

Abstract
Guillain–Barré syndrome (GBS) and transverse myelitis (TM) both represent immunologically mediated polyneuropathies of major clinical importance. Both are thought to have a genetic predisposition, but as of yet no specific genetic risk loci have been clearly defined. Both are considered autoimmune, but again the etiologies remain enigmatic. Both may be induced via molecular mimicry, particularly from infectious agents and vaccines, but clearly host factor and co-founding host responses will modulate disease susceptibility and natural history. GBS is an acute inflammatory immune-mediated polyradiculoneuropathy characterized by tingling, progressive weakness, autonomic dysfunction, and pain. Immune injury specifically takes place at the myelin sheath and related Schwann-cell components in acute inflammatory demyelinating polyneuropathy, whereas in acute motor axonal neuropathy membranes on the nerve axon (the axolemma) are the primary target for immune-related injury. Outbreaks of GBS have been reported, most frequently related to Campylobacter jejuni infection, however, other agents such as Zika Virus have been strongly associated. Patients with GBS related to infections frequently produce antibodies against human peripheral nerve gangliosides. In contrast, TM is an inflammatory disorder characterized by acute or subacute motor, sensory, and autonomic spinal cord dysfunction. There is interruption of ascending and descending neuroanatomical pathways on the transverse plane of the spinal cord similar to GBS. It has been suggested to be triggered by infectious agents and molecular mimicry. In this review, we will focus on the putative role of infectious agents as triggering factors of GBS and TM.
Introduction
Guillain–Barré syndrome (GBS) is an acute inflammatory immune-mediated polyradiculoneuropathy presenting typically with tingling, progressive weakness, autonomic dysfunction and pain.1,2 Immune injury specifically takes place at the myelin sheath and related Schwann-cell components in acute inflammatory demyelinating polyneuropathy (AIDP), whereas in acute motor axonal neuropathy (AMAN) membranes on the nerve axon (the axolemma) are the primary target for immune-related injury.3 Incidence of GBS ranges from 0.8 to 1.9 cases per 100 000 people per year.4 It increases with age and is slightly more frequent in males than in females.3 Several outbreaks of GBS have been reported, most frequently related to Campylobacter jejuni (C. jejuni) infections, however, other agents such as Zika Virus (ZIKV) have been strongly associated.5,6
Patients with GBS related to infections frequently produce antibodies against human peripheral nerve gangliosides through molecular mimicry.7 A bacterial cross-reactive antigen recognized by macrophages and T cells induces B cells to produce an anti-ganglioside response, which penetrates blood–nerve barrier and activates complement. These antibodies bind both to gangliosides from nerves and to antigens from microbes. Activated endoneurial macrophages release cytokines and free radicals (for example, nitric oxide), invade compact myelin, periaxonal space, and occasionally block nerve conduction or cause axonal degeneration. Activated T cells release proinflammatory cytokines, fix complement, damage Schwann cells and ultimately produce dissolution of myelin.8 On the other hand, transverse myelitis (TM) is an inflammatory disorder characterized by acute or subacute motor, sensory and autonomic (for example, bladder, bowel and sexual symptoms) spinal cord dysfunction.9 The clinical signs are caused by an interruption of ascending and descending neuroanatomical pathways on the transverse plane of the spinal cord.10 It has been suggested to be triggered by infectious agents and to be associated with autoimmune diseases (ADs). Nevertheless, its etiology remains unknown in a substantial portion of cases, which are classified as idiopathic.11 Vaccinations have been incriminated in both of these disorders but the focus herein will be only on infectious diseases.
Herein, a comprehensive review on infections associated with GBS and TM and the proposed molecular mechanisms of their pathogenesis are offered.
Bacterial infections and GBS
About 75% of patients with GBS have a history of preceding infection, usually of the respiratory and gastrointestinal tract, within 6 weeks prior to onset (Figure 1).12,13 Some bacteria have been reported as GBS triggers and known molecular mechanisms will be discussed (Supplementary Table).
Figure 1.
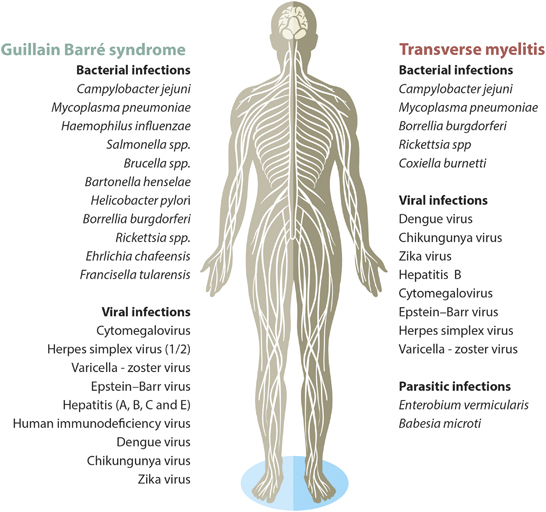
Infectious agents as causative of Guillain–Barré syndrome and transverse myelitis.
Campylobacter jejuni and GBS
C. jejuni is a frequent food-borne pathogen. Human infections caused by C. jejuni are a leading cause of food-borne enteritis, usually transmitted by the ingestion of undercooked poultry or contact with farm animals. New advances in the field of human campylobacteriosis include an increased appreciation of the role of C. jejuni in post-infectious sequelae, a broadened understanding of Campylobacter-associated disease burden and the interplay between host immunity and bacterial factors.14,15 In 30–40% of GBS cases are preceded by C. jejuni infection. Moreover, it has been estimated that 1.17/1000 C. jejuni infections result in GBS.16
Campylobacter jejuni factors and molecular mimicry
C. jejuni presents different epitopes capable of modulating host immunity. Most of them are glycoconjugates that are formed through glycosylation, including lipooligosacharides (LOS) and capsule polysaccharides.17 Protein glycosylation is the most common post-translational modification. It is essential to define protein activity and function, as well as to coordinate intra- and intercellular communications.17
Glycans include every mono-, oligo- or polysaccharide linked to another molecule, either free or covalently attached. Most cells possess a dense covering layer on their surface, called glycocalix, which is composed of glycoconjugates. A glycoprotein is defined as a glycoconjugate comprising a polypeptide backbone with one or more glycans covalently attached, either via N- or O-linkages. N-linkage is due to the attachment of glycans to the polypeptide via an asparagine residue, in contrast to O-linkage that is via a serine or threonine residue.18 Further, a glycosphingolipid or glycolipid is a glycan attached to lipid species, which may be anionic or neutral. A ganglioside is an anionic glycolipid containing one or more residues of sialic acid, which are considered the most common glycans in eukaryotic cells.18 Glycosylation may yield virtually an infinite diversity to its final compounds. However, few combinations are present in nature. In any case, certain structures are usually shared among different glycans, such as common structures between glycolypids and N- and O-glycans.18
GBS secondary to C. jejuni was the first confirmed case of molecular mimicry.19 C. jejuni LOS and lipopolysaccharides (LPS) display molecular mimicry with gangliosides. Production of antibodies against these structures induces a cross-reactive immune response to nerve structures, triggering autoimmunity and neural damage (Figure 2).20 Gangliosides attacked by the immune system are located as follows: GM1, GD1a and GM1/GD1 complex situated at the terminal nerves and anterior roots, closely related to AIDP and AMAN. GQ1b is located on oculomotor nerves (that is, III, IV and VI) and primary sensory neurons, and is correlated with Miller Fisher syndrome (MFS).19 Serum antibodies against gangliosides are found in one third of GBS patients secondary to C. jejuni.21
Figure 2.
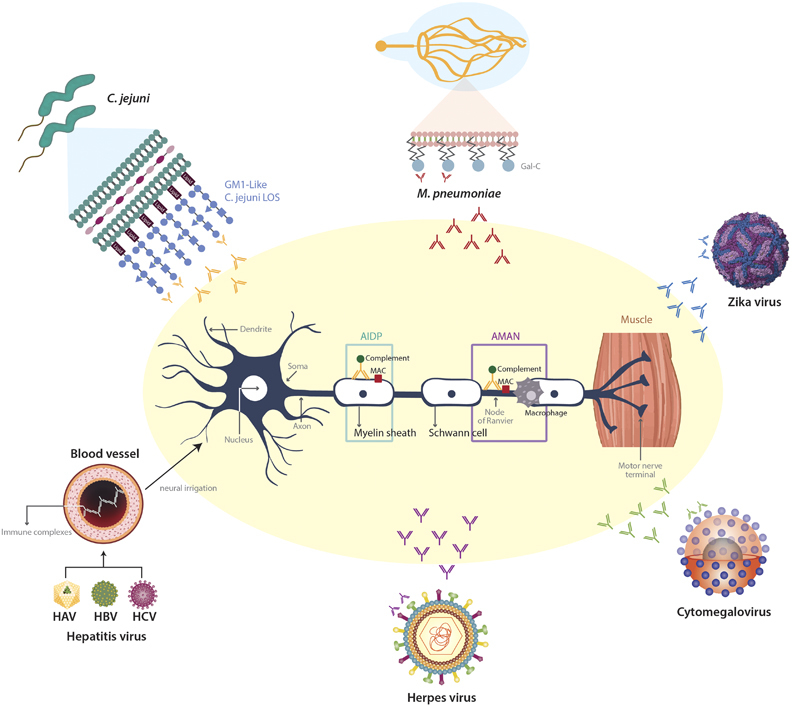
Pathogenesis of post-infectious Guillain-Barré syndrome (see text for details). GM1, monosialotetrahexosylganglioside; LOS, lipooligosacharide; Gal-C, Galactocerebroside; MAC, membrane-attack complex.
C. jejuni genes such as LOS loci (that is, A, B and C) involved in LOS biosynthesis are crucial for the induction of ganglioside like structures.20,22 Class A and B encode for C. jejuni sialyltransferase gene (cst-II).23 The remaining classes (that is, D or E) did not exhibit these epitopes, which is consistent with the fact that classes D and E are unable to synthesize or transfer sialic acid. In addition, unclassified strains also express ganglioside-like epitopes.20 Cst-II (Thr51) strain has a mono-functional α-2,3-sialyltransferase, which produces GM1-like, GM2-like and GD1a-like LOS. On the other hand, cst-II (Asn51) strain has both α-2,3- and α-2,8-sialyltransferase activity, which allow to produce GT1a-like and GD1c-like LOS that can mimic GQ1b.24,25
There was a predominance of class B locus in strains isolated from patients with MFS compared with controls and GBS. However, strains isolated from MFS patients displayed more frequently cst-II (Asn51), regardless of the class thus suggesting that the cst-II polymorphism, rather than the class, is important for MFS.20 Further, Koga et al. 26 showed that cst-II (Asn51) strains often present GQ1b-like epitope, in contrast to cst-II (Thr51), which express GM1-like and GD1a-like epitope. However, half of patients with enteric C. jejuni infection without GBS disclosed positive cst-II, which suggests that this gene is necessary but not sufficient to trigger GBS. However, these studies are controversial since a further study was not able to reproduce the results. Noteworthy, HS:19 strain (classically associated with GBS) displayed more often Thr51.27
In addition, the C. jejuni strain CF90–26 was isolated from AMAN patients with anti-GM1 IgG antibodies, which portrayed an oligosaccharide structure similar to the GM1 antigen.28 Interestingly, C. jejuni strains knocked down for Orf11, which codes for sialate O-acetyltransferase disclosed a reduced reactivity to anti-GM1 sera in patients with GBS, and did not induce anti-GD1a IgG antibody response in mice.29 Recently, it was described that GM1-like together with GD1a-like LOS may form a GM1b-like epitope thus inducing the development of anti-GM1b antibodies. Those antibodies bind to either GM1b itself or to a heteromeric complex of GM1 and GD1a at the nodes of Ranvier thus activating complement in the peripheral motor nerves.30 As peripheral nerve axonal membranes containing high levels of GD1a are frequently attacked by anti-GD1a antibodies,31 LPS from some C. jejuni strains, such as HS:19 are commonly associated with GBS and have shown to contain GM1, GD1a, GD3 and GT1a-like saccharide motifs.32
Other C. jejuni genes have been involved in molecular mimicry. NeuB1 gene, which encodes NeuAc-synthetase and is required for the synthesis of NeuAc of C. jejuni LOS, proved to be necessary for the induction of anti-GM1 antibodies and pathological changes in peripheral nerves.33 Further, wlaN gene product, which encodes for β-1,3-galactosyltransferase is responsible for converting the GM2-like LOS structure to a GM1-like structure.34 In addition, LOS from C. jejuni strain 81–176 expresses cores containing GM2 and GM3-like gangliosides.35
In this line, Godshalk et al. 36 found that two genes orf11 and orf10, this last encoding for a CMP-sialic acid synthetase, were more frequent in strains from patients with ophthalmoplegia. Previously, they had demonstrated that orf10 is necessary for the synthesis of ganglioside-like epitopes and anti-ganglioside autoantibodies.20 These genes are unique for classes A and B. Further, these two genes, as well as orf7ab (cst-II), were associated with the occurrence of GQ1b-like epitopes on the bacterial LOS.20 Taboada et al. 37 reported high conservation rates in genes associated with LOS loci classes A and B in neuropathic strains despite a wide genomic heterogeneity within this group. One further gene was highly conserved in neuropathic strains, namely Cj1411c, which appear to be involved in capsule biosynthesis.38 In addition, C. jejuni 81116 galE gene, encoding an UDP-galactose-4-epimerase is indispensable for synthesize ganglioside-like structures in the LOS core oligosaccharide.39 Animals sensitized with C. jejuni HB9313 (HS:19) strain mutated for galE, showed high titers of anti-GM1 IgG and axonal degeneration.40 The lack of neuB1 in C. jejuni resulted in the loss of epitope of N-acetylneuraminic acid synthetase, which is critical for the peripheral neuropathy induction.33
Host factors: HLA and non-HLA
Different approaches have been used to determine the possible role of host immunogenetic background in the development of GBS and its variants. Several reports have associated HLA alleles with GBS risk (Table 1).41,42,43,44,45,46,47,48,49,50,51,52,53,54,55 However, to date, there are no robust and powerful data concerning the association between GBS and HLA typing, probably due to different population, sample size or technique used for HLA typing.23,56
Table 1.
