

Abstract
Targeted therapies for ER+/PR+ and HER2-amplified breast cancers have improved patient survival, but there are no therapies for triple negative breast cancers (TNBC) that lack expression of estrogen and progesterone receptors (ER/PR), or amplification or overexpression of HER2. Gene expression profiling of TNBC has identified molecular subtypes and representative cell lines. An extract of the Texas native plant Amyris texana was found to have selective activity against MDA-MB-453 cells, a model of the luminal androgen receptor (LAR) subtype of TNBC. Bioassay-guided fractionation identified two oxazole natural products with selective activity against this cell line. Analog synthesis and structure-activity relationship studies conducted provided analogs with more potent and selective activity against two LAR subtype cell line models, culminating in the discovery of compound 30 (CIDD-0067106). Lead compounds discovered have potent and selective antiproliferative activities, and mechanisms of action studies show they inhibit the activity of the mTORC1 pathway.
Keywords: triple-negative breast cancer, natural products, structure-activity relationship, Amyris texana, androgen receptor
INTRODUCTION
Triple-negative breast cancers (TNBC) represent approximately 15% of breast cancers and are defined by the lack of expression of estrogen/progesterone receptors (ER/PR) and human epidermal growth factor receptor 2 (HER2).1,2 However, the classification of these cancers as triple-negative only provides information as to what these cancers are not, rather than what they are. Patients with TNBC typically have poorer prognoses than patients with ER+/PR+ and HER2-amplified tumors; the median survival is a mere 13 months for patients with metastatic TNBC.1,3,4 There are no targeted therapies or validated pharmacological targets for TNBC. Therefore, patients receive chemotherapy with cytotoxic and genotoxic agents, which include taxanes, eribulin, anthracyclines, carboplatin, gemcitabine and/or cyclophosphamide.1,3 However, drug-resistance invariably develops leaving few effective treatment options. Patients with ER+/PR+ and/or HER2-amplified breast cancers are now surviving longer as a direct result of the development of targeted therapies that inhibit the molecular drivers of these cancers. However, the highly heterogeneous nature of TNBC, which encompasses diverse cancers with different molecular and phenotypic characteristics, has thus far precluded targeted therapies for these patients.5,6
In 2011, Lehmann and Bauer et al conducted a large meta-analysis of gene expression profiles for TNBC patient tumor samples and identified 6 distinct molecular subtypes of TNBC, each with defined molecular alterations and phenotypic characteristics.7 More recently, Lehmann et al refined these subtypes based on new analyses, identifying four molecular subtypes of TNBC.8 These include two basal-like (BL1 and BL2), a mesenchymal-like (M) and a luminal androgen receptor (LAR) subtype. They also identified cell lines representing these subtypes, thus allowing us to initiate a screening program to identify natural product extracts with selective activities against cell lines representing the different TNBC subtypes. In this study, a crude extract of the Texas native plant Amyris texana was found to have selective activity against MDA-MB-453 cells, a model of the LAR subtype of TNBC. The LAR subtype accounts for approximately 16% of all TNBCs, or roughly 3% of all breast cancers.8 These tumors express high levels of androgen receptors (AR), making it the TNBC subtype with the clearest biomarker. Bioassay-guided fractionation identified one new and one known oxazole with selective activity in this cell line. Based on these data, 32 analogs of the natural products were synthesized and structure-activity relationship studies were performed to identify a lead candidate for in vivo efficacy experiments and investigations into the mechanisms of action for this class of compounds against LAR cells.
RESULTS AND DISCUSSION
Isolation and identification of novel cytotoxic oxazoles from Amyris texana
We initiated a screening program to identify crude extracts of plants and fungal cultures with selective activity against cell lines modeling five molecular subtypes of TNBC. It was hypothesized that based on the different molecular characteristics and drivers of each subtype, extracts with selective activity against particular cell lines could be identified from novel natural product libraries. The plant and fungal extract libraries screened for this project were generated by the Mooberry and Cichewicz laboratories and previously yielded both new and known compounds with selective in vitro activities against different TNBC molecular subtypes in vitro.9,10 The crude supercritical CO2 extract of the leaves and branches of Amyris texana, commonly known as the Texas torchwood, displayed modest selective antiproliferative activity against MDA-MB-453 cells, a model of the LAR subtype, relative to cells representing other molecular subtypes of TNBC. A second sample of the plant material was collected, extracted, and also found to have selective antiproliferative activity against this cell line (Figure 1A). The crude plant extract was subjected to bioassay-guided fractionation on silica gel VLC and HP20ss columns, followed by C18 preparative HPLC and C18 semi-preparative HPLC. Fractions F2 and F3 showed selective antiproliferative activity against MDA-MB-453 cells relative to HCC70 cells which model the BL2 subtype (Figure 1B, 1C), and were selected for further purifications to identify the active constituents. From these fractions, one new diaryloxazole (1), which we named texazole A, and one known diaryloxazole (2) were purified (Figure 2). Isolation of 2 from Amyris plumieri was first reported by Philip, Burke and Jacobs in 1984, but its cytotoxic activity against cancer cell lines was not investigated.11 The chemical structures of both 1 and 2 were established unequivocally through analysis of the spectroscopic data of both the naturally sourced material (see Supplementary Figures S1, S2 and S3-S8), as well as the spectroscopic data for both 1 and 2 from materials produced through two different and independent synthesis routes.
Figure 1.
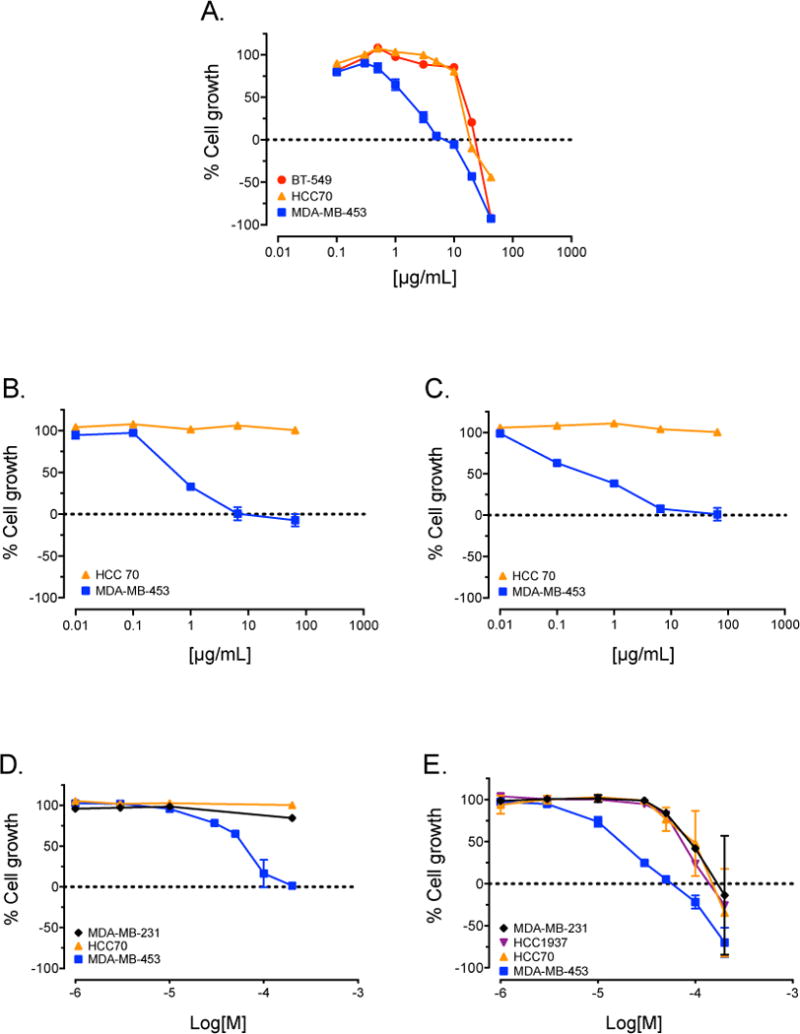
Amyris texana, extract and isolated compounds selectively inhibit growth of LAR cells. SRB assay concentration-response curves for a crude supercritical CO2 extract of Amyris texana (A); fractions F2 (B) and F3 (C); and isolated pure compounds 1 (D) and 2 (E) against TNBC cell lines representing different molecular subtypes.
Figure 2.
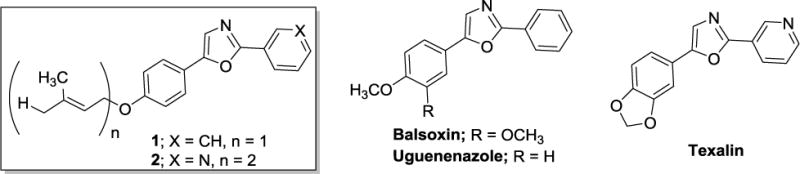
Chemical structures of isolated natural products 1 and 2 and similar diaryloxazoles reported from other plant species.
The structure of compounds 1 and 2 were of interest to us not only because of their selectivity for cells representing the LAR subtype, but also the opportunity they represent as structural leads for further medicinal chemistry investigation based on multiple structural moieties for SAR studies and favorable drug-like physicochemical properties.12 Compounds 1 and 2 are both comprised of an oxazole ring core substituted at the 2- and 5-positions. The phenyl substituent at the 5-position either bears a 4-substituted O-prenyl group (compound 1) or a 4-substituted O-geranyl group (compound 2). The aryl substituent at the 2-position is either phenyl (compound 1) or 3-pyridyl (compound 2). Compounds 1 and 2 are also similar to the diaryloxazoles texalin (Figure 2), previously isolated from Amyris elemifera,13 and balsoxin (Figure 2), isolated from Amyris balsamifera.14,15 Additionally, Cheplogoi and colleagues reported the isolation of uguenenazole (Figure 2) from Vepris uguenensis,16 suggesting similar compounds may be present in multiple plant genera. Although other diaryloxazoles with algicidal and fungicidal activities have been isolated from A. texana,17 to our knowledge there are no reports of these compounds having cytotoxic activity against cancer cells.
Evaluation of in vitro antiproliferative and cytotoxic activities of isolated natural products
The in vitro antiproliferative and cytotoxic activities of the natural products 1 and 2 were evaluated using a sulforhodamine B (SRB) assay.18,19 Both compounds displayed modest selective antiproliferative and cytotoxic activity against MDA-MB-453 cells, a model of the LAR subtype, compared to other TNBC cell lines (Figure 1D, 1E). Since our goal is to identify targeted therapies for the subtypes of TNBC, we sought to identify compounds that have potent activities in a cell line representing one TNBC subtype, as compared to cells representing the other subtypes. The discovery of such natural products is predicated on the hypothesis that these compounds are likely to specifically target a molecular driver of the sensitive cells. The results with 1 and 2 showed that both of these compounds had relatively low cytotoxic potency. The concentration of 1 that inhibited growth by 50% (GI50) was approximately 40 μM in MDA-MB-453 cells, but greater than 200 μM in HCC70 and MDA-MB-231 cells (Table 1). The GI50 of 2 was approximately 17 μM in MDA-MB-453 cells, but ranged from 77 – 100 μM in HCC70, HCC1937, and MDA-MB-231 cells. These results demonstrate that 1 and 2 have at least 5- and 4.5-fold selectivity for MDA-MB-453 cells. However, we expected that more potent analogs would be optimal for in vivo efficacy studies. Additionally, both compounds, but particularly 1, have low aqueous solubility and analogs with improved physicochemical properties will be needed for consideration of this class of compounds for more advanced in vivo profiling.
Table 1.
