

Abstract
Accurate assessment of genetic variation in human DNA sequencing studies remains a nontrivial challenge in clinical genomics and genome informatics. Ascribing functional roles and/or clinical significances to single nucleotide variants identified from a next-generation sequencing study is an important step in genome interpretation. Experimental characterization of all the observed functional variants is yet impractical; thus, the prediction of functional and/or regulatory impacts of the various mutations using in silico approaches is an important step toward the identification of functionally significant or clinically actionable variants. The relationships between genotypes and the expressed phenotypes are multilayered and biologically complex; such relationships present numerous challenges and at the same time offer various opportunities for the design of in silico variant assessment strategies. Over the past decade, many bioinformatics algorithms have been developed to predict functional consequences of single nucleotide variants in the protein coding regions. In this review, we provide an overview of the bioinformatics resources for the prediction, annotation and visualization of coding single nucleotide variants. We discuss the currently available approaches and major challenges from the perspective of protein sequence, structure, function and interactions that require consideration when interpreting the impact of putatively functional variants. We also discuss the relevance of incorporating integrated workflows for predicting the biomedical impact of the functionally important variations encoded in a genome, exome or transcriptome. Finally, we propose a framework to classify variant assessment approaches and strategies for incorporation of variant assessment within electronic health records.
Keywords: human genome, functional variant, human variation, variant interpretation, mutation, human proteome, non-synonymous mutations, prediction algorithms, functional genomics, sequence analysis, structure analysis
Introduction
Genomic technologies are redefining the understanding of genotype–phenotype relationships. In the early 2000s, array-based genomic technologies enabled gene expression analysis using microarrays, followed by single nucleotide polymorphism (SNP, genetic variation observed in a population) genotyping platforms in mid-2000s, and subsequently by low-cost, high-throughput, massively parallel sequencing platforms in the late 2000s [ 1–3 ]. Genomic sequencing data offer insights into the relationships between the human genomic variations and various molecular and disease phenotypes. Sequencing a biological or clinical sample to characterize genomic, exomic or transcriptomic variations using next-generation sequencing (NGS) technologies helps to identify genomic variations underlying complex diseases. Moreover, approaches such as targeted sequencing of disease-susceptible genomic regions, whole exome sequencing (WES), whole genome sequencing (WGS) and RNA sequencing (RNA-Seq) provide deeper insights into the genetic bases of familial diseases and a better understanding of the biological processes underlying disease phenotypes such as tumors.
Over the past decade, the genetic basis of several complex diseases and clinically relevant quantitative traits were examined using SNP genotyping array-based genome-wide association studies (GWAS). Subsequently, sequencing [ 4–6 ] studies have improved the understanding of the associations between SNPs with clinically relevant traits and human diseases (See http://www.ebi.ac.uk/fgpt/gwas/ ). SNP arrays have uncovered the more common variations, while sequencing has unravelled much of the rare variants spectrum. The frequency of SNPs in the human genome is approximately 1/300 base pair (bp). SNPs are generally categorized as common variants [in which the minor allele frequency (MAF) = 1–5%] or rare variants (MAF < 1%) according to their population frequencies. Both GWAS and targeted WES and WGS studies have expanded the catalog of genotype–phenotype associations and offered insights into the roles of previously uncharacterized genetic regions in complex diseases [ 7–12 ].
The rapid increase in the high-quality sequencing data generation using low-cost NGS experimental platforms coupled with speedy bioinformatics algorithms have enhanced the identification of a large number of sequence and structural variants variations (characterized by genomic DNA > 1 kb in size). SNPs are associated with medically relevant phenotypes, as well as diseases [ 13–15 ] and are often observed on a population scale, whereas single nucleotide variations (SNVs) are specific polymorphisms observed in an individual. Recent studies have highlighted the roles of the different types of structural variants (copy number variations, insertions and deletions (indels), inversions, translocations, linking, anchored split mapping, gain/loss) in the genetic bases of several complex diseases [ 16–19 ]. Furthermore, sequencing studies have helped to identify rare personal variants and variants of unknown significance (VUS).
Public repositories of genomic sequencing and variation data have experienced an exponential growth in the past decade. For example Single Nucleotide Polymorphism database (dbSNP) [ 20 ] and Ensembl Variation database [ 21 ] that archive short genetic variants and structural variants and Database of Genomic Variants [ 22 ] that archives a catalog of curated, large-scale genomic structural variants in the human genome are expanding. Since the first GWAS study reported in 2005, so far genetic basis of 1251 traits were discovered. These investigations also led phenotypic annotations for 15 396 SNPs. A large number of clinical-grade genomes, exomes or transcriptomes sequenced for individualized medicine [ 23–25 ] and population-scale sequencing [ 26 ] projects such as 1000 genomes [ 27 ], Genome10K ( http://www.genome10k.org/ ) and UK10K ( http://www.uk10k.org/ ) will further add to the size of variant-centric databases in the future. Analysis of the data from the sequencing experiments can be broadly divided into four major tasks: (i) quality assessment of the sequencing reads, (ii) alignment of the sequencing reads with the reference genome, (iii) variant calling and (iv) functional and/or clinical assessment and prioritization of variants.
The variants identified from the NGS studies present several data interpretation challenges in bioinformatics. The initial data inference is a key filtering step in the identification and prioritization of a subset of variants for functionally important cues and validation studies. The following sections describe a simple framework to classify the available and widely used bioinformatics resources for prediction, annotation and interpretation of coding SNVs. We also discuss 10 different analytical themes that can potentially be investigated from a bioinformatics perspective to obtain a better understanding of coding SNVs.
Landscape of genetic variants
The mutation spectrum of the human genome is complex and has been classified based on diverse criteria such as the mode of inheritance, heterozygosity pattern, impact on chromosome or alleles, impact on protein sequence, structure and function, impact on the population or evolutionary role and penetrance ( Figure 1 ). On the other hand, SNVs may be broadly classified as coding SNVs (functional variations) that perturb the function of a transcript or a protein, or variants may be classified as noncoding SNVs (regulatory variations) that are located in the regulatory elements [promoters, untranslated regions (UTRs), enhancers, human accelerated regions, noncoding RNA genes, transcription factor binding sites (TFBS)] that regulate the expression levels of a transcript or a protein. Regulatory variants may also perturb gene or protein functions via complex cis or trans -activation mechanisms and may, thus, play important roles in influencing the expression and functions of other genes. An example of a functional variant (P33S) in the ribonucleoside-diphosphate reductase subunit M2 B ( RRM2B ) gene associated with autosomal recessive progressive external ophthalmoplegia is provided in Figure 2 (i) and 2 (ii). Another example of a missense variant R363H (rs11556924) in zinc finger, C3HC-type containing 1 ( ZC3HC1 ) gene, is associated with coronary heart disease [ 28 ]. P33S in RRM2B is part of a conserved haribonucleotide reductase domain. The variant R363H in ZC3HC1 does not lie within a known functional domain, and thus, the variation is located in an unassigned region of the protein. The domain architectures of RRM2B and ZC3HC1 along with the location of functional variants are highlighted in Figure 2 (ii). Regulatory variants may influence the expression levels of cis - or trans -acting genes through gene regulation networks. For example, an intergenic variant (rs10811661) in the 9p21 locus, which is harbored near CDKN2A and CDKN2B on chromosome 9, was associated with myocardial infarction [ 29 ] and coronary heart disease [ 9 ]. A targeted deletion study in mice that investigated the 9p21 region, revealed a regulatory role of the variant in perturbing the expression of two genes (Cdkn2a and Cdkn2b) via a cis -acting mechanism [ 30 ]. Another regulatory variant rs342293 (intergenic variant between FLJ36031-PIK3CG ), which influences a quantitative trait (mean platelet volume) [ 31 ], perturbed a TFBS of ecotropic viral integration site-1 ( EVI1 ) and influenced the expression levels of PIK3CG [ 32 ]. The genomic locations of the regulatory variants rs10811661 and rs342293 that were generated using the Ensembl Genome Browser are highlighted in Figure 2 (iii). Regulatory variants too impact the biological function by altering the level of transcription that may influence protein levels as well. The majority of regulatory variants are not within the protein coding regions, but may indirectly influence gene/protein function via alternative splicing mechanisms [ 33 ]. See the recent reviews that summarized the impact of regulatory variation of protein expression and function for a detailed account of regulatory variants [ 34 , 35 ].
Figure 1.

The human mutation spectrum.
Figure 2.

Examples of the coding (functional) and noncoding (regulatory) variants. (i) Functional variant (Pro33Ser) in RRM2B associated with autosomal recessive progressive external ophthalmoplegia visualized on a protein structure (PDB ID: 2vux; Quaternary assembly is generated using PISA/PDBe). Functional variant (Pro33Ser) is highlighted in red color inside the green circle on chains A (part of loop) and B (part of helix). Visualization was created using UCSF Chimera ( www.cgl.ucsf.edu/chimera ). (ii) Protein domain architectures and functional variants mapped to ii (a) RRM2B and ii (b) ZC3HC1. Ribonuc_red_sm = Ribonucleotide reductase domain; zf = C3HC zinc finger-like domain; Rsm1 = Rsm1-like domain. Functional variants are highlighted using red vertical line. Figure was generated using MyDomains ( http://prosite.expasy.org/mydomains/ ). (iii) Genomic localization of the regulatory variants (a) rs10811661 and (b) rs342293. The location of the variants are highlighted using a vertical red line. Regulatory variant rs10811661 regulated the expression of nearby genes CDKN2A and CDKN2B (highlighted in red boxes). Intergenic variant rs342293 located between FLJ36031 (CCDC71L)-PIK3CG is located in a TFBS of EVI1 that regulates the expression (repress) of PIK3CG. Genomic regions were visualized using Ensembl Genome Browser v. 67.
Prediction, annotation and visualization of coding SNVs
Scientific literature often refers to variants using diverse sets of terms including recommended nomenclature [ 36 ] or standard terms from Sequence Ontology (SO) [ 37 ]. In this review, we broadly classify genomic variants as ‘functional variants’ and ‘regulatory variants', and we primarily focus on coding SNVs and computational approaches for the prediction, annotation, visualization and interpretation of coding SNVs. The different terms used to define variants in the Ensembl Variation resources and dbSNP and their corresponding SO identifiers and descriptions are summarized in Table 1 . The interpretation of variants can be broadly divided into three steps: ‘annotation’, ‘prediction’ and ‘visualization’.
Table 1.
The naming convention used to describe the sequence variants in the Ensembl Variation resources and dbSNP
| Ensembl variant consequences | dbSNP functional classes | SO ID | SO: Definition |
|---|---|---|---|
| Essential splice site | splice-3 | SO:0001574; SO:0001575 | A splice variant that changes the two base regions at the 3′ end of an intron; a splice variant that changes the two base regions at the 5′ end of an intron. |
| Stop gained | splice-5 | SO:0001587 | A sequence variant whereby at least one base of a codon is changed, resulting in a premature stop codon, leading to a shortened transcript. |
| Stop lost | nonsense | SO:0001578 | A sequence variant where at least one base of the terminator codon (stop) is changed, resulting in an elongated transcript. |
| Complex in/del | NA | SO:0001577 | A transcript variant with a complex INDEL—Insertion or deletion that spans an exon/intron border or a coding sequence/UTR border. |
| Frameshift coding | frameshift | SO:0001589 | A sequence variant, which causes a disruption of the translational reading frame, because the number of nucleotides inserted or deleted is not a multiple of three. |
| Non-synonymous coding | missense | SO:0001582; SO:0001652; SO:0001651; SO:0001583 | A codon variant that changes at least one base of the first codon of a transcript; an inframe non-synonymous variant that deletes bases from the coding sequence; an inframe non-synonymous variant that inserts bases into in the coding sequence; a sequence variant, where the change may be longer than three bases, and at least one base of a codon is changed, resulting in a codon that encodes for a different amino acid. |
| Splice site | NA | SO:0001630 | A sequence variant in which a change has occurred within the region of the splice site, either within 1–3 bases of the exon or 3–8 bases of the intron. |
| Partial codon | NA | SO:0001626 | A sequence variant where at least one base of the final codon of an incompletely annotated transcript is changed. |
| Synonymous coding | cds-synon | SO:0001567; SO:0001588 | A sequence variant where at least one base in the terminator codon is changed, but the terminator remains; a sequence variant where there is no resulting change to the encoded amino acid. |
| Coding unknown | NA | SO:0001580 | A sequence variant that changes the coding sequence. |
| Within mature miRNA | NA | SO:0001620 | A transcript variant located with the sequence of the mature miRNA. |
| 5′ UTR | untranslated_5 /UTR-5 | SO:0001623 | A UTR variant of the 5′ UTR. |
| 3′ UTR | untranslated_3 /UTR-3 | SO:0001624 | A UTR variant of the 3′ UTR. |
| Intronic | intron | SO:0001627 | A transcript variant occurring within an intron. |
| NMD transcript | SO:0001621 | A variant in a transcript that is the target of NMD. | |
| Within non-coding gene | ncRNA | SO:0001619 | A transcript variant of a non-coding RNA gene. |
| Upstream | near-gene-5 | SO:0001636; SO:0001635 | A sequence variant located within 2 KB 5′ of a gene; a sequence variant located within 5 KB 5′ of a gene. |
| Downstream | near-gene-3 | SO:0001634; SO:0001633 | A sequence variant located within a half KB of the end of a gene; a sequence variant located within 5 KB of the end of a gene. |
| Regulatory region | NA | SO:0001566 | A sequence variant located within a regulatory region. |
| Transcription factor binding motif | NA | SO:0001782 | A sequence variant located within a transcription factor-binding site. |
| Intergenic | intergenic | SO:0001628 | A sequence variant located in the intergenic region, between genes. |
SO identifiers and descriptions were obtained from MISO ( www.sequenceontology.org/miso/ ).