Associations between HLA and Guillain–Barré syndrome
| Author | Population | Commentary |
|---|---|---|
| Schirmer L et al. 41 | Germany | HLA-DQB1*05:01 allele associated with severe GBS |
| Hasan ZN et al. 42 | Iraq | HLA-DRB alleles associated with GBS risk and HLA-DR6 with protection |
| Fekih-Mrissa N et al. 43 | Tunisia | HLA-DRB1*14 and DRB1*13 associated with GBS |
| Blum et al. 44 | Australia | HLA ligands HLA-C2 and HLA Bw4+Thr80 were more frequently seen in patients with GBS than in controls |
| Kaslow et al. 45 | USA | Slight reduction in HLA-A11 frequency in GBS than in controls |
| Sinha et al. 46 | India | HLA-DRB1*0701 associated with GBS with preceding infection |
| Magira et al. 47 | China | DQ beta/DR beta epitopes were associated with AIDP. |
| Guo et al. 48 | China | HLA-A33, DR15 and DQ5 associated with AIDP; HLA-B15, B35 associated with AMAN |
| Ma et al. 49 | Japan | A trend to a higher frequency of HLA-DRB1*0803 in GBS patients with previous C. jejuni infection and positive anti-GM1 antibodies |
| Monos et al. 50 | China | Increased HLA-DRB1*13 frequencies in patients with AIDP |
| Rees et al. 51 | England | Association between HLA-DQB1*0301 and GBS preceded by C. jejuni infection as compared with GBS patients without previous infection, but not with controls |
| Yuki et al. 52 | Japan | HLA-B39 associated with MFS |
| Yuki et al. 53 | Japan | HLA-B35 associated with GBS and anti-GM1 antibodies. |
| Hafez et al. 54 | Egypt | HLA-A3 and B8 were more frequent in GBS patients |
| Gorodezky et al. 55 | Mexico | Increased HLA-DR3 in GBS patients |
Abbreviations: GBS, Guillain-Barré syndrome; AIDP, acute inflammatory demyelinating polyneuropathy; AMAN, acute motor axonal neuropathy; MFS, Miller Fisher syndrome.
Regarding non-HLA, some authors have assessed polymorphisms in other immune receptors. For instance, Geleijns et al. 23 did not find any association between the SNPs in CD14 or TLR4 and GBS. Further, no relation was observed regarding C. jejuni serology, neurological deficit or anti-gangliosides antibodies. In contrast, Nyati et al. 57 showed in Indian patients that Asp299Gly and Thr399Ile TLR4 polymorphisms are associated with a higher risk for GBS, particularly for AMAN subtype.
Caporale et al. 58 showed that subjects with CD1E*01/01 genotype were 2.5 times more likely to develop GBS, whereas subjects with CD1A*01/02 or CD1E*01/02 had a reduced relative risk by 3.6 and 2.3 times, respectively. No relationship was found with anti-ganglioside seropositivity or recent C. jejuni infection.
Polymorphisms in the genes for Fc receptors for immunoglobulin G (IgG) have been associated with less disease severity in GBS, probably due to a more aggressive response induced by those IgG or because of better clearance of circulating antibodies.46 In addition, SNPs in the Fas promoter in particular at position −670, have been associated with the presence of anti-ganglioside antibodies in GBS patients thus suggesting that Fas-FasL interaction is involved in the production of cross-reactive antibodies in GBS.59 TNF −308 G/A or −863C/A polymorphisms are also associated with GBS risk in AMAN and AMSAN subtypes, but not AIDP.60,61 Finally, IL17 (Glu126Gly) and ICAM1 (Gly241Arg) polymorphisms have been associated with GBS and could be genetic susceptibility markers.62
Innate immune response
C. jejuni induces activation of dendritic cells involving cooperative signaling through TLR4-MyD88, TLR4-TRIF axes and NF-kB activation.63 Interestingly, it has been proven that sialylation of C. jejuni LOS is needed to induce an innate immune response through CD14-driven production of IFNβ and TNFα and it is needed to promote B-cell response.64
It has been demonstrated that C. jejuni strains expressing α2–3-linked sialylated surface structures are able to bind to sialoadhesin (Sn, Siglec-1, or CD169), a sialic acid receptor found on a subset of macrophages.65 Siglecs (sialic acid-binding immunoglobulin-like lectin) are expressed on microglial cells, oligodendrocytes and Schwann cells, and can bind to gangliosides containing sialic acid residues.66 Avril et al. 67 demonstrated the potential interaction of siglec-7 with C. jejuni LOS expressing ganglioside-like structures. Different strains that present ganglioside-like structures on their surface were evaluated, and only HS:19 strain with GM1- and GT1a-like epitopes were associated with GBS.68 When sialic acid structures were removed using a sialidase, the observed binding was abrogated, which proved that these interactions were sialic acid-dependent. Further, when siglec-7 was blocked using a monoclonal antibody, no interaction was observed.67
Siglec-7 is considered to have an inhibitory function through the immunoreceptor tyrosine-based inhibitory motifs, present in its cytoplasmic tail and located within particular signaling domain configurations, thus being able to modulate NK-cells response.69 Regarding GBS, in which nerve damage is seen, this model of molecular mimicry may be contradictory. In fact, siglec-7 appears to be usually masked that may regulate its inhibitory activity. However, another mechanism has been proposed in which immunomodulation is inhibited through SOCS3.70
Humoral immune response
Given the high production of IgG against myelin peptides, it has been proposed that memory B cells play a key role in the pathogenesis of GBS.71 Wang et al. 72 found that patients with GBS portrayed higher percentage of memory B cells than healthy controls, and were positively correlated with clinical severity. The humoral immune response is driven by γδ T cells that promotes class switching and enhancement of GM1 antibodies secretion by B cells in patients with GBS associated with C. jejuni infection.73
Tolerance to self-gangliosides is the major factor restricting the antibody response to ganglioside-mimicking LPS.74 However, this is disrupted by C. jejuni-LOS that disturbs gastrointestinal tract immune tolerance and allows an exaggerated peripheral immune response against myelin-like molecules.75
IgG autoantibodies bind effectively to self-antigens and can activate complement system thus promoting demyelination and axonal degeneration.19 IgG, C3 and membrane attack complex were found to be deposited in the nodes of Ranvier in autopsied patients with axonal GBS,76 and in animal models immunized with a bovine brain ganglioside mixture including GM1.77 Interestingly, it has been described that anti-ganglioside antibody complexes activate the complement more frequently than antibodies to single gangliosides.78
The conformation of the oligosaccharide moieties in C. jejuni LOS and the recognition of these complex moieties by the adaptive immune system cannot be predicted by its biochemical structure, since several antibodies to ganglioside complexes are associated with specific clinical subphenotypes.79 The configuration of the oligosaccharide moieties is highly influenced by the microenvironment, and depends on the epitope density and the proximity of other oligosaccharides, and possibly on the nature of the lipid carrier.80 This may partly explain why C. jejuni strains expressing ganglioside-like structures have also been isolated in patients with uncomplicated enteritis, without the production of anti-ganglioside antibodies and subsequent development of GBS.79,81 Recently, in vivo models have shown that C. jejuni DNA-binding protein from starved cells (C-Dps) may cause direct nodal axolemmal damage interfering sulfatide functions. This leads to paranodal myelin detachment and unclustering of sodium channels.81 Further, C. jejuni-related GBS patients disclosed higher serum levels of both C-Dps and anti-C-Dps antibodies than controls,82 thus suggesting that C-Dps could invade the host and induce peripheral nerve damage. Thus, membrane attack complex is not the only mechanism associated with GBS pathogenesis.
Cellular immune response
Perinodal and/or patchy demyelination, perivascular focal lymphocytic infiltration of T cells, myelin swelling and the presence of macrophages within the nerve fibers are typical findings in GBS.83 γδ T cells participate in the early immune response against infections and have been reported in several ADs. They may induce neural injury either by secreting cytokines, activating B cells and macrophages or by a loss of immunological tolerance to self-antigens.84 Further, these cells have shown to be reactive to myelin proteins (for example, P0, P2, PMP22),85 to trigger a response to non-protein antigens through CD1b receptor,86 and to be associated with immunomodulatory responses.87 In vitro stimulation with C. jejuni have proved to enhance production of γδ TCR+ T lymphocytes. Interestingly, Vγ5/Vδ1+ subpopulation is the most common subset involved in nerve cells infiltration.88 Other studies have shown TCR+ T lymphocytes expressing Vβ at GBS onset.89 In a case control study, GBS patients disclosed high levels of Vδ1/CD8+, suggesting that cytotoxicity following infection by C. jejuni might be associated with GBS development.84
Mycoplasma pneumoniae and GBS
Mycoplasma pneumoniae (M. pneumoniae) is a common cause of respiratory tract infection. Respiratory illness due to M. pneumoniae can be followed by neurological complications, such as GBS (AIDP and AMAN) probably associated with anti-glycolipid antibodies.90 The main mechanisms related to the presence of GBS and M. pneumoniae can be summarized in molecular mimicry, cellular, humoral and innate immune response.
Molecular mimicry
Galactocerebroside (Gal-C) is a major glycolipid antigen in the myelin of both the central and peripheral nervous systems.91 Sensitization to Gal-C causes antibody-mediated demyelinating neuropathy, and anti-Gal-C antibody by itself is considered a demyelinating factor (Figure 2). Kusunoki et al. 91 showed that 12% of GBS patients with previous M. pneumoniae infection presented anti-Gal-C antibodies. Further, rabbit anti-Gal-C antibody bound to several glycolipids from M. pneumoniae as well as Gal-C. These data are indicative of molecular mimicry between the myelin glycolipid Gal-C and M. pneumoniae.92,93 Molecular mimicry between GM1 ganglioside and M. pneumoniae has also been reported.90 Regarding GBS variants, associations between M. pneumoniae and Bickerstaff brainstem encephalitis (BBE), MFS and anti-GQ1b, -GM1,-GD1b and -GA1 antibodies production have been observed.94,95 Nevertheless, in a cross-sectional analytical study MFS was not associated with M. pneumoniae infection.95
Cellular immune response
Although molecular mimicry can play an important role in the pathogenesis of GBS related to M. pneumoniae, other factors may be necessary for the development of demyelination, since anti-Gal-C antibody-positive subjects do not always develop neurological disorders.93,96 Indeed, the role of auto-reactive T cells in central nervous system (CNS) disease caused by M. pneumoniae has been suggested.97 Both T-cell activation and anti-Gal-C antibodies are necessary to enhance demyelination in experimental allergic neuritis (EAN) model,96 a murine model of GBS considered an acute T-cell-mediated disease, as it presents inflammation and demyelination of peripheral nerves with an acute monophasic course. T-cell activation and cytokines along with anti-Gal-C antibodies may be associated with GBS pathogenesis.98
Humoral and innate immune response
Some data have emerged to involve B cells abnormalities as the main mechanism of damage in GBS induced by M. pneumoniae. It has been described that patients with GBS following infection by M. pneumoniae disclosed a high immune complex production, and this phenomenon could lead to peripheral nerve ischemia thus producing clinical features and symptoms of disease.99 Additionally, the binding of antibodies to self-antigens elicits complement stimulation and membrane attack complex formation against myelin, yielding demyelination.100
Haemophilus influenzae and GBS
Haemophilus influenzae (H. influenzae) has been associated with GBS development. Koga et al. 101 describe that 9% GBS were previously infected by H. influenzae. It has been described that patients infected with H. influenzae portrayed upper respiratory tract infections, better outcomes, GM1-like ganglioside structure and an AMAN variant.102 Interestingly, both H. influenzae infection and GQ1b antibodies production have been related to MFS, GBS and BBE.103
Mori et al. 104 described a patient with H. influenzae infection and axonal-GBS, in whom a high production of anti-GM1 antibody was observed. Interestingly, the result of ELISA inhibition and cytochemical staining studies evidenced the presence of a GM1-like structure on the surface of H. influenza isolated from one patient.104 Also, in a non-typeable H. influenzae strain, its LOS disclosed similarity with GQ1b.105 This suggests that molecular mimicry plays a role in GBS associated with H. influenzae. Further, patients with GBS associated with H. influenzae infection revealed a higher production of Vβ5.2 T cells than controls. These cells could mediate immune response and eventually could drive cytotoxicity in peripheral nerves.89
Viral infections and GBS
Similar to bacterial infections, GBS-associated viral infection discloses similar mechanisms to bacterial-triggered GBS. However, given the wide variety of viral antigens that can be associated with GBS, the pathophysiology, the clinical course and the outcomes may be different. The most common viruses associated with GBS are cytomegalovirus (CMV), herpes simplex virus (HSV), varicella zoster virus (VZV), Epstein-Barr virus (EBV), hepatitis (A, B and E), human immunodeficiency virus (HIV), and arbovirus, including Dengue virus (DENV), Chikungunya virus (CHIKV), and ZIKV (Figure 1). Given the recent outbreaks of DENV, CHIKV and ZIKV, and the related increase in GBS cases, GBS should be considered as a sentinel surveillance for these arboviruses.106
Cytomegalovirus and GBS
CMV presents four basic structural elements, namely an outer lipid envelope, tegument, a nucleocapsid and an internal nucleoprotein core, where genome resides. Seroprevalence in the general population in the USA reaches 50%,107 although in developing countries may be over 80%.108 Nonetheless, the prevalence increases with age.108 Primary CMV infection is usually asymptomatic and it presents latency in different cells such as endothelial cells, smooth-muscle cells, fibroblasts and monocytes, where replication takes place. Reactivation in immunocompetent hosts occurs intermittently throughout life, which is controlled effectively by the immune system.109
The first description of GBS associated with CMV infection was made by Klemola et al. in 1967.110 It is considered the second most frequent infectious etiology of GBS and the first viral cause.111 In the largest prospective study on GBS and CMV (n=506 patients), the incidence of CMV-GBS was estimated to be between 0.6 and 2.2 cases per 1000 cases of primary CMV infection.111 Steininger et al. 112 provided evidence of both primary CMV infection and endogenous reactivation and reinfection as a cause of GBS. CMV-related GBS may disclose a different phenotype as compared with C. jejuni-related GBS.113 Although both infection-related GBS appear to present a delayed recovery,114 patients with CMV-GBS tend to be younger, to present more frequently cranial nerve involvement and sensory impairment than C. jejuni-related GBS.114 Further, respiratory insufficiency,115 and liver abnormalities,111 appear to be more common. Demyelinating disease is observed in up to 70% of cases.111 A relationship with anti-GM2 ganglioside antibodies has been described and it will be discussed in the following section. Furthermore, there is no solid evidence of CMV cytopathic mechanisms in GBS pathogenesis.116
Molecular mimicry
Anti-GM2 was first reported by Irie et al. 117 in three patients with CMV-GBS. Some authors have found anti-GM2 antibodies in acute CMV infections without GBS thus suggesting that this antibody is not sufficient to elicit neurologic compromise.118 In addition, both IgM anti-GM2 and anti-GalNAc-GD1a, which share GalNACAcβ1–4 (NeuAcα2–3) Gal, have been found simultaneously in CMV-GBS patients.119 Most anti-gangliosides tend to be IgG, but IgM is the most frequent anti-GM2 antibody (Figure 2).120
As CMV envelope contains a number of glycoproteins,121 and GM2 is present in peripheral nerves,122 molecular mimicry has been examined. Irie et al. 117 performed immunoabsorption using AD169 strain CMV-infected cells and reported a decrease in titers of both IgM and IgG GM2 from sera of CMV-GBS patients, supporting the interaction of anti-GM2 with CMV-infected cells. Further, as a strong bound with GM2 was seen, the authors examined reactivity to gangliosides having a terminal GalNAc-Gal structure. Weak bound was observed to those gangliosides. GM2 may recognize GalNAc-Gal terminal of gangliosides, however, sialic acid residues may affect the binding of antibodies. Further, as patient’s serum did not bind to GalNAc-GD1a the authors suggested that the antibodies may recognize internal residues and not only terminal sugar residues.117 Tsukaguchi et al. 123 suggested cross-reactivity between GM2 and GalNAc-GD1a and the possible molecular mimicry of CMV with GalNAc-GD1a.123
Further, Nakamura et al. 124 reported a strong reactivity of serum from CMV-GBS patients, positive for anti-GM2 and/or GD2, to CMV proteins and peripheral nerve proteins. Additionally, Ang et al. 125 demonstrated that fibroblasts infected with CMV express ganglioside-like epitopes that recognize anti-GM2 antibodies. These findings suggest the existence of carbohydrate structures similar to GM2 and/or GD2 on CMV that may cross-react with peripheral nerves.124,125
Regarding potential mimicry with other epitopes, two targets have been identified in EAN, the animal model of GBS: P0 and P2. Adelmann et al. 126 found through bioinformatics homology between P0 and EBV, CMV, VZV and HIV-I. However, Irie et al. 117 did not find reactivity of anti-GM2 with P0 glycoprotein. Nevertheless, evidence for P2 molecular mimicry remains controversial.127
On the other hand, the membrane-organizing extension spike protein moesin is a member of the ERM family cytoskeletal proteins, which comprises ezrin, radixin and moesin. Schwann cells express ERM proteins in microvilli and a pivotal role on myelination has been suggested.128 Further, they are found close to the nodal axolemma.129 As ERM complex is mainly intracellular, its role as a major target for autoantibodies has been questioned.130 Nonetheless, (1) moesin has been detected on the cell surface of lymphocytes and macrophages, indicating a role of moesin in cytokine production during infection or inflammation,131 and (2) it has been hypothesized that an antibody-mediated degradation of nodal microvilli would explain nodal dysfunction, instead of moesin itself.132 Further, moesin classically links the cell membrane and cytoskeleton, and mediates the formation of microtubules and cell adhesion sites.133
In this line, Sawai et al. 134 applied a proteomic-based approach in extracted proteins from schwannoma cells line YST-1, using sera from GBS, inflammatory disease and controls. They found that CMV-GBS patients present serum antibodies against moesin. Antibodies to moesin were found in 5 out of 6 patients, and none of the controls (that is, active CMV infection without neurologic impairment and GBS without CMV). The authors ascertained that levels of serum anti-moesin antibodies precisely discriminates CMV-related AIDP from non-CMV-related AIDP (sensitivity 83%, specificity 93%). Further, by alignment analysis they found homology of 6 consecutive aminoacids (HRGMLR) between moesin and a CMV protein.134 Nevertheless, other authors failed to reproduce Sawai findings.130
Cellular responses
T lymphocyte subpopulations in CMV-GBS patients vary throughout the disease.135 During the progressive phase, T-naive subset was diminished, whereas T-helper was increased, and normalized during the course of the disease. On the other hand, T-cytotoxic cells were increased during progressive and plateau phases, and diminished during the recovery phase. The same behavior was observed for all activated T cells. This supports T-cell activation during GBS, which declined with clinical recovery.135
The presence of HLA-G on immune cells of a CMV-GBS patient suggests the expression of immunotolerant molecules on myelomonocytic cells during CMV infection, which elicits a persistent viral load that may trigger cross-reactive immune response in susceptible individuals. Additionally, HLA-G affects myelomonocytic cells cytokine expression, and shifts it to Th2 profile, yielding an increase in antibody production. This imbalance has been involved in autoimmune peripheral nerve diseases.136
Herpes and GBS
Herpes simplex virus and varicella zoster virus
Besides CMV, HSV-1, 2, 6 have also been associated with axonal degeneration and demyelination.137 It is hypothesized that inflammatory nerve injury caused by cross-reactive antibodies to HSV-1 and 2 through molecular mimicry is the most likely mechanism in GBS (Figure 2).138 Interestingly, titers of HSV-IgM decreased as GBS convalescence progressed, and patients seemed to benefit of plasmapheresis thus suggesting the possible role of auto-antibodies in the HSV-GBS pathogenesis.137,138 It has been suggested that some cases of GBS associated with HSV (EBV, CMV) could have been misdiagnosed due to viral cross-reactivity.139
Other sub-phenotypes of GBS associated with HSV have been described. Yuki et al. 140 reported a patient with BBE, anti-HSV-1 IgM antibody and high anti-GQ1b antibody titers. Interestingly, ophthalmoplegia has been reported in patients following HSV infection and isolation of anti-GQ1b antibodies,141 thus suggesting a possible misdiagnosis of BBE and MFS.140 HSV-1-infected human fibroblasts can express the GQ1b epitope suggesting a possible role for molecular mimicry in BBE and MFS.125
Miyaji et al. 142 concluded that HSV-1 infection was associated with both decreased and increased ganglioside-related gene expression (that is, increased: β3-galactosyltransferase-IV, α2,8-sialyltransferase-I; decreased: β-galactoside α2,3-sialyltransferase-II) whereas HSV-2 induced a higher gene expression than controls (that is, β3-galactosyltransferase-IV, α2,8-sialyltransferase-I) in neuroblastoma and astrocytoma cells. Thus suggesting that HSV could enhance the production of self-antigens.
Since the prevalence of VZV is higher in immunosuppressed patients, it has been stated that many cases of GBS secondary to VZV infection could be underdiagnosed.143 Interestingly, patients with VZV infection and GBS portrayed a reduced CD8+ T cells count during illness and helper/suppressor ratio was increased.144 Further, GBS development following VZV infection have been recognized to emerge acute or subacutely and short latent periods of infection were associated with more severe illness.145 Although a pathogenic mechanism of VZV and GBS has not been clearly elucidated, a population-based study disclosed that patients with VZV infection have 18.37 times higher risk for GBS development.146
Conflicting evidence has emerged regarding HSV-6 and GBS.147 Although it has been associated with GBS development,148 recent reports indicate that the incidence of HSV-6 infection in GBS patients is low.149 Since HSV-6 structure is similar to CMV, some authors have suggested the possible role of cross-reactivity test between these two viruses.148 Lack of experimental studies hinders any conclusion regarding HSV-6 and the development of GBS.
Epstein–Barr virus
In the classical study of Jacobs et al.,141 infections with C. jejuni (32%), CMV (13%) and EBV (10%) were significantly more frequent in GBS patients than in controls.141 EBV can directly infiltrate the peripheral nervous system, usually resulting in focal or multifocal neuritis rather than polyneuropathy.150 Neuropathological studies have revealed perivascular lymphocytic infiltrate, parenchymal edema, microglial proliferation and inflammatory demyelinating lesions.151 Further, in situ hybridization using EBV-encoded small RNA-1 (EBER1) did not disclose positive signals, suggesting that demyelination with secondary axonal degeneration occurs without infiltration of EBV-infected lymphocytes.
The role of humoral immune response has been suggested by a case report of a patient with anti-GQ1b antibodies following EBV infection and negative C. jejuni serology.152 Bioinformatics analysis disclosed that microbial sequence (that is, AKGQP) shares 54–58 homologous residues with myelin P0 protein.126 However, in a cross-sectional study no anti-gangliosides antibodies were identified in patients with GBS and EBV infection.153
Vascular damage has been proposed as a possible mechanism of GBS secondary to EBV. EBV infection may cause vasculitis by direct invasion of endothelial cells or by immune complex-mediated vessel inflammation.151 After vascular damage, ischemia develops and neural damage may occur.150
Hepatitis virus and GBS
Regarding hepatitis B virus (HBV), some authors suggest that deposition of circulating hepatitis B surface antigen containing immune complexes (HBsAg-ICs) along nerve structures could have resulted in angitic or ischemic lesions leading to the neuropathy of GBS. Interestingly, high levels of HBsAg-ICs have been found in patients with GBS and HBV infection.154 Regarding hepatitis A and hepatitis C viruses, immune complex deposition along the vascular endothelium with a resulting vasculitis around the nerve, may explain the association with GBS (Figure 2).155
Additionally, the hypothesis of an imbalance between T-cell subsets, with decreased T suppressor activity induced by circulating immune complexes, have been proposed.156 Lymphocytes from GBS patients were incubated with bone-marrow-derived lymphoblastoid cell line PGLC-33H. A significant decrease in a subpopulation of peripheral blood T lymphocytes that form ‘PGLC rosettes’ (PGR) with the PGLC-33H cells was observed.157 Then, HBV polymerase was found to share six consecutive aminoacids with the encephalitogenic site of rabbit myelin basic protein. Thus, viral infection may trigger the production of antibodies and mononuclear cells activation through molecular mimicry.158
Tropism by hepatitis E virus (HEV) for nervous system has been reported.159 HEV might acquire host RNA sequences that confer the ability to infect multiple cell types and, thus potentially infect and damage the CNS.160 However, this mechanism has not been suggested for GBS. Although positive serology has been detected in patients with GBS, lack of circulating HEV RNA strengthen the hypothesis of an immune-mediated process.161 Several patients with GBS and HEV-related infection may develop antibodies to gangliosides GM1 and GM2, thus suggesting a possible role for molecular mimicry.162,163
Human immunodeficiency virus and GBS
GBS has been observed during the acute phase of HIV and recurrence of viremia with recover after intravenous immunoglobulin.164 In murine models, sciatic nerves were exposed to HIV-1 envelope protein gp120, disclosing axonal swelling and increased TNFα within the nerve trunk, thus suggesting that HIV can cause nerve damage.165 In addition, a patient with HIV infection and GBS portrayed anti-GM1 IgG-positive antibodies. However, after improvement of CD4+ T-cell count, neither anti-ganglioside antibody titer were increased nor tetraparesis were worsened,166 suggesting a limited role of T-cell immunity in pathogenesis of GBS associated with HIV infection. It has been proposed that at lower T cells CD4+ count, the CMV infection could be the cause of GBS associated with HIV.167
Dengue virus and GBS
Dengue is a mosquito-borne viral disease caused by one of four closely related dengue virus serotypes (DENV 1–4). From the time DENV was recognized as a clinical entity, neurological manifestations have been described.168 Neurologic involvement occurs in 5% of DENV cases. The major mechanisms of the disease may be related to direct viral infection or post-infectious autoimmune process.169 Since the end of the 1990s, evidence of DENV neurotropism has increased and the number of reports on dengue patients with virus isolation from CSF or brain tissue has risen.170
The pathogenesis of dengue neurological involvement and underlying virus–host interactions remain to be elucidated. However, DENV infection might result in the release of mediators, including cytokines, chemokines and complement-associated viral particles, with vasoactive or pro-coagulant properties, capillary leakage, increased fibrinolysis and bleeding.171 Furthermore, DENV could trigger a complex immune response, resulting in IL-2, IFNγ, and TNFα overproduction and inversion of the CD4:CD8 ratio.172
In GBS patients, IgM antibodies against DENV in CSF have been detected together with virus isolates from brain tissue and CSF thus suggesting a direct virus invasion of the CNS and neurotropism of DENV.170 Then, neurological involvement can be related to the neurotropic effect of the virus and the systemic complication of infection and can also be immune mediated.173 As other infections, DENV may trigger an abnormal immune response, which can cross-react with the peripheral nerve via molecular mimicry.
Chikungunya virus and GBS
CHIKV is an arthropod-borne alphavirus that belongs to the family Togaviridae and mainly transmitted to humans by the Aedes mosquito. The clinical presentation comprises a flu-like syndrome including fever, headache, arthralgia, myalgia and maculopapular rash that occur after 10 days of incubation.174 Neurologic manifestations of CHIKV infection are unusual; however, in the last years nervous system involvement associated to this arbovirus has been described.175
Neurological symptoms start during the invasion phase, prior to seroconversion. The presence of CSF abnormalities and CHIKV-specific IgM intrathecal synthesis were highly suggestive of CHIKV-induced pathology in the nervous system.176 Tropism for brain tissue has been validated in murine models of CHIKV neuroinfection.177 However, animal models have focused on musculoskeletal symptoms since arthropathy is one of the main consequences of CHIKV infection.
Zika virus and GBS
ZIKV belongs to the Flaviviridae family, an enveloped, positive stranded RNA virus. ZIKV relies mainly upon mosquito vectors for viral spread. Recently, ZIKV has rapidly emerged and is now widespread across the Americas.178 While the infection is generally asymptomatic or limited to flu-like symptoms, ZIKV is increasingly being implicated in severe neurological diseases including congenital microcephaly and GBS.6,179
Recent evidence has described how ZIKV is able to counter host IFN signaling through degradation of STAT2 or inhibition of RIG-I-mediated IFN induction.180,181 Identification of the host cell receptors required for ZIKV infection has also been a focus of research, with cell culture reports initially identifying AXL receptor tyrosine kinase as a potential candidate for modulating entry and cellular immune responses.182 Further studies comparing infection between BalBc WT and Axl−/− mice revealed no significant difference in recovered virus titers from mice brains.182 These conflicting results might be due to ZIKV NS5 expression results in proteasomal degradation of the IFN-regulated transcriptional activator STAT2 from humans, but not mice, which may explain the requirement for IFN deficiency to observe ZIKV-induced disease in mice.180 These results highlight the necessary care that must be taken when translating cell culture results to organism level conclusions. Additionally, the Asian strains of ZIKV indicates that a combination of factors, such as genetic variation in the NS5 gene as a result of mutations or recombination events and the reacquisition of a E-154 glycosylation motif, could contribute to neurovirulence.183
Recent in vitro studies have shown that axonal changes occurred in the absence of overt infection or neuronal death. This suggests that axons could be injured by soluble or contact-dependent factors in their environment. Previously, it has been shown that ZIKV-infected neural crest cells produce cytokines at levels that kill or cause aberrant differentiation of neural progenitors.184 Moreover, in myelinating CNS cultures, a reduction of myelin 6 days post infection with ZIKV was observed. This could reflect a failure of myelin to form normally or loss of previously formed myelin. The myelin that remained often appeared fragmented and many infected positive oligodendrocyte cell bodies lacked myelin sheaths. These observations tend to exclude the possibility that myelination is simply arrested. Rather, myelin changes probably represent a combination of the arrest of myelination and the degradation of previously established sheaths.185
Following reports linking ZIKV infection to GBS, it has been analyzed the peptide sharing between the virus and human proteins potentially related to GBS related proteins.186 The immunological potential of the peptide sharing was described, which appeared to favor an autoimmune cross-reactive connection between ZIKV and GBS. Indeed, a conspicuous number of peptides shared between the virus and proteins associated, when altered, with (de)myelination and axonal neuropathy are present in >500 epitopes that have been cataloged as immunopositive in the human host.186
The cytopathic effects of ZIKV are poorly characterized. Innate immunity controls ZIKV infection and disease in most infected patients through mechanisms that remain to be understood. The failure of any of those mechanisms could be the clue to the triggering of GBS after ZIKV infection. It has been recently reported that ZIKV induces massive vacuolization, followed by implosive cell death in human epithelial cells, primary skin fibroblasts and astrocytes. This phenomenon is exacerbated when IFN-induced transmembrane protein (IFITM3) levels are low. It is reminiscent of paraptosis, a caspase-independent programmed cell death, associated with the formation of large cytoplasmic vacuoles.187 IFITM3 is a major component of host innate control of ZIKV and is likely implicated in the low pathogenicity observed in most infected individuals.188 IFITM3 is required to prevent pathogenesis of Influenza A virus or west Nile virus infections in mice. SNP rs12252-C in human IFITM3 is predicted to produce a truncated form of IFITM3, which is confined to the plasma membrane.189 Since this polymorphism may be associated with severe outcomes following Influenza A virus infection,190 it could be a plausible genetic variant to be evaluated in ZIKV infection.187 Another candidate genes to be evaluated could be those incriminated into the innate anti-viral immunity (for example, APOBEC3G).
Studies suggest that the expression of pre-membrane protein (prM) resulted in cell cycle G1 accumulation, whereas membrane-anchored capsid (anaC), membrane protein (M), envelope protein (E) and nonstructural protein 4A (NS4A) caused cell cycle G2/M accumulation. A mechanistic study revealed that NS4A-induced cellular hypertrophy and growth restriction were mediated specifically through the target of rapamycin (TOR) cellular stress pathway involving Tor1 and type 2A phosphatase activator Tip41.191 Moreover, the appearance of neurologic syndromes associated with ZIKV could be related to the persistence of the virus, which can persist for a substantially long period, likely as a result of activation of mTOR, proinflammatory, and anti-apoptotic pathways that promote survival of infected cells, as well as exclusion of virus-specific antibodies from CSF. If persistent or occult neurologic and lymphoid disease similarly occurs following clearance of peripheral virus in ZIKV-infected humans, such a finding would have important clinical implications. It is likely that mTOR activation and persistent virus in the CNS contribute to the hallmark neuropathology of ZIKV infection.192
Furthermore, it is conceivable that antibody-dependent enhancement (ADE) of ZIKV infection could have resulted in high viral titers, and hence an unusually vigorous immune response based on high levels of pro-inflammatory cytokines,193 which could trigger the emergence of GBS in some people even with low genetic susceptibility to the illness. The phenomenon of ADE has been demonstrated in experiments involving the ZIKV.194 Furthermore, it was reported that DENV immune sera cross-react with ZIKV without neutralizing the virions. In fact, this response enhanced the replication of the ZIKV. The use of a panel of anti-DENV monoclonal antibodies revealed that the vast majority also reacted with the ZIKV.194 Noteworthy, the majority of GBS patients had evidence of prior DENV infection,6 which makes the phenomenon of ADE a prime suspect, as far as the development of GBS in these patients is concerned.183 In addition to DENV, a previous or coexistant infection with M. pneumoniae has been suggested as cofactor to GBS development in patients with ZIKV infection.6
However, the above does not fully explain the apparent capacity of Asian lineages of ZIKV to trigger GBS in some patients, which appears to be lacking or greatly attenuated in the African strains. In order to propose such an explanation, we will revisit the phenomenon of NS1 codon adaptation initially described by Freire et al. 195 Hence, codon preferences of the Asian and African lineages are distinct, and codon usage adaptation in the NSI gene for human host housekeeping genes was detected.195 Codon usage adaptation to human hosts displayed by the NS1 gene of the Asian strains also seems to be an important factor, as this phenomenon is known to increase translational efficiency leading to increased fitness survival and higher titers, as well as enhancing immune evasion strategies. Changes in NS1 structure between the African and Asian strains may also contribute to increased neuro-invasiveness and a distinct pattern of host gene activation.183
Transverse myelitis
Acute transverse myelitis is a subgroup of various conditions characterized by focal inflammation of the spinal cord and resultant neural injury.196 The etiologies of myelopathies are varied and can be subdivided into compressive and non-compressive causes. While compressive myelopathies stem from trauma and intra- or extraspinal tumors, the etiologies of non-compressive myelopathies can be classified as delayed radiation effects, ischemic, paraneoplastic, systemic ADs and infectious or parainfectious.11
Among the latter, microbial agents play an important role in the pathogenesis of this syndrome. Indeed, infectious diseases frequently precede the onset of TM (Figure 1). Four main mechanisms can explain how infection can trigger TM: bystander effect, molecular mimicry, microbial superantigen-mediated inflammation and humoral response.197 Regarding bystander effect, the neurologic injury may be associated with direct or indirect microbial infection with immune-mediated damage against the agent; the infectious agent is required to be in the CNS.11 As in GBS, molecular mimicry has long been thought to play a primary role in triggering TM, being C. jejuni one of the most important agents involved in this mechanism.198 Some case reports of TM developed in close temporal association with C. jejuni showed high titers of anti-GM1 ganglioside, that could cross-react with LOS from a C. jejuni strain.199
Additionally, some cases of TM after M. pneumoniae infection have been also described,200 and although one mechanism could be molecular mimicry between M. pneumoniae antigens and another class of nervous system glycosphingolipids, as Gal-C, the production of anti-Gal-C antibodies in these patients has not been explored.11
Immune complex deposition, a prominent feature of several ADs, has been an additional mechanism observed in patients with TM. For instance, patients following HBV booster immunization present extremely high serum and CSF levels of anti-HbSAg antibodies.201 Circulating antibodies may form immune complexes that deposit in focal areas of the spinal cord.197 Such a mechanism was proposed in a patient with recurrent TM and high titers of HbSAg, in whom circulating immune complexes containing anti-HbSAg were detected in the serum and CSF during the acute phase and the disappearance of these complexes following treatment correlated with functional recovery.202
Other possible link between an antecedent infection and the development of TM is microbial super-antigens, which can trigger a fulminant activation of lymphocytes.203 Super-antigens activate T-lymphocytes by interacting with the more conserved Vβ region.204 Stimulation of large numbers of lymphocytes may trigger ADs by activating autoreactive T-cell clones.205 The production of abnormal antibodies may activate other components of the immune system and/or recruit additional cellular elements to the spinal cord, as observed in patients with neuromyelitis optica.120,206 Finally, even though parasite and viral infections such as Enterobium vermicularis, DENV, ZIKV, have been related to TM, the mechanisms to explain these associations have not been elucidated.6,207,208
Conclusions and perspectives
Infectious diseases play an important role in the development of both GBS and TM. GBS is a post-infectious autoimmune syndrome that could act as sentinel surveillance for several infectious outbreaks, such as arboviruses. The fact that only some individuals develop GBS and TM after an infection demonstrates the complex host–guest interaction triggering these conditions. Therefore, studies aimed at simultaneously evaluating the genetic of both the guest and the host are warranted to reveal risk or protective factors at the population level in order to implement preventive measures and find targets for therapeutic interventions.
Electronic supplementary material
Acknowledgements
We express our gratitude to Julián Barahona-Correa for his help in the earliest stages of the preparation of the manuscript. This work was supported by Universidad del Rosario (ABN011), and Colciencias (747–2016), Bogotá, Colombia.
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2017.142
References
- 1.Wijdicks EF, Klein CJ. Guillain–Barré Syndrome. Mayo Clin Proc. 2017;92:467–479. doi: 10.1016/j.mayocp.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Anandan C, Khuder SA, Koffman BM. Prevalence of autonomic dysfunction in hospitalized patients with Guillain-Barré syndrome. Muscle Nerve. 2017;56:331–333. doi: 10.1002/mus.25551. [DOI] [PubMed] [Google Scholar]
- 3.Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barré syndrome. Lancet. 2016;388:717–727. doi: 10.1016/S0140-6736(16)00339-1. [DOI] [PubMed] [Google Scholar]
- 4.Sejvar JJ, Baughman AL, Wise M, Morgan OW. Population incidence of Guillain-Barré syndrome: a systematic review and meta-analysis. Neuroepidemiology. 2011;36:123–133. doi: 10.1159/000324710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heikema AP, Islam Z, Horst-Kreft D, Huizinga R, Jacobs BC, Wagenaar JA, et al. Campylobacter jejuni capsular genotypes are related to Guillain-Barré syndrome. Clin Microbiol Infect. 2015;21:1–24. doi: 10.1016/j.cmi.2015.05.031. [DOI] [PubMed] [Google Scholar]
- 6.Anaya JM, Rodríguez Y, Monsalve DM, Vega D, Ojeda E, González-Bravo D, et al. A comprehensive analysis and immunobiology of autoimmune neurological syndromes during the Zika virus outbreak in Cúcuta, Colombia. J Autoimmun. 2017;77:123–138. doi: 10.1016/j.jaut.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Ang CW. Structure of Campylobacter jejuni lipopolysaccharides determines antiganglioside specificity and clinical features of Guillain-Barre and Miller Fisher patients. Infect Immun. 2002;70:1202–1208. doi: 10.1128/IAI.70.3.1202-1208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nyati KK, Nyati R. Role of Campylobacter jejuni infection in the pathogenesis of Guillain-Barré syndrome: an update. Biomed Res Int. 2013;2013:852195. doi: 10.1155/2013/852195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes G, Benjamin S, Bowen JD, Cutter N, De Lateur BJ, Dietrich WD, et al. Proposed diagnostic criteria and nosology of acute transverse myelitis. Neurology. 2002;59:499–505. doi: 10.1212/wnl.59.4.499. [DOI] [PubMed] [Google Scholar]
- 10.Frohman EM, Wingerchuk DM. Transverse myelitis. N Engl J Med. 2010;363:564–572. doi: 10.1056/NEJMcp1001112. [DOI] [PubMed] [Google Scholar]
- 11.Borchers AT, Gershwin ME. Transverse myelitis. Autoimmun Rev. 2012;11:231–248. doi: 10.1016/j.autrev.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 12.Islam Z, Jacobs BC, van Belkum A, Mohammad QD, Islam MB, Herbrink P, et al. Axonal variant of Guillain-Barre syndrome associated with Campylobacter infection in Bangladesh. Neurology. 2010;74:581–587. doi: 10.1212/WNL.0b013e3181cff735. [DOI] [PubMed] [Google Scholar]
- 13.Hughes RA, Rees JH. Clinical and epidemiologic features of Guillain-Barré syndrome. J Infect Dis. 1997;176:92–98. doi: 10.1086/513793. [DOI] [PubMed] [Google Scholar]
- 14.Gardner SP, Kendall KJ, Taveirne ME, Olson JW. Complete genome sequence of Campylobacter jejuni subsp. jejuni ATCC 35925. Genome Announc. 2017;5:1–2. doi: 10.1128/genomeA.00743-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirkpatrick BD, Tribble DR. Update on human Campylobacter jejuni infections. Curr Opin Gastroenterol. 2011;27:1–7. doi: 10.1097/MOG.0b013e3283413763. [DOI] [PubMed] [Google Scholar]
- 16.Tam CC, Rodrigues LC, Petersen I, Islam A, Hayward A, O’Brien SJ. Incidence of Guillain-Barré syndrome among patients with campylobacter infection: a general practice Research Database Study. J Infect Dis. 2006;194:95–97. doi: 10.1086/504294. [DOI] [PubMed] [Google Scholar]
- 17.Phongsisay V. The immunobiology of Campylobacter jejuni: Innate immunity and autoimmune diseases. Immunobiology. 2016;221:535–543. doi: 10.1016/j.imbio.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 18.Varki A, Sharon NHistorical background and overview In:Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR et al 3 eds. Essentials of Glycobiology. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY. 2009 pp 1–22.
- 19.Yuki N. Ganglioside mimicry and peripheral nerve disease. Muscle Nerve. 2007;35:691–711. doi: 10.1002/mus.20762. [DOI] [PubMed] [Google Scholar]
- 20.Godschalk PCR, Heikema AP, Gilbert M, Komagamine T, Wim Ang C, Glerum J, et al. The crucial role of Campylobacter jejuni genes in anti-ganglioside antibody induction in Guillain-Barré syndrome. J Clin Invest. 2004;114:1659–1665. doi: 10.1172/JCI15707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moran AP, Prendergast MM. Molecular mimicry in Campylobacter jejuni and Helicobacter pylori lipopolysaccharides: contribution of gastrointestinal infections to autoimmunity. J Autoimmun. 2001;16:241–256. doi: 10.1006/jaut.2000.0490. [DOI] [PubMed] [Google Scholar]
- 22.Phongsisay V, Perera VN, Fry BN. Exchange of lipooligosaccharide synthesis genes creates potential Guillain-Barre syndrome-inducible strains of Campylobacter jejuni. Infect Immun. 2006;74:1368–1372. doi: 10.1128/IAI.74.2.1368-1372.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geleijns K, Schreuder GMT, Jacobs BC, Sintnicolaas K, van Koningsveld R, Meulstee J, et al. HLA class II alleles are not a general susceptibility factor in Guillain-Barre syndrome. Neurology. 2005;64:44–49. doi: 10.1212/01.WNL.0000148727.02732.01. [DOI] [PubMed] [Google Scholar]
- 24.Nachamkin I, Liu J, Li M, Ung H, Moran AP, Prendergast MM, et al. Campylobacter jejuni from patients with Guillain-Barre syndrome preferentially expresses a GD1a-like epitope. Infect Immun. 2002;70:5299–5303. doi: 10.1128/IAI.70.9.5299-5303.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilbert M, Karwaski MF, Bernatchez S, Young NM, Taboada E, Michniewicz J, et al. The genetic bases for the variation in the lipo-oligosaccharide of the mucosal pathogen, Campylobacter jejuni. Biosynthesis of sialylated ganglioside mimics in the core oligosaccharide. J Biol Chem. 2002;277:327–337. doi: 10.1074/jbc.M108452200. [DOI] [PubMed] [Google Scholar]
- 26.Koga M, Takahashi M, Masuda M, Hirata K, Yuki N. Campylobacter gene polymorphism as a determinant of clinical features of Guillain-Barré syndrome. Neurology. 2005;65:1376–1381. doi: 10.1212/01.wnl.0000176914.70893.14. [DOI] [PubMed] [Google Scholar]
- 27.Koga M, Yuki N. Campylobacter jejuni cst-II polymorphisms and association with development of Guillain-Barré syndrome. Neurology. 2007;69:1727–1728. doi: 10.1212/01.wnl.0000285517.52448.e8. [DOI] [PubMed] [Google Scholar]
- 28.Yuki N, Susuki K, Koga M, Nishimoto Y, Odaka M, Hirata K, et al. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barre syndrome. Proc Natl Acad Sci USA. 2004;101:11404–11409. doi: 10.1073/pnas.0402391101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Houliston RS, Endtz HP, Yuki N, Li J, Jarrell HC, Koga M, et al. Identification of a sialate O-acetyltransferase from Campylobacter jejuni: demonstration of direct transfer to the C-9 position of terminalalpha-2, 8-linked sialic acid. J Biol Chem. 2006;281:11480–11486. doi: 10.1074/jbc.M512183200. [DOI] [PubMed] [Google Scholar]
- 30.Koga M, Gilbert M, Li J, Yuki N. Complex of GM1- and GD1a-Like Lipo-oligosaccharide mimics GM1b, inducing anti-GM1b antibodies. PLoS ONE. 2015;10:e0124004. doi: 10.1371/journal.pone.0124004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodfellow JA. Overexpression of GD1a ganglioside sensitizes motor nerve terminals to anti-GD1a antibody-mediated injury in a model of acute motor axonal neuropathy. J Neurosci. 2005;25:1620–1628. doi: 10.1523/JNEUROSCI.4279-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aspinall GO, Fujimoto S, McDonald AG, Pang H, Kurjanczyk LA, Penner JL. Lipopolysaccharides from Campylobacter jejuni associated with Guillain-Barré syndrome patients mimic human gangliosides in structure. Infect Immun. 1994;62:2122–2125. doi: 10.1128/iai.62.5.2122-2125.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiang SL, Zhong M, Cai FC, Deng B, Zhang XP. The sialic acid residue is a crucial component of C. jejuni lipooligosaccharide ganglioside mimicry in the induction Guillain–Barré syndrome. J Neuroimmunol. 2006;174:126–132. doi: 10.1016/j.jneuroim.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Linton D, Gilbert M, Hitchen PG, Dell A, Morris HR, Wakarchuk WW, et al. Phase variation of a β-1,3 galactosyltransferase involved in generation of the ganglioside GM1-like lipo-oligosaccharide of Campylobacter jejuni. Mol Microbiol. 2000;37:501–514. doi: 10.1046/j.1365-2958.2000.02020.x. [DOI] [PubMed] [Google Scholar]
- 35.Guerry P. Phase variation of Campylobacter jejuni 81-176 lipooligosaccharide affects ganglioside mimicry and invasiveness in vitro. Infect Immun. 2002;70:787–793. doi: 10.1128/iai.70.2.787-793.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Godschalk PCR, van Belkum A, van den Braak N, van Netten D, Ang CW, Jacobs BC, et al. PCR-restriction fragment length polymorphism analysis of Campylobacter jejuni genes involved in lipooligosaccharide biosynthesis identifies putative molecular markers for Guillain-Barré syndrome. J Clin Microbiol. 2007;45:2316–2320. doi: 10.1128/JCM.00203-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taboada EN, van Belkum A, Yuki N, Acedillo RR, Godschalk PC, Koga M, et al. Comparative genomic analysis of Campylobacter jejuni associated with Guillain-Barré and Miller Fisher syndromes: neuropathogenic and enteritis-associated isolates can share high levels of genomic similarity. BMC Genomics. 2007;8:359. doi: 10.1186/1471-2164-8-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alvarez LAJ, Bourke B, Pircalabioru G, Georgiev AY, Knaus UG, Daff S, et al. Cj1411c encodes for a cytochrome P450 involved in Campylobacter jejuni 81-176 pathogenicity. PLoS ONE. 2013;8:e75534. doi: 10.1371/journal.pone.0075534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fry BN, Feng S, Chen YY, Newell DG, Coloe PJ, Korolik V. The galE gene of Campylobacter jejuni is involved in lipopolysaccharide synthesis and virulence. Infect Immun. 2000;68:2594–2601. doi: 10.1128/iai.68.5.2594-2601.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shu XM, Cai FC, Zhang XP. Carbohydrate mimicry of Campylobacter jejuni lipooligosaccharide is critical for the induction of anti-GM1 antibody and neuropathy. Muscle Nerve. 2006;33:225–231. doi: 10.1002/mus.20457. [DOI] [PubMed] [Google Scholar]
- 41.Schirmer L, Worthington V, Solloch U, Loleit V, Grummel V, Lakdawala N, et al. Higher frequencies of HLA DQB1*05:01 and anti-glycosphingolipid antibodies in a cluster of severe Guillain-Barré syndrome. J Neurol. 2016;263:2105–2113. doi: 10.1007/s00415-016-8237-6. [DOI] [PubMed] [Google Scholar]
- 42.Hasan ZN, Zalzala HH, Mohammedsalih HR, Mahdi BM, Abid LA, Shakir ZN, et al. Association between human leukocyte antigen-DR and demylinating Guillain-Barre syndrome. Neurosci. 2014;19:301–305. [PMC free article] [PubMed] [Google Scholar]
- 43.Fekih-Mrissa N, Mrad M, Riahi A, Sayeh A, Zaouali J, Gritli N, et al. Association of HLA-DR/DQ polymorphisms with Guillain-Barré syndrome in Tunisian patients. Clin Neurol Neurosurg. 2014;121:19–22. doi: 10.1016/j.clineuro.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 44.Blum S, Csurhes P, Reddel S, Spies J, McCombe P. Killer immunoglobulin-like receptor and their HLA ligands in Guillain-Barré Syndrome. J Neuroimmunol. 2014;267:92–96. doi: 10.1016/j.jneuroim.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 45.Kaslow RA, Sullivan-Bolyai JZ, Hafkin B, Schonberger LB, Kraus L, Moore MJ, et al. HLA antigens in Guillain-Barre syndrome. Neurology. 1984;34:240–242. doi: 10.1212/wnl.34.2.240. [DOI] [PubMed] [Google Scholar]
- 46.Sinha S, Prasad KN, Jain D, Nyati KK, Pradhan S, Agrawal S. Immunoglobulin IgG Fc-receptor polymorphisms and HLA class II molecules in Guillain-Barré syndrome. Acta Neurol Scand. 2010;122:21–26. doi: 10.1111/j.1600-0404.2009.01229.x. [DOI] [PubMed] [Google Scholar]
- 47.Magira EE, Papaioakim M, Nachamkin I, Asbury AK, Li CY, Ho TW, et al. Differential distribution of HLA-DQ /DR epitopes in the two forms of Guillain-Barre syndrome, acute motor axonal neuropathy and acute inflammatory demyelinating polyneuropathy (AIDP): identification of DQ epitopes associated with susceptibility to and pro. J Immunol. 2003;170:3074–3080. doi: 10.4049/jimmunol.170.6.3074. [DOI] [PubMed] [Google Scholar]
- 48.Guo L, Wang W, Li C, Liu R, Wang G. The association between HLA typing and different subtypes of Guillain Barré syndrome. Zhonghua nei ke za zhi. 2002;41:381–383. [PubMed] [Google Scholar]
- 49.Ma JJ, Nishimura M, Mine H, Kuroki S, Nukina M, Ohta M, et al. HLA and T-cell receptor gene polymorphisms in Guillain-Barré syndrome. Neurology. 1998;51:379–384. doi: 10.1212/wnl.51.2.379. [DOI] [PubMed] [Google Scholar]
- 50.Monos DS, Papaioakim M, Ho TW, Li CY, McKhann GM. Differential distribution of HLA alleles in two forms of Guillain-Barre syndrome. J Infect Dis. 1997;176((Suppl 2)):S180–S182. doi: 10.1086/513786. [DOI] [PubMed] [Google Scholar]
- 51.Rees JH, Vaughan RW, Kondeatis E, Hughes RAC. HLA-class II alleles in Guillain-Barré syndrome and Miller Fisher syndrome and their association with preceding Campylobacter jejuni infection. J Neuroimmunol. 1995;62:53–57. doi: 10.1016/0165-5728(95)00102-8. [DOI] [PubMed] [Google Scholar]
- 52.Yuki N, Sato S, Tsuji S, Ogawa K, Miyatake T. Human leukocyte antigens in Fisher’s syndrome. Ann Neurol. 1993;33:655–657. doi: 10.1002/ana.410330617. [DOI] [PubMed] [Google Scholar]
- 53.Yuki N, Sato S, Itoh T, Miyatake T. HLA-B35 and acute axonal polyneuropathy following Campylobacter infection. Neurology. 1991;41:1561–1563. doi: 10.1212/wnl.41.10.1561. [DOI] [PubMed] [Google Scholar]
- 54.Hafez M, Nagaty M, Al-Tonbary Y, El-Shennawy FA, El-Mongui A, El-Sallab S, et al. HLA-antigens in Guillain-Barre syndrome. J Neurogenet. 1985;2:285–290. doi: 10.3109/01677068509102324. [DOI] [PubMed] [Google Scholar]
- 55.Gorodezky C, Varela B, Castro-Escobar LE, Chávez-Negrete A, Escobar-Gutiérrez A, Martínez-Mata J. HLA-DR antigens in Mexican patients with Guillain-Barré syndrome. J Neuroimmunol. 1983;4:1–7. doi: 10.1016/0165-5728(83)90058-9. [DOI] [PubMed] [Google Scholar]
- 56.McCombe PA, Csurhes PA, Greer JM. Studies of HLA associations in male and female patients with Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) J Neuroimmunol. 2006;180:172–177. doi: 10.1016/j.jneuroim.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 57.Nyati KK, Prasad KN, Verma A, Singh AK, Rizwan A, Sinha S, et al. Association of TLR4 Asp299Gly and Thr399Ile polymorphisms with Guillain-Barré syndrome in Northern Indian population. J Neuroimmunol. 2010;218:116–119. doi: 10.1016/j.jneuroim.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 58.Caporale CM, Papola F, Fioroni MA, Aureli A, Giovannini A, Notturno F, et al. Susceptibility to Guillain-Barré syndrome is associated to polymorphisms of CD1 genes. J Neuroimmunol. 2006;177:112–118. doi: 10.1016/j.jneuroim.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 59.Geleijns K, Laman JD, van Rijs W, Tio-Gillen AP, Hintzen RQ, van Doorn PA, et al. Fas polymorphisms are associated with the presence of anti-ganglioside antibodies in Guillain–Barré syndrome. J Neuroimmunol. 2005;161:183–189. doi: 10.1016/j.jneuroim.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 60.Jiao H, Wang W, Wang H, Wu Y, Wang L. Tumor necrosis factor alpha 308 G/A polymorphism and Guillain-Barré syndrome risk. Mol Biol Rep. 2012;39:1537–1540. doi: 10.1007/s11033-011-0892-1. [DOI] [PubMed] [Google Scholar]
- 61.Jahan I, Ahammad RU, Farzana KS, Khalid MM, Islam MB, Rahman MI, et al. Tumor necrosis factor-alpha -863C/A polymorphism is associated with Guillain-Barré syndrome in Bangladesh. J Neuroimmunol. 2017;310:46–50. doi: 10.1016/j.jneuroim.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 62.Kharwar NK, Prasad KN, Singh K, Paliwal VK, Modi DR. Polymorphisms of IL-17 and ICAM-1 and their expression in Guillain-Barré syndrome. Int J Neurosci. 2017;127:680–687. doi: 10.1080/00207454.2016.1231186. [DOI] [PubMed] [Google Scholar]
- 63.Rathinam VAK, Appledorn DM, Hoag KA, Amalfitano A, Mansfield LS. Campylobacter jejuni-induced activation of dendritic cells involves cooperative signaling through toll-like receptor 4 (TLR4)-MyD88 and TLR4-TRIF axes. Infect Immun. 2009;77:2499–2507. doi: 10.1128/IAI.01562-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chatzipanagiotou S, Michalopoulou M, Marinou I, Boufidou F, Papavasileiou E, Trikka-Graphakos E, et al. Investigation of possible cytokine induction in peripheral blood mononuclear cells by heat-stable serotypes of Campylobacter jejuni. Clin Microbiol Infect. 2005;11:63–65. doi: 10.1111/j.1469-0691.2004.01038.x. [DOI] [PubMed] [Google Scholar]
- 65.Heikema AP, Bergman MP, Richards H, Crocker PR, Gilbert M, Samsom JN, et al. Characterization of the specific interaction between sialoadhesin and sialylated Campylobacter jejuni lipooligosaccharides. Infect Immun. 2010;78:3237–3246. doi: 10.1128/IAI.01273-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.von Gunten S, Bochner BS. Basic and clinical immunology of Siglecs. Ann N Y Acad Sci. 2008;1143:61–82. doi: 10.1196/annals.1443.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Avril T, Wagner ER, Willison HJ, Crocker PR. Sialic acid-binding immunoglobulin-like lectin 7 mediates selective recognition of sialylated glycans expressed on Campylobacter jejuni lipooligosaccharides. Infect Immun. 2006;74:4133–4141. doi: 10.1128/IAI.02094-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aspinall GO, McDonald AG, Pang H, Kurjanczyk LA, Penner JL. Lipopolysaccharides of Campylobacter jejuni serotype o-19: structures of core oligosaccharide regions from the serostrain and two bacterial isolates from patients with the Guillain-Barré Syndrome. Biochemistry. 1994;33:241–249. doi: 10.1021/bi00167a032. [DOI] [PubMed] [Google Scholar]
- 69.Avril T, Floyd H, Lopez F, Vivier E, Crocker PR. The membrane-proximal immunoreceptor tyrosine-based inhibitory motif is critical for the inhibitory signaling mediated by siglecs-7 and -9, CD33-related siglecs expressed on human monocytes and NK Cells. J Immunol. 2004;173:6841–6849. doi: 10.4049/jimmunol.173.11.6841. [DOI] [PubMed] [Google Scholar]
- 70.Orr SJ, Morgan NM, Buick RJ, Boyd CR, Elliott J, Burrows JF, et al. SOCS3 targets Siglec 7 for proteasomal degradation and blocks siglec 7-mediated responses. J Biol Chem. 2007;282:3418–3422. doi: 10.1074/jbc.C600216200. [DOI] [PubMed] [Google Scholar]
- 71.Yuki N, Ichihashi Y, Taki T. Subclass of IgG antibody to GM1 epitope-bearing lipopolysaccharide of Campylobacter jejuni in patients with Guillain-Barré syndrome. J Neuroimmunol. 1995;60:161–164. doi: 10.1016/0165-5728(95)00052-4. [DOI] [PubMed] [Google Scholar]
- 72.Wang Q, Xing C, Hao Y, Shi Q, Qi Z, Lv Z, et al. Memory B cells in Guillain-Barré syndrome. J Neuroimmunol. 2017;305:1–4. doi: 10.1016/j.jneuroim.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 73.Van Rhijn I, Logtenberg T, Ang CW, Van den Berg LH. Gammadelta T cell non-responsiveness in Campylobacter jejuni-associated Guillain-Barre syndrome patients. Neurology. 2003;61:994–996. doi: 10.1212/01.wnl.0000083986.53792.90. [DOI] [PubMed] [Google Scholar]
- 74.Bowes T, Wagner ER, Boffey J, Nicholl D, Cochrane L, Benboubetra M, et al. Tolerance to self gangliosides is the major factor restricting the antibody response to lipopolysaccharide core oligosaccharides in Campylobacter jejuni strains associated with Guillain-Barré syndrome. Infect Immun. 2002;70:5008–5018. doi: 10.1128/IAI.70.9.5008-5018.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jung S, Zimmer S, Lüneberg E, Frosch M, Karch H, Korn T, et al. Lipooligosaccharide of Campylobacter jejuni prevents myelin-specific enteral tolerance to autoimmune neuritis—a potential mechanism in Guillain-Barré syndrome? Neurosci Lett. 2005;381:175–178. doi: 10.1016/j.neulet.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 76.Hafer-Macko C, Hsieh S-T, Ho TW, Sheikh K, Cornblath DR, Li CY, et al. Acute motor axonal neuropathy: An antibody-mediated attack on axolemma. Ann Neurol. 1996;40:635–644. doi: 10.1002/ana.410400414. [DOI] [PubMed] [Google Scholar]
- 77.Susuki K, Rasband MN, Tohyama K, Koibuchi K, Okamoto S, Funakoshi K, et al. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J Neurosci. 2007;27:3956–3967. doi: 10.1523/JNEUROSCI.4401-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Notturno F, Luciani M, Caporale CM, Ciarelli A, Uncini A. Antibodies to ganglioside complexes in Guillain-Barré syndrome: clinical correlates, fine specificity and complement activation. Int J Immunopathol Pharmacol. 2009;22:437–445. doi: 10.1177/039463200902200220. [DOI] [PubMed] [Google Scholar]
- 79.Yuki N, Yoshino H, Sato S, Miyatake T. Acute axonal polyneuropathy associated with anti-GM1 antibodies following Campylobacter enteritis. Neurology. 1990;40:1900–1902. doi: 10.1212/wnl.40.12.1900. [DOI] [PubMed] [Google Scholar]
- 80.Willison HJ. Ganglioside complexes: new autoantibody targets in Guillain–Barré syndromes. Nat Clin Pract Neurol. 2005;1:2–3. doi: 10.1038/ncpneuro0001. [DOI] [PubMed] [Google Scholar]
- 81.Piao H, Minohara M, Kawamura N, Li W, Matsushita T, Yamasaki R, et al. Tissue binding patterns and in vitro effects of campylobacter jejuni DNA-binding protein from starved cells. Neurochem Res. 2011;36:58–66. doi: 10.1007/s11064-010-0263-7. [DOI] [PubMed] [Google Scholar]
- 82.Kawamura N, Piao H, Minohara M, Matsushita T, Kusunoki S, Matsumoto H, et al. Campylobacter jejuni DNA-binding protein from starved cells in Guillain-Barré syndrome patients. J Neuroimmunol. 2011;240–241:74–78. doi: 10.1016/j.jneuroim.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 83.Ben-Smith A, Gaston JS, Barber PC, Winer JB. Isolation and characterisation of T lymphocytes from sural nerve biopsies in patients with Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. 1996;61:362–368. doi: 10.1136/jnnp.61.4.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scelsa SN, Ghali V, Herskovitz S, Bieri P, Shank DL, MacGowan DDJ, et al. Blood γδ T cells, Campylobacter jejuni, and GM1 titers in Guillain-Barré syndrome. Muscle Nerve. 2004;30:423–432. doi: 10.1002/mus.20105. [DOI] [PubMed] [Google Scholar]
- 85.Khalili-Shirazi A, Hughes RAC, Brostoff SW, Linington C, Gregson N. T cell responses to myelin proteins in Guillain-Barré syndrome. J Neurol Sci. 1992;111:200–203. doi: 10.1016/0022-510x(92)90069-w. [DOI] [PubMed] [Google Scholar]
- 86.Cooper JC, Hughes S, Ben-Smith A, Savage COS, Winer JB. T cell recognition of a non-protein antigen preparation of Campylobacter jejuni in patients with Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 2002;72:413–414. doi: 10.1136/jnnp.72.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Born W, Cady C, Jones-Carson J, Mukasa A, Lahn M, O’Brien R. Immunoregulatory functions of gamma delta T cells. Adv Immunol. 1999;71:77–144. [PubMed] [Google Scholar]
- 88.Ben-Smith A, Goodall JC, Gaston JS, Winer JB. Stimulation of peripheral blood lymphocytes with Campylobacter jejuni generates a gammadelta T cell response in patients with Guillain-Barré syndrome. Clin Exp Immunol. 1997;109:121–126. doi: 10.1046/j.1365-2249.1997.4221318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Koga M, Yuki N, Tsukada Y, Hirata K, Matsumoto Y. CDR3 spectratyping analysis of the T cell receptor repertoire in Guillain-Barre and Fisher syndromes. J Neuroimmunol. 2003;141:112–117. doi: 10.1016/s0165-5728(03)00212-1. [DOI] [PubMed] [Google Scholar]
- 90.Susuki K, Odaka M, Mori M, Hirata K, Yuki N. Acute motor axonal neuropathy after Mycoplasma infection: Evidence of molecular mimicry. Neurology. 2004;62:949–956. doi: 10.1212/01.wnl.0000115123.42929.fd. [DOI] [PubMed] [Google Scholar]
- 91.Kusunoki S, Chiba A, Hitoshi S, Takizawa H, Kanazawa I. Anti-gal-C antibody in autoimmune neuropathies subsequent to mycoplasma infection. Muscle Nerve. 1995;18:409–413. doi: 10.1002/mus.880180407. [DOI] [PubMed] [Google Scholar]
- 92.Kusunoki S, Shiina M, Kanazawa I. Anti-Gal-C antibodies in GBS subsequent to mycoplasma infection: evidence of molecular mimicry. Neurology. 2001;57:736–738. doi: 10.1212/wnl.57.4.736. [DOI] [PubMed] [Google Scholar]
- 93.Ang CW, Tio-Gillen AP, Groen J, Herbrink P, Jacobs BC, Van Koningsveld R, et al. Cross-reactive anti-galactocerebroside antibodies and Mycoplasma pneumoniae infections in Guillain-Barre syndrome. J Neuroimmunol. 2002;130:179–183. doi: 10.1016/s0165-5728(02)00209-6. [DOI] [PubMed] [Google Scholar]
- 94.Yoshino H, Inuzuka T, Miyatake T. IgG antibody against GM1, GD1b and asialo-GM1 in chronic polyneuropathy following Mycoplasma pneumoniae infection. Eur Neurol. 1992;32:28–31. doi: 10.1159/000116872. [DOI] [PubMed] [Google Scholar]
- 95.Koga M, Gilbert M, Li J, Koike S, Takahashi M, Furukawa K, et al. Antecedent infections in Fisher syndrome: a common pathogenesis of molecular mimicry. Neurology. 2005;64:1605–1611. doi: 10.1212/01.WNL.0000160399.08456.7C. [DOI] [PubMed] [Google Scholar]
- 96.Kusunoki S, Takizawa H, Kanazawa I. Infection by Mycoplasma pneumoniae induces serum antibody against Gal-C. Muscle Nerve. 1996;19:257–258. [PubMed] [Google Scholar]
- 97.Nishimura M, Saida T, Kuroki S, Kawabata T, Obayashi H, Saida K, et al. Post-infectious encephalitis with anti-galactocerebroside antibody subsequent to Mycoplasma pneumoniae infection. J Neurol Sci. 1996;140:91–95. doi: 10.1016/0022-510x(96)00106-2. [DOI] [PubMed] [Google Scholar]
- 98.Kuwahara M, Samukawa M, Ikeda T, Morikawa M, Ueno R, Hamada Y, et al. Characterization of the neurological diseases associated with Mycoplasma pneumoniae infection and anti-glycolipid antibodies. J Neurol. 2017;264:467–475. doi: 10.1007/s00415-016-8371-1. [DOI] [PubMed] [Google Scholar]
- 99.Guleria R, Nisar N, Chawla TC, Biswas NR. Mycoplasma pneumoniae and central nervous system complications: a review. J. Lab. Clin. Med. 2005;146:55–63. doi: 10.1016/j.lab.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 100.Meyer Sauteur PM, Jacobs BC, Spuesens EBM, Jacobs E, Nadal D, Vink C, et al. Antibody responses to Mycoplasma pneumoniae: role in pathogenesis and diagnosis of encephalitis? PLoS Pathog. 2014;10:e1003983. doi: 10.1371/journal.ppat.1003983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Koga M, Yuki N, Tai T, Hirata K. Miller Fisher syndrome and Haemophilus influenzae infection. Neurology. 2001;57:686–691. doi: 10.1212/wnl.57.4.686. [DOI] [PubMed] [Google Scholar]
- 102.Koga M, Yuki N, Hirata K, Morimatsu M, Mori M, Kuwabara S. Anti-GM1 antibody IgG subclass: a clinical recovery predictor in Guillain-Barre syndrome. Neurology. 2003;60:1514–1518. doi: 10.1212/01.wnl.0000061615.77865.83. [DOI] [PubMed] [Google Scholar]
- 103.Yuki N, Shahrizaila N. How do we identify infectious agents that trigger Guillain–Barré syndrome, Fisher syndrome and Bickerstaff brainstem encephalitis? J Neurol Sci. 2011;302:1–5. doi: 10.1016/j.jns.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 104.Mori M, Kuwabara S, Miyake M, Dezawa M, Adachi-Usami E, Kuroki H, et al. Haemophilus influenzae has a GM1 ganglioside-like structure and elicits Guillain-Barré syndrome. Neurology. 1999;52:1282–1284. doi: 10.1212/wnl.52.6.1282. [DOI] [PubMed] [Google Scholar]
- 105.Houliston RS, Koga M, Li J, Jarrell HC, Richards JC, Vitiazeva V, et al. A Haemophilus influenzae strain associated with fisher syndrome expresses a novel disialylated ganglioside mimic. Biochemistry. 2007;46:8164–8171. doi: 10.1021/bi700685s. [DOI] [PubMed] [Google Scholar]
- 106.Vieira MADCES, Cruz ACR, Barros ANM, Costa DL, Silva EVPD, Batista FMA, et al. Guillain-Barré syndrome and dengue-like disease in 2015: temporal relationship in Piauí state and implications on Zika virus surveillance. Rev Inst Med Trop Sao Paulo. 2017;59:e22. doi: 10.1590/S1678-9946201759022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bate SL, Dollard SC, Cannon MJ. Cytomegalovirus Seroprevalence in the United States: The National Health and Nutrition Examination Surveys, 1988–2004. Clin Infect Dis. 2010;50:1439–1447. doi: 10.1086/652438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Arias-Murillo YR, Osorio-Arango K, Cortés JA, Beltrán M. Seroprevalencia de citomegalovirus en donantes de órganos y receptores de trasplante renal, Colombia, 2010-2014. Biomédica. 2016;36:187. doi: 10.7705/biomedica.v36i0.2938. [DOI] [PubMed] [Google Scholar]
- 109.Limaye AP, Kirby KA, Rubenfeld GD, Leisenring WM, Bulger EM, Neff MJ, et al. Cytomegalovirus reactivation in critically Ill immunocompetent patients. JAMA. 2008;300:413. doi: 10.1001/jama.300.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Klemola E, Weckman N, Haltia K, Kääriäinen L. The Guillain-Barré syndrome associated with acquired Cytomegalovirus infection. Acta Med Scand. 2009;181:603–607. doi: 10.1111/j.0954-6820.1967.tb07283.x. [DOI] [PubMed] [Google Scholar]
- 111.Orlikowski D, Porcher R, Sivadon-Tardy V, Quincampoix JC, Raphaël JC, Durand MC, et al. Guillain-barré syndrome following primary cytomegalovirus infection: A prospective cohort study. Clin Infect Dis. 2011;52:837–844. doi: 10.1093/cid/cir074. [DOI] [PubMed] [Google Scholar]
- 112.Steininger C, Seiser A, Gueler N, Puchhammer-Stöckl E, Aberle SW, Stanek G, et al. Primary cytomegalovirus infection in patients with Guillain-Barré syndrome. J Neuroimmunol. 2007;183:214–219. doi: 10.1016/j.jneuroim.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 113.Lunn M, Hughes R. The relationship between Cytomegalovirus infection and Guillain-Barre syndrome. Clin Infect Dis. 2011;52:845–847. doi: 10.1093/cid/cir082. [DOI] [PubMed] [Google Scholar]
- 114.Visser LH, van der Meché FG, Meulstee J, Rothbarth PP, Jacobs BC, Schmitz PI, et al. Cytomegalovirus infection and Guillain-Barré syndrome: the clinical, electrophysiologic, and prognostic features. Dutch Guillain-Barré Study Group. Neurology. 1996;47:668–673. doi: 10.1212/wnl.47.3.668. [DOI] [PubMed] [Google Scholar]
- 115.Kobori S, Kubo T, Otani M, Muramatsu K, Fujino Y, Adachi H, et al. Coexisting infectious diseases on admission as a risk factor for mechanical ventilation in patients with Guillaine-Barré syndrome. J Epidemiol. 2017;27:311–316. doi: 10.1016/j.je.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hughes R, Atkinson P, Coates P, Hall S, Leibowitz S. Sural nerve biopsies in Guillain-Barre syndrome: axonal degeneration and macrophage-associated demyelination and absence of cytomegalovirus genome. Muscle Nerve. 1992;15:568–575. doi: 10.1002/mus.880150506. [DOI] [PubMed] [Google Scholar]
- 117.Irie S, Saito T, Nakamura K, Kanazawa N, Ogino M, Nukazawa T, et al. Association of anti-GM2 antibodies in Guillain-Barre syndrome with acute cytomegalovirus infection. J Neuroimmunol. 1996;68:19–26. doi: 10.1016/0165-5728(96)00059-8. [DOI] [PubMed] [Google Scholar]
- 118.Yuki N, Tagawa Y. Acute cytomegalovirus infection and IgM anti-GM2 antibody. J Neurol Sci. 1998;154:14–17. doi: 10.1016/s0022-510x(97)00174-3. [DOI] [PubMed] [Google Scholar]
- 119.Kaida K, Kusunoki S, Kamakura K, Motoyoshi K, Kanazawa I. Guillain-Barré syndrome with IgM antibody to the ganglioside GalNAc-GD1a. J Neuroimmunol. 2001;113:260–267. doi: 10.1016/s0165-5728(00)00451-3. [DOI] [PubMed] [Google Scholar]
- 120.Haase CG, Schmidt S. Detection of brain-specific autoantibodies to myelin oligodendrocyte glycoprotein, S100beta and myelin basic protein in patients with Devic’s neuromyelitis optica. Neurosci Lett. 2001;307:131–133. doi: 10.1016/s0304-3940(01)01949-8. [DOI] [PubMed] [Google Scholar]
- 121.Britt W Virus Entry Into Host, Establishment Of Infection, Spread in Host, Mechanisms of Tissue Damage. Cambridge 2007. [PubMed]
- 122.Svennerholm L, Fredman P. Antibody detection in Guillain-Barre syndrome. Ann Neurol. 1990;27((Suppl)):36–40. doi: 10.1002/ana.410270710. [DOI] [PubMed] [Google Scholar]
- 123.Tsukaguchi M, Tagawa Y, Takeuchi H, Yuki N. IgM anti-GM2 antibody in a patient with Guillain-Barré syndrome subsequent to cytomegalovirus hepatitis cross reacts with N-acetylgalactosaminyl GD1a. J Neurol Neurosurg Psychiatry. 1998;65:407–408. doi: 10.1136/jnnp.65.3.407a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nakamura K, Irie S, Kanazawa N, Saito T, Tamai Y. Anti-GM2 antibodies in Guillain-Barré syndrome with acute cytomegalovirus infection. Ann N Y Acad Sci. 1998;845:423. doi: 10.1111/j.1749-6632.1998.tb09710.x. [DOI] [PubMed] [Google Scholar]
- 125.Ang CW, Jacobs BC, Brandenburg AH, Laman JD, van der Meché FG, Osterhaus AD, et al. Cross-reactive antibodies against GM2 and CMV-infected fibroblasts in Guillain-Barré syndrome. Neurology. 2000;54:1453–1458. doi: 10.1212/wnl.54.7.1453. [DOI] [PubMed] [Google Scholar]
- 126.Adelmann M, Linington C. Molecular mimicry and the autoimmune response to the peripheral nerve myelin P0 glycoprotein. Neurochem Res. 1992;17:887–891. doi: 10.1007/BF00993264. [DOI] [PubMed] [Google Scholar]
- 127.Milner P, Lovelidge CA, Taylor WA, Hughes RAC. P0 myelin protein produces experimental allergic neuritis in Lewis rats. J Neurol Sci. 1987;79:275–285. doi: 10.1016/0022-510x(87)90235-8. [DOI] [PubMed] [Google Scholar]
- 128.Gatto CL, Walker BJ, Lambert S. Local ERM activation and dynamic growth cones at schwann cell tips implicated in efficient formation of nodes of Ranvier. J Cell Biol. 2003;162:489–498. doi: 10.1083/jcb.200303039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Scherer SS, Xu T, Crino P, Arroyo EJ, Gutmann DH. Ezrin, radixin, and moesin are components of Schwann cell microvilli. J Neurosci Res. 2001;65:150–164. doi: 10.1002/jnr.1138. [DOI] [PubMed] [Google Scholar]
- 130.Miyaji K, Devaux J, Yuki N. WriteClick Editor’s Choice: Moesin is a possible target molecule for cytomegalovirus-related Guillain-Barré syndrome. Neurology. 2014;83:2314–2315. doi: 10.1212/WNL.0000000000001090. [DOI] [PubMed] [Google Scholar]
- 131.Iontcheva I, Amar S, Zawawi KH, Kantarci A, Van Dyke TE. Role for moesin in lipopolysaccharide-stimulated signal transduction. Infect Immun. 2004;72:2312–2320. doi: 10.1128/IAI.72.4.2312-2320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Willison H, Scherer SS. Ranvier revisited: novel nodal antigens stimulate interest in GBS pathogenesis. Neurology. 2014;83:106–108. doi: 10.1212/WNL.0000000000000581. [DOI] [PubMed] [Google Scholar]
- 133.Tsukita S, Yonemura S. Cortical actin organization: lessons from ERM (Ezrin/Radixin/Moesin) proteins. J Biol Chem. 1999;274:34507–34510. doi: 10.1074/jbc.274.49.34507. [DOI] [PubMed] [Google Scholar]
- 134.Sawai S, Satoh M, Mori M, Misawa S, Sogawa K, Kazami T, et al. Moesin is a possible target molecule for cytomegalovirus-related Guillain-Barre syndrome. Neurology. 2014;83:113–117. doi: 10.1212/WNL.0000000000000566. [DOI] [PubMed] [Google Scholar]
- 135.Sindern E, Oreja-Guevara C, Raulf-Heimsoth M, Baur X, Pierre Malin J. A longitudinal study of circulating lymphocyte subsets in the peripheral blood during the acute stage of Guillain-Barré syndrome. J Neurol Sci. 1997;151:29–34. doi: 10.1016/s0022-510x(97)00082-8. [DOI] [PubMed] [Google Scholar]
- 136.Pangault C, Le Tulzo Y, Minjolle S, Le Page E, Sebti Y, Guilloux V, et al. HLA-G Expression in Guillain-Barré Syndrome Is Associated with Primary Infection with Cytomegalovirus. Viral Immunol. 2004;17:123–125. doi: 10.1089/088282404322875520. [DOI] [PubMed] [Google Scholar]
- 137.Gerken G, Trautmann F, Köhler H, Falke D, Bohl J, Nix W, et al. Rare association of herpes simplex virus IgM-specific antibodies and Guillain-Barré syndrome successfully treated with plasma exchange and immunosuppression. Klin Wochenschr. 1985;63:468–474. doi: 10.1007/BF01731495. [DOI] [PubMed] [Google Scholar]
- 138.Dilena R, Strazzer S, Esposito S, Paglialonga F, Tadini L, Barbieri S, et al. Locked-in-like fulminant infantile Guillain-Barré syndrome associated with herpes simplex virus 1 infection. Muscle Nerve. 2016;53:140–143. doi: 10.1002/mus.24908. [DOI] [PubMed] [Google Scholar]
- 139.Ntziora F, Euthimiou A, Tektonidou M, Andreopoulos A, Konstantopoulos K. Guillain-Barre syndrome presenting with sensory disturbance following a herpes virus infection: a case report. J Med Case Rep. 2011;5:563. doi: 10.1186/1752-1947-5-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yuki N, Susuki K, Odaka M, Hirata K. Overlapping Guillain-Barre syndrome and Bickerstaff’s brainstem encephalitis associated with anti-GQ1b IgG antibody after herpes simplex virus infection. Acta Neurol Scand. 2001;104:57–60. doi: 10.1034/j.1600-0404.2001.00288.x. [DOI] [PubMed] [Google Scholar]
- 141.Jacobs BC, Rothbarth PH, van der Meché FG, Herbrink P, Schmitz PI, de Klerk MA, et al. The spectrum of antecedent infections in Guillain-Barré syndrome: a case-control study. Neurology. 1998;51:1110–1115. doi: 10.1212/wnl.51.4.1110. [DOI] [PubMed] [Google Scholar]
- 142.Miyaji K, Furukawa J, Suzuki Y, Yamamoto N, Shinohara Y, Yuki N. Altered gene expression of glycosyltransferases and sialyltransferases and total amount of glycosphingolipids following herpes simplex virus infection. Carbohydr Res. 2016;434:37–43. doi: 10.1016/j.carres.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 143.Hart IK, Kennedy PG. Guillain-Barre syndrome associated with herpes zoster. Postgrad Med J. 1987;63:1087–1088. doi: 10.1136/pgmj.63.746.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sanders EA, Peters AC, Gratana JW, Hughes RA. Guillain-Barré syndrome after varicella-zoster infection. Report of two cases. J Neurol. 1987;234:437–439. doi: 10.1007/BF00314094. [DOI] [PubMed] [Google Scholar]
- 145.Rabbani MU, Gupta D. Guillain Barré syndrome following herpes zoster: a case report and review of literature. Jpn J Med. 1990;29:397–398. doi: 10.2169/internalmedicine1962.29.397. [DOI] [PubMed] [Google Scholar]
- 146.Kang J-H, Sheu J-J, Lin H-C. Increased Risk of Guillain-Barré syndrome following recent herpes zoster: A Population-Based Study across Taiwan. Clin Infect Dis. 2010;51:525–530. doi: 10.1086/655136. [DOI] [PubMed] [Google Scholar]
- 147.Galvan M, Rotola A, Govoni V, Granieri E, Cassai E, Di Luca D. Simultaneous Guillain-Barré syndrome and active human herpesvirus 6 infection in the central nervous system. J Clin Virol. 2007;38:271–272. doi: 10.1016/j.jcv.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 148.Merelli E, Sola P, Faglioni P, Poggi M, Montorsi M, Torelli G. Newest human herpesvirus (HHV-6) in the Guillain-Barré syndrome and other neurological diseases. Acta Neurol Scand. 2009;85:334–336. doi: 10.1111/j.1600-0404.1992.tb04054.x. [DOI] [PubMed] [Google Scholar]
- 149.Gustafsson R, Reitsma R, Strålfors A, Lindholm A, Press R, Fogdell-Hahn A. Incidence of human herpesvirus 6 in clinical samples from Swedish patients with demyelinating diseases. J Microbiol Immunol Infect. 2014;47:418–421. doi: 10.1016/j.jmii.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 150.Corssmit EP, Leverstein-van Hall MA, Portegies P, Bakker P. Severe neurological complications in association with Epstein-Barr virus infection. J Neurovirol. 1997;3:460–464. doi: 10.3109/13550289709031193. [DOI] [PubMed] [Google Scholar]
- 151.Nikkels AF, Debrus S, Sadzot-Delvaux C, Piette J, Delvenne P, Rentier B, et al. Comparative immunohistochemical study of herpes simplex and varicella-zoster infections. Virchows Arch A Pathol Anat Histopathol. 1993;422:121–126. doi: 10.1007/BF01607163. [DOI] [PubMed] [Google Scholar]
- 152.Schnorf H, Rathgeb JP, Kohler A. Anti-GQ1b-positive Miller Fisher syndrome in a patient with acute Epstein-Barr virus infection and negative Campylobacter serology. Eur Neurol. 1998;40:177. [PubMed] [Google Scholar]
- 153.Caudie C, Quittard Pinon A, Taravel D, Sivadon-Tardy V, Orlikowski D, Rozenberg F, et al. Preceding infections and anti-ganglioside antibody profiles assessed by a dot immunoassay in 306 French Guillain–Barré syndrome patients. J Neurol. 2011;258:1958–1964. doi: 10.1007/s00415-011-6042-9. [DOI] [PubMed] [Google Scholar]
- 154.Penner E, Maida E, Mamoli B, Gangl A. Serum and cerebrospinal fluid immune complexes containing hepatitis B surface antigen in Guillain-Barré syndrome. