (2011) 2795–7. [DOI] [PubMed] [Google Scholar]
- [35].Park C, Marqusee S. Pulse proteolysis: A simple method for quantitative determination of protein stability and ligand binding. Nat Methods. 2 (2005) 207–12. [DOI] [PubMed] [Google Scholar]
- [36].Watanabe H, Tanase S, Nakajou K, Maruyama T, Kragh-Hansen U, Otagiri M. Role of Arg-410 and Tyr-411 in human serum albumin for ligand binding and esterase-like activity. Biochem. J. 349 (2000) 813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jes´us C´ordova JDR, Boonchai B. Boonyaratanak ornkit, Clark Douglas S.. Esterase activity of bovine serum albumin up to 160 °C: A new benchmark for biocatalysis. Enzyme Microb Technol. 42 (2008) 278–83. [Google Scholar]
- [38].Benson K, Cramer S, Galla HJ. Impedance-based cell monitoring: barrier properties and beyond. Fluids Barriers CNS. 10 (2013) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 24 (1989) 148–54. [DOI] [PubMed] [Google Scholar]
- [40].Mehrara E, Forssell-Aronsson E, Ahlman H, Bernhardt P. Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res. 67 (2007) 3970–5. [DOI] [PubMed] [Google Scholar]
- [41].Anand U, Lu J, Loh D, Aabdin Z, Mirsaidov U. Hydration Layer-Mediated Pairwise Interaction of Nanoparticles. Nano lett. 16 (2016) 786–90. [DOI] [PubMed] [Google Scholar]
- [42].Cardellini A, Fasano M, Chiavazzo E, Asinari P. Interfacial water thickness at inorganic nanoconstructs and biomolecules: Size matters. Phys Lett A 380 (2016) 1735–40. [Google Scholar]
- [43].Park J, Sun B, Yeo Y. Albumin-coated nanocrystals for carrier-free delivery of paclitaxel. J Control Release. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Thiele L, Diederichs JE, Reszka R, Merkle HP, Walter E. Competitive adsorption of serum proteins at microparticles affects phagocytosis by dendritic cells. Biomaterials. 24 (2003) 1409–18. [DOI] [PubMed] [Google Scholar]
- [45].Cedervall T, Lynch I, Lindman S, Berggård T, T hulin E, Nilsson H, et al. Understanding the nanoparticle–protein corona using methods to quanti fy exchange rates and affinities of proteins for nanoparticles. Proc Natl Acad Sci U S A. 104 (2007) 2050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lusis AJ, Taylor BA, Quon D, Zollman S, LeBoeuf RC. Genetic factors controlling structure and expression of apolipoproteins B and E in mice. J Biol Chem. 262 (1987) 7594–604. [PubMed] [Google Scholar]
- [47].Mahley RW, Innerarity TL, Rall SC Jr., Weisgraber KH. Plasma lipoproteins: apolipoprotein structure and function. Journal of lipid research. 25 (1984) 1277–94. [PubMed] [Google Scholar]
- [48].Pavlides S, Gutierrez-Pajares JL, Iturrieta J, Lisanti MP, Frank PG. Endothelial caveolin-1 plays a major role in the development of atherosclerosis. Cell Tissue Res. 356 (2014) 147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Montrucchio G, Lupia E, De Martino A, Silvestro L, Savu SR, Cacace G, et al. Plasmin promotes an endothelium-dependent adhesion of neutrophils. Involvement of platelet activating factor and P-selectin. Circulation. 93 (1996) 2152–60. [DOI] [PubMed] [Google Scholar]
- [50].Ye D, Raghnaill MN, Bramini M, Mahon E, Aberg C, Salvati A, et al. Nanoparticle accumulation and transcytosis in brain endothelial cell layers. Nanoscale. 5 (2013) 11153–65. [DOI] [PubMed] [Google Scholar]
- [51].Minshall RD, Tiruppathi C, Vogel SM, Niles WD, Gilchrist A, Hamm HE, et al. Endothelial cell-surface gp60 activates vesicle formation and trafficking via G(i)-coupled Src kinase signaling pathway. J Cell Biol. 150 (2000) 1057–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Schnitzer JE. gp60 is an albumin-binding glycoprotein expressed by continuous endothelium involved in albumin transcytosis. Am. J. Physiol 262 (1992) H246–54. [DOI] [PubMed] [Google Scholar]
- [53].Liu Y, Zhong R, Zhang P, Ma Y, Yun X, Gong P, et al. Understanding the Robust Physisorption between Bovine Serum Albumin and Amphiphilic Polymer Coated Nanoparticles. ACS Appl Mater Interfaces. 8 (2016) 2478–85. [DOI] [PubMed] [Google Scholar]
- [54].Chen J, Braet F, Brodsky S, Weinstein T, Romanov V, Noiri E, et al. VEGF-induced mobilization of caveolae and increase in permeability of endothelial cells. Am J Physiol Cell Physiol. 282 (2002) C1053–63. [DOI] [PubMed] [Google Scholar]
- [55].Liu X, Xue Y, Ding T, Sun J. Enhancement of proinflammatory and procoagulant responses to silica particles by monocyte-endothelial cell interactions. Part Fibre Toxicol. 9 (2012) 36-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liu X, Sun J. Potential proinflammatory effects of hydroxyapatite nanoparticles on endothelial cells in a monocyte–endothelial cell co culture model. International Journal of Nanomedicine. 9 (2014) 1261–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhou Y, Hofstetter WL, He Y, Hu W, Pataer A, Wang L, et al. KLF4 inhibition of lung cancer cell invasion by suppression of SPARC expression. Cancer Biol. Ther. 9 (2010) 507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hoang B, Ernsting MJ, Roy A, Murakami M, Undzys E, Li SD. Docetaxel-carboxymethylcellulose nanoparticles target cells via a SPARC and albumin dependent mechanism. Biomaterials. 59 (2015) 66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sheng J, Wang Y, Turesky RJ, Kluetzman K, Zhang Q-Y, Ding X. Novel Transgenic Mouse Model for Studying Human Serum Albumin as A Biomarker of Carcinogenic Exposure. Chem Res Toxicol. 29 (2016) 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Choi CH, Hao L, Narayan SP, Auyeung E, Mirkin CA. Mechanism for the endocytosis of spherical nucleic acid nanoparticle conjugates. Proc Natl Acad Sci U S A. 110 (2013) 7625–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gu J, Chen X, Fang X, Sha X. Retro-inverso d-peptide-modified hyaluronic acid/bioreducible hyperbranched poly(amido amine)/pDNA core-shell ternary nanoparticles for the dual-targeted delivery of short hairpin RNA-encoding plasmids. Acta Biomater. 57 (2017) 156–69. [DOI] [PubMed] [Google Scholar]
- [62].Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinyl alcohol associated with poly (D,L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release. 82 (2002) 105–14. [DOI] [PubMed] [Google Scholar]
- [63].Ryan M Pearson VVJaSH. Biomolecular corona on nanoparticles: A survey of recent literature and its implications in targeted drug delivery. In: João Conde PVB, De La Fuente Jesús M,Tian Furong, editor. Cancer Nanotheranostics: What Have We Learned So Far? Front Chem. (2016) p. 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Perez A, Wright MB, Maugeais C, Braendli-Baiocco A, Okamoto H, Takahashi A, et al. MARCO, a macrophage scavenger receptor highly expressed in rodents, mediates dalcetrapib-induced uptake of lipids by rat and mouse macrophages. Toxicol In Vitro. 24 (2010) 745–50. [DOI] [PubMed] [Google Scholar]
- [65].Chen PY, Seabrook SA, Epa VC, Kurabayashi K, Barnard AS, Winkler DA, et al. Contrasting Effects of Nanoparticle Binding on Protein Denaturation. J Phys Chem C. 118 (2014) 22069–78. [Google Scholar]
- [66].Deng ZJ, Liang MT, Monteiro M, Toth I, Minchin RF. Nanoparticle-induced unfolding of fibrinogen promotes Mac-1 receptor activation and inflammation. Nat Nanotechnol. 6 (2011) 39–44. [DOI] [PubMed] [Google Scholar]
- [67].Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 243 (2011) 206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Shamaa OR, Mitra S, Gavrilin MA, Wewers MD. Monocyte Caspase-1 Is Released in a Stable, Active High Molecular Weight Complex Distinct from the Unstable Cell Lysate-Activated Caspase-1. PloS One. 10 (2015) e0142203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kilarski WW, Güç E, Teo JCM, Oliver SR, Lund AW, Swartz MA. Intravital Immunofluorescence for Visualizing the Microcirculatory and Immune Microenvironments in the Mouse Ear Dermis. PloS One. 8 (2013) e57135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kostrominova TY. Application of WGA lectin staining for visualization of the connective tissue in skeletal muscle, bone and ligament/tendon studies. Microsc Res Tech. 74 (2011) 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Robertson RT, Levine ST, Haynes SM, Gutierrez P, Baratta JL, Tan Z, et al. Use of labeled tomato lectin for imaging vasculature structures. Histochem Cell Biol. 143 (2015) 225–34. [DOI] [PubMed] [Google Scholar]
- [72].Schmieder AH, Caruthers SD, Zhang H, Williams TA, Robertson JD, Wickline SA, et al. Three-dimensional MR mapping of angiogenesis with α5β1(ανβ3)-targeted theranostic nanoparticles in the MDA-MB-435 xenograft mouse model. FASEB J. 22 (2008) 4179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Thurston G, McLean JW, Rizen M, Baluk P, Haskell A, Murphy TJ, et al. Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. J Clin Invest. 101 (1998) 1401–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 156 (2000) 1363–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Nakamura-Ishizu A, Morikawa S, Shimizu K, Ezaki T. Characterization of sinusoidal endothelial cells of the liver and bone marrow using an intravital lectin injection method. J Mol Histol. 39 (2008) 471–9. [DOI] [PubMed] [Google Scholar]
- [76].Paal K, Muller J, Hegedus L. High affinity binding of paclitaxel to human serum albumin. FEBS. 268 (2001) 2187–91. [DOI] [PubMed] [Google Scholar]
- [77].Kim WR, Flamm SL, Di Bisceglie AM, Bodenheimer HC. Serum activity of alanine aminotransferase (ALT) as an indicator of health and disease. Hepatology. 47 (2008) 1363–70. [DOI] [PubMed] [Google Scholar]
- [78].Meng J, Guo F, Xu H, Liang W, Wang C, Yang X-D. Combination Therapy using Co-encapsulated Resveratrol and Paclitaxel in Liposomes for Drug Resistance Reversal in Breast Cancer Cells in vivo. Sci Rep. 6 (2016) 22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wu Z, Zhang B, Yan B. Regulation of Enzyme Activity through Interactions with Nanoparticles. Int J Mol Sci. 10 (2009) 4198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Liu F, Wang L, Wang H, Yuan L, Li J, Brash JL, et al. Modulating the Activity of Protein Conjugated to Gold Nanoparticles by Site-Directed Orientation and Surface Density of Bound Protein. ACS Appl Mater Interfaces. 7 (2015) 3717–24. [DOI] [PubMed] [Google Scholar]
- [81].Fleischer CC, Payne CK. Nanoparticle-cell interactions: molecular structure of the protein corona and cellular outcomes. Acc Chem Res. 47 (2014) 2651–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Schaffler M, Semmler-Behnke M, Sarioglu H, Takenaka S, Wenk A, Schleh C, et al. Serum protein identification and quantification of the corona of 5, 15 and 80 nm gold nanoparticles. Nanotechnology. 24 (2013) 265103. [DOI] [PubMed] [Google Scholar]
- [83].Neyen C, Plüddemann A, Roversi P, Thomas B, Cai L, van der Westhuyzen DR, et al. Macrophage Scavenger Receptor A Mediates Adhesion to Apolipoproteins A-I and E. Biochemistry. 48 (2009) 11858–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Hughes DA, Fraser IP, Gordon S. Murine macrophage scavenger receptor: in vivo expression and function as receptor for macrophage adhesion in lymphoid and non-lymphoid organs. Eur J Immunol. 25 (1995) 466–73. [DOI] [PubMed] [Google Scholar]
- [85].Kim SC, Kim DW, Shim YH, Bang JS, Oh HS, Kim SW, et al. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 72 (2001) 191–202. [DOI] [PubMed] [Google Scholar]
- [86].Neesse A, Frese KK, Chan DS, Bapiro TE, Howat WJ, Richards FM, et al. SPARC independent drug delivery and antitumour effects of nab-paclitaxel in genetically engineered mice. Gut. 63 (2014) 974–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.