Antiproliferative potency of natural products and first generation analogs in TNBC cell lines
| GI50 ± SD (μM)
|
|||||
|---|---|---|---|---|---|
| Cmpd | MDA-MB-453 | MDA-MB-231 | HCC1937 | HCC70 | BT-549 |
| Natural 1 | 40 ± 30 | >200 | n.d. | >200 | n.d. |
| Synthetic 1 | 60 ± 40 | n.d. | n.d. | n.d. | n.d. |
| Natural 2 | 17 ± 4 | 92 | 77 | 100 ± 50 | n.d. |
| Synthetic 2 | 11 ± 7 | 29 ± 4 | 13.7 | 16.8 | 27 ± 3 |
| 3 | 13 ± 2 | 19 ± 6 | 8.7 | 11 | 20 ± 8 |
| 4 | 29 ± 9 | n.d. | n.d. | n.d. | n.d. |
| 10 | 10 ± 3 | 13 ± 1 | 16 ± 4 | 19 ± 5 | 20 ± 8 |
| 11 | 8 ± 2 | 30 ± 4 | 35.0 ± 0.4 | 40 ± 10 | 25.3 ± 0.4 |
| 12 | 12 ± 4 | 15 ± 1 | 17 ± 2 | 20 ± 10 | 30 ± 6 |
| 13 | 17 ± 4 | 95 | 66 | 152 | 50 ± 10 |
| 14 | 35 ± 6 | 50 ± 20 | 40 ± 10 | 32 | 52 |
| 15 | >100 | >100 | >100 | >100 | >100 |
| 16 | 40 ± 10 | 53 ± 8 | 41 ± 8 | 42 ± 8 | 43 ± 5 |
| 17 | 2 ± 1 | 9.0 ± 0.6 | 6 ± 2 | 8 ± 2 | 9 ± 1 |
| 18 | 12 ± 6 | 21 ± 3 | 21 ± 3 | 24 ± 2 | 25 ± 3 |
| 19 | 31 ± 6 | 50 ± 30 | 51.2 | 70 ± 30 | 80 ± 50 |
| 20 | >100 | >100 | >100 | >100 | >100 |
Synthesis of natural products 1 and 2 and structural analogs
Having isolated, characterized and screened natural products 1 and 2, we aimed to prepare the two compounds via independent synthesis to confirm structure, purity and the antiproliferative and cytotoxic activities with synthesized samples. To this end, we utilized two different synthesis strategies to prepare 1 and 2, such that the regiochemistry of the diaryloxazole substitution could be established, and two different synthesis routes would be available to prepare analogs for SAR studies. Within our synthesis strategy, we focused on developing a synthesis route that would allow us to easily incorporate structural changes at the 2-position of the oxazole core as well as the 4-phenolic ether side chain moieties to support preliminary SAR studies. In the first route to obtain 1 and 2 (Scheme 1), para-anisaldehyde was subjected to Van Leusen oxazole forming conditions to provide the desired oxazole intermediate 5 in excellent yield. With this common intermediate in hand, two different synthesis methods were employed to access the desired natural products 1 and 2. First, functionalization of the 2-position of the oxazole was carried out using a palladium mediated cross coupling reaction with an aryl bromide in the presence of CuI and K2CO3 under microwave conditions to provide the desired coupled product 6 in low to moderate yields, depending on the aryl bromide substrate employed.20 Multiple coupling conditions were screened for this transformation and we ultimately settled on a palladium-free, copper-mediated cross coupling reaction reported by the Miura group, which proved to be more synthetically useful and reproducible.19 Under these conditions (CuI, PPh3, Na2CO3, DMF), compound 6 could be produced in moderate yield with a wide range of aryl and heteroaryl bromide coupling partners. Thus, the forward synthesis involved treatment of compound 5 with excess BBr3 which cleanly produced the corresponding phenol (not depicted), which was alkylated under standard conditions to produce the geranyl or prenyl derivatives represented by compound 7. Compound 7 was subjected to the CuI/PPh3 coupling conditions with either bromobenzene or 3-bromopyridine to provide compounds 1 and 2. Alternatively, compound 6 could undergo the BBr3 demethylation/alkylation procedure to provide the final compounds 1 and 2 directly. In addition to the two natural products 1 and 2 prepared using these routes, the corresponding structural congeners 3 and 4 were also prepared. Analytical characterization and spectral data of the synthetically-derived samples of compounds 1 and 2 supported the desired structures and were spectroscopically identical to those samples isolated from the natural source (See Supplementary Figures S1-S14). To further confirm the regiochemistry of the oxazole substitution in 6 and develop an alternative route to analogs, we employed a similar route to that described by Copp and co-workers during their reported synthesis of the oxazole texalin (Figure 2).21 Thus, p-methoxyacetophenone was carried through a 3-step sequence involving bromination, azide formation and reduction to form the desired amine 8. Coupling of 8 and benzoic acid under HATU/DIPEA conditions afforded the desired benzamide 9, which was subjected to Ac2O/H2SO4 cyclization conditions to afford oxazole 6. The 1H and 13C NMR, as well as the HPLC/MS data for both samples of 6 from the two different synthetic routes were found to be identical, thus confirming the regiochemistry of oxazole 6 (See Supplementary Figures S15 and S16). Utilizing the synthesis methods described in Scheme 1, compounds 1-4 and additional structural analogs 10-20, highlighted in Chart 1, were also prepared for SAR studies.
Scheme 1. Synthesis of natural products 1 and 2 and analogs 3 and 4.
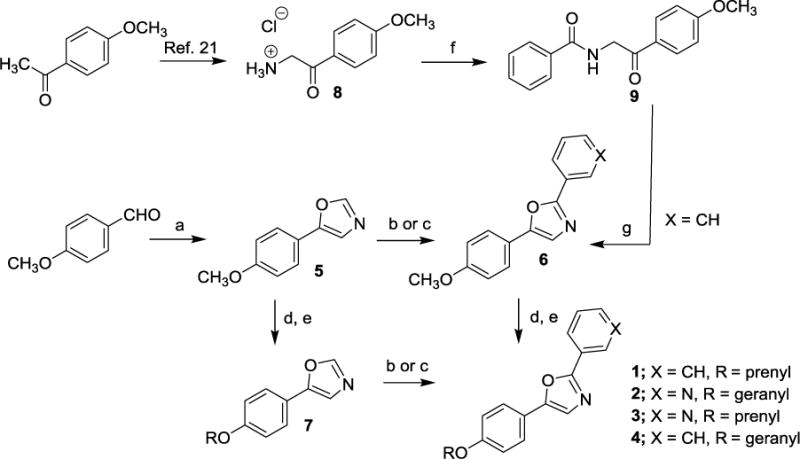
Reagent and conditions: (a) p- Toluenesulfonylmethylisocyanide (1.0 equiv.), K2CO3 (1.7 equiv.), MeOH (0.2M), reflux, 97%; (b) Pd(OAc)2 (0.2 equiv.), Cui (1 equiv.), K2C03 (2 equiv.), Ar-Br (1.2 equiv.), DMF (0.85M), mW, 150 °C, 41-55%; (c) Ar-Br (1.2 equiv.), Cui (1 equiv., PPh3 (1 equiv.), Na2CO3 (2 equiv.), DMF (1.0M), 30-65% yield; (d) BBr3 (6 equiv.), DCM (0.2M), −78 °C to r.t., 96% yield; (e) NaH (1.1 equiv.), R-Br (1.1 equiv.), THF/DMF (0.1M), 0 °C to r.t, 28-63%; (f) benzoic acid, HATU, DIPEA, DMF, 57% yield; (g) Cone. H2SO4, AC2O, 90 °C, 85% yield.
Chart 1.
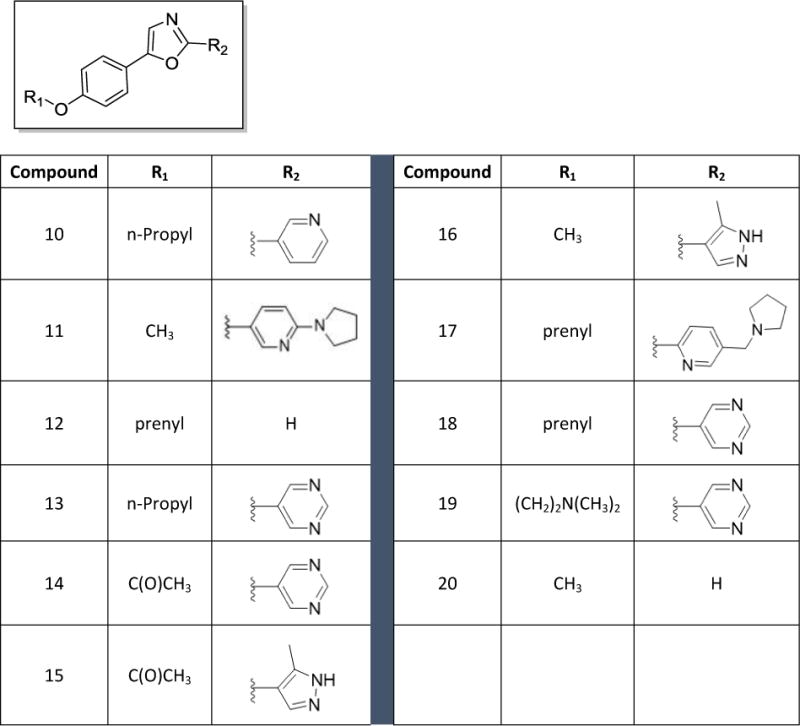
Structures of first generation analogs 10 – 20
In vitro evaluation and SAR
The antiproliferative and cytotoxic activities of compounds 1 - 4 were evaluated in a panel of TNBC cells lines with the SRB assay. All four compounds inhibited growth and were cytotoxic to MDA-MB-453 cells. The GI50 values of synthetically-derived 1 and 2 were 60 and 11 μM, respectively (Table 1). These potencies were comparable to the natural products, further confirming the determined structures. Compounds 1 and 4, which have a phenyl group at the 2-position of the oxazole, were less potent than compounds 2 and 3, which have a 3-pyridine at the 2-position of the oxazole. (Table 1). This suggested that a 3-pyridine group at the 2-position of the oxazole is advantageous for potency in this cell line and also offers physicochemical property advantages over the corresponding phenyl derivatives by providing lower lipophilicity and higher topological polar surface area. Interestingly, while 2 retained the selectivity observed with the natural product, compound 3 did not have the same selectivity for MDA-MB-453 cells as compared to cells representing other TNBC molecular subtypes. This suggested that the longer geranyl side chain, together with the 3-pyridine moiety, are important for LAR subtype specificity, however further SAR studies were required.
Based on the preliminary data obtained from evaluation of the first four oxazoles, SAR studies focused on changing both the heteroaryl group at the 2-position of the oxazole, as well as the structure and electronic nature of the phenol side chain, as represented by analogs 10-20 (Chart 1). The antiproliferative activities of each compound were evaluated and nine of these compounds showed antiproliferative and/or cytotoxic activities at concentrations below 100 μM (Table 1). Compounds 15 and 20 did not have any activity at 100 μM so their biological effects were not evaluated further. The GI50 values for the other nine compounds ranged from 2 – 40 μM in MDA-MB-453 cells (Table 1). First, modification of the phenolic side chain to n-propyl (compound 10) produced an analog with similar potency to the natural product 2. Replacement of the 3-pyridine moiety with other 6-member heteraromatics such as pyrimidine provided potent activity as well, assuming the prenyl phenolic side chain was present (compound 13 and 18). Five-membered heterocyclic analogs such as 1H-pyrrazole showed a marked decrease in potency (compound 16). A phenolic ester functionality such as acetate also decreased potency as represented by compound 14. Installation of a 4-pyrrolidine moiety (compound 11) produced potency in the MDA-MB-453 cell line virtually equivalent to natural product 2; even though compound 11 possesses the methyl group at the R1 position. This result suggests that some structural modifications at both positions are tolerated and can produce analogs with good potency. Further investigation into pyridine substitution patterns and the substituent effects lead to the discovery of compound 17, which was the most potent analog identified during initial SAR studies. Compound 17 showed selectivity for MDA-MB-453 cells, consistent with the activities of the natural products (Figure 3). The activity of this compound also indicated that the relative position of the pyridine nitrogen could be changed while still maintaining the potency and selectivity for MDA-MB-453 cells observed with the natural products. The GI50 for 17 was found to be 2 μM in MDA-MB-453 cells, but ranged from 6 – 9 μM in the other TNBC cells, indicating 3- to 4.5-fold selectivity for MDA-MB-453 cells compared to other TNBC cell lines (Figure 3, Table 1). The structural nature and physicochemical properties of compound 17 were also greatly improved relative to the natural products by lowering LogD, increasing tPSA and providing a tertiary amine handle for salt formation, all of which likely contributed to the improved aqueous solubility observed for 17 in comparison to earlier compounds. These results indicated that analogs with improved pharmacokinetic properties could be synthesized without compromising potency for MDA-MB-453 cells, prompting further efforts into the synthesis of LAR subtype-selective oxazoles with improved pharmaceutical properties. Based on the outcomes of the brief SAR study described in Table 1 and the identification of lead compound 17, we focused our medicinal chemistry efforts to study the impact other structural modifications to the (pyridin-3-yl)methanamine moiety would have on potency, selectivity and physicochemical properties. To this end, we designed a synthesis and SAR study holding the prenyl-phenolic side chain constant, while modifying the pyridyl-methanamine moiety. These analogs were prepared as shown in Scheme 2 starting from 4-hydroxybenzaldehyde. Treatment of 4-hydroxybenzaldehyde with NaH/prenylbromide in CH3CN provided prenyl ether 21, which was reacted under the standard Van Leusen conditions to form oxazole 22 in near quantitative yield. Coupling of 22 with 2- or 3-bromonicotinaldehyde under the CuI/PPh3 conditions described previously produced the desired intermediate represented by 23. Reductive amination22 with an amine (HNR1R2) and NaHB(OAc)3, followed by HCl salt formation afforded the desired analogs 24-34. The analogs 24-34 prepared in this study highlighted in Scheme 2 are a small group of acyclic and cyclic aliphatic amine derivatives that offer changes in tPSA, LogD, hydrogen bond donors (HBD) and acceptors (HBA), while maintaining MW<500. As before, these compounds were evaluated with the SRB assay against the panel of TNBC cell lines representing different molecular subtypes, and all eleven displayed antiproliferative activity and were selective for MDA-MB-453 cells (Table 2). These analogs were substantially more potent overall than our previous compounds, with GI50 values ranging from approximately 0.2 μM to 2 μM in MDA-MB-453 cells. A direct comparison of the 3-pyridine analog 24 and the 2-pyridine analog 26 bearing the piperidine amine suggested that the position of the pyridine-nitrogen atom was not a major factor for potency or selectivity, thus the 2-pyridine template was used for the remaining analogs. Both secondary and tertiary amine derivatives showed potent activity as represented by compounds 25, 27, 31 and 32. Amine groups bearing an additional heteroatom or polar functional groups such as 28, 29, 30, 33 and 34 offered compounds with further improvements in potency and selectivity. While 1,2-diamine type analogs such as 28 and 33 show good potency, compounds bearing an additional carbonyl group such as compound 30 offer improved potency and selectively for MDA-MB-453 cells as compared to the other TNBC cells. Thus, compound 30 showed the greatest combination of potency and selectivity for MDA-MB-453 cells (Figure 4, Table 2). The GI50 of 30 was 0.8 μM in MDA-MB-453 cells and 30 – 50 μM in the TNBC cell lines representing four different molecular subtypes. This indicated that 30 has approximately 37.5- to 62.5-fold antiproliferative selectivity for MDA-MB-453 cells (Figure 4, Table 2). As outlined in our medicinal chemistry strategy, SAR studies leading to the identification of compound 30 provided simultaneous improvements in both potency against MDA-MB-453 cells and physicochemical properties. Interestingly, Compounds 26, 27, 28 and 30 had biphasic concentration-response curves in MDA-MB-453 cells but not in other TNBC cell lines (Figure 4), suggesting these compounds may have multiple mechanisms of action. These differences might explain the improved selectivity for MDA-MB-453 cells.
Figure 3.
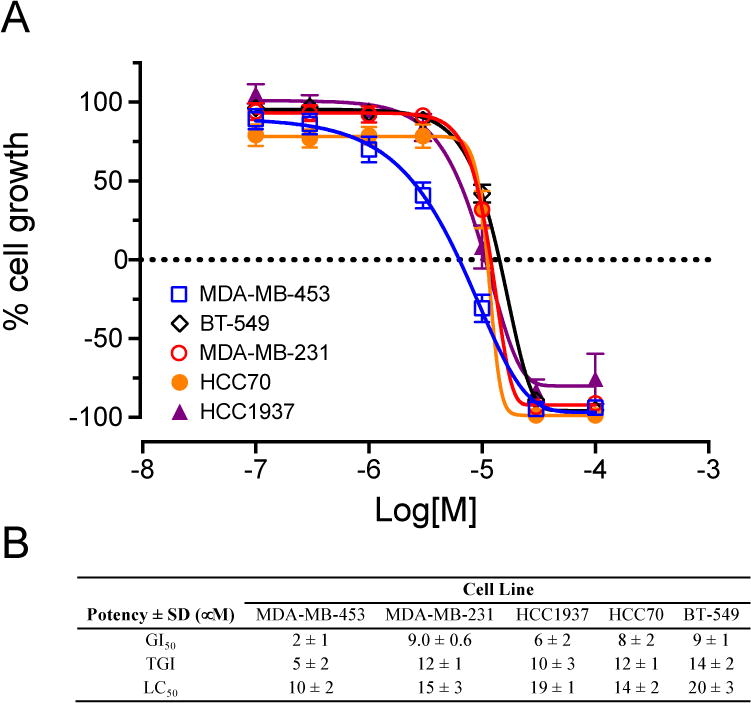
Compound 17 more potently inhibits growth of MDA-MB-453 cells than natural product hits. SRB assay concentration-response curves for 17 (A) and potency measurements (B) in five TNBC cell lines representing different molecular subtypes. Results represent mean ± SE.
Scheme 2. Synthesis and structures of analogs 24-34.
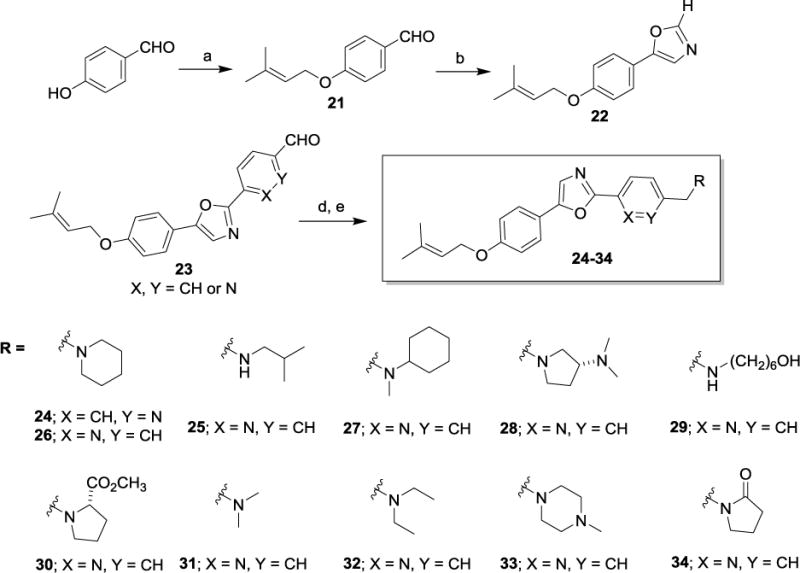
Reagent and conditions: (a) NaH (1.1 equiv.), prenylbromide (1 equiv.), ACN (1.0M), 0 °C, 61% yield; (b) p-Toluenesulfonylmethylisocyanide (1.0 equiv.), K2CO3 (1.7 equiv.), MeOH (0.2M), reflux, 98%; (c) 2-bromonicotinaldehyde or 3- bromonicotinaldehyde, Cui (1 equiv.), PPh3 (1 equiv.), Na2CO3 (2 equiv.), DMF (1.0M), 160 °C, 47% yield; (d) HNR, R2 (1.1 equiv.), NaHB(OAc)3, DCE, r.t., 21-71% yield; (e) HC1 (1.0 equiv.), ether (1.0M), r.t.
Table 2.
Antiproliferative potency of second generation analogs in TNBC cell lines
| GI50 ± SD (μM)
|
|||||||
|---|---|---|---|---|---|---|---|
| Cmpd | MDA-MB-453 | SUM-185PE | MDA-MB-231 | HCC1937 | HCC1806 | BT-549 | A10 |
| 24 | 2 ± 2 | 3 ± 2 | 18 ± 3 | 7 ± 1 | 16 ± 7 | 20 ± 10 | 10 ± 1 |
| 25 | 0.40 | n.d. | 2.0 | n.d. | 1.9 | 1.6 | n.d |
| 26 | 2 ± 1 | 2.6 ± 0.5 | 16 ± 3 | 10 ± 1 | 17 ± 5 | 20 ± 10 | 8.5 ± 0.6 |
| 27 | 0.8 ± 0.3 | 2.1 ± 0.9 | 27 ± 5 | 11 ± 2 | 22 ± 9 | 23 ± 8 | 19 ± 6 |
| 28 | 0.2 ± 0.3 | 1 ± 1 | 6 ± 3 | 3.6 ± 0.2 | 5 ± 2 | 6 ± 4 | 3.3 ± 0.1 |
| 29 | 0.28 | n.d. | 1.5 | n.d. | 0.33 | 1.3 | n.d |
| 30 | 0.8 ± 0.7 | 1.6 ± 0.4 | 50 ± 9 | 35 ± 1 | 30 ± 20 | 40 ± 30 | 35 ± 1 |
| 31 | 1.4 ± 0.9 | 2.8 ± 0.8 | 12 ± 1 | 10 ± 1 | 20 ± 20 | 14 ± 7 | 10.5 ± 0.2 |
| 32 | 1.1 | n.d. | 2.5 | n.d. | n.d | 8.2 | n.d |
| 33 | 0.48 | n.d. | 2.5 | n.d. | 6.0 | 4.2 | n.d |
| 34 | 0.90 | n.d. | 5.4 | n.d. | n.d. | 8.8 | n.d |
Figure 4.
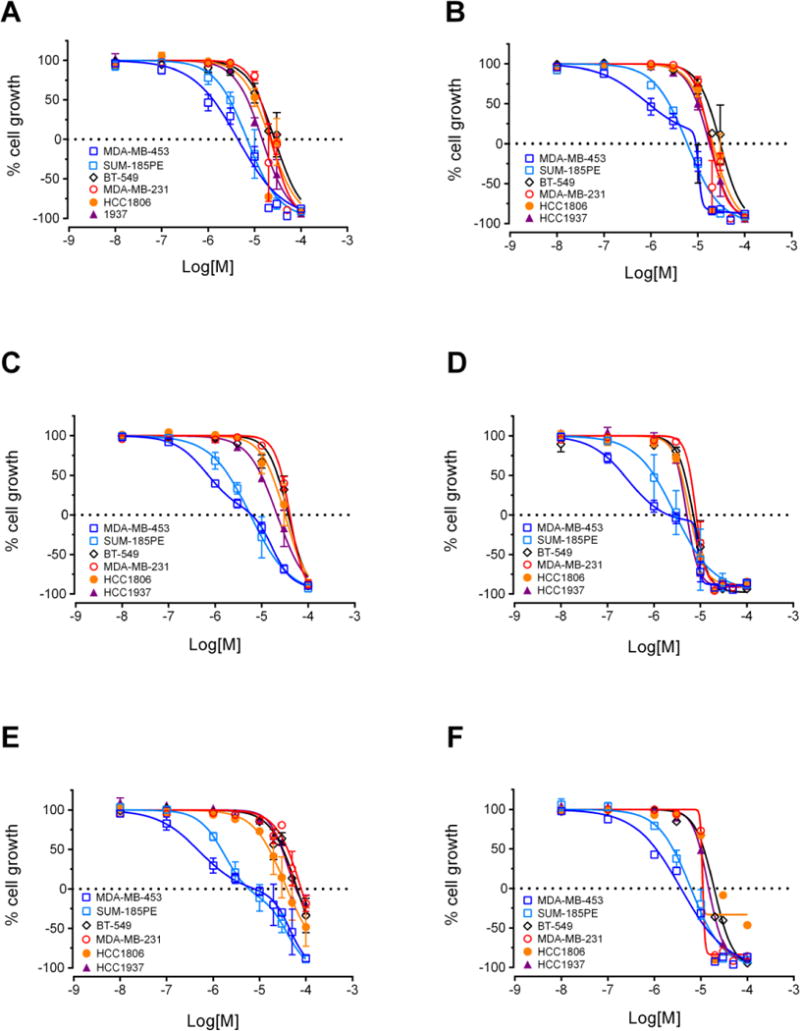
Second generation analogs selectively inhibit growth of LAR cell lines MDA-MB-453 and SUM-185PE. SRB assay concentration-response curves for 24 (A), 26 (B), 27 (C), 28 (D), 30 (E) and 31 (F) in six TNBC cell lines representing five different molecular subtypes. Results represent mean ± SE.
To further evaluate the selectivity of this second series of compounds for LAR models, the six compounds with the greatest selectivity for MDA-MB-453 cells were evaluated against a second LAR cell line, SUM-185PE. Compounds 24, 26, 27, 28, 30 and 31 potently inhibited growth of this cell line as well, with compound 30 again demonstrating the best combination of potency and selective activity in this cell line compared to cells modeling other TNBC molecular subtypes (Figure 4, Table 2). The GI50 of 30 was 1.6 μM in SUM-185PE cells, indicating this compound has 18.8 to 31.3-fold selectivity against these cells as compared to cells representing other TNBC subtypes (Table 2). The other five compounds had GI50 values ranging from 1 to 3 μM in SUM-185PE cells. However, these compounds did not show the same degree of selectivity for both LAR cell lines as compound 30. Compounds 24, 26, 27, 28, 30 and 31 were also tested against the A-10 rat smooth muscle cell line to measure their selectivity compared to non-cancerous cells. The GI50 values in A-10 cells were comparable to those calculated for non-LAR cells, further demonstrating their selectivity.
Table 3 captures the key physicochemical property data on lead compounds 17 and 30 in comparison to the original natural products 1 and 2. Decreases in lipophilicity (LogD) and increases to tPSA were realized by the introduction of more polar functional groups, structural modifications which simultaneously improved MDA-MB-453 cell potency >100-fold compared to our starting natural products, and generated increased selectivity for LAR cells over other TNBC cells. These results, taken together, suggested that 30 is a suitable candidate for future in vivo efficacy studies against LAR xenograft models and investigations into this class of compounds’ mechanisms of action.
Table 3.
Calculated physicochemical properties of selected compounds
| Cmpd | MW | LogP | LogD | tPSA | HBD | HBA |
|---|---|---|---|---|---|---|
| 1 | 305.4 | 4.71 | 4.71 | 35.3 | 0 | 2 |
| 2 | 374.5 | 5.15 | 5.15 | 48.15 | 0 | 3 |
| 17 | 389.5 | 4.23 | 3.02 | 51.39 | 0 | 4 |
| 30 | 447.5 | 4.06 | 4.03 | 77.69 | 0 | 5 |
Mechanism of Action Studies
Prior to evaluation of our lead molecule against an in vivo LAR model, we sought to gain insight into the potential mechanisms of action of these compounds and why they have selectivity for LAR cells and potentially identify a molecular targets, which might have therapeutic value for the LAR subtype of TNBC. Cell cycle analyses were performed to determine if these compounds produced changes in cell cycle distribution characteristic of other anticancer agents and to give an initial indication of their mechanism of action. Compounds 1, 2, 17, 27 and 30, representing various potencies and selectivity for MDA-MB-453 cells, were evaluated for their effects on cell cycle distribution of MDA-MB-453 cells after 18 h of treatment. A range of concentrations spanning the GI50 to LC50 values for each compound were tested, but no significant changes in cell cycle distribution were observed for any of the compounds (data not shown). Accumulation of a sub-G1 cell population was observed with compounds 17, 27 and 30, consistent with the cytotoxicity of these compounds. Next the effects of compound 17 were evaluated using a fluorescence-based intracellular signaling array (data not shown). These data suggested that 17 affected PI3K/Akt/mTORC1 signaling in MDA-MB-453 cells, so further studies were conducted to evaluate the effects of the compounds on this signaling pathway.
The mechanisms of action of 30 were investigated because it shows the highest degree of selectivity for MDA-MB-453 cells. Interestingly, Lehmann and Bauer et al initially demonstrated that LAR cells are not only sensitive to antiandrogens, but are also more sensitive to PI3K inhibitors than cells representing other TNBC subtypes in vitro and in vivo.23 Additionally, we previously reported that LAR cells are particularly sensitive to inhibition of mTORC1 signaling.10 Based on this knowledge and our preliminary mechanistic data, we hypothesized that the highly selective activity of 30 against LAR cells results from inhibition of mTORC1 signaling. An immunoblotting time-course experiment was conducted to measure the activation of Akt, an upstream mediator of mTORC1 signaling, after treatment with 30 (Figure 5). Akt is phosphorylated at T308 by PDK1, which is phosphorylated by PI3Ks. Akt is also phosphorylated at S473 by mTORC2. Treatment of MDA-MB-453 cells with 30 resulted in a nearly two-fold increase in phosphorylation of Akt at T308, which was noticeable 1 – 6 h after treatment. No significant differences in Akt phosphorylation at S473 were observed relative to vehicle-treated cells over the same time-course. These data suggest that 30 caused an increase in upstream signaling from PI3K/PDK1 to Akt. Signaling downstream of Akt was then evaluated to investigate the effects of 30 on the entire PI3K/PDK1/Akt/mTORC1 signaling pathway in MDA-MB-453 cells. At one hour post-treatment, we observed an approximate 49% decrease in the relative phosphorylation of mTOR at S2448 in compound 30-treated cells compared to vehicle-treated controls. This effect was transient and phosphorylation at S2448 increased over the next 5 h and by 6 h post-treatment the relative phosphorylation at S2448 was approximately 60% higher than in vehicle-treated controls. mTOR is phosphorylated at S2448 via PI3K/Akt signaling, so these data suggest that 30 causes an initial inhibition of mTORC1 activation. We then investigated the activity of signaling proteins downstream of mTORC1. Interestingly, treatment with 30 resulted in complete loss of S6 kinase (S6K) phosphorylation at T389, as well as loss of S6 phosphorylation at S235/S236. Collectively, these results suggested that 30 inhibits downstream signaling through mTORC1. Although the precise point of inhibition could not be established from these results, it is likely the observed increase in phosphorylation of Akt at T308 is a result of compensatory upstream activation of PI3K-mediated signaling, which can occur as a result of mTORC1 inhibition.24
Figure 5.
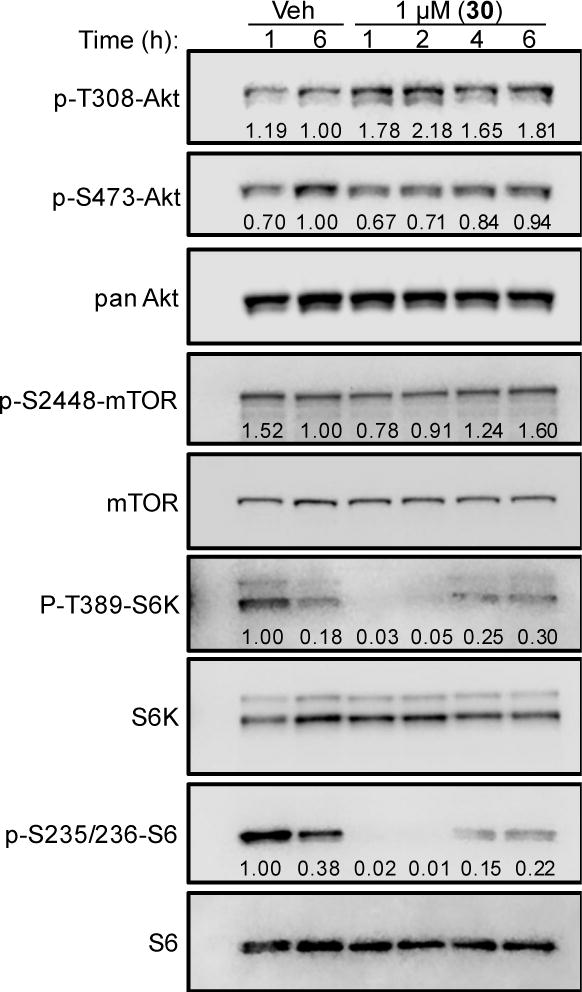
Compound 30 rapidly decreases abundance of activated mTORC1 signaling proteins in LAR cells. Immunoblotting of compound 30-treated MDA-MB-453 whole-cell lysates for phosphorylated and total PI3K/Akt/mTORC1 signaling proteins. Cells were treated with vehicle (DMSO) or 1 μM 30 for 1 – 6 h, as indicated. Quantification of phosphorylated protein relative to total protein is indicated below each band.
CONCLUSION
Evidence suggests that the molecular characteristics of the LAR subtype are significantly different than those of other TNBC subtypes and these tumors respond poorly to current chemotherapeutic agents. Masuda et al found that patients with the LAR subtype had one of the poorest rates of pathological complete response (10%) to neoadjuvant chemotherapy with taxanes and anthracyclines, suggesting that this subtype may be inherently resistant to these agents.25 These findings highlight the need for the development of better, more rationally targeted therapies for AR-expressing breast cancers.
Our results, obtained by initially evaluating extracts of Texas-native plants for selective activity against TNBC molecular subtypes, identified a new class of synthetically-tractable, potent molecules with selective effects against cells modeling the LAR subtype of TNBC. Although the natural products that we initially procured had low potency and only moderate selectivity, SAR studies and medicinal chemistry efforts led to the identification of substantially more potent and selective analogs with improved physicochemical properties. Many of the compounds described herein are new chemical structures that may be useful scaffolds for developing additional compounds with efficacy against AR-expressing breast cancer cells, or form the basis for the development of new chemical tools to be used for mechanism of action studies. Additionally, these studies identified lead molecules such as compound 30 with more favorable drug-like physicochemical properties, which can be used to evaluate the potential in vivo antitumor efficacy of this class of molecules in AR-expressing TNBC models.
EXPERIMENTAL SECTION
Materials and Instrumentation
All compounds for cell treatments were dissolved in DMSO (Sigma-Aldrich) and stored at -20°C. Absorbance (A560) of SRB solutions was measured with a Spectramax 384 Plus spectrophotometer (Molecular Devices). NMR spectra were collected on a Varian 400 MHz NMR spectrometer. LCESIMS data were obtained on a Shimadzu LC-MS 2020 system (ESI quadrupole) coupled to a photodiode array detector, with a Phenomenex Kintex column (2.6 μm C18 column, 100 Å, 75 × 3.0 mm). The preparative HPLC system utilized SCL-10A VP pumps and system controller with Luna 5 μm C18 columns (110 Å, 250 × 21.2 mm, 10 mL/min). The analytical and semi-preparative HPLC system utilized 1525 binary pumps (Waters) with 2998 photodiode array detectors (Waters), and Luna 5 μm C18 columns (110 Å, 250 × 4.6 mm, 1 mL/min and 110 Å, 250 × 10 mm, 4 mL/min). All solvents were of ACS grade or better.
Compound Isolation
A supercritical CO2 extract (5.0 g) was obtained from approximately 106 g of lyophilized Amyris texana (collected at San Antonio Botanical Gardens). A 5 mg sample of HP20ss and C18 processed crude extract was applied to a HPLC-based microtiter plate fractionation process, as previously described.26 Fractions F2 and F3 were identified as having selective activity and became our target fractions. To generate greater quantities of the compounds found in these fractions, the remaining crude extract was fractionated over silica gel and eluted with hexane-CH2Cl2-MeOH (hexane, hexane-CH2Cl2 50:50, CH2Cl2, CH2Cl2-MeOH 90:10, MeOH). Fractions F2 and F3 contained the targeted chemical signatures (mass spectrometric and UV profiles from LCMS) and these fractions were combined and applied to an HP20ss column, and eluted with MeOH-H2O. The fifth subfraction (100% MeOH) was further separated over C18 preparative HPLC (gradient elution with MeOH-H2O from 30:70 to 100% organic phase in 30 min.), followed by C18 semi-preparative HPLC (MeOH-H2O 90:10) to obtain 1 (4.0 mg) and 2 (10.0 mg), which corresponded to the target peaks from fraction F2. Balsoxin,14,15 O-isopentenyl-halfordinol,27 Texamine,28 Uguenenazole,16 and 3-[5-(3,4-dimethoxyphenyl)-2-oxazolyl]-Pyridine,29 which displayed similar UV profiles were purified from the fourth subfraction (eluted with MeOH-H2O 90:10) of HP20ss column via C18 preparative HPLC (gradient elution with MeOH-H2O from 30:70 to 100% organic phase in 30 min) followed by C18 semi-preparative HPLC (MeCN-H2O 75:25). These compounds were obtained from fractions that did not demonstrate selective activity against MDA-MB-453 cells and their cytotoxic activities were not determined.
5-(4-((3-methylbut-2-en-1-yl)oxy)phenyl)-2-phenyloxazole (1)
White amorphous powder; UV (from HPLC PDA detector) λmax 200, 314 nm; 1H NMR (CDCl3, 400 MHz): 8.02 (2H, dd, 7.9, 1.7), 7.64 (2H, brd, 8.7), 7.48 (3H, m), 7.32 (1H, s), 6.98 (2H, brd, 8.7), 5.51 (1H, t, 6.7), 4.56 (2H, d, 6.7), 1.81 (3H, s), 1.77 (3H, s); 13C NMR (CDCl3, 100 MHz): 160.9, 159.1, 151.4, 138.6, 130.1, 128.8 (×2), 127.6, 126.1 (×2), 125.7 (×2), 121.9, 120.8, 119.3, 115.1 (×2), 64.9, 25.8, 18.2; ESIMS [M + H]+ m/z 306.
Chemistry Experimentals
General procedures
Unless otherwise indicated all reactions were conducted in standard commercially available glassware using standard synthetic chemistry methods and setup. All air- and moisture-sensitive reactions were performed under nitrogen atmosphere with dried solvents and glassware under anhydrous conditions. Starting materials and reagents were commercial compounds of the highest purity available and were used without purification. Solvents used for reactions were indicated as of commercial dry or extra-dry or analytical grade. Analytical thin layer chromatography was performed on aluminium plates coated with Merck Kieselgel 60F254 and visualized by UV irradiation (254 nm) or by staining with a solution of potassium permanganate. Flash column chromatography was performed on Biotage Isolera One 2.2 using commercial columns that were pre-packed with Merck Kieselgel 60 (230– 400 mesh) silica gel. Final compounds for biological testing are all ≥96% purity as determined by HPLC-MS and 1H NMR. 1H NMR experiments were recorded on Agilent DD2 400MHz spectrometers at ambient temperature. Samples were dissolved and prepared in deuterated solvents (CDCl3, CD3OD and DMSOd6) with TMS used as the internal standard in all cases. All deuterated solvent peaks were corrected to the standard chemical shifts (CDCl3, dH = 7.26 ppm; CD3OD, dH = 3.31 ppm; DMSO-d6, dH = 2.50 ppm). Spectra were all manually integrated after automatic baseline correction. Chemical shifts (d) are given in parts per million (ppm), and coupling constants (J) are given in Hertz (Hz). The proton spectra are reported as follows: d (multiplicity, coupling constant J, number of protons). The following abbreviations were used to explain the multiplicities: app = apparent, b = broad, d = doublet, dd = doublet of doublets, ddd = doublet of doublet of doublets, dddd = doublet of doublet of doublet of doublets, m = multiplet, s = singlet, t = triplet. All samples were analyzed on Agilent 1290 series HPLC system comprised of binary pumps, degasser and UV detector, equipped with an auto-sampler that is coupled with Agilent 6150 mass spectrometer. Purity was determined via UV detection with a bandwidth of 170nm in the range from 230-400nm. The general LC parameters were as follows: Column - Zorbax Eclipse Plus C18, size 2.1 × 50 mm; Solvent A: 0.10 % formic acid in water, Solvent B: 0.00 % formic acid in CH3CN; Flow rate – 0.7 mL/min; Gradient: 5 % B to 95 % B in 5 min and hold at 95 % B for 2 min; UV detector – channel 1 = 254 nm, channel 2 = 254 nm. Mass detector Agilent Jet Stream – Electron Ionization (AJS-ES).
General Procedure for Pd/Cu Mediated Cross Coupling
In a dried microwave vial equipped with a stir bar, oxazole (5.70 mmol), aryl bromide (11.25 mmol), anhydrous potassium carbonate (1.58 g, 11.4 mmol), copper (I) iodide (1.08 g, 5.70 mmol), and palladium (II) acetate (65 mg, 0.29 mmol) were dissolved in DMF (7 mL). The vial was purged with N2 and the vial heated to 150 °C for 13 minutes under microwave irradiation. The crude reaction was then filtered over a pad of celete, pushing with DCM and concentrated under reduced pressure. The crude oil was then diluted with EtOAc (100 mL) and extracted with water (70 mL × 2) and brine (70 mL × 3). The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude mixtures were then purified on a silica gel column, eluting with EtOAc/Hexanes.
General Phenol Alkylation Procedure
In a dried round bottom flask equipped with a stir bar, sodium hydride (1.50 mmol., 60% dispersion in mineral oil) was added and the atmosphere exchanged for N2. The sodium hydride is then washed with Hexanes (3 mL) before the addition of THF (3 mL) and cooled to 0 °C. In a separate vial, the phenol (1.0 mmol) was dissolved in DMF (2 mL) and added to the sodium hydride solution mixture slowly. The reaction was allowed to stir for 5 minutes before the slow addition of the alkyl bromide (1.1 mmol). Once the reaction was deemed complete by TLC, the reaction was quenched with saturated aqueous sodium bicarbonate (5 mL), diluted with EtOAc (50 mL) and washed with water (30 mL) and brine (30 mL × 3). The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude mixture was then purified on a silica column, eluting with EtOAc/Hexanes.
5-(4-Propoxyphenyl)oxazole
Brown solid (155 mg, 31% yield), LC-MS m/z 204.2 (APCI+) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 7.87 (s, 1H), 7.57 (d, J = 9.2 Hz, 2H), 7.22 (s, 1H), 6.94 (d, J = 9.2 Hz, 2H), 3.96 (t, J = 6.4 Hz, 2H), 1.83 (sxt. J = 6.8 Hz, 2H), 1.05 (t, J = 7.2 Hz, 3H).
General Procedure for Phenol Deprotection
In a dry round bottom flask equipped with a stir bar, the methoxy phenol (2.0 mmol) is dissolved in DCM (20 mL). The solution was then cooled to −78 °C and the headspace exchanged for N2. Boron tribromide (12 mL, 12.0 mmol, 1.0 M in DCM) was then added slowly. The reaction was then allowed to reach RT over 18 hours before cooling to −78 °C and slowly quenching with methanol (30 mL). The crude reaction mixture was then warmed to RT and concentrated under reduced pressure. The crude mixture was then dissolved EtOAc (300 mL) and extracted with brine (200 mL × 3). The organic layer was then dried over sodium sulfate, filtered and concentrated under reduced pressure.
4-(2-Phenyloxazol-5-yl)phenol
Tan solid (423 mg, 91% yield), LC-MS m/z 328.1 (APCI+) 1H NMR (500 MHz, d6-DMSO): δ 9.89 (s, 1H), 8.08 (d, J = 6.3 Hz, 2H), 7.69 (d, J = 7.8 Hz, 2H), 7.62 (s, 1H), 7.56-7.54 (m, 3H), 6.93 (d, J = 7.8 Hz, 2H).
4-(Oxazol-5-yl)phenol
Brown solid (777 mg, 96% yield), LC-MS m/z 162.1 (APCI+) (M+H)+. 1H NMR (500 MHz, d6-DMSO): δ 9.82 (s, 1H), 8.34 (s, 1H), 7.55 (d, J = 7.3 Hz, 2H), 7.46 (s, 1H), 6.88 (d, J = 6.9, 2 H).
4-(2-(Pyrimidin-5-yl)oxazol-5-yl)phenol
Light yellow solid (42 mg, 95% yield), LC-MS m/z 240.1 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 9.40 (s, 2H), 9.24 (s, 1H), 7.68 (d, J = 9.0 Hz, 2H), 7.54 (s, 1H), 6.90 (d, J = 8.0 Hz, 2H).
5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)-2-phenyloxazole (1)
White Solid (296 mg, 81% yield), LC-MS m/z 306.2 (APCI+) (M+H)+. 1H NMR (500 MHz, CDCl3): δ 8.07 (d, J = 7.4 Hz, 2H), 7.60 (d, J = 8.3 Hz, 2H), 7.45-7.38 (m, 3H), 7.29 (s, 1H), 6.95 (d, J = 6.3 Hz, 2H), 5.48 (t, J = 6.4 Hz, 1H), 4.51 (d, J = 6.9 Hz, 2H), 1.78 (s, 3H), 1.73 (s, 3H). 13C (500 MHz, CDCl3): δ 160.53, 159.18, 151.42, 138.46, 130.10, 128.81, 127.68, 126.16, 125.73, 121.97, 120.80, 119.48, 115.16, 64.93, 25.88, 18.28. HRMS (ESI+) Calculated m/z 306.1489, Found 306.1502 (M+H)+.
(E)-5-(4-[(3,7-Dimethylocta-2,6-dien-1-yl)oxy]phenyl)-2-(pyridin-3-yl)oxazole (2)
Yellow oil (95 mg, 20%), LC-MS m/z 375.3 (APCI+) (M+H)+. 1H NMR (500 MHz, CDCl3): δ 9.31 (s, 1H), 8.66 (d, J = 4.4 Hz, 1H), 8.32 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 8.8 Hz, 2H), 7.40 (dd, J = 6.8 Hz, 4.8 Hz, 1 H), 7.34 (s, 1H), 6.98 (d, J = 8.8 Hz, 2H), 5.49 (t, J = 6.3 Hz, 1 H), 5.09 (t, J = 5.8 Hz, 1H), 4.58 (d, J = 6.3 Hz, 2H), 2.09-2.14 (m, 4H), 1.75 (s, 3H), 1.67 (s, 3H), 1.60 (s, 3H). 13C (500 MHz, CDCl3): δ 159.60, 158.24, 152.34, 150.86, 147.58, 141.79, 133.34, 132.03, 126.02, 123.99, 123.89, 123.73, 122.25, 120.42, 119.28, 115.42, 65.20, 39.70, 26.44, 25.84, 17.87, 16.87. HRMS (ESI+) Calculated m/z 375.2067, Found 375.2062 (M+H)+.
5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)-2-(pyridin-3-yl)oxazole (3)
Pale yellow solid (82 mg, 40%), LC-MS m/z 307.2 (APCI+) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 9.32 (s, 1H), 8.68 (d, J = 3.9 Hz, 1H), 8.35 (dt, J = 8.2 Hz, 2.0 Hz, 1H), 7.65 (d, J = 9.0 Hz, 2H), 7.42 (dd, J = 7.4 Hz, 4.7 Hz, 1H), 7.36 (s, 1H), 6.99 (d, J = 9.0 Hz, 2H), 5.51 (t, J = 5.1 Hz, 1H), 5.50 (d, J = 1.6 Hz, 2H), 1.82 (s, 3H), 1.77 (s, 3H). 13C NMR (400 MHz, CDCl3): δ 159.60, 158.28, 152.34, 150.90, 147.62, 138.81, 133.35, 126.05, 124.01, 123.73, 122.27, 120.46, 119.45, 65.11, 26.00, 18.41. HRMS (ESI+) Calculated m/z 307.1441, Found 307.1442 (M+H)+.
(E)-5-(4-[(3,7-Dimethylocta-2,6-dien-1-yl)oxy]phenyl)-2-phenyloxazole (4)
White solid (152 mg, 45% yield), LC-MS m/z 374.2 (APCI+) (M+H)+. 1H NMR (500 MHz, CDCl3): δ 8.09 (d, J = 7.8 Hz, 2H), 7.65 (d, J = 8.3 Hz, 2H), 7.49-7.43 (m, 3H), 7.32 (s, 1h), 6.99 (d, J = 8.8 Hz, 2H), 5.50 (t, J = 6.4 Hz, 1H), 5.10 (t, J = 6.1 Hz, 1H), 4.59 (d, J = 6.3 Hz, 2H), 2.17-2.10 (m, 4H), 1.76 (s, 3H), 1.68 (s, 3H), 1.61 (s, 3H). 13C (500 MHz, CDCl3): δ 160.73, 159.35, 151.59, 141.77, 132.07, 130.27, 128. 97, 127.80, 127.80, 126.32, 125.90, 123.93, 122.10, 120.96, 119.38, 115.38, 65.21, 39.74, 26.48, 25.88, 17.90, 16.90. HRMS (ESI+) Calculated m/z 374.2115, Found 374.2121 (M+H)+.
General Procedure for the Van Leusen Oxazole Synthesis
5-(4-Methoxyphenyl)oxazole (5)
In a round bottom flask equipped with a stir bar, p-toluenesulfonylmethyl isocyanide (10.04 g, 51.41 mmol), anhydrous potassium carbonate (7.00 g, 51.4 mmol), and p-anisaldehyde (6.25 mL, 84.8 mmol) were dissolved in MeOH (257 mL). The solution was then heated to reflux, monitoring by TLC. Once complete, the reaction was cooled to room temperature and concentrated under reduced pressure. The crude mixture was then dissolved in EtOAc (200 mL) and washed with water (100 mL × 3). The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure to yield 5-(4-Methoxyphenyl)oxazole as a light yellow solid (8.7 g, 97% yield). LC-MS: 176.1 m/z (APCI) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 7.87 (s, 1H), 7.59 (d, J = 9.0 Hz, 2H), 7.23 (s, 1H), 6.96 (d, J = 8.6 Hz, 2H), 3.85 (s, 3H).
[Coupling route] 5-(4-Methoxyphenyl)-2-phenyloxazole (6)
Yellow solid (580 mg, 41% yield), LC-MS m/z 252.2 (APCI) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 8.09 (d, J = 7.8 Hz, 2H), 7.64 (d, J = 8.0 Hz, 2H), 7.41-7.50 (m, 3H), 7.31 (s, 1H), 6.97 (d, J = 8.0 Hz, 2H), 3.84 (s, 3H).
[Cyclization route] 5-(4-methoxyphenyl)-2-phenyloxazole (6)
In a round bottom flask equipped with a stir bar, N-(2-(4-methoxyphenyl)-2-oxoethyl)benzamide 9 was dissolved in acetic anhydride (1 mL) followed by dropwise addition of conc. H2SO4 (10 μL, 0.18 mmol). The reaction was heated to 90°C and monitored by TLC. Upon completion, the reaction was cooled to RT and diluted with 50 mL water and 25 mL DCM. The organic layer was then washed with 25 mL sat. sodium bicarbonate (x1), brine (x3), and then dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude mixture was purified on a silica gel column, eluting with EtOAc/hexanes to yield 5-(4-methoxyphenyl)-2-phenyloxazole as an off-yellow solid (31.5 mg, 85% yield). LC-MS m/z 252.2 (APCI) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 8.09 (d, J = 7.8 Hz, 2H), 7.64 (d, J = 8.0 Hz, 2H), 7.41-7.50 (m, 3H), 7.31 (s, 1H), 6.97 (d, J = 8.0 Hz, 2H), 3.84 (s, 3H).
N-(2-(4-methoxyphenyl)-2-oxoethyl)benzamide (9)
In a round bottom flask equipped with a stir bar, benzoic acid (101.5 mg, 0.83 mmol), and HATU (381.5 mg, 1 mmol) were dissolved in DMF and allowed to stir for ten minutes before slow addition of 2-amino-4’-methoxyacetophenone HCl 8 (177.8 mg, 0.91 mmol). Once homogenous, DIPEA (0.36 mL, 2 mmol) was added dropwise to the solution. The reaction was monitored by TLC and upon completion, taken into 20 mL EtOAc and washed with 50 mL water (x2) and brine (x3). The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude mixture was purified on a silica gel column, eluting with EtOAc/hexanes to yield N-(2-(4-methoxyphenyl)-2-oxoethyl)benzamide as a white solid (139.1 mg, 57% yield). LC-MS m/z 270.1 (APCI+) (M+H)+. 1H NMR (400 MHz, Methanol-d4) δ 8.05 (dd, 2H), 7.90 (dd, J = 8.3, 1.3 Hz, 2H), 7.59 – 7.45 (m, 3H), 7.06 (dd, 2H), 3.89 (s, 3H), 3.31 (s, 2H).
5-(4-Propoxyphenyl)-2-(pyridin-3-yl)oxazole (10)
Yellow solid (69 mg, 30% yield), LC-MS m/z 281.2 (APCI+) (M+H)+. 1H NMR (500 MHz, CDCl3): δ 9.32 (s, 1H), 8.67 (d, J = 3.4 Hz, 1H), 8.34 (d, J = 7.8 Hz, 1H), 7.64 (d, J = 8.8 Hz, 2H), 7.41 (dd, J = 7.8 Hz, 5.1 Hz, 1H), 7.35 (s, 1H), 6.97 (d, J = 8.8 Hz, 2H), 3.97 (t, J = 6.8 Hz, 2H), 1.84 (sxt, J = 7.3 Hz, 2H), 1.06 (t, J = 7.8 Hz, 3H). HRMS (ESI+) Calculated m/z 281.1285, Found 281.1293 (M+H)+.
5-(4-Methoxyphenyl)-2-(6-(pyrrolidin-1-yl)pyridin-3-yl)oxazole (11)
Yellow solid (148 mg, 70% yield). LC-MS m/z 322.2 (APCI+) (M+H)+. 1H NMR (500 MHz, CDCl3): δ 8.87 (s, 1H), 8.05 (dd, J = 8.8 Hz, 1.5 Hz, 1H), 7.61 (d, J = 8.3 Hz, 2 H), 7.25 (s, 1 H), 6.95 (d, J = 8.3 Hz, 2H), 6.41 (d, J = 8.8 Hz, 2H), 3.84 (s, 3H), 3.53 (broad s, 4H), 2.02 (broad s, 4H). HRMS (ESI+) Calculated m/z 322.1550, Found 322.1551 (M+H)+.
(E)-5-(4-[(3,7-dimethylocta-2,6-dien-1-yl)oxy]phenyl)oxazole (12)
Yellow solid (259 mg, 28% yield), LC-MS m/z 298.2 (APCI+) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 7.78 (s, 1H), 7.77 (d, J = 8.7 Hz, 2H), 7.23 (s, 1H), 6.96 (d, J = 9.0 Hz, 2H), 5.49 (t, J = 5.4 Hz, 1H), 5.09 (t, J = 5.1 Hz, 1H), 4.57 (d, J = 6.3 Hz, 2H), 2.17-2.07 (m, 4H), 1.75 (s, 3H), 1.68 (s, 3H), 1.60 (s, 3H). HRMS (ESI+) Calculated m/z 282.0873, Found 282.0874 (M+H)+.
5-(4-Propoxyphenyl)-2-(pyrimidin-5-yl)oxazole (13)
Yellow solid (6 mg, 15% yield), LC-MS m/z 282.2 (APCI+) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 9.40 (s, 2H), 9.28 (s, 1H), 7.65 (d, J = 9.0 Hz, 2H), 7.39 (s, 1H), 6.99 (d, J = 8.6 Hz, 2H), 3.98 (t, J = 6.7 Hz, 2H), 1.84 (sxt, J = 6.7 Hz, 2H), 1.07 (t, J = 7.4 Hz, 3H). HRMS (ESI+) Calculated m/z 282.1237, Found 282.1243 (M+H)+.
4-(2-(Pyrimidin-5-yl)oxazol-5-yl)phenyl acetate (14)
Pale yellow solid (11 mg, 22 % yield), LC-MS m/z 282.1 (APCI+) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 9.38 (s, 2H), 9.27 (s, 1H), 7.73 (d, J = 9.0 Hz, 2H), 7.48 (s, 1H), 7.20 (d, J = 8.6 Hz, 2H), 2.14 (s, 3H).
4-(2-(5-methyl-1H-pyrazol-4-yl)oxazol-5-yl)phenyl acetate (15)
Pale yellow solid (9 mg, 51% yield). LC-MS m/z 284.1 (APCI+) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 9.2 Hz, 2H), 7.68 (s, 1H), 7.31 (s, 1H), 7.17 (d, J = 8.0 Hz, 2H), 6.24 (s, 1H), 3.49 (s, 3H), 2.67 (s, 3H).
5-(4-Methoxyphenyl)-2-(5-methyl-1H-pyrazol-4-yl)oxazole (16)
Pale yellow solid (8 mg, 20% yield). LC-MS m/z 256.2 (APCI+) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 1.6 Hz, 1H), 7.62 (d, J = 8.8 Hz, 2H), 7.21 (s, 1H), 6.95 (d, J = 9.2 Hz, 2H), 6.23 (s, 1H), 3.85 (s, 3H), 2.66 (s, 3H). HRMS (ESI+) Calculated m/z 256.1081, Found 256.1081 (M+H)+.
5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)-2-(5-(pyrrolidin-1-ylmethyl)pyridin-2-yl)oxazole (17)
Yellow oil (65 mg, 49% yield). LC-MS m/z 390.3 (APCI+) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 8.67 (d, J = 1.9 Hz, 1H), 8.10 (d, J = 8.3 Hz, 1H), 7.82 (dd, J = 8.2 Hz, 1.9 Hz, 1H), 7.71 (d, J = 9.0 Hz, 2H), 7.38 (s, 1H), 6.97 (d, J = 8.6 Hz, 2H), 5.50 (tt, J = 6.6 Hz, 1.1 Hz, 1H), 4.56 (d, J = 6.7 Hz, 2H), 3.73 (s, 1H), 3.70 (s, 1H), 2.54 (t, J = 5.1 Hz, 4H), 1.81 (broad s, 6H), 1.76 (broad s, 4H).
5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)-2-(5-(pyrrolidin-1-ylmethyl)pyridin-2-yl)oxazole hydrochloride (17-HCl salt)
Yellow solid (61 mg). LC-MS m/z 390.3 (APCI+) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 8.84 (s, 1H), 8.29 (d, J = 8.2 Hz, 1H), 8.16 (dd, 8.2 Hz, 2.3 Hz, 1H), 7.80 (d, J = 9.0 Hz, 2H), 7.61 (s, 1H), 7.04 (d, J = 9.0 Hz, 2H), 5.48 (tt, J = 6.6 Hz, 1.6 Hz, 1H), 4.60 (d, J = 6.7 Hz, 2H), 4.55 (s, 2H), 3.58 (broad s, 2H), 3.26 (broad s, 2H), 2.23 (broad s, 2H), 2.05 (broad s, 2H), 1.80 (d, J = 1.2 Hz, 3H), 1.77 (d, J = 0.8 Hz, 3H). 13C (400 MHz, CDCl3): δ 159.92, 158.33, 151.05, 146.38, 139.55, 137.66, 125.92, 121.96, 121.74, 199.68, 119.40, 115.01, 64.57, 54.71, 53.77, 24.41, 22.43, 17.83, 16.78. HRMS (ESI): Calculated: 390.2176 m/z, Found: 390.2193 m/z (M+H)+.
5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)-2-(pyrimidin-5-yl)oxazole (18)
Yellow Solid (6 mg, 13% yield). LC-MS m/z 308.2 (APCI+) (M+H)+. 1H NMR (400 MHz, CDCl3): δ 9.38 (s, 2H), 9.28 (s, 1H), 7.65 (d, J = 9.0 Hz, 2H), 7.39 (s, 1H), 7.00 (d, J = 9.0 Hz, 2H), 5.51 (t, J = 6.6 Hz, 1H), 4.57 (d, J = 7.1 Hz, 2H), 1.82 (s, 3H), 1.77 (s, 3H). HRMS (ESI+) Calculated m/z 308.1394, Found 308.1394 (M+H)+.
N,N-Dimethyl-2-(4-(2-(pyrimidin-5-yl)oxazol-5-yl)phenoxy)ethanamine (19)
Modification to the general procedures as follows. In this reaction 3-chloro-N,N-dimethylpropan-1-amine (36 mg, 0.25 mmol) and sodium iodide (4 mg, 0.03 mmol) was used and the reaction was heated 100 °C After deemed complete by TLC, the reaction was diluted with DCM (5 mL) and extracted with NaOH (1.0 M, 5 mL x2). The organic layer was then extracted with HCl (1.0 M, 5 mL × 2). The combined acidic aqueous extracts were extracted with DCM (5 mL × 2). The combined DCM layers were combined, dried over sodium sulfate, filtered and concentrated under reduced pressure. Amber oil (14 mg, 36% yield), LC-MS m/z 311.2 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 9.42 (s, 2H), 9.27 (s, 1H), 7.79 (d, J = 9.0 Hz, 2H), 7.61 (s, 1H), 7.09 (d, J = 9.0 Hz, 2H), 3.35 (t, J = 1.6 Hz, 2H), 2.84 (t, J = 5.5 Hz, 2H), 2.40 (s, 6H).
N,N-Dimethyl-2-(4-(2-(pyrimidin-5-yl)oxazol-5-yl)phenoxy)ethanamine hydrochloride (19-HCl salt)
Yellow solid (6 mg, Quant.). LC-MS m/z 311.2 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 9.42 (s, 2H), 9.26 (s, 1H), 7.85 (d, J = 9.0 Hz, 2H), 7.64 (s, 1H), 7.18 (d, J = 9.0 Hz, 2H), 4.44 (t, J = 5.1 Hz, 2H), 3.64 (t, J = 5.1 Hz, 2H), 3.01 (s, 6H).
4-[(3-methylbut-2-en-1-yl)oxy]benzaldehyde (21)
Pale yellow oil (475 mg, 61% yield), LC-MS m/z 123.1 (APCI+) (M + H - CH2CHC(CH3)2)+. 1H NMR (400 MHz, CDCl3): δ 9.87 (s, 1H), 7.81 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.3 Hz, 2H), 5.48 (t, J = 6.3 Hz, 1H), 4.58 (d, J = 6.3 Hz, 2H), 1.80 (s, 3H), 1.75 (s, 3H).
5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)oxazole (22)
LC-MS m/z 230.1 m/z (APCI+) (M+H)+. 1H NMR (500 MHz, CDCl3): δ 7.86 (s, 1H), 7.56 (d, J = 8.3 Hz, 2H), 7.22 (s, 1H), 6.96 (d, J = 8.3 Hz, 2H), 5.49 (t, J = 5.8 Hz, 1H), 4.54 (d, J = 6.3 Hz, 2H), 1.80 (s, 3H), 1.75 (s, 3H). HRMS (ESI+) Calculated m/z 230.1176, Found 230.1179 (M+H)+.
6-(5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)nicotinaldehyde (23)
In a dried flask equipped with a stir bar, 22 (700 mg, 3.05 mmol), 6-bromonicotinaldehyde (685 mg, 3.66 mmol), anhydrous sodium carbonate (647 mg, 6.11 mmol), copper (I) iodide (581 mg, 5.05 mmol), and triphenylphosphine (800 mg, 3.05 mmol) were dissolved in dry, deoxygenated DMF (3 mL). The reaction was purged with N2 and heated to reflux for 4.5 hours. The reaction was then cooled to RT and poured into a 7% solution of ammonium hydroxide in water (100 mL). The solution was then diluted with DCM (100 mL) and washed with sat. ammonium chloride (70 mL × 1) and sat. sodium bicarbonate (70 mL × 3). The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude mixture was then purified on a silica column using EtOAc/Hexanes. Red Solid (484 mg, 47% yield). 1H NMR (400 MHz, CDCl3): δ 10.14 (s, 1H), 9.16 (t, J = 1.4 Hz, 1H), 8.28-8.27 (m, 2H), 7.71 (d, J = 9.0 Hz, 2H), 7.45 (s, 1H), 6.97 (d, J = 8.7 Hz, 2H), 5.48 (t, J = 6.6 Hz, 1H), 4.55 (d, J = 6.7 Hz, 2H), 1.79 (s, 3H), 1.75 (s, 3H).
General Procedure for Reductive Amination
In a vial equipped with a stir bar, aldehyde 23 (30 mg, 0.089 mmol) and amine (0.099 mmol) were dissolved in DCE (1 mL) and allowed to stir for 5 min. Sodium triatoxyborohyride (57 mg, 0.27 mmol) was then added. The reaction was then allowed to stir at RT. Once complete, as deemed by TLC, the reaction was quenched with saturated aqueous sodium bicarbonate (1 mL). The reaction was then extracted with DCM (1 mL × 2). The combined organic layers were then concentrated under reduced pressure to produce a crude oil. The crude oil was the purified on a silica gel column using a combination of EtOAc and hexanes followed by 1M NH3 in MeOH/DCM.
General Procedure for Salt Formation
In a vial equipped with stir bar, the amine (1 equiv.) is dissolved in ether (1 mL) and HCl (1-2 equiv., 1M in ether) was added drop-wise. The reaction was then allowed to stir for 10 minutes before concentrating under reduced pressure.
5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)-2-(6-(piperidin-1-ylmethyl)pyridin-3-yl)oxazole (24)
Red Solid (21 mg, 36% yield). LC-MS m/z 404.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 9.16 (s, 1H), 8.42 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 7.1 Hz, 2H), 7.69 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 1.9 Hz, 1H), 7.02 (d, J = 6.7 Hz, 2H), 5.48 (t, J = 6.0 Hz, 1H), 4.59 (d, J = 7.0 Hz, 1H), 3.70 (d, J = 3.1 Hz, 2H) 2.51 (broad s, 4H), 1.80 (s, 3H), 1.77 (s, 3H), 1.63 (t, J = 3.6 Hz, 4H), 1.49 (broad s, 2H).
5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)-2-(6-(piperidin-1-ylmethyl)pyridin-3-yl)oxazole hydrochloride (24-HCl salt)
Yellow solid (24 mg, Quant.) LC-MS m/z 404.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d6-DMSO): δ 10.64 (s, 1H), 9.30 (d, J = 1.5 Hz, 1H), 8.53 (dd, J = 7.8 Hz, 1.9 Hz, 1H), 7.89 (d, J = 8.3 Hz, 1H), 7.83-7.79 (m, 3H), 7.08 (d, J = 8.6 Hz, 2H), 5.45 (t, J = 5.1 Hz, 1H), 4.60 (d, J = 6.7 Hz, 2H), 4.51 (s, 2H), 3.42 (broad s, 2H), 3.00 (broad s, 2H), 1.81 (broad s, 4H), 1.76 (s, 3H), 1.70 (s, 3H), 1.08-1.03 (m, 2H). HRMS (ESI+) Calculated m/z 404.2333, Found 404.2352 (M+H)+.
2-Methyl-N-[(5-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-2-yl)methyl]propan-1-amine (25)
Yellow oil (28 mg, 80% yield). LC-MS m/z 392.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.67 (s, 1H), 8.15 (d, J = 8.2 Hz, 1H), 7.99 (d, J = 7.9 Hz, 1H), 7.79 (d, J = 8.6 Hz, 2H), 7.54 (s, 1H), 7.02 (d, J = 8.6 Hz, 2H), 5.48 (t, J = 5.6 Hz, 1H), 4.59 (d, J = 6.6 Hz, 2H), 3.87 (s, 2H), 2.43 (d, J = 7.1 Hz, 2H), 2.02-1.92 (m, 1H), 1.80 (s, 3H), 1.77 (s, 3H), 0.94 (d, J = 6.7 Hz, 6H).
2-Methyl-N-[(5-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-2-yl)methyl]propan-1-amine hydrochloride (25-HCl salt)
Yellow solid (35 mg, Quant.). LC-MS m/z 392.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.85 (broad s, 1H), 8.29 (broad s, 1H), 8.17 (d, J = 8.2 Hz, 1H), 7.80 (d, J = 8.6 Hz, 2H), 7.62 (s, 1H), 7.04 (d, J = 9.0 Hz, 1H), 5.51 (t, J = 5.0 Hz, 1H), 4.60 (d, J = 6.7 Hz, 2H), 4.38 (s, 2H), 2.99 (d, J = 7.1 Hz, 2H), 2.13-2.04 (m, 1H), 1.80 (s, 3H), 1.78 (s, 3H), 1.07 (d, J = 6.7 Hz, 6H).
5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)-2-(5-(piperidin-1-ylmethyl)pyridin-2-yl)oxazole (26)
Yellow oil (7 mg, 19% yield). LC-MS m/z 404.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.64 (s, 1H), 8.16 (d, J = 8.3 Hz, 1H), 7.97 (d, J = 7.9 Hz, 1H), 7.80 (d, J = 9.0 Hz, 2H), 7.55 (s, 1H), 7.03 (d, J = 8.2 Hz, 2H), 5.48 (t, J = 6.0 Hz, 1H), 4.60 (d, J = 7.0 Hz, 1H), 3.61 (s, 2H), 3.27 (broad s, 4H), 1.80 (s, 3H), 1.77 (s, 3H), 1.61 (d, J = 5.9 Hz, 4H), 1.51 (t, J = 9.0 Hz, 2H).
5-(4-[(3-Methylbut-2-en-1-yl)oxy)phenyl)-2-(5-(piperidin-1-ylmethyl]pyridin-2-yl)oxazole hydrochloride (26-HCl salt)
Yellow solid (12 mg, Quant.) LC-MS m/z 404.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d6-DMSO): δ 10.38 (s, 1H), 8.87 (s, 1H), 8.25-8.18 (m, 2H), 7.81 (s, 1H), 7.77 (d, J = 8.6 Hz, 2H), 7.19 (d, J = 9.0 Hz, 2H), 5.45 (t, J = 1.6 Hz, 1H), 4.60 (d, J = 6.7 Hz, 2H), 4.40 (d, J = 5.1 Hz, 2H), 2.90 (q, J = 11.3 Hz, 4H), 1.79 (broad s, 4H), 1.76 (s, 3H), 1.73 (s, 3H), 1.38 (bs, 2H). HRMS (ESI+) Calculated m/z 404.2333, Found 404.2346 (M+H)+.
N-Methyl-N-[(6-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-3-yl)methyl]cyclohexanamine (27)
Yellow oil (44%). LCMS: 432.3 m/z (APCI) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.63 (s, 1H), 8.14 (d, J = 7.8 Hz, 1H), 7.95 (d, J = 8.6 Hz, 2H), 7.54 (s, 1H), 7.03 (d, J = 8.6 Hz, 2H), 5.48 (d, J = 6.6 Hz, 1H), 4.59 (d, J = 6.7 Hz, 2H), 3.72 (s, 2H), 2.48 (t, J = 1.3 Hz, 1H), 2.24 (s, 3H), 1.93 (d, J = 10.9 Hz, 2H), 1.84 (d, J = 11.4 Hz, 2H), 1.80 (s, 3H), 1.77 (s, 3H) 1.65 (d, J = 14.0 Hz, 1H), 1.42-1.18 (m, 5H).
N-Methyl-N-[(6-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-3-yl)methyl]cyclohexanamine hydrochloride (27-HCl salt)
Yellow solid (20 mg, Quant.). LC-MS m/z 432.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.87 (bs, 1H), 8.34 (bs, 1H), 8.19 (d, J = 8.2 Hz, 1H), 7.80 (d, J = 8.6 Hz, 2H), 7.65 (s, 1H), 7.04 (d, J = 8.6 Hz, 2H), 5.49 (t, J = 4.3 Hz, 1H), 4.60 (d, J = 6.3 Hz, 2H), 4.37 (d, J = 12.9 Hz, 1H), 2.78 (s, 3H), 2.23-2.14 (m, 2H), 2.03-1.97 (m, 2H), 1.80 (s, 3H), 1.78 (s, 3H), 1.64-1.61 (m, 2H), 1.50-1.42 (m, 2H), 1.30 (t, J = 13.7 Hz, 1H), 1.17 (td, J = 7.0 Hz, 1.1 Hz, 1H). HRMS (ESI+) Calculated m/z 432.2646, Found 432.2657 (M+H)+.
(R)-N,N-Dimethyl-1-[(6-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-3-yl)methyl]pyrrolidin-3-amine (28)
Yellow oil (22 mg, 57% yield). LC-MS m/z 433.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.63 (d, J = 1.5 Hz, 1H), 8.14 (d, J = 8.2 Hz, 1H), 7.95 (dd, J = 8.2 Hz, 1.9 Hz, 1H), 7.78 (d, J = 9.0 Hz, 2H), 7.53 (s, 1H), 7.01 (d, J = 9.0 Hz, 2H), 5.46 (t, J = 5.1 Hz, 1H), 4.58 (d, J = 6.2 Hz, 2H), 4.55 (bs, 2H), 3.74 (q, J = 13.7 Hz, 2H), 2.85 (t, J = 7.8 Hz, 1H), 2.64-2.61 (m, 1H), 2.28-2.50 (m, 8H), 2.02 (d, J = 9.0 Hz, 1H), 1.78 (s, 3H), 1.76 (s, 3H).
(R)-N,N-Dimethyl-1-[(6-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-3-yl)methyl]pyrrolidin-3-amine dihydrochloride (28-HCl salt)
Two equivalents of HCl was used. Yellow solid (35 mg, Quant.) LC-MS m/z 433.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d6-DMSO): δ 8.93 (s, 1H), 8.45-8.21 (m, 2H), 7.81-7.76 (m, 3H), 7.08 (d, J = 8.6 Hz, 2H), 5.46 (t, J = 6.4 Hz, 1H), 4.56 (bs, 2H) 4.60 (d, J = 6.7 Hz, 2H), 3.34 (s, 1H), 2.99 (s, 1H), 2.80-2.72 (m, 9H), 2.09 (s, 1H), 1.96 (d, J = 10.6 Hz, 1H), 1.76 (s, 3H), 1.74 (s, 3H). HRMS (ESI+) Calculated m/z 433.2598, Found 433.2600 (M+H)+.
6-[((6-(5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-3-yl)methyl)amino]hexan-1-ol (29)
Yellow oil (17 mg, 44% yield). LC-MS m/z 436.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.67 (d, J = 1.5 Hz, 1H), 8.16 (d, J = 7.4 Hz, 1H), 7.99 (dd, J = 8.2 Hz, 2.3 Hz, 1H), 7.80 (d, J = 8.6 Hz, 2H), 7.55 (s, 1H), 7.03 (d, J = 9.0 Hz, 2H), 5.48 (t, J = 1.6 Hz, 1H), 4.60 (d, J = 6.6 Hz, 2H), 3.89 (s, 2H), 3.54 (t, J = 6.7 Hz, 2H), 1.92 (t, J = 6.6 Hz, 2H), 1.80 (s, 3H), 1.78 (s, 3H), 1.60-1.49 (m, 4H), 1.42-1.35 (m, 4H). HRMS (ESI+) Calculated m/z 436.2595, Found 436.2598 (M+H)+.
6-[((6-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-3-yl)methyl)amino]hexan-1-ol hydrochloride (29-HCl salt)
Yellow solid (29 mg, Quant.) LC-MS m/z 436.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.83 (bs, 1H), 8.30 (bs, 1H), 8.17 (d, J = 7.9 Hz, 1H), 7.80 (d, J = 9.0 Hz, 2H), 7.63 (bs, 1H), 7.04 (d, J = 8.6 Hz, 2H), 5.48 (t, J = 5.1 Hz, 1H), 4.60 (d, J = 6.3 Hz, 2H), 4.37 (s, 2H), 3.57 (t, J = 6.3 Hz, 4H), 1.80 (s, 4H), 1.78 (s, 4H), 1.61-1.53 (m, 2H), 1.49-1.42 (m, 4H). HRMS (ESI+) Calculated m/z 436.2595, Found 436.2598 (M+H)+.
(S)-Methyl-1-[(6-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-3-yl)methyl]pyrrolidine-2-carboxylate (30)
Yellow oil (16 mg, 40% yield). LC-MS m/z 448.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.65 (d, J = 2.0 Hz, 1H), 8.14 (d, J = 8.2 Hz, 1H), 7.98 (dd, J = 8.2 Hz, 2.0 Hz, 1H), 7.80 (d, J = 2.3 Hz, 2H), 7.79 (s, 1H), 7.03 (d, J = 9.0 Hz, 2H), 5.48 (t, J = 1.6 Hz, 1H), 4.59 (d, J = 6.6 Hz, 2H), 3.99 (d, J = 3.3 Hz, 1H), 3.71 (d, J = 13.7 Hz, 1H), 3.64 (s, 3H), 3.37-3.32 (m, 1H), 3.06-3.01 m, 1H), 2.49 (q, J = 8.2 Hz, 1H), 2.23-2.15 (m, 1H), 1.96-1.83 (m, 3H), 1.80 (s, 3H), 1.77 (s, 3H).
(S)-Methyl-1-[(6-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-3-yl)methyl]pyrrolidine-2-carboxylate hydrochloride (30-HCl salt)
Yellow solid (20 mg, Quant.) LC-MS m/z 448.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d6-DMSO): δ 8.85 (s, 1H), 8.23 (t, J = 8.2 Hz, 1H), 8.16 (d, J = 6.6 Hz, 1H), 7.81-7.76 (m, 3H), 7.09 (d, J = 8.7 Hz, 2H), 5.44 (t, J = 6.2 Hz, 1H), 4.60 (d, J = 6.7 Hz, 3H), 4.53 (bs, 1H), 3.68 (s, 3H), 3.57 (bs, 1H), 3.44 (q, J = 7.1 Hz, 2H), 3.34 (s, 2H), 1.76 (s, 3H), 1.74 (s, 3H), 1.08-1.03 (m, 2H). HRMS (ESI): Calculated: 448.2231 m/z, Found: 448.2236 m/z (M+H)+.
N,N-Dimethyl-1-(6-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-3-yl)methanamine (31)
Yellow oil (17 mg, 49% yield). LC-MS m/z 364.2 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.64 (d, J = 1.9 Hz, 1H), 8.17 (d, J = 7.8 Hz, 1H), 7.96 (dd, J = 8.2 Hz 2.0 Hz, 1H), 7.80 (d, J = 9.0 Hz, 2H), 7.55 (s, 1H), 7.03 (d, J = 9.0 Hz, 2H), 5.47 (t, J = 5.6 Hz, 1H), 4.60 (d, J = 6.6 Hz, 2H), 3.60 (s, 2H), 2.30 (s, 6H), 1.80 (s, 3H), 1.78 (s, 3H).
N,N-Dimethyl-1-(5-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-2-yl)methanamine hydrochloride (31-HCl salt)
Tan solid (21 mg, Quant.). LC-MS m/z 364.2 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.79 (s, 1H), 8.28 (d, J = 8.2 Hz, 1H), 8.11 (dd, J = 8.2 Hz, 2.4 Hz, 1H), 7.80 (d, J = 9.0 Hz, 2H), 7.60 (s, 1H), 7.04 (d, J = 9.0 Hz, 2H), 5.49 (t, J = 7.2 Hz, 1H), 4.60 (d, J = 6.7 Hz, 2H), 4.33 (s, 2H), 2.84 (s, 6H), 1.80 (s, 3H), 1.78 (s, 3H). HRMS (ESI): Calculated: 364.2020 m/z, Found: 364.2024 m/z (M+H)+. HRMS (ESI+) Calculated m/z 448.2231, Found 448.2236 (M+H)+.
N-Ethyl-N-[(6-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-3-yl)methyl]ethanamine (32)
Yellow oil (25 mg, 71% yield). LC-MS m/z 392.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.65 (s, 1H), 8.14 (dd, J = 8.3 Hz, 3.6 Hz, 1H), 7.97 (dd, J = 8.2 Hz, 2.0 Hz, 1H), 7.79 (d, J = 9.0 Hz, 2H), 7.54 (s, 1H), 7.03 (d, J = 9.0 Hz, 2H), 5.48 (t, J = 2.3 Hz, 1H), 4.59 (s, J = 6.6 Hz, 2H), 3.72 (s, 2H), 2.59 (q, J = 7.0 Hz, 4H), 1.80 (s, 3H), 1.77 (s, 3H), 1.10 (t, J = 7.0 Hz, 6H).
N-Ethyl-N-[(6-(5-(4-[(3-methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl)pyridin-3-yl)methyl]ethanamine hydrochloride (32-HCl salt)
Orange oil (29 mg, Quant.). LC-MS m/z 392.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.85 (s, 1H), 8.30 (d, J = 8.2 Hz, 1H), 8.18 (dd, J = 8.2 Hz, 1.9 Hz, 1H), 7.80 (d, J = 8.6 Hz, 2H), 7.62 (s, 1H), 7.04 (d, J = 8.6 Hz, 2H), 5.49 (t, J = 8.4 Hz, 1H), 4.60 (d, J = 6.6 Hz, 2H), 4.52 (s, 2H), 3.31 (quin, J = 1.5 Hz, 4H), 1.80 (s, 3H), 1.78 (s, 3H), 1.40 (t, 7.4 Hz, 6H). HRMS (ESI+) Calculated m/z 392.2333, Found 392.2317 (M+H)+.
5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)-2-(5-[(4-methylpiperazin-1-yl)methyl]pyridin-2-yl)oxazole (33)
Yellow oil (8 mg, 21% yield). LC-MS m/z 419.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.65 (s, 1H), 8.16 (d, J = 8.2 Hz, 1H), 7.97 (d, J = 8.7 Hz, 1H), 7.79 (d, J = 7.8 Hz, 2H), 7.55 (s, 1H), 7.03 (d, J = 7.9 Hz, 2H), 5.48 (t, J = 6.4 Hz, 1H), 4.60 (d, J = 6.2 Hz, 2H), 3.66 (s, 2H), 3.60-3.53 (m, 4H), 2.45 (t, J = 5.1 Hz, 2H), 2.40 (t, J = 5.0 Hz, 2H), 2.30 (d, J = 1.2 Hz, 3H), 2.29 (s, 2H), 1.80 (s, 3H), 1.77 (s, 3H).
5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)-2-(5-[(4-methylpiperazin-1-yl)methyl]pyridin-2-yl)oxazole dihydrochloride (33-HCl salt)
Two equivalents of HCl were used. Tan solid (12 mg, Quant.). LC-MS m/z 419.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.64 (s, 1H), 8.12 (d, J = 8.3 Hz, 1H), 8.06 (s, 1H), 8.02 (d, J = 7.8 Hz, 1H), 7.92 (d, J = 8.2 Hz, 2H), 6.94 (d, J = 9.0 Hz, 2H), 5.47 (t, J = 9.0 Hz, 1H), 4.58 (d, J = 6.6 Hz, 2H), 3.87 (s, 2H), 3.46 (t, J = 7.7 Hz, 4H), 2.91-2.89 (m, 6H), 2.13 (s, 1H), 1.77 (s, 3H), 1.74 (s, 3H), 1.23 (s, 1H). HRMS (ESI+) Calculated m/z 419.2442, Found 419.2430 (M+H)+.
1-[(6-(5-(4-[(3-Methylbut-2-en-1-yl)oxy]phenyl)oxazol-2-yl]pyridin-3-yl)methyl)pyrrolidin-2-one (34)
Yellow oil (30 mg, 83% yield). LC-MS m/z 405.3 (APCI+) (M+H)+. 1H NMR (400 MHz, d4-MeOD): δ 8.61 (d, J = 1.9 Hz, 1H), 8.17 (d, J = 8.2 Hz, 1H), 7.89 (dd, J = 8.2 Hz, 2.3 Hz, 1H), 7.79 (d, J = 9.0 Hz, 2H), 7.55 (s, 1H), 7.03 (d, J = 8.6 Hz, 2HL), 5.48 (t, J = 1.6 Hz, 1H), 4.61-4.58 (m, 4H), 3.43 (t, J = 7.1 Hz, 2H), 2.47 (t, J = 7.8 Hz, 2H), 2.08 (quin, J = 7.4 Hz, 2H), 1.80 (s, 3H), 1.77 (s, 3H). HRMS (ESI+) Calculated m/z 404.1969, Found 404.1979 (M+H)+.
Biological Experiments
Cell Culture
MDA-MB-468, MDA-MB-453, MDA-MB-231, HCC1937, HCC70 and A-10 cell lines were purchased directly from the American Type Culture Collection (Manassas, VA, USA). The BT-549 cell line was obtained from Lombardi Comprehensive Cancer Center of Georgetown University (Washington, DC, USA) and the identity was validated by Promega (Fitchburg, WI, USA). MDA-MB-453 and MDA-MB-231 cells were cultured in IMEM (Gibco) containing 25 μg/mL gentamicin (Gibco) and 10% FBS. MDA-MB-468, HCC70, HCC1937, and BT-549 cells were cultured in RPMI-1640 medium (Sigma-Aldrich) containing 50 μg/mL gentamicin and 10% FBS. A-10 cells were cultured in basal medium eagle (Sigma-Aldrich) containing 50 μg/mL gentamicin and 10% FBS. Cells were maintained in humidified incubators at 37°C with 5% CO2. All cell lines were initially expanded and frozen as stocks in liquid nitrogen. Cells were passaged for no more than 3 months after revival from liquid nitrogen.
Sulforhodamine B (SRB) Assay
The antiproliferative and cytotoxic effects of all compounds were evaluated with the SRB assay as previously described.18,19 Cells were treated with the compounds indicated for 48 – 96 h and cell density at the time of treatment (T0) was measured to determine cytotoxic activity. Concentration-response curves were plotted and the concentrations causing 50% inhibition of proliferation compared to vehicle control (GI50), total growth inhibition (TGI) and 50% cell death compared to T0 (LC50) were interpolated from nonlinear regressions of the data using Prism 6 (GraphPad Software).
Cell cycle analysis by flow cytometry
The effects of selected compounds on cell cycle distribution were evaluated by flow cytometry. MDA-MB-453 cells were treated with vehicle (DMSO) or predetermined concentrations of the test compound for 18 h. Cells were then harvested, stained with Krishan’s reagent,30 and their DNA content was measured using a Muse Cell Analyzer (EMD Millipore).
Intracellular signaling array
Cells were treated with vehicle or the test compound, harvested by scraping and lysed in Cell Extraction Buffer (Life Technologies, Waltham, MA, USA) containing protease inhibitor cocktail, PMSF and Na3VO4 (Sigma-Aldrich). Protein concentrations were quantified with a Pierce Coomassie Plus Assay Kit (Life Technologies) and equal amounts of protein were evaluated with a PathScan® intracellular signaling array (fluorescent readout; Cell Signaling Technology) according to the manufacturer’s protocol. Near-infrared fluorescent signals were captured with an Odyssey CLx (LI-COR Biosciences, Lincoln, NE, USA) and quantified with ImageStudio (LI-COR Biosciences).
Cell Lysis and Immunoblotting
Cells were treated with vehicle or the test compound, harvested by scraping and lysed as described for the intracellular signaling array. Protein concentrations were quantified with a Pierce Coomassie Plus Assay Kit (Life Technologies) and equal amounts of protein were separated by SDS-PAGE on NuPAGE Bis-Tris gels (Life Technologies). Proteins were transferred to PVDF membranes (EMD Millipore, Billerica, MA, USA) which were subsequently blocked with Odyssey Blocking Buffer (LI-COR Biosciences) in tris-buffered saline. Membranes were probed with antibodies for P-mTOR, mTOR, P-S6K, S6K, P-RPS6, RPS6, P-S473-Akt, P-T308-Akt or pan Akt (Cell Signaling Technology, Danvers, MA, USA) diluted in Odyssey Blocking Buffer. Membranes were incubated with appropriate IRDye 680 or IRDye 800 secondary antibodies (LI-COR Biosciences) and near-infrared fluorescence signals were captured on an Odyssey FC (LI-COR Biosciences). Signal intensities were quantified with ImageStudio (LI-COR Biosciences).
Supplementary Material
Acknowledgments
The authors would like to thank the San Antonio Botanical Gardens for allowing us to collect samples of Amyris texana for these studies.
Funding Sources
This study was funded by grants to S.L.M. and R.H.C. from the National Cancer Institute (UO1CA182740), the UTHSCSA President’s Council Excellence Award (S.L.M), and the Greehey Distinguished Chair in Targeted Molecular Therapeutics endowment (S.L.M.). Support of the Flow Cytometry, Macromolecular Structure and Mass Spectrometry Shared Resources of the CTRC Cancer Center Support Grant (P30 CA054174) are gratefully acknowledged. A.J.R was partially supported by the IMSD program of the NIGMS (1R25GM095480-01). Medicinal chemistry support from the Cancer Prevention Research Institute of Texas (CPRIT) grant RP160844 (S.F.M) is gratefully acknowledged.
ABBREVIATIONS
- TNBC
triple-negative breast cancer
- ER
estrogen receptor
- PR
progesterone receptor
- HER2
human epidermal growth factor receptor 2
- BL1
basal-like 1
- BL2
basal-like 2
- M
mesenchymal-like
- LAR
luminal androgen receptor
- SRB
sulforhodamine B. SAR, structure-activity relationships
- DCM
dichloromethane
- EtOAc
ethyl acetate
- DCE
1,2-dichloroethane
- DMF
N,N-dimethylformamide
- TLC
thin layer chromatography
- DIPEA
diisopropylethylamine
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- Ac2O
acetic anhydride
- HPLC-MS
high-performance liquid chromatography-mass spectrometry
- UV, NMR
nuclear magnetic resonance
Footnotes
Supporting Information. The following files are available free of charge. Supplemental information [1H, 13C, gCOSY and NOESY2D NMR spectra (pdf); Molecular strings file (csv)]
Author Contributions
A.J.R. and S.L.M. designed the biological experiments and A.J.R. conducted them. S.F.M. and S.M. designed all analogs and planned all chemistry experiments and compound synthesis. S.M. synthesized and characterized all compounds and analogs. M.G. developed alternative synthesis routes. S.C. and R.H.C. planned the isolation of the natural products. S.C. isolated the natural products and determined their structures. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
NA
References
- 1.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 2.Irvin WJ, Carey LA. What Is Triple-Negative Breast Cancer? Eur J Cancer. 2008;44(18):2799–2805. doi: 10.1016/j.ejca.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 3.Vaz-Luis I, Ottesen RA, Hughes ME, Mamet R, Burstein HJ, Edge SB, Gonzalez-Angulo AM, Moy B, Rugo HS, Theriault RL, Weeks JC, Winer EP, Lin NU. Outcomes by Tumor Subtype and Treatment Pattern in Women with Small, Node-Negative Breast Cancer: A Multi-Institutional Study. J Clin Oncol. 2014;32(20):2142–2150. doi: 10.1200/JCO.2013.53.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brewster AM, Chavez-MacGregor M, Brown P. Epidemiology, Biology, and Treatment of Triple-Negative Breast Cancer in Women of African Ancestry. Lancet Oncol. 2014;15(13):e625–e634. doi: 10.1016/S1470-2045(14)70364-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abramson VG, Mayer IA. Molecular Heterogeneity of Triple Negative Breast Cancer. Curr Breast Cancer Rep. 2014;6(3):154–158. doi: 10.1007/s12609-014-0152-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayer IA, Abramson VG, Lehmann BD, Pietenpol JA. New Strategies for Triple-Negative Breast Cancer-Deciphering the Heterogeneity. Clin Cancer Res. 2014;20(4):782–790. doi: 10.1158/1078-0432.CCR-13-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy Ba, Shyr Y, Pietenpol Ja. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J Clin Invest. 2011;121(7):2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehmann BD, Jovanović B, Chen X, Estrada MV, Johnson KN, Shyr Y, Moses HL, Sanders ME, Pietenpol JA. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS One. 2016;11(6):e0157368. doi: 10.1371/journal.pone.0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du L, Robles AJ, King JB, Powell DR, Miller AN, Mooberry SL, Cichewicz RH. Crowdsourcing Natural Products Discovery to Access Uncharted Dimensions of Fungal Metabolite Diversity. Angew Chem Int Ed Engl. 2014;53(3):804–809. doi: 10.1002/anie.201306549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robles AJ, Cai S, Cichewicz RH, Mooberry SL. Selective Activity of Deguelin Identifies Therapeutic Targets for Androgen Receptor-Positive Breast Cancer. Breast Cancer Res Treat. 2016;157(3):475–488. doi: 10.1007/s10549-016-3841-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Philip SA, Burke BA, Jacobs H. Amyris of Jamaica. 2,5-Diaryloxazoles and a Chromane from Amyris Plumieri D.C (Rutaceae) Heterocycles. 1984;22(1):9. [Google Scholar]
- 12.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 13.Rastogi N, Abaul J, Goh KS, Devallois A, Philogène E, Bourgeois P. Antimycobacterial Activity of Chemically Defined Natural Substances from the Caribbean Flora in Guadeloupe. FEMS Immunol Med Microbiol. 1998;20(4):267–273. doi: 10.1111/j.1574-695X.1998.tb01136.x. [DOI] [PubMed] [Google Scholar]
- 14.Burke BA, Burke B, Parkins H, Marie Talbot A. An Oxazole and Its Precursor in Amyris Balsamifera. Heterocycles. 1979;12(3):349. [Google Scholar]
- 15.Yoshizumi T, Satoh T, Hirano K, Matsuo D, Orita A, Otera J, Miura M. Synthesis of 2,5-Diaryloxazoles through van Leusen Reaction and Copper-Mediated Direct Arylation. Tetrahedron Lett. 2009;50:3273–3276. [Google Scholar]
- 16.Cheplogoi PK, Mulholland DA, Coombes PH, Randrianarivelojosia M. An Azole, an Amide and a Limonoid from Vepris Uguenensis (Rutaceae) Phytochemistry. 2008;69(6):1384–1388. doi: 10.1016/j.phytochem.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 17.Meepagala KM, Schrader KK, Burandt CL, Wedge DE, Duke SO. New Class of Algicidal Compounds and Fungicidal Activities Derived from a Chromene Amide of Amyris Texana. Journal of Agricultural and Food Chemistry. 2010;58:9476–9482. doi: 10.1021/jf101626g. [DOI] [PubMed] [Google Scholar]
- 18.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J Natl Cancer Inst. 1990;82(13):1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 19.Boyd MR, Paull KD, Rubinstein LR. Data Display and Analysis Strategies for the NCI Disease-Oriented in Vitro Antitumor Drug Screen. In: Valeriote FA, Corbett TH, Baker LH, editors. Cytotoxic Anticancer Drugs: Models and Concepts for Drug Discovery and Development: Proceedings of the Twenty-Second Annual Cancer Symposium Detroit, Michigan, USA --- April 26--28, 1990. Springer US; Boston, MA: 1992. pp. 11–34. [Google Scholar]
- 20.Besselièvre F, Mahuteau-Betzer F, Grierson DS, Piguel S. Ligandless Microwave-Assisted Pd/Cu-Catalyzed Direct Arylation of Oxazoles. J Org Chem. 2008;73(8):3278–3280. doi: 10.1021/jo7027135. [DOI] [PubMed] [Google Scholar]
- 21.Giddens AC, Boshoff HIM, Franzblau SG, Barry CE, Copp BR. Antimycobacterial Natural Products: Synthesis and Preliminary Biological Evaluation of the Oxazole-Containing Alkaloid Texaline. Tetrahedron Lett. 2005;46:7355–7357. [Google Scholar]
- 22.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures. J Org Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 23.Lehmann BD, Bauer JA, Schafer JM, Pendleton CS, Tang L, Johnson KC, Chen X, Balko JM, Gómez H, Arteaga CL, Mills GB, Sanders ME, Pietenpol JA. PIK3CA Mutations in Androgen Receptor-Positive Triple Negative Breast Cancer Confer Sensitivity to the Combination of PI3K and Androgen Receptor Inhibitors. Breast Cancer Res. 2014;16(4):406. doi: 10.1186/s13058-014-0406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Reilly KE, Rojo F, She Q-B, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Res. 2006;66(3):1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, Valero V, Lehmann BD, Pietenpol JA, Hortobagyi GN, Symmans WF, Ueno NT. Differential Response to Neoadjuvant Chemotherapy among 7 Triple-Negative Breast Cancer Molecular Subtypes. Clin Cancer Res. 2013;19(19):5533–5540. doi: 10.1158/1078-0432.CCR-13-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai S, Risinger AL, Nair S, Peng J, Anderson TJC, Du L, Powell DR, Mooberry SL, Cichewicz RH. Identification of Compounds with Efficacy against Malaria Parasites from Common North American Plants. J Nat Prod. 2016;79:490–498. doi: 10.1021/acs.jnatprod.5b00874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasbum C, Castro O. A New Benzamide from Bark of Amyris Brenesii. J Nat Prod. 1988;51(4):817–818. doi: 10.1021/np50058a035. [DOI] [PubMed] [Google Scholar]
- 28.Lee JJ, Kim J, Jun YM, Lee BM, Kim BH. Indium-Mediated One-Pot Synthesis of Benzoxazoles or Oxazoles from 2-Nitrophenols or 1-Aryl-2-Nitroethanones. Tetrahedron. 2009;65(43):8821–8831. [Google Scholar]
- 29.Mahuteau-Betzer F, Piguel S. Synthesis and Evaluation of Photophysical Properties of Series of π-Conjugated Oxazole Dyes. Tetrahedron Lett. 2013;54(24):3188–3193. [Google Scholar]
- 30.Krishan A. Rapid Flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Bio. 1975;66:188–193. doi: 10.1083/jcb.66.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.