A given set of variant(s) identified from a sequencing study will be segregated into various classes of genomic variants in the first step in variant annotation ( Table 1 ). Cross-referencing the variants with reference databases and clinical annotation databases like ClinVar [ 38 ] can be used to assess the novelty of genomic variants. Fully automated pipelines can be used to annotate variants based on coordinate specific mapping to genomic regions using proprietary and/or public databases, and various features associated with variant can be derived from such annotation mapping and variant-specific layering approach. For example, a variant can be mapped to a protein-coding region, junction regions or noncoding regions of the genome.
Based on the location of the variant in the protein-coding or noncoding regions, variants (non-synonymous, missense, nonsense or frameshift variants) can be further examined to understand their impact on protein functions. Tools like Combined Annotation-Dependent Depletion (CADD) [ 39 ], SIFT (as tolerated or damaging variants) [ 40 ], PolyPhen2 (as benign, possibly damaging or probably damaging variants) [ 41 ] or Condel (meta-predictor that combines prediction scores from multiple tools) [ 42 ] can be leveraged for predictive assessment of genetic variants. Annotation primarily provides localization of a genetic variant using genome coordinates; prediction aims to hypothesize the probable impact of a particular genetic variant on function or regulation. The combination of annotation and prediction provides an integrated view of genomic variants. Tools such as snpEff or the Ensembl Variant Predictor can be used to predict the impact of a variation in comparison with the reference sequence. The impact of a sequence variant with respect to the evolutionary conservation can also be predicted or derived from GRANTHAM score [ 43 ], genomic evolutionary rate profiling (GERP) score [ 44 ], phylogenetic P -value (PhyloP) score [ 45 ] and PhastCons score [ 46 ]. The development of substitution matrices in the early 1970s was instrumental in fueling the design and implementation of computational approaches to infer the functional impact of genomic variants [ 43 , 47 ]. Matrices such as Point Accepted Mutations (PAM1 matrix: substitution probabilities for sequences with a mutation rate of 1/100 amino acids; PAM250 matrix: 250 mutations/100 amino acids), BLOCK SUbstitution Matrix (BLOSUM) matrices [ 48 ] and sequence search algorithms designed to identify the similarity between any two sequences (pairwise sequence alignment) and the evolutionary relationships between two or more sequences (multiple sequence alignments) have spawned the development of improved heuristic homology search tools [ 49 , 50 ]. The early 2000s witnessed the development of several predictive methods based on sequence conservation, amino acid substitutions and perturbation of the local structural environment to assess the impact of functional variants in proteins [ 51–57 ]. The ka/ks ratio (or dN/dS) test, which estimates the ratio of the total number of non-synonymous substitutions per non-synonymous sites (dN) to the total number of synonymous substitutions to synonymous sites (dS), is widely used to quantify the selection pressure on functional genes [ 52 ]. Furthermore, bioinformatics tools, databases and statistical methods for the identification, annotation and analysis of SNPs from genotyping and sequencing data have been amply surveyed in literature [ 58–66 ].
Integrative approaches can provide additional annotations for variants such as the location of the variants within the conserved protein sequence/structure domains (contiguous unit in protein sequence or structure with evidence of functional annotation from experimental or computational function association methods), within known functional sites. Gene Ontology (GO) terms and pathways associated with the variant-containing genes, associations mapped within genetic databases such as dbSNP, Ensembl, Online Mendelian Inheritance in Man [ 67 ], Human Gene Mutation Database [ 68 ], Catalogue of Somatic Mutations in Cancer (COSMIC) [ 69 ] and other clinically relevant genomic regions also enable enhanced variant annotation. Together, integrative data analysis platforms (such as TargetMine [ 70 ]) and integrated annotation tools such as ANNOVAR [ 71 ] or webservers that can handle Variant Call Format (VCF) [ 72 ] files, facilitate the annotations for a large number of sequence variants in a small amount of time. Adopting a standard set of terms from the SO to define the type of variants would also help in streamlining the interoperability of results from the different prediction tools. A comprehensive list of tools for rapid variant annotation is provided in Table 2 .
Table 2.
Top-10 terms from a gene-disease enrichment analyses performed using list of genes with shared polymorphism, disease and unclassified variants using disease ontology
| DO term | No. of genes | P -values (Bonferroni corrected) |
|---|---|---|
| Congenital abnormality | 38 | 9.13E-34 |
| Cancer | 64 | 7.222E-33 |
| Diabetes mellitus | 39 | 8.913E-23 |
| Breast cancer | 36 | 3.56E-17 |
| Atherosclerosis | 26 | 1.478E-16 |
| Retinal disease | 16 | 2.044E-16 |
| Adenovirus infection | 15 | 8.816E-14 |
| Hypertension | 21 | 2.072E-13 |
| Alzheimer's disease | 22 | 7.548E-13 |
The visualization of a variant in the context of multiple layers of biological information is helpful to interpret and prioritize variants for functional studies. A growing number of genome browsers and data visualization libraries enable the interactive and static visualizations of variants in the context of the human genome or transcriptome with biological, clinical and population scale annotation data compiled from multiple resources. Genome browsers [Integrative Genomics Viewer (IGV), UCSC Genome Browsers, NCBI Sequence Viewer, etc.] enable the visualization of variants in the context of a reference genome. Resources such as Distributed Annotation System (DAS) and DASTY (a protein-centric DAS client) can be leveraged for interactive visualization of coding variants in a protein with rich annotations. A list of genome browsers and visualization libraries capable of visualizing variants and multiple annotation components is provided in Table 3 .
Table 3.
Rapid variant annotation tools
| Name | Description | URL |
|---|---|---|
| ANNOVAR | Efficient software tool to use up-to-date information to functionally annotate genetic variants detected from diverse genomes. | http://www.openbioinformatics.org/annovar/ |
| AnnTools | Comprehensive and versatile annotation toolkit for genomic variants. | http://anntools.sourceforge.net/ |
| dbNSFP | A lightweight database of human non-synonymous SNPs and their functional predictions. | https://sites.google.com/site/jpopgen/dbNSFP |
| EVA | An efficient and versatile tool for filtering strategies in medical genomics. | http://plateforme-genomique-irib.univ-rouen.fr/EVA/index.php |
| Exome Variant Server | Provides different calculated values (GERP, GRANTHAM, etc.) and annotations for SNPs. | http://evs.gs.washington.edu/EVS/ |
| gSearch | gSearch compares sequence variants in the Genome Variation Format (GVF) or VCF with a pre-compiled annotation or with variants in other genomes. | http://ml.ssu.ac.kr/gSearch/index.html |
| HugeSeq | A pipeline for detection and annotation of genetic variations. | http://hugeseq.snyderlab.org/ |
| MuSiC | Comprehensive mutational analysis pipeline to segregate passenger and driver mutations from cancer genomes. | http://gmt.genome.wustl.edu/genome-music/current/ |
| NGS-SNP | Collection of command-line scripts for providing rich annotations for SNPs. | http://stothard.afns.ualberta.ca/downloads/NGS-SNP/ |
| SeattleSeq Annotation | Provides annotation of known and novel SNPs. | http://snp.gs.washington.edu/SeattleSeqAnnotation/ |
| snpEff | Variant annotation and effect prediction tool. | http://snpeff.sourceforge.net/ |
| SVA | Software system designed for annotation and visualization of genetic variants. | http://www.svaproject.org/ |
| STORMSeq | Cloud computing solution for read mapping, read cleaning, and variant calling and annotation. | https://github.com/konradjk/stormseq |
| TREAT | Targeted RE-sequencing Annotation Tool. | http://ndc.mayo.edu/mayo/research/biostat/stand-alone-packages.cfm |
| VAAST | Probabilistic search tool for identifying damaged genes and their disease-causing variants in personal genome sequences. | http://www.yandell-lab.org/software/vaast.html |
| VARIANT | VARIANT (VARIant ANalysis Tool) can report the functional properties of any variant in all the human, mouse or rat genes. | http://variant.bioinfo.cipf.es/ |
| Variant Reporter | Generate a report of known variants and functional consequences. | www.ncbi.nlm.nih.gov/variation/tools/reporter |
| Variant Tools | Tool for the annotation, selection and analysis of variants in the context of next-gen sequencing analysis. | http://varianttools.sourceforge.net/ |
Text mining and natural language processing for knowledge aggregation for SNVs
Biomedical knowledge about relationships between genes, diseases, phenotypes and genetic variations are scattered across a large number of unstructured literature databases. Application of natural language processing and text-mining, therefore, offers a useful approach for function assignment of coding SNVs. Further text mining of large biomedical literature databases like PubMed and Medline helps to provide clues for further investigations and leads to hypothesis [ 73 ]. For example, PhenGen [ 74 ] offers links to a variety of literature evidence to support genotype–phenotype connections. Integrated databases like T-HOD database [ 75 ] and PolySearch [ 76 ] provide text-mining tools to derive meaningful biological inferences to interpret coding SNVs.
Emerging challenges in annotation and interpretation of coding SNVs
The growing number of tools for the prediction, annotation and visualization of coding SNVs can address several gaps in the current state of knowledge on variant interpretation. The development of new algorithms for variant interpretation could be considered for several emerging themes of the protein sequence-structure-function paradigm. Several tools [See Tables 4 , 5 and 6 ] are currently available to assess sequence and structure-based features; yet, a reliable interpretation on how a genomic variant could perturb a protein or a protein network is often a challenging task.
Table 4.
Genome browsers and biological data visualization libraries
| Name | Description | URL |
|---|---|---|
| 1000 Genomes browser | Genome browser to access data from 1000 Genomes project. | http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/ |
| AnnoJ | Web 2.0 application designed for visualizing sequencing and annotation data. | http://www.annoj.org/ |
| Artemis | Genome Browser and Annotation Tool. | http://www.sanger.ac.uk/resources/software/artemis/ |
| Bio::Graphics | Perl modules for biological data visualization. | http://search.cpan.org/dist/Bio-Graphics/ |
| Bio::Graphics (Ruby) | Ruby library for drawing overviews of genomic regions. | http://bio-graphics.rubyforge.org/ |
| Bluejay | Java-based integrated computational environment for the exploration of genomic data. | http://bluejay.ucalgary.ca/ |
| CGView | Circular Genome Viewer. | http://wishart.biology.ualberta.ca/cgview/ |
| Circos | Circos is a software package for visualizing genomic data and annotations in circular layout. | http://circos.ca/ |
| Dalliance | Interactive genome viewer, which runs directly in a modern web browser. | http://www.biodalliance.org/ |
| DAS | DAS is an integrated visualization toolkit to share and collate genomic annotation information. | www.biodas.org |
| DASTY | Web client for visualizing protein sequence feature information using DAS. | http://www.ebi.ac.uk/dasty/ |
| DNAPlotter | DNAPlotter can be used to generate images of circular and linear DNA maps to display regions and annotations of interest. | http://www.sanger.ac.uk/resources/software/dnaplotter/ |
| Ensembl Genome Browser | Ensembl Genome Browser enables the visualization of genomic and transcriptomic sequence and related information for several vertebrate and non-vertebrate species. | http://useast.ensembl.org/index.html |
| GASV | Geometric Analysis of Structural Variants. | http://compbio.cs.brown.edu/software.html |
| Gbrowse | Generic Genome Browser (GBrowse) is a genome viewer developed as part of Generic Model Organism Database (GMOD) project. | http://gmod.org/wiki/GBrowse |
| GENBOREE | Customizable genome browser. | http://genboree.org/java-bin/login.jsp |
| GeneViTo | JAVA-based workbench for genome-wide analysis through visual interaction. | http://athina.biol.uoa.gr/bioinformatics/GENEVITO/ |
| GenomeGraphs | R-based interface to plot genomic information from Ensembl. | http://www.bioconductor.org/packages/release/bioc/html/GenomeGraphs.html |
| GenomePixelizer | Tool to generate custom images of genomes out of the given set of genes. | http://www.atgc.org/GenomePixelizer/ |
| GenomeTools | Versatile genome analysis software. | http://genometools.org/ |
| GenomeView | Next-generation stand-alone genome browser and editor. | http://genomeview.org |
| Gremlin | Interactive visualization model for analyzing genomic rearrangements. | http://compbio.cs.brown.edu/software.html |
| IGB | Integrated genome browser. | http://bioviz.org/igb/index.html |
| IGV | Integrative Genomics Viewer. | http://www.broadinstitute.org/igv/ |
| Jbrowse | Genome browser with a fully dynamic AJAX interface. | http://gmod.org/wiki/JBrowse |
| jsDAS | JavaScript client library for the DAS. | http://www.ebi.ac.uk/dasty/ebi/html/jsdas.html |
| MagicViewer | Integrated solution for NGS data visualization and genetic variation detection and annotation. | http://bioinformatics.zj.cn/magicviewer/index.php |
| NCBI Graphical Sequence Viewer | Graphical display for the Nucleotide and Protein sequences. | http://www.ncbi.nlm.nih.gov/projects/sviewer/ |
| Rover | Genome browser framework to build custom genomic tools. | http://chmille4.github.com/Rover/site/home.html |
| Rviewer | Interactive online tool for comparing and prioritizing genomic regions. | http://rviewer.lbl.gov/rviewer/ |
| Savant | Genome Browser for high-throughput sequencing data. | http://genomesavant.com/ |
| Scribl | HTML5 Canvas-based graphics library for visualization of genomic data and annotations. | http://chmille4.github.com/Scribl/ |
| UCSC Genome Browser | Interactive genome browser that provide access to sequence data from different species, integrated with a large collection of layered annotations from experiments and prediction algorithms. | http://genome.ucsc.edu/cgi-bin/hgGateway |
| VISTA Browser | Visualization of pairwise and multiple alignments of whole genome assemblies. | http://pipeline.lbl.gov/cgi-bin/gateway2?selector=vista |
| Whole Genome rVISTA | Visualization of TFBS that are conserved between species and overrepresented in upstream regions of groups of genes. | http://genome.lbl.gov/vista/index.shtml |
Table 5.
Examples of the functional variants located in sequence features perturbing diverse functional and structural effects in proteins
| Variant location | Description | URL |
|---|---|---|
| Protein domain | DMDM: A database that compiles domain mapping of disease mutations have information about 202 507 mutations associated with 10 919 domains (compiled from CDD, Pfam, COG and SMART databases). | http://bioinf.umbc.edu/dmdm/ |
| Phosphorylation site | Mutation of an AKT phosphorylation site of human B-raf. | http://www.ncbi.nlm.nih.gov/pubmed/15791648 |
| Propeptide | Mutation in the von Willebrand factor (VWF) propeptide affects the oligomerization. | http://www.ncbi.nlm.nih.gov/pubmed/20335223 |
| Signal peptide | Mutation in signal peptide of ADAMTS10 influence secretion of full-length enzyme. | http://www.ncbi.nlm.nih.gov/pubmed/18567016 |
| Active site | Mutation in the active site of human deoxycytidine kinase affects the substrate specificity. | http://www.ncbi.nlm.nih.gov/pubmed/18361501 |
| Linear motif | Linear motifs mediate functional diversity of transcript variants. | http://www.ncbi.nlm.nih.gov/pubmed/22638587 |
| Structural motif | Heterozygous missense mutation of a spatially distributed structural motif in human connexin ( GJB3 ) gene cause erythrokeratodermia variabilis. | http://www.ncbi.nlm.nih.gov/pubmed/9843209 |
| Subcellular localization | Missense mutations in the NPHS2 gene altering the trafficking of nephrin to the plasma membrane. | www.ncbi.nlm.nih.gov/pubmed/15496146 |
Table 6.
Tools for predicting various sequence and structural features
| Name | Description | URL |
|---|---|---|
| 3dswap-pred | Classify a protein sequence as domain-swapping or non-domain swapping using an SVM model. | http://caps.ncbs.res.in/3dswap-pred/index.html |
| AAIndex | An amino acid index is a set of 20 numerical values representing various physico-chemical and biochemical properties of amino acids. | http://www.genome.jp/aaindex/ |
| Bioinformatics Link Directory (Protein) | Extensive list of tools for prediction of protein sequence features, structure features and function. | http://bioinformatics.ca /links_directory/category/protein |
| dbPTM | Comprehensive resource for protein PTMs. | http://dbptm.mbc.nctu.edu.tw/ |
| DISOPRED | Dynamically disordered protein chains do not have stable secondary structures and have high flexibility in solution. Disordered regions also play critical roles in protein function. | http://bioinf.cs.ucl.ac.uk/disopred/ |
| ELM | Eukaryotic Linear Motif server. | http://elm.eu.org/ |
| Eris | Eris server computes the change of the protein stability induced by mutations using structural data. | http://dokhlab.unc.edu/tools/eris/index.html |
| FoldAmyloid | Method for predicting of amyloidogenic regions from protein sequence. | http://bioinfo.protres.ru/fold-amyloid/oga.cgi |
| FoldX | FoldX can be used to find interactions contributing to the stability of proteins and protein complexes using structural data. | http://foldx.crg.es/ |
| Globplot | Globplot can predict disordered regions in protein sequence. | http://globplot.embl.de/ |
| H-Predictor | Predict hinge regions involved in protein oligomerization via the domain-swapping mechanism from structural data. | http://troll.med.unc.edu/dokhlab/index.php/Special:Hpredictor |
| Harmony | Substitution and propensity score-based protein structure assessment algorithm. | http://caps.ncbs.res.in/harmony/ |
| HORI | Webserver for prediction of higher order residue interactions in protein structures. | http://caps.ncbs.res.in/hori |
| InterPro | Integrated database of predictive protein signatures used for the classification and automatic annotation of proteins and genomes. | http://www.ebi.ac.uk/interpro/ |
| IUPred | Prediction of intrinsically unstructured proteins. | http://iupred.enzim.hu/ |
| LIMBO | Predicts the amylogenic regions in a protein. | http://limbo.vib.be |
| MUPRO | Prediction of protein stability changes for single-site mutations from sequences. | http://mupro.proteomics.ics.uci.edu/ |
| NCBI-CDD | Extensive protein domain and family annotation database. | http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml |
| Pfam | Database of conserved protein domain families. | http://pfam.sanger.ac.uk/ |
| PFILT | Program to filter various sequence regions including low-complexity regions. | http://bioinfadmin.cs.ucl.ac.uk/downloads/pfilt/ |
| PIC | Protein interactions calculator. | http://pic.mbu.iisc.ernet.in/ |
| ProtParam | Compute biochemical features like Molecular Weight, Theoretical pI, Grand Average of Hydropathy (GRAVY), instability index, etc. | http://web.expasy.org/protparam/ |
| PSIPRED | Secondary structure prediction. | http://bioinf.cs.ucl.ac.uk/psipred/ |
| PURE | Prediction of unassigned regions in proteins. | http://caps.ncbs.res.in/pure |
| SABBLE | Relative solvent accessibility prediction. | http://sable.cchmc.org/ |
| ScanProsite | Report the functional motifs/patterns encoded in the sequence. Helps to assess the gain/loss of functional sites owing to mutation. | http://prosite.expasy.org/scanprosite/ |
| SignalP | Predicts the presence and location of signal peptide cleavage sites in amino-acid sequences. | http://www.cbs.dtu.dk/services/SignalP/ |
| SMART | Simple modular architecture research tool for assigning domains to protein. | http://smart.embl-heidelberg.de/ |
| TANGO | Predicts the aggregation-prone regions in a protein. | http://tango.crg.es/ |
| TargetP | Predicts the subcellular location of eukaryotic proteins. | http://www.cbs.dtu.dk/services/TargetP/ |
| UniProtKB | Catalog of information on proteins. | http://www.uniprot.org/ |
| WALTZ | Predicts the aggregation-prone regions in a protein. | http://waltz.vub.ac.be/ |
VUS (also known as incidental variations or secondary variations) are a class of variants hitherto unknown in known disease genes, but the influence of these variants on the disease phenotype is largely unknown. Pilot data from 1000 Genomes project on exon capture sequencing of 1092 individuals selected from 14 populations across Europe, East Asia, sub-Saharan Africa and the Americas reported an observed frequency of 2500 non-synonymous variants at conserved positions; 20–40 variants identified as damaging; 24 at conserved sites and about 150 loss-of-function (LOF) variants that includes stop-gains, frameshift insertions and deletions (indels) in a coding sequence and disruptions to essential splice sites. Emerging evidence suggests that rare variants may have a higher collective impact on disease incidence rate than common variants, and considering the allele frequency as a metric to assess the clinical impact of the variant may help to assess the clinical impact of VUS. Majority of the variants identified in individuals in 1000 Genomes project are common with >5% of MAF or low frequency (MAF: 0.5–5%) than rare variants (MAF: <0.5%). Rare variant frequencies estimated as 130–400 non-synonymous variants per individual that includes 10–20 LOF variants, 2–5 damaging mutations, and 1–2 variants identified from cancer genome sequencing projects [ 77–79 ]. As an ever-increasing number of VUS are being characterized, their annotation and interpretation is becoming more challenging. With the advent of WGS and WES in clinical settings, the repertoire of VUS associated with complex, chronic and rare diseases is rapidly expanding. We surveyed the Human polymorphisms and disease mutations index (humsavar.txt) to gather unclassified variants reported in UniProtKB, a curated database of functional information on proteins. The current release (2014_05) of the humsavar lists 6564 variants as ‘Unclassified variants’; these variants were mapped to 1910 protein coding genes. A manually curated annotation of the variants and disease ontology-based disease term-gene enrichment analyses indicated that the genes were largely from cancer patients (322 genes tested; P < 0.05). We noted that 497 genes encoded polymorphism, disease and unclassified variants, suggesting that VUS-labeled variants may directly influence the genes that are relevant to human diseases and clinically relevant phenotypes. A proportional Venn diagram of genes mapped to variants labeled as ‘Polymorphism’, ‘Disease’ and ‘Unclassified’ is illustrated in Figure 3 . The top 10 disease ontology terms enriched among the genes within the three classes of variants are summarized in Table 2 . We observed that certain diseases were represented by genes that share multiple classes of variants, thereby suggesting that unclassified variants may play a key role in certain clinical conditions such as cancers, chronic diseases (diabetes, atherosclerosis, hypertension and kidney disease), neurodegenerative disease (Alzheimer’s disease) and infection ( P < 0.05; Bonferroni corrected). Annotating functional coding SNVs including VUS validated using an orthogonal method also requires detailed sequence, structure and interaction-based analyses. In this context, we discuss some of the predictive features and analytical approaches that once incorporated in coding variant annotation algorithms, may potentially enhance the interpretation of the functional variants on a proteome-wide scale.
Figure 3.
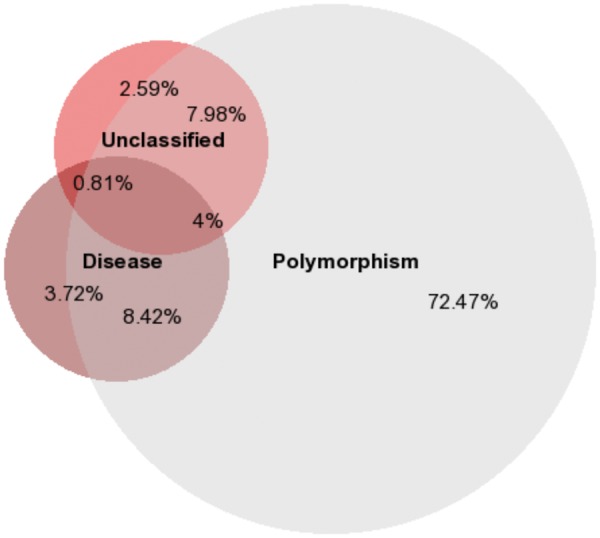
Proportional Venn diagram of genes with coding variants annotated as polymorphism (Polymorphism, n = 11 527), disease (Disease, n = 2105) and unclassified (Unclassified, n = 1910) in Human polymorphisms and disease mutations index. Percentages of genes shared between the three groups are provided.
Sequence and structural properties perturbed by coding SNVs
A protein performs its defined function after attaining a specific tertiary or quaternary structure [ 80 , 81 ]. This is often mediated by cross-links of inter-chain and intra-chain amino-acid residue interactions within a protein. These interactions (hydrogen bond, disulphide bonds, salt bridges, ionic interactions, electrostatic interactions, hydrophobic interactions, etc.) stabilize the fold of individual protein chains and the overall quaternary assembly. Individual amino acids within a protein are under the influence of the varied elements of the sequence-structure-function paradigm that modulate their biochemical role [ 82 ]. Primarily, a residue may be located within an evolutionarily conserved compact globular domain [ 83 ] or in an unassigned region with no known protein domain association [ 84 ]. A residue can be a part of a motif which can be functional [ 85 ], that of a linear motif [ 86 ], propeptide [ 87 ], signaling peptide [ 88 ] or a part of structural [ 89 ] and spatially interacting sites, which participate in higher order interactions [ 90 ], catalytic sites [ 91 ], ligand binding sites [ 92 ] and allosteric sites [ 93 ] associated with extensive inter-chain and intra-chain interactions [ 90 ] based on the oligomeric property of the proteins. Amino acids can also be part of specific sub-cellular localization signals that direct the proteins to specific locations in the cell [ 94 ]. The lengths of the proteins [ 62 ], sequential localization (N-terminal, C-terminal or other region of protein sequence) and the location of a coding SNV with respect to the surface—interior or interface of the protein structure—could influence disease manifestation [ 95 ].
Previously, studies have established ( Table 4 ) that functional variants may impact the overall structural conformation and function of proteins (ligand binding, substrate specificity, protein–protein interaction, transcription factor-target binding, etc.). Residue-specific interactions can play a key role in such variations. Sequence-based investigations will further help to identify linear sequence motifs that confer diversity in the splice variants [ 6 ], but the identification and analysis of the impact of point mutations on spatially distributed structural motifs may require structural data [ 55 ]. However, the limited availability of structural data compared with sequence data may pose additional challenges for incorporating structural analysis of the functional variants [ 96 ]. Moreover, additional challenges arise owing to the dynamic protein conformations and allosteric or other long-distance effects (including higher order interactions) of few mutations on the activity of the protein.
UniProtKB provides extensive information about residue properties and curated information about globular domains (via annotations derived from Pfam, SMART or InterPro), posttranslational modifications (PTMs) and several bioinformatics tools and webservers are available to investigate residue properties and structural properties. (See Table 5 for a list of tools to predict various sequence features. See URL http://bioinformatics.ca /links_directory/category/protein for list of tools available for different protein-centric analyses.)
Occasionally, multiple sequence or structural features can be annotated onto a single variant: for example, a residue could be part of a functional motif and/or a structural motif, and coding SNVs may perturb either or both features. Quantifying multiple features perturbed by variants using an objective-scoring schema will help to capture the entire set of features perturbed by a functional variant. Comparative analysis of the sequence and structural features of the wild type and the variant sequences will also enhance the understanding of the impact of multiple coding variants within a protein.
Gain and loss of PTMs owing to SNVs
Amino acids in a mature peptide chain are targets of varied PTM [ 97 ] events. PTMs include phosphorylation, methylation, acetylation, amidation, hydroxylation, sulfation, lipidation, glycosylation and palmitoylation [ 98 ]. Several studies have now compiled and assessed the impact of phosphorylation in cancer [ 97 ] and have reported mutational landscapes of various PTM events including gain or loss of particular PTMs owing to SNVs (for example, gain of glycosylation [ 99 ], gain and loss of phosphorylation in cancer [ 100 ]). Pan-cancer [ 101 ] and proteome wide studies [ 102 ] have also assessed the impact of variety of PTMs. Several tools and databases are currently available to assess impact of individual PTMs owing to SNVs [refs?]. However, tools that can provide complete in silico profiling of mutations will help the researchers to identify PTM-relevant mutations and use the information to assess therapeutic stratifications.
Coding SNVs in unassigned regions of proteins
An unassigned region refers to the segments in proteins with no known functional domain assignments [ 84 ]. Human proteins have a variable degree of unassigned regions, and small-unassigned regions are often defined as the linker regions between two domains ( Figure 2 i). We surveyed the human proteome using Pfam-based domain annotations to understand the unassigned regions in the human proteome. We computed assigned and unassigned regions (in percentage) for 20 242 protein sequences from the SwissProt database (reviewed sequences). Pfam-A-based domain assignments were retrieved for 20 137 sequences. A subset of 105 proteins was excluded from the analysis owing to overlapping domain assignments. Of the 20 137 sequences that were analyzed ( Figure 4 A), one protein (haloacid dehalogenase-like hydrolase domain-containing protein 2) was assigned with a conserved domain over its entire length, 3234 sequences were completely unassigned and the rest of the proteins had varying segments of unassigned regions (mean: 53.87%; SD:±30.94%). Current approaches in variant annotation and interpretation are primarily focused on highly conserved globular domains. Given that domains are presently assigned to only ∼50% of the human proteome, analytical methods such as Prediction of Unassigned Regions (PURE) that use intermediate sequence search techniques for domain assignments [ 103 ] or similar approaches that use sensitive sequence search protocols ( Table 5 ) may potentially help investigate the evolutionary and functional roles of variants in unassigned regions.
Figure 4.

Histograms depicting the distribution (in percentage) of: ( A ) unassigned regions and ( B ) LCRs in the human proteome.
Impact of coding SNVs in low-complexity regions
Low-complexity regions (LCRs) in the protein universe refer to a stretch of amino acids with low Shannon entropy (leucine-rich domains or poly-alanine tracts). Unlike linear motifs, which have a specific function and sequence signature, the individual functions of LCRs are poorly characterized. LCRs do not adopt a definite secondary structure but may exist as solvent-exposed amino acids in the coiled or disordered regions in proteins. LCRs are observed in functionally diverse proteins and in both eukaryotes and prokaryotes. The predominant functions of LCRs include promoting mRNA stability and mediating a diverse set of protein–protein interactions [ 104 ]. We scanned the reference human proteome (reviewed subset of 20 237 sequences) using PFILT [ 105 ] and observed that 14.32% ( n = 2899) protein sequences have LCRs with a median length of 16 amino acids ( Figure 4 B). A recent study showed that a functional variant localized to an LCR in Nance-Horan Syndrome (NHS) gene was associated with clinical features of NHS including cardiac anomaly [ 106 ]. In the absence of direct functional information to interpret the impact of functional variants, scanning protein sequences with an LCR prediction tool like PFILT is highly recommended to investigate the probable gain or loss of LCRs owing to the functional variations.
Coding SNVs and intrinsically disordered regions in proteins
Proteins are believed to be functional when the structure attains its definite globular fold [ 107 ]. Recent studies, however, suggest that proteins may perform their functions even when not in a fully folded state. Such proteins or regions within proteins that exist in a stable conformation without attaining a definite structural conformation are generally referred to as intrinsically disordered proteins (IDPs) or proteins with intrinsically disordered regions (IDRs) or simply disordered proteins [ 108 , 109 ]. Disprot is a database that catalogs a curated list of disordered proteins that includes 248 experimentally validated human proteins with disordered regions ( http://www.disprot.org/actionsearch.php?keyword=human&criterion =organism). Prediction models suggest that 30–40% of human proteins are considered to be IDPs or have IDRs and approximately 25% of eukaryotic proteins are predicted to be fully disordered [ 108 , 109 ].
Regions of the intrinsically disordered segments in proteins have been found to be functionally important ( Figure 5 ). Disordered regions mediate various functional roles including protein binding and protein–protein interactions. Previous studies have shown that IDPs can attain a definite structure on binding to their interacting partner and may thus exist in an intermediate stage of disordered (unfolded) to ordered (folded) stage [ 110 , 111 ]. Irrespective of the structural plasticity, recent studies have shown the presence of evolutionarily conserved functional motifs within IDPs [ 112 , 113 ].
Figure 5.

Examples of IDRs in human proteins: ( A ) Breast cancer type 1 susceptibility protein (BRCA1), ( B ) Cellular tumor antigen p53 (Oncoprotein p53), ( C ) T-cell surface glycoprotein CD4 (Disprot), ( D ) Secondary structure information (UniProtKB) and ( E ) mutation histogram (COSMIC) of BRCA1 is provided to illustrate mutations in the unstructured regions.
Recently, the influence of disease mutations in the disordered regions was extensively surveyed, and the crucial roles of disorder-promoting amino acids in imparting variations in the protein structures were proposed [ 114 , 115 ]. Disorder-promoting amino acids are a potential cause of variations in protein structures and structural models. Disorder-to-order transitions are enriched among disease mutations compared with neutral polymorphisms [ 116 ]. Currently, protein modeling or variant effect prediction algorithms do not take the intrinsic disorder characteristics of a protein into consideration. Integrating tools such as Disopred [ 117 ] for disorder region prediction into the variant annotation pipelines will improve our understanding of the impact of coding SNVs in the disordered regions of a protein.
Influence of coding SNVs on protein misfolding, domain swapping, aggregation, macromolecular crowding and degradation
Diseases have multiple environmental, as well as genetic, causes and structural biology is an active area of research to understand how changes in individual protein structures or protein complexes may play a key role in disease manifestations. Understanding the structural bases of human diseases would help to identify better targets and design better ligands for more effective therapeutic interventions.
Protein misfolding and aggregation
Protein folding pathways play a crucial role in mediating cellular homeostasis. Defective folding of a protein product is the mechanistic basis of several disease phenotypes like Alzheimer’s disease and Parkinson’s disease [ 118 ]. When non-synonymous mutations lead to the production of misfolded proteins with aberrant function, several gate-keeping pathways act on such misfolded proteins to clear them from the cellular environment [ 119 ]. Misfolded proteins targeted for ubiquitylation may also lead to protein aggregation. Protein aggregation [ 120–123 ] is defined as a molecular phenomenon where a protein is not cleared from the cellular environment by the normal pathways ( Figure 6 A). This leads to the aggregation of proteins in the cellular environment, which in turn leads to cellular toxicity and is considered to be the mechanistic basis of various human diseases such as prion diseases [ 123 , 124 ].
Figure 6.

Schematic representation of the impact of functional mutation on protein misfolding, folding, aggregation, domain swapping, macromolecular crowding and protein degradation pathways. Structure of misfolded rat CD2 structure (PDB ID: 1A6P) and normal CD2 (PDB ID: 1HNG) is used for representing misfolded and folded structures. Functional variant is represented using red asterisk.
Domain swapping
3D domain swapping ( Figure 6 B) is a phenomenon observed in a subset of proteins where intermolecular interactions are replaced by intramolecular interactions [ 125 , 126 ]. Domain swapping is also recognized as a mechanism for forming protein aggregates via open-ended mode. Domain swapping is observed in a variety of therapeutically important proteins and is considered to be the mechanism mediating deposition diseases such as neurodegenerative diseases and Alzheimer’s disease that are caused by conformational perturbations [ 127 , 128 ]. Missense mutations in the human phenylalanine hydroxylase have been shown to infleunce aggregation and degradation properties [ 129 ]. Functional mutations are also considered to be a causative factor for proteins to adopt swapped conformation from monomers to higher oligomers [ 130–132 ]. A recent study [ 128 ] that surveyed a subset of human proteins involved in 3D domain swapping suggested that swapping is not only confined to conformational diseases; it is also associated with several key biological pathways and also plays a role in mediating diverse diseases in humans. Another study [ 133 ] that investigated the structural properties of proteins involved in 3D domain swapping suggested that 10% of protein folds and 5% of protein families include domain-swapped structures.
Macromolecular crowding
Proteins interact with a variety of small molecules and other macromolecules to perform a specific functional task. The quaternary assembly of a protein is influenced by the important, yet understudied, phenomenon of macromolecular crowding (117, 118). Protein function is driven by interactions mediated by inter- and intra-chain amino acids, and mutations in the surface amino acids may affect macromolecular homeostasis. Mutagenesis studies have shown the impact of functional mutations on protein–protein interfaces (119), oligomerization (120) and stability (121). Mutations that influence the intra-chain interactions may influence the crowding effect in the cellular environment. Probing the impact of functional mutations on such effects using theoretical models and molecular dynamic simulation studies is likely to enhance the understanding of the relationship between coding SNVs and macromolecular crowding. A list of selected tools and databases available for the prediction of misfolding, protein aggregation and domain swapping is summarized in Table 5 .
Protein degradation
Targeted biochemical studies have revealed that the protein degradation pathway plays an important role in the clearance of the mutated proteins to reduce cellular toxicity. For example, mutant Cu, Zn-superoxide dismutase associated with amyotrophic lateral sclerosis was cleared by macroautophagy pathway that includes proteasomal cleavage; phenylalanine hydroxylase (109) and NAD(P)H: quinone oxidoreductase 1 (156) were also cleared by similar mechanisms. Functional variants play a crucial role in aggregation and protein degration pathways [ 134 , 135 ]. Computational approaches that can predict the degradation properties of mutated proteins using sequence or structural information will be useful for rapid characterization of functionally active proteins. Integrated models that combine folding, misfolding, aggregation, 3D domain swapping, macromolecular crowding and degradation pathways ( Figure 6 ) in a systems biology approach are likely to provide additional insights into the regulation of these important mechanisms and the role of coding SNVs in mediating such mechanisms.
Coding SNVs and metamorphic proteins
The molecular paradigm of sequence-structure-function suggests that a diverse set of sequences could attain a similar structural fold that may lead to a functional convergence. While this is generally true for the protein universe, occasionally deviations are observed. Metamorphic proteins refer to a relatively new class of proteins in which a given sequence has been shown to attain different folded conformations under native conditions while performing distinct functions [ 136–138 ]. Metamorphic proteins such as human chemokine lymphotactin have been shown to influence evolutionary transitions of structure and coding SNVs, which could play an important role in switching between two or more folds and functions for a single sequence [ 139 ]. Computational approaches to catalog and predict the metamorphic properties and the impact of coding SNVs on metamorphic proteins would help us understand how mutations drive functional plasticity at the proteome level.
Impact of coding SNVs on the transcriptomic diversity
Dynamic features of the human proteome are driven by transcript diversity, and the average number of characterized transcripts per gene is rapidly expanding. The recent adoption of RNA-Seq as a tool for expression profiling has led to the characterization of a large number of novel transcripts including fusion transcripts [ 140 ]. Several novel tissue-specific or cell-type-specific transcripts were reported from RNA-Seq experiments. The cellular compartment-based functional roles of such transcripts are determined by alternative splicing events [ 141 ]. RNA editing is a phenomenon where a pre-mRNA molecule is altered through a chemical change in the base makeup, thereby adding to the diversity of the transcriptome. RNA editing events occur via two distinct mechanisms of substitution editing and insertion/deletion editing, leading to functional diversity. Fusion transcripts [ 142 ] and RNA editing events are also associated with various diseases including prostate cancer and amyotrophic lateral sclerosis [ 143 ]. Computational approaches are available for the identification of fusion transcripts [ 144 ] and RNA editing events [ 145–147 ] from sequencing data. Understanding the precise roles and the impact of variants on such a diverse set of transcripts using computational approaches is a challenging task. Ascribing the functional role and assessing the impact of coding SNVs for mediating novel and fusion transcripts using bioinformatics approaches is an emerging problem and being addressed in recent studies [ 148 ]. Recently, we designed an integrated method to simultaneous analyses of genome and transcriptome from RNA-seq data. The eSNV-Detect method can precisely capture genetic variation (genotypes) from RNA-seq data and helps to design cost-effective and sustainable experimental strategies [ 149 ]. Analytic and interpretation strategies that rely on multiple data-types would provide greater confidence for variant calling, annotation and interpretation and thus actionability for clinical interventions.
Functional impact of synonymous variations
Synonymous mutations are defined as mutations that result in a variation at the DNA level that code for the same amino acid in the protein level owing to codon degeneracy. Synonymous variants may either be coding or noncoding and the functional roles of such variants are emerging. A recent study [ 150 ] suggests that introns are involved in functional mechanisms, and introns with positional conservation across eukaryotic lineage are classified as functional introns. A comparative analysis of synonymous and non-synonymous variants associated with complex diseases has shown similar likelihood and effect size with disease association [ 151 ]. Synonymous mutations may also influence the introns regulating gene expression [ 152 ]. A recent report also suggests that the coding exons function as tissue-specific enhancers and synonymous variations in such enhancer sites may influence the expression level of certain genes [ 153 ]. Synonymous variants could influence the expression of intronic noncoding RNAs [ 154 ], perturb the transient protein–DNA [ 155 , 156 ] and protein–RNA interactions in the cell. Such perturbations can lead to diseases such as cancer, neurological disorders and cardiac disease [ 157 ]. Developing bioinformatics approaches to explore the putative impact of synonymous mutations on different layers of the coding and noncoding genome and their relationships will be an important aspect in the detailed interpretation of the genomic variants. The detailed structural exploration of protein–DNA [ 158 ] and protein–RNA interactions [ 159 ] will help to precisely map the mechanistic bases of the loss or gain of interactions owing to synonymous variations.
Impact of coding SNVs on the interactome of a protein
The individual interactome of proteins varies to a great extent [ 160 ]. Proteins often interact with nucleotides (protein–DNA or protein–RNA interactions), proteins (protein complexes, obligate or non-obligate) [ 161 ] and protein–protein interactions (transient or permanent) [ 107 ]) and small molecules (molecular reactions, enzymatic reactions, metabolic pathways) [ 162 , 163 ]. Coding SNVs play an important role in mediating such interactions. An interactome of a protein can be defined using information gathered from high-throughput experiments that systematically identify interactants of proteins. Public protein-protein databases like BioGRID [ 164 ] and STRING [ 165 ] provide large data sets for deriving a protein interactome. Network-level investigations to understand the category of interactors that are perturbed owing to coding SNVs will help delineate the impact of such mutations on the protein interactomes. Using network measures (centrality, degree, stress, betweenness, closeness, cliques, radiality, transitivity, reciprocity, assortativity, structural equivalence, network heterogeneity, network density, clustering coefficients, neighborhood connectivity, shared neighbors, network topology, etc.) as quantitative parameters in variant annotation pipelines will help the researchers gain better insights into the impact of coding SNVs on the protein interactome. Biologically relevant network properties such as network modularity [ 166 ], network fragility [ 166 ] and lethality [ 166 ] can also be derived from an interactome. Incorporating a comparative network analysis framework that compares wild-type and variant interactomes will help quantify the impact of coding SNVs on a network scale. Methods such as VCF2Networks employ genotype networks (i.e. all genotypes associated with a single phenotype) to understand the relationships between the genotype space and clinical or biological phenotypes [ 167 ]. Table 7 summarizes the available tools to investigate gene lists and to derive global functional trends and network properties.
Table 7.
Software libraries and tools for biological network analysis
| Name | Description | URL |
|---|---|---|
| Bio4J | Bio4j is a bioinformatics graph-based database | http://www.bio4j.com/ |
| BioConductor (Graphs and Networks view) | Collection of BioConductor modules for biological network analysis and visualization | http://www.bioconductor.org/packages/release/BiocViews.html#___GraphsAndNetworks |
| Cytoscape | Open source platform for complex network analysis and visualization with a large collection of plug-ins for biological network analysis | http://www.cytoscape.org/ |
| DAVID | Integrated functional annotation tool | http://david.abcc.ncifcrf.gov/home.jsp |
| FunDO | Functional Disease Ontology server | http://django.nubic.northwestern.edu/fundo/ |
| gene2pathway | R package for prediction of KEGG pathway membership for individual genes based on InterPro domain signatures | http://www.bioconductor.org/packages/release/bioc/html/gene2pathway.html |
| GeneAnswers | R package for biological or medical interpretation of the given one or more groups of genes by means of statistical test | http://www.bioconductor.org/packages/release/bioc/html/GeneAnswers.html |
| GO Tools | Tools for analysis of GO T term enrichment, statistical analysis, semantic similarity and functional similarity using GO terms derived from gene lists | http://www.geneontology.org/GO.tools.shtml |
| Gephi | Open-source graph visualization and analysis software | http://www.gephi.org |
| Gremlin | Graph-based programming language | https://github.com/tinkerpop/gremlin/wiki/ |
| iGraph | Network analysis and visualization library in C. Also available R package and a Python extension | http://igraph.sourceforge.net/ |
| KEGGgraph | R package for analysis of KEGG pathways | http://www.bioconductor.org/packages/2.10/bioc/html/KEGGgraph.html |
| LEDA | A broad-spectrum C++ class library for efficient data types and algorithms including large-scale network analysis | http://www.algorithmic-solutions.com/leda/about/index.htm |
| Neat | Web-based network analysis tools | http://gephi.org/ |
| Ontology Analysis plugins for Cytoscape | http://chianti.ucsd.edu/cyto_web/plugins/ | Plugins for functional enrichment analysis using network data |
| PANTHER | Classification of genes and proteins | http://pantherdb.org/ |
| Reactome | Curated knowledgebase of molecular events and pathways | http://www.reactome.org |
| TargetMine | Integrates different types of biological data and enable flexible queries, export results and analyze lists of data. | http://targetmine.mizuguchilab.org |
Pathway-level impact of coding SNVs
Multiple functional genomics studies have investigated the perturbations of pathways as a result of mutations in diseases such as adenocarcinoma (MAPK signaling, p53 signaling, Wnt signaling, cell cycle and mammalian target of rapamycin pathways) [ 168 ], childhood acute lymphoblastic leukemia (RAS pathway) [ 169 ], colorectal carcinoma (WNT signaling pathway) [ 170 ], etc. Several SNP-centric approaches for pathway-level inference have been designed for the interpretation of SNPs identified from GWA studies or differentially expressed genes from gene-expression studies [ 171–175 ]. Coding SNVs may also influence the cross talk between various signaling pathways. Current variant interpretation algorithms strive to identify pathways associated with genes harboring coding SNVs; yet, a detailed understanding of the impact of variants on biological pathways and pathway cross talks [ 176 ] is often lacking. Incorporating analytical routines to quantify the effect of functional mutations on pathways and pathway cross talk will be useful in interpreting functional variants from a biological perspective.
A classification schema for annotating coding SNVs
Identifying the entire spectrum of molecular perturbations owing to coding variations at the level of sequence, structure, interaction and function of proteins is considered the basis of variant interpretation. Multiple automated prediction tools that can assess the functional effect of mutations are currently available [See Table 6 ]. Several variant annotation tools focus on the task of predicting the type or effect of mutations, provide extended annotations from precomputed databases or lookup tables and map a coding variant to its corresponding gene, protein, functional domain or signaling pathway. These approaches have several limitations because additional layers of protein-centric information that can be derived from the prediction or computation of sequence-based features with coding variants are lacking. To deal with this challenge and to design broad-spectrum tools for deep variant interpretation, we recommend a three-level annotation schema for the interpretation of coding variants ( Figure 7 ). Primary level annotation provides an overview of the types of mutations and association annotations. Secondary level annotation enables the systematic investigation of multiple variants in a single gene. Tertiary-level annotations help to find the global characteristics of genes harboring coding variants and various properties in a network-scale.
Figure 7.

Three-level schema for annotation of functional variants.
Level 1: Primary annotation of coding SNVs
Tools that use position information from VCF files to derive the type of mutation (synonymous or non-synonymous), the location of the corresponding gene and its mapping onto several annotation resources using positional data may be viewed as ‘primary annotations’. See Table 2 for a list of tools that use minimum input data to gather a diverse set of annotations using database lookup tables and identifier mapping.
Level 2: Secondary annotation of coding SNVs:- Impact of multiple variants in a single gene
Sequence and structural explorations of coding variants are referred to as secondary annotations. VCF files can have multiple variants of the same gene associated with a given phenotype or disease. A systematic investigation of multiple variants in a single gene and an assessment of the relative impact of coding variants could help in classifying multiple variants in a gene and help to delineate the variants as pathogenic, moderately pathogenic or VUS. Comparative analysis of wild-type and variant sequences can be performed and quantified using the total number of gains or losses in the sequence features. Based on the availability of the structural data, a given variant can be modeled in a protein structure using in-silico mutagenesis experiments. In the absence of experimental protein structural data, a protein structure model can be obtained from the homology model database or a new model can be built using homology modeling approaches. Once the wild type and mutant type structures are obtained, the impact of variation can be rapidly computed using structural feature analyses or using computationally expensive molecular dynamics simulations. Table 5 summarizes key sequence and structure feature prediction tools.
Level 3: Tertiary annotation of coding SNVs:- Impact of coding SNVs on multiple genes, pathways and interactome
The global impact of multiple coding variants on a genome or exome can be assessed using a combination of both knowledge-based enrichment or depletion analysis and network or interactome analyses. Functional profiling using GO terms have been used as an effective approach for characterizing collective functional characteristics of a perturbed set of genes [ 177 , 178 ]. Gene-list-based analyses can provide biologically relevant information about the variants. Enrichment analysis is not just confined to a priori defined gene sets or GO annotations; enrichment can be performed using several types of annotations and could provide insights into the probable functional associations of genes with coding variants. Enrichment analysis using protein annotations would help to identify functionally significant protein domains, protein classes, families (membrane proteins or kinases, etc.), protein superfamilies (angiotensin receptors or G-protein coupled receptors) or protein folds (Rossman fold, beta-propeller) associated with proteins harboring coding variants. Furthermore, it is possible to identify enriched molecular events mediated by genes, enriched TFBS in the upstream of genes and biological pathways mediated by the genes using annotations from Reactome, UCSC and KEGG repositories. Disease ontology can be used to find known gene–disease associations. Following the knowledge-based analysis, a network-level analysis of genes harboring coding SNVs can be performed using publicly available network analysis tools such as Cytoscape and Gephi.
Future directions
The rapidly increasing availability of sequence, structure, functional and interaction information offers an attractive means to obtain a detailed characterization of coding variants. However, the use of large data sets to systematically explore the functional variants is currently limited owing to the gaps in the available sequence–structure–function interaction data. Sequence data are available for the entire human proteome, but the availability of protein structural data, experimentally verified functional associations and biomolecular interaction data is limited. The availability of structural data of homologous protein structures is a prerequisite for structural investigations of functional variants and its impact on the structural environment. Availability of high-quality interaction data will also help analyze the cellular networks modulated by proteins harboring coding variants. The expansion of structural data sets using structural genomics [ 179 ], homology modeling [ 180 ] and the application of computational approaches such as genomics-aided structure prediction [ 181 ] is likely to lower the increasing chasm between sequence, structure, function and interaction data and may help to ascertain the impact of variants at the biomolecular level [ 163 , 182 , 183 ]. Improving the annotation databases using high-quality, curated data and integration with cloud-based or stand-alone pipelines [ 184–189 ] can also help facilitate such efforts.
The current version of the Bioinformatics Link Directory enlists >900 bioinformatics tools that are capable of processing different types of protein data (sequence, structure, interaction, quantification and annotation). Unifying multiple resources and enabling programmatic access via application program interfaces and web services for rapid integration will significantly enhance the efficacy of variant annotation pipelines. The development of novel statistical methods that can quantify various residue-specific properties would enable a comparative analysis of wild-type and variant sequences and can thereby help to facilitate the identification and prioritization of functional mutations. The inclusion of a diverse set of sequence, structure and network features into a graphically depicted database and an assessment of the impact of various data types (sequence, structure, function and interaction) using probabilistic or machine learning models would also help automate variant annotation interpretation.
From a sequence perspective, understanding the precise functional role of LCRs, unassigned regions and disordered regions as well as the relationship between these features and coding SNVs is a key step in variant annotation and interpretation. Identifying deviations in protein structural space that are likely to lead to misfolding, domain swapping, aggregation, degradation or deviations that are metamorphic in nature owing to the impact of coding SNVs is an emerging challenge. Several algorithms are available to predict sequence or structural domains, assign distantly related domains to unassigned regions, functional motifs, structural motifs and structural features, protein disorder and aggregation propensities. However, algorithms and tools to predict 3D domain swapping or to analyze metamorphic properties, degradation pathways and the network-level impact of coding SNVs are less abundant. This may be attributed to the fact that these concepts are continually evolving in the protein universe and concerted efforts are needed to understand the impact of coding variants of such unique features from both experimental and computational biologists alike.
This review summarizes the major bioinformatics challenges involved in gaining a deeper understanding of coding SNVs. Coding SNVs may impart a molecular or disease phenotype by interacting with noncoding, regulatory parts of the genome and the structural variants may provide an important contribution to this phenomenon. Analyzing functional and regulatory variants in a single analytical framework may further enhance the interpretation of the genomic variants.
Conclusions
A rapid decline in sequencing costs using NGS technologies has led to an exponential increase in the frequency of the sequencing projects over the past decade. A large number of personal genomes and exomes, clinical samples and cancer genomes are being sequenced as part of large-scale collaborative projects. The identification of a plethora of sequence variants associated with diverse molecular and disease phenotypes. Scalable computational approaches that integrate annotations using sequence, structure, functional and interaction data are necessary for the rapid interpretation of coding SNVs. We have discussed the bioinformatics resources available for the prediction, annotation and visualization of coding SNVs, summarized the major bioinformatics challenges into 10 different themes for a deeper interpretation of coding SNVs and recommended a three-level schema to assess the phenotypic impact of functional variations on individual protein sequences, structures, different functional categories, biological pathways and interactomes. We envisage that addressing the key challenges discussed in this review and adopting a comprehensive annotation schema for variant annotation could improve genomic reports that are generated as part of genomic medicine investigations and experimental studies to better understand the variations implicated in rare, common and complex disease manifestations.
Key Points
Interpretations of variants identified from next-generation sequencing pose several challenges.
Systems-level experimental investigation of the functional variants is expensive and time-consuming; efficient computational techniques are required to identify the impact of functional variants.
We recommended an integrated approach that combines multiple data types and tools for the prediction, annotation and visualization of functional variants and we have proposed a systematic approach for functional variant annotation and interpretation.
Significant challenges that need to be negotiated during the interpretation of coding single nucleotide variants are presented with the help of various examples.
A three-level annotation approach that combines the information at the level of an individual variant, multiple variants in a single protein and global trends of multiple genes harboring multiple variants is proposed for an effective interpretation of coding single nucleotide variants.
Acknowledgements
K.S. and J.T.D would like to thank Icahn Institute for Genomics and Multiscale Biology ( http://icahn.mssm.edu/departments-and-institutes/genomics ), Mount Sinai Health System for infrastructural support. K.S. would like to acknowledge Michael J. Zimmermann and Jean-Pierre Kocher (Mayo Clinic, Rochester) for useful discussions. The Mayo Clinic Bioinformatics Core provided infrastructure and analytical support to K.R.K. R.S. would like to thank National Centre for Biological Sciences (TIFR) for infrastructural support.
Funding
K.S. and J.T.D was supported by grant R01-DK098242-03 from the National Institute of Diabetes and Digestive and Kidney Diseases. K.R.K. is funded in part by the Mayo Clinic Center for Individualized Medicine, the Pharmacogenomics Research Network (PGRN) and the Mayo Clinic Breast Specialized Program of Research Excellence (SPORE).
References
- 1. Lander ES . Initial impact of the sequencing of the human genome . Nature 2011. ; 470 : 187 – 97 . [DOI] [PubMed] [Google Scholar]
- 2. Mardis ER . A decade's perspective on DNA sequencing technology . Nature 2011. ; 470 : 198 – 203 . [DOI] [PubMed] [Google Scholar]
- 3. Green ED, Guyer MS . Charting a course for genomic medicine from base pairs to bedside . Nature 2011. ; 470 : 204 – 13 . [DOI] [PubMed] [Google Scholar]
- 4. Maxmen A . Exome sequencing deciphers rare diseases . Cell 2011. ; 144 : 635 – 7 . [DOI] [PubMed] [Google Scholar]
- 5. Wheeler DA, Srinivasan M, Egholm M, et al. . The complete genome of an individual by massively parallel DNA sequencing . Nature 2008. ; 452 : 872 – 6 . [DOI] [PubMed] [Google Scholar]
- 6. Weatheritt RJ, Davey NE, Gibson TJ . Linear motifs confer functional diversity onto splice variants . Nucleic Acids Res 2012. ; 40 : 123 – 31 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldstein DB . Common genetic variation and human traits . N Engl J Med 2009. ; 360 : 1696 – 8 . [DOI] [PubMed] [Google Scholar]
- 8. Manolio TA, Collins FS, Cox NJ, et al. . Finding the missing heritability of complex diseases . Nature 2009. ; 461 : 747 – 53 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Genome-wide association study of 14 000 cases of seven common diseases and 3 000 shared controls . Nature 2007. ; 447 : 661 – 78 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kullo IJ, Ding K, Shameer K, et al. . Complement receptor 1 gene variants are associated with erythrocyte sedimentation rate . Am J Hum Genet 2011. ; 89 : 131 – 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Welter D, MacArthur J, Morales J, et al. . The NHGRI GWAS Catalog, a curated resource of SNP-trait associations . Nucleic Acids Res 2014. ; 42 : D1001 – 6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leslie R, O'Donnell CJ, Johnson AD . GRASP: analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database . Bioinformatics 2014. ; 30 : i185 – 94 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nelson MR, Wegmann D, Ehm MG, et al. . An abundance of rare functional variants in 202 drug target genes sequenced in 14 002 people . Science 2012. ; 337 : 100 – 4 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. MacArthur DG, Balasubramanian S, Frankish A, et al. . A systematic survey of loss-of-function variants in human protein-coding genes . Science 2012. ; 335 : 823 – 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raychaudhuri S . Mapping rare and common causal alleles for complex human diseases . Cell 2011. ; 147 : 57 – 69 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stankiewicz P, Lupski JR . Structural variation in the human genome and its role in disease . Annu Rev Med 2010. ; 61 : 437 – 455 . [DOI] [PubMed] [Google Scholar]
- 17. McCarroll SA, Altshuler DM . Copy-number variation and association studies of human disease . Nat Genet 2007. ; 39 : S37 – 42 . [DOI] [PubMed] [Google Scholar]
- 18. Zhang F, Gu W, Hurles ME, et al. . Copy number variation in human health, disease, and evolution . Annu Rev Genomics Hum Genet 2009. ; 10 : 451 – 81 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Medvedev P, Stanciu M, Brudno M . Computational methods for discovering structural variation with next-generation sequencing . Nat Methods 2009. ; 6 : S13 – 20 . [DOI] [PubMed] [Google Scholar]
- 20. Sherry ST, Ward MH, Kholodov M, et al. . dbSNP: the NCBI database of genetic variation . Nucleic Acids Res 2001. ; 29 : 308 – 11 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rios D, McLaren WM, Chen Y, et al. . A database and API for variation, dense genotyping and resequencing data . BMC Bioinformatics 2010. ; 11 : 238 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iafrate AJ, Feuk L, Rivera MN, et al. . Detection of large-scale variation in the human genome . Nat Genet 2004. ; 36 : 949 – 51 . [DOI] [PubMed] [Google Scholar]
- 23. Ashley EA, Butte AJ, Wheeler MT, et al. . Clinical assessment incorporating a personal genome . Lancet 2010. ; 375 : 1525 – 35 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen R, Mias GI, Li-Pook-Than J, et al. . Personal omics profiling reveals dynamic molecular and medical phenotypes . Cell 2012. ; 148 : 1293 – 307 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. . Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy . N Engl J Med 2010. ; 362 : 1181 – 91 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Y, Vinckenbosch N, Tian G, et al. . Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants . Nat Genet 2010. ; 42 : 969 – 72 . [DOI] [PubMed] [Google Scholar]
- 27. Clarke L, Zheng-Bradley X, Smith R, et al. . The 1000 Genomes Project: data management and community access . Nat Methods 2012. ; 9 : 459 – 462 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schunkert H, Konig IR, Kathiresan S, et al. . Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease . Nat Genet 2011. ; 43 : 333 – 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Helgadottir A, Thorleifsson G, Manolescu A, et al. . A common variant on chromosome 9p21 affects the risk of myocardial infarction . Science 2007. ; 316 : 1491 – 3 . [DOI] [PubMed] [Google Scholar]
- 30. Visel A, Zhu Y, May D, et al. . Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice . Nature 2010. ; 464 : 409 – 12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Soranzo N, Rendon A, Gieger C, et al. . A novel variant on chromosome 7q22.3 associated with mean platelet volume, counts, and function . Blood 2009. ; 113 : 3831 – 7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paul DS, Nisbet JP, Yang TP, et al. . Maps of open chromatin guide the functional follow-up of genome-wide association signals: application to hematological traits . PLoS Genet 2011. ; 7 : e1002139 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hull J, Campino S, Rowlands K, et al. . Identification of common genetic variation that modulates alternative splicing . PLoS Genet 2007. ; 3 : e99 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Albert FW, Kruglyak L . The role of regulatory variation in complex traits and disease . Nat Rev Genet 2015. ; 16 : 197 – 212 . [DOI] [PubMed] [Google Scholar]
- 35. Battle A, Khan Z, Wang SH, et al. . Genomic variation. Impact of regulatory variation from RNA to protein . Science 2015. ; 347 : 664 – 7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. den Dunnen JT, Antonarakis SE . Nomenclature for the description of human sequence variations . Hum Genet 2001. ; 109 : 121 – 4 . [DOI] [PubMed] [Google Scholar]
- 37. Eilbeck K, Lewis SE, Mungall CJ, et al. . The sequence ontology: a tool for the unification of genome annotations . Genome Biol 2005. ; 6 : R44 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Landrum MJ, Lee JM, Riley GR, et al. . ClinVar: public archive of relationships among sequence variation and human phenotype . Nucleic Acids Res 2014. ; 42 : D980 – 5 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kircher M, Witten DM, Jain P, et al. . A general framework for estimating the relative pathogenicity of human genetic variants . Nat Genet 2014. ; 46 : 310 – 15 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kumar P, Henikoff S, Ng PC . Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm . Nat Protoc 2009. ; 4 : 1073 – 81 . [DOI] [PubMed] [Google Scholar]
- 41. Adzhubei I, Schmidt S, Peshkin L, et al. . A method and server for predicting damaging missense mutations . Nat Meth 2010. ; 7 : 248 – 9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gonzalez-Perez A, Lopez-Bigas N . Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel . Am J Hum Genet 2011. ; 88 : 440 – 9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grantham R . Amino acid difference formula to help explain protein evolution . Science 1974. ; 185 : 862 – 4 . [DOI] [PubMed] [Google Scholar]
- 44. Cooper GM, Stone EA, Asimenos G, et al. . Distribution and intensity of constraint in mammalian genomic sequence . Genome Res 2005. ; 15 : 901 – 13 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pollard KS, Hubisz MJ, Rosenbloom KR, et al. . Detection of nonneutral substitution rates on mammalian phylogenies . Genome Res 2010. ; 20 : 110 – 21 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Siepel A, Bejerano G, Pedersen JS, et al. . Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes . Genome Res 2005. ; 15 : 1034 – 50 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dayhoff MO, Eck RV, Park CM . Model of evolutionary change in proteins . In: Dayhoff MO. (ed). Atlas of Protein Sequence and Structure , Washington, DC: : National Biomedical Research Foundation; , 1972. , 89 – 99 . [Google Scholar]
- 48. Henikoff S, Henikoff JG . Amino acid substitution matrices from protein blocks . Proc Natl Acad Sci USA 1992. ; 89 : 10915 – 19 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Altschul SF, Gish W, Miller W, et al. . Basic local alignment search tool . J Mol Biol 1990. ; 215 : 403 – 10 . [DOI] [PubMed] [Google Scholar]
- 50. Altschul SF, Madden TL, Schaffer AA, et al. . Gapped BLAST and PSI-BLAST: a new generation of protein database search programs . Nucleic Acids Res 1997. ; 25 : 3389 – 402 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sunyaev S, Ramensky V, Koch I, et al. . Prediction of deleterious human alleles . Hum Mol Genet 2001. ; 10 : 591 – 7 . [DOI] [PubMed] [Google Scholar]
- 52. Yang Z, Bielawski JP . Statistical methods for detecting molecular adaptation . Trends Ecol Evol 2000. ; 15 : 496 – 503 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ng PC, Henikoff S . Predicting deleterious amino acid substitutions . Genome Res 2001. ; 11 : 863 – 74 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang Z, Moult J . SNPs, protein structure, and disease . Hum Mutat 2001. ; 17 : 263 – 70 . [DOI] [PubMed] [Google Scholar]
- 55. Thomas PD, Kejariwal A . Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects . Proc Natl Acad Sci USA 2004. ; 101 : 15398 – 403 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stone EA, Sidow A . Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity . Genome Res 2005. ; 15 : 978 – 86 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miller MP, Kumar S . Understanding human disease mutations through the use of interspecific genetic variation . Hum Mol Genet 2001. ; 10 : 2319 – 28 . [DOI] [PubMed] [Google Scholar]
- 58. Johnson AD . Single-nucleotide polymorphism bioinformatics: a comprehensive review of resources . Circ Cardiovasc Genet 2009. ; 2 : 530 – 6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marjoram P, Tavare S . Modern computational approaches for analysing molecular genetic variation data . Nat Rev Genet 2006. ; 7 : 759 – 70 . [DOI] [PubMed] [Google Scholar]
- 60. Excoffier L, Heckel G . Computer programs for population genetics data analysis: a survival guide . Nat Rev Genet 2006. ; 7 : 745 – 58 . [DOI] [PubMed] [Google Scholar]
- 61. Peterson TA, Nehrt NL, Park D, et al. . Incorporating molecular and functional context into the analysis and prioritization of human variants associated with cancer . J Am Med Inform Assoc 2012. ; 19 : 275 – 83 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kann MG . Advances in translational bioinformatics: computational approaches for the hunting of disease genes . Brief Bioinform 2010. ; 11 : 96 – 110 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cooper GM, Shendure J . Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data . Nat Rev Genet 2011. ; 12 : 628 – 40 . [DOI] [PubMed] [Google Scholar]
- 64. Stitziel NO, Kiezun A, Sunyaev S . Computational and statistical approaches to analyzing variants identified by exome sequencing . Genome Biol 2011. ; 12 : 227 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Alexander RP, Fang G, Rozowsky J, et al. . Annotating non-coding regions of the genome . Nat Rev Genet 2010. ; 11 : 559 – 71 . [DOI] [PubMed] [Google Scholar]
- 66. Mort M, Evani US, Krishnan VG, et al. . In silico functional profiling of human disease-associated and polymorphic amino acid substitutions . Hum Mutat 2010. ; 31 : 335 – 46 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hamosh A, Scott AF, Amberger JS, et al. . Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders . Nucleic Acids Res 2005. ; 33 : D514 – 17 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stenson PD, Mort M, Ball EV, et al. . The human gene mutation database: 2008 update . Genome Med 2009. ; 1 : 13 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Forbes SA, Bindal N, Bamford S, et al. . COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer . Nucleic Acids Res 2011. ; 39 : D945 – 50 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen YA, Tripathi LP, Mizuguchi K . TargetMine, an integrated data warehouse for candidate gene prioritisation and target discovery . PLoS One 2011. ; 6 : e17844 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data . Nucleic Acids Res 2010. ; 38 : e164 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Danecek P, Auton A, Abecasis G, et al. . The variant call format and VCFtools . Bioinformatics 2011. ; 27 : 2156 – 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Macintyre G, Jimeno, Yepes A, Ong CS, et al. . Associating disease-related genetic variants in intergenic regions to the genes they impact . Peer J 2014. ; 2 : e639 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Javed A, Agrawal S, Ng PC . Phen-Gen: combining phenotype and genotype to analyze rare disorders . Nat Methods 2014. ; 11 : 935 – 7 . [DOI] [PubMed] [Google Scholar]
- 75. Dai HJ, Wu JC, Tsai RT, et al. . T-HOD: a literature-based candidate gene database for hypertension, obesity and diabetes . Database (Oxford) 2013. ; 2013 : bas061 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cheng D, Knox C, Young N, et al. . PolySearch: a web-based text mining system for extracting relationships between human diseases, genes, mutations, drugs and metabolites . Nucleic Acids Res 2008. ; 36 : W399 – 405 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Do R, Kathiresan S, Abecasis GR . Exome sequencing and complex disease: practical aspects of rare variant association studies . Hum Mol Genet 2012. ; 21 : R1 – 9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wagner MJ . Rare-variant genome-wide association studies: a new frontier in genetic analysis of complex traits . Pharmacogenomics 2013. ; 14 : 413 – 24 . [DOI] [PubMed] [Google Scholar]
- 79. Genomes Project C, Abecasis GR, Auton A, et al. . An integrated map of genetic variation from 1 092 human genomes . Nature 2012. ; 491 : 56 – 65 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Anfinsen CB . The formation and stabilization of protein structure . Biochem J 1972. ; 128 : 737 – 49 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Anfinsen CB . Principles that govern the folding of protein chains . Science 1973. ; 181 : 223 – 30 . [DOI] [PubMed] [Google Scholar]
- 82. Fay J, Wu C . Sequence divergence, functional constraint, and selection in protein evolution . Annu Rev Genomics Hum Genet 2003. ; 4 : 213 – 35 . [DOI] [PubMed] [Google Scholar]
- 83. Ponting CP, Russell RR . The natural history of protein domains . Annu Rev Biophys Biomol Struct 2002. ; 31 : 45 – 71 . [DOI] [PubMed] [Google Scholar]
- 84. Ekman D, Bjorklund AK, Frey-Skott J, et al. . Multi-domain proteins in the three kingdoms of life: orphan domains and other unassigned regions . J Mol Biol 2005. ; 348 : 231 – 43 . [DOI] [PubMed] [Google Scholar]
- 85. Sigrist CJ, Cerutti L, Hulo N, et al. . PROSITE: a documented database using patterns and profiles as motif descriptors . Brief Bioinform 2002. ; 3 : 265 – 74 . [DOI] [PubMed] [Google Scholar]
- 86. Dinkel H, Michael S, Weatheritt RJ, et al. . ELM–the database of eukaryotic linear motifs . Nucleic Acids Res 2012. ; 40 : D242 – 51 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Haberichter SL, Budde U, Obser T, et al. . The mutation N528S in the von Willebrand factor (VWF) propeptide causes defective multimerization and storage of VWF . Blood 2010. ; 115 : 4580 – 7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kutz WE, Wang LW, Dagoneau N, et al. . Functional analysis of an ADAMTS10 signal peptide mutation in Weill-Marchesani syndrome demonstrates a long-range effect on secretion of the full-length enzyme . Hum Mutat 2008. ; 29 : 1425 – 34 . [DOI] [PubMed] [Google Scholar]
- 89. Pugalenthi G, Suganthan PN, Sowdhamini R, et al. . SMotif: a server for structural motifs in proteins . Bioinformatics 2007. ; 23 : 637 – 8 . [DOI] [PubMed] [Google Scholar]
- 90. Sundaramurthy P, Shameer K, Sreenivasan R, et al. . HORI: a web server to compute Higher Order Residue Interactions in protein structures . BMC Bioinformatics 2010. ; 11 ( Suppl 1 ): S24 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Porter CT, Bartlett GJ, Thornton JM . The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data . Nucleic Acids Res 2004. ; 32 : D129 – 33 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Stuart AC, Ilyin VA, Sali A . LigBase: a database of families of aligned ligand binding sites in known protein sequences and structures . Bioinformatics 2002. ; 18 : 200 – 01 . [DOI] [PubMed] [Google Scholar]
- 93. Swapna LS, Mahajan S, de Brevern A, et al. . Comparison of tertiary structures of proteins in protein-protein complexes with unbound forms suggests prevalence of allostery in signalling proteins . BMC Struct Biol 2012. ; 12 : 6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mathivanan S, Ahmed M, Ahn NG, et al. . Human Proteinpedia enables sharing of human protein data . Nat Biotechnol 2008. ; 26 : 164 – 7 . [DOI] [PubMed] [Google Scholar]
- 95. Bhaskara RM, Srinivasan N . Stability of domain structures in multi-domain proteins . Sci Rep 2011. ; 1 : 40 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Furnham N, de Beer TA, Thornton JM . Current challenges in genome annotation through structural biology and bioinformatics . Curr Opin Struct Biol 2012. ; 22 : 594 – 601 . [DOI] [PubMed] [Google Scholar]
- 97. Wang Y, Cheng H, Pan Z, et al. . Reconfiguring phosphorylation signaling by genetic polymorphisms affects cancer susceptibility . J Mol Cell Biol 2015. ; 7 : 187 – 202 . [DOI] [PubMed] [Google Scholar]
- 98. Gnad F, Gunawardena J, Mann M . PHOSIDA 2011: the posttranslational modification database . Nucleic Acids Res 2011. ; 39 : D253 – 60 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Nicolaou N, Margadant C, Kevelam SH, et al. . Gain of glycosylation in integrin alpha3 causes lung disease and nephrotic syndrome . J Clin Invest 2012. ; 122 : 4375 – 87 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Radivojac P, Baenziger PH, Kann MG, et al. . Gain and loss of phosphorylation sites in human cancer . Bioinformatics 2008. ; 24 : i241 – 7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pan Y, Karagiannis K, Zhang H, et al. . Human germline and pan-cancer variomes and their distinct functional profiles . Nucleic Acids Res 2014. ; 42 : 11570 – 88 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mazumder R, Morampudi KS, Motwani M, et al. . Proteome-wide analysis of single-nucleotide variations in the N-glycosylation sequon of human genes . PLoS One 2012. ; 7 : e36212 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Reddy CC, Shameer K, Offmann BO, et al. . PURE: a webserver for the prediction of domains in unassigned regions in proteins . BMC Bioinformatics 2008. ; 9 : 281 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Coletta A, Pinney JW, Solis DY, et al. . Low-complexity regions within protein sequences have position-dependent roles . BMC Syst Biol 2010. ; 4 : 43 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Jones DT, Swindells MB . Getting the most from PSI-BLAST . Trends Biochem Sci 2002. ; 27 : 161 – 4 . [DOI] [PubMed] [Google Scholar]
- 106. Chograni M, Rejeb I, Jemaa LB, et al. . The first missense mutation of NHS gene in a Tunisian family with clinical features of NHS syndrome including cardiac anomaly . Eur J Hum Genet 2011. ; 19 : 851 – 6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jones S, Thornton JM . Principles of protein-protein interactions . Proc Natl Acad Sci USA 1996. ; 93 : 13 – 20 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Babu MM, van der Lee R, de Groot NS, et al. . Intrinsically disordered proteins: regulation and disease . Curr Opin Struct Biol 2011. ; 21 : 432 – 40 . [DOI] [PubMed] [Google Scholar]
- 109. Dunker AK, Silman I, Uversky VN, et al. . Function and structure of inherently disordered proteins . Curr Opin Struct Biol 2008. ; 18 : 756 – 64 . [DOI] [PubMed] [Google Scholar]
- 110. Wright PE, Dyson HJ . Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm . J Mol Biol 1999. ; 293 : 321 – 31 . [DOI] [PubMed] [Google Scholar]
- 111. Dyson HJ, Wright PE . Coupling of folding and binding for unstructured proteins . Curr Opin Struct Biol 2002. ; 12 : 54 – 60 . [DOI] [PubMed] [Google Scholar]
- 112. Nguyen Ba AN, Yeh BJ, van Dyk D, et al. . Proteome-wide discovery of evolutionary conserved sequences in disordered regions . Sci Signal 2012. ; 5 : rs1 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Das RK, Mao AH, Pappu RV . Unmasking functional motifs within disordered regions of proteins . Sci Signal 2012. ; 5 : pe17 . [DOI] [PubMed] [Google Scholar]
- 114. Hu Y, Liu Y, Jung J, et al. . Changes in predicted protein disorder tendency may contribute to disease risk . BMC Genomics 2011. ; 12 ( Suppl 5 ): S2 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Vacic V, Iakoucheva LM . Disease mutations in disordered regions–exception to the rule? . Mol BioSyst 2012. ; 8 : 27 – 32 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Srivastava SK, Gayathri S, Manjasetty BA, et al. . Analysis of conformational variation in macromolecular structural models . PLoS One 2012. ; 7 : e39993 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ward JJ, Sodhi JS, McGuffin LJ, et al. . Prediction and functional analysis of native disorder in proteins from the three kingdoms of life . J Mol Biol 2004. ; 337 : 635 – 45 . [DOI] [PubMed] [Google Scholar]
- 118. Thomas PJ, Qu BH, Pedersen PL . Defective protein folding as a basis of human disease . Trends Biochem Sci 1995. ; 20 : 456 – 9 . [DOI] [PubMed] [Google Scholar]
- 119. Welch W . Role of quality control pathways in human diseases involving protein misfolding . Semin Cell Dev Biol 2004. ; 15 : 31 – 8 . [DOI] [PubMed] [Google Scholar]
- 120. Fink AL . Protein aggregation: folding aggregates, inclusion bodies and amyloid . Fold Des 1998. ; 3 : R9 – 23 . [DOI] [PubMed] [Google Scholar]
- 121. Thirumalai D, Klimov DK, Dima RI . Emerging ideas on the molecular basis of protein and peptide aggregation . Curr Opin Struct Biol 2003. ; 13 : 146 – 59 . [DOI] [PubMed] [Google Scholar]
- 122. Khare SD, Dokholyan NV . Molecular mechanisms of polypeptide aggregation in human diseases . Curr Protein Pept Sci 2007. ; 8 : 573 – 9 . [DOI] [PubMed] [Google Scholar]
- 123. Soto C, Estrada L, Castilla J . Amyloids, prions and the inherent infectious nature of misfolded protein aggregates . Trends Biochem Sci 2006. ; 31 : 150 – 5 . [DOI] [PubMed] [Google Scholar]
- 124. Prusiner SB . Molecular biology and pathogenesis of prion diseases . Trends Biochem Sci 1996. ; 21 : 482 – 7 . [DOI] [PubMed] [Google Scholar]
- 125. Shameer K, Shingate PN, Manjunath SC, et al. . 3DSwap: curated knowledgebase of proteins involved in 3D domain swapping . Database (Oxford) 2011. ; 2011 : bar042 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bennett MJ, Choe S, Eisenberg D . Domain swapping: entangling alliances between proteins . Proc Natl Acad Sci USA 1994. ; 91 : 3127 – 31 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bennett MJ, Sawaya MR, Eisenberg D . Deposition diseases and 3D domain swapping . Structure 2006. ; 14 : 811 – 24 . [DOI] [PubMed] [Google Scholar]
- 128. Shameer K, Sowdhamini R . Functional repertoire, molecular pathways and diseases associated with 3D domain swapping in the human proteome . J Clin Bioinform 2012. ; 2 : 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Waters PJ, Parniak MA, Hewson AS, et al. . Alterations in protein aggregation and degradation due to mild and severe missense mutations (A104D, R157N) in the human phenylalanine hydroxylase gene (PAH) . Hum Mutat 1998. ; 12 : 344 – 54 . [DOI] [PubMed] [Google Scholar]
- 130. O'Neill JW, Kim DE, Johnsen K, et al. . Single-site mutations induce 3D domain swapping in the B1 domain of protein L from Peptostreptococcus magnus . Structure 2001. ; 9 : 1017 – 27 . [DOI] [PubMed] [Google Scholar]
- 131. Seeliger MA, Spichty M, Kelly SE, et al. . Role of conformational heterogeneity in domain swapping and adapter function of the Cks proteins . J Biol Chem 2005. ; 280 : 30448 – 59 . [DOI] [PubMed] [Google Scholar]
- 132. Kirsten Frank M, Dyda F, Dobrodumov A, et al. . Core mutations switch monomeric protein GB1 into an intertwined tetramer . Nat Struct Biol 2002. ; 9 : 877 – 85 . [DOI] [PubMed] [Google Scholar]
- 133. Huang Y, Cao H, Liu Z . Three-dimensional domain swapping in the protein structure space . Proteins 2012. ; 80 : 1610 – 19 . [DOI] [PubMed] [Google Scholar]
- 134. Gidalevitz T, Wang N, Deravaj T, et al. . Natural genetic variation determines susceptibility to aggregation or toxicity in a C. elegans model for polyglutamine disease . BMC Biol 2013. ; 11 : 100 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ross CA, Poirier MA . Protein aggregation and neurodegenerative disease . Nat Med 2004. ; 10 ( Suppl ): S10 – 17 . [DOI] [PubMed] [Google Scholar]
- 136. Murzin A . Metamorphic proteins . Science 2008. ; 320 : 1725 – 6 . [DOI] [PubMed] [Google Scholar]
- 137. Shortle D . One sequence plus one mutation equals two folds . Proc Natl Acad Sci USA 2009. ; 106 : 21011 – 12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yadid I, Kirshenbaum N, Sharon M, et al. . Metamorphic proteins mediate evolutionary transitions of structure . Proc Natl Acad Sci USA 2010. ; 107 : 7287 – 92 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yadid I, Kirshenbaum N, Sharon M, et al. . Metamorphic proteins mediate evolutionary transitions of structure . Proc Natl Acad Sci USA 2010. ; 107 : 7287 – 92 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Pickrell J, Marioni J, Pai A, et al. . Understanding mechanisms underlying human gene expression variation with RNA sequencing . Nature 2010. ; 464 : 768 – 72 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Matlin AJ, Clark F, Smith CW . Understanding alternative splicing: towards a cellular code . Nat Rev Mol Cell Biol 2005. ; 6 : 386 – 98 . [DOI] [PubMed] [Google Scholar]
- 142. Rickman DS, Pflueger D, Moss B, et al. . SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer . Cancer Res 2009. ; 69 : 2734 – 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Flomen R, Makoff A . Increased RNA editing in EAAT2 pre-mRNA from amyotrophic lateral sclerosis patients: involvement of a cryptic polyadenylation site . Neurosci Lett 2011. ; 497 : 139 – 43 . [DOI] [PubMed] [Google Scholar]
- 144. Asmann YW, Hossain A, Necela BM, et al. . A novel bioinformatics pipeline for identification and characterization of fusion transcripts in breast cancer and normal cell lines . Nucleic Acids Res 2011. ; 39 : e100 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Bundschuh R . Computational prediction of RNA editing sites . Bioinformatics 2004. ; 20 : 3214 – 20 . [DOI] [PubMed] [Google Scholar]
- 146. Eisenberg E, Li JB, Levanon EY . Sequence based identification of RNA editing sites . RNA Biol 2010. ; 7 : 248 – 52 . [DOI] [PubMed] [Google Scholar]
- 147. Levanon EY, Eisenberg E . Algorithmic approaches for identification of RNA editing sites . Brief Funct Genomics Proteomic 2006. ; 5 : 43 – 5 . [DOI] [PubMed] [Google Scholar]
- 148. McCarthy DJ, Humburg P, Kanapin A, et al. . Choice of transcripts and software has a large effect on variant annotation . Genome Med 2014. ; 6 : 26 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Tang X, Baheti S, Shameer K, et al. . The eSNV-detect: a computational system to identify expressed single nucleotide variants from transcriptome sequencing data . Nucleic Acids Res 2014. ; 42 : e172 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chorev M, Carmel L . The function of introns . Front Genet 2012. ; 3 : 55 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Chen R, Davydov EV, Sirota M, et al. . Non-synonymous and synonymous coding SNPs show similar likelihood and effect size of human disease association . PLoS One 2010. ; 5 : e13574 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Rose AB . Intron-mediated regulation of gene expression . Curr Top Microbiol Immunol 2008. ; 326 : 277 – 90 . [DOI] [PubMed] [Google Scholar]
- 153. Birnbaum RY, Clowney EJ, Agamy O, et al. . Coding exons function as tissue-specific enhancers of nearby genes . Genome Res 2012. ; 22 : 1059 – 68 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Nakaya HI, Amaral PP, Louro R, et al. . Genome mapping and expression analyses of human intronic noncoding RNAs reveal tissue-specific patterns and enrichment in genes related to regulation of transcription . Genome Biol 2007. ; 8 : R43 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Rohs R, Jin X, West SM, et al. . Origins of specificity in protein-DNA recognition . Ann Rev Biochem 2010. ; 79 : 233 – 69 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Karczewski KJ, Dudley JT, Kukurba KR, et al. . Systematic functional regulatory assessment of disease-associated variants . Proc Natl Acad Sci USA 2013. ; 110 : 9607 – 12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Khalil AM, Rinn JL . RNA-protein interactions in human health and disease . Semin Cell Dev Biol 2011. ; 22 : 359 – 65 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Jones S, van Heyningen P, Berman HM, et al. . Protein-DNA interactions: a structural analysis . J Mol Biol 1999. ; 287 : 877 – 96 . [DOI] [PubMed] [Google Scholar]
- 159. Jones S, Daley DT, Luscombe NM, et al. . Protein-RNA interactions: a structural analysis . Nucleic Acids Res 2001. ; 29 : 943 – 54 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Barabasi AL, Oltvai ZN . Network biology: understanding the cell's functional organization . Nat Rev Genet 2004. ; 5 : 101 – 13 . [DOI] [PubMed] [Google Scholar]
- 161. Nooren IM, Thornton JM . Diversity of protein-protein interactions . EMBO J 2003. ; 22 : 3486 – 92 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Caspi R, Altman T, Dreher K, et al. . The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases . Nucleic Acids Res 2012. ; 40 : D742 – 53 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Yamada T, Bork P . Evolution of biomolecular networks: lessons from metabolic and protein interactions . Nat Rev Mol Cell Biol 2009. ; 10 : 791 – 803 . [DOI] [PubMed] [Google Scholar]
- 164. Stark C, Breitkreutz BJ, Chatr-Aryamontri A, et al. . The BioGRID interaction database: 2011 update . Nucleic Acids Res 2011. ; 39 : D698 – 704 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Szklarczyk D, Franceschini A, Kuhn M, et al. . The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored . Nucleic Acids Res 2011. ; 39 : D561 – 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Bader GD, Hogue CW . An automated method for finding molecular complexes in large protein interaction networks . BMC Bioinformatics 2003. ; 4 : 2 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Dall'Olio GM, Vahdati AR, Bertranpetit J, et al. . VCF2Networks: applying Genotype Networks to Single Nucleotide Variants data . Bioinformatics 2015. ; 31 : 438 – 9 . [DOI] [PubMed] [Google Scholar]
- 168. Ding L, Getz G, Wheeler DA, et al. . Somatic mutations affect key pathways in lung adenocarcinoma . Nature 2008. ; 455 : 1069 – 75 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Case M, Matheson E, Minto L, et al. . Mutation of genes affecting the RAS pathway is common in childhood acute lymphoblastic leukemia . Cancer Res 2008. ; 68 : 6803 – 9 . [DOI] [PubMed] [Google Scholar]
- 170. Thorstensen L, Lind GE, Lovig T, et al. . Genetic and epigenetic changes of components affecting the WNT pathway in colorectal carcinomas stratified by microsatellite instability . Neoplasia 2005. ; 7 : 99 – 108 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Weng L, Macciardi F, Subramanian A, et al. . SNP-based pathway enrichment analysis for genome-wide association studies . BMC Bioinformatics 2011. ; 12 : 99 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Wang K, Li M, Bucan M . Pathway-based approaches for analysis of genomewide association studies . Am J Hum Genet 2007. ; 81 : 1278 – 83 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. O'Dushlaine C, Kenny E, Heron EA, et al. . The SNP ratio test: pathway analysis of genome-wide association datasets . Bioinformatics 2009. ; 25 : 2762 – 3 . [DOI] [PubMed] [Google Scholar]
- 174. Tarca AL, Draghici S, Khatri P, et al. . A novel signaling pathway impact analysis . Bioinformatics 2009. ; 25 : 75 – 82 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ramanan VK, Shen L, Moore JH, et al. . Pathway analysis of genomic data: concepts, methods, and prospects for future development . Trends Genet 2012. ; 28 : 323 – 2 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Li Y, Agarwal P, Rajagopalan D . A global pathway crosstalk network . Bioinformatics 2008. ; 24 : 1442 – 7 . [DOI] [PubMed] [Google Scholar]
- 177. Rivals I, Personnaz L, Taing L, et al. . Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics 2007. ; 23 : 401 – 7 . [DOI] [PubMed] [Google Scholar]
- 178. Rhee SY, Wood V, Dolinski K, et al. . Use and misuse of the gene ontology annotations . Nat Rev Genet 2008. ; 9 : 509 – 15 . [DOI] [PubMed] [Google Scholar]
- 179. Chandonia JM, Brenner SE . The impact of structural genomics: expectations and outcomes . Science 2006. ; 311 : 347 – 51 . [DOI] [PubMed] [Google Scholar]
- 180. Johnson MS, Srinivasan N, Sowdhamini R, et al. . Knowledge-based protein modeling . Crit Rev Biochem Mol Biol 1994. ; 29 : 1 – 68 . [DOI] [PubMed] [Google Scholar]
- 181. Sulkowska JI, Morcos F, Weigt M, et al. . Genomics-aided structure prediction . Proc Natl Acad Sci USA 2012. ; 109 : 10340 – 5 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Lee D, Redfern O, Orengo C . Predicting protein function from sequence and structure . Nat Rev Mol Cell Biol 2007. ; 8 : 995 – 1005 . [DOI] [PubMed] [Google Scholar]
- 183. Worth CL, Gong S, Blundell TL . Structural and functional constraints in the evolution of protein families . Nat Rev Mol Cell Biol 2009. ; 10 : 709 – 20 . [DOI] [PubMed] [Google Scholar]
- 184. Karczewski KJ, Fernald GH, Martin AR, et al. . STORMSeq: an open-source, user-friendly pipeline for processing personal genomics data in the cloud . PLoS One 2014. ; 9 : e84860 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Kalari KR, Rossell D, Necela BM, et al. . Deep sequence analysis of non-small cell lung cancer: integrated analysis of gene expression, alternative splicing, and single nucleotide variations in lung adenocarcinomas with and without oncogenic KRAS mutations . Front Oncol 2012. ; 2 : 12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Patil S, Upadhayay A, Arya D, et al. . A high throughput exome sequencing approach to analyse events within a good responder CML patient under imatinib at diagnosis and under remission . Blood 2013. ; 122 : 5161 . [Google Scholar]
- 187. Kullo IJ, Shameer K, Jouni H, et al. . The ATXN2-SH2B3 locus is associated with peripheral arterial disease: an electronic medical record-based genome-wide association study . Front Genet 2014. ; 5 : 166 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Shameer K, Denny JC, Ding K, et al. . A genome- and phenome-wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects . Hum Genet 2014. ; 133 : 95 – 109 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Sabarinathan R, Wenzel A, Novotny P, et al. . Transcriptome-wide analysis of UTRs in non-small cell lung cancer reveals cancer-related genes with SNV-induced changes on RNA secondary structure and miRNA target sites . PLoS One 2014. ; 9 : e82699 . [DOI] [PMC free article] [PubMed] [Google Scholar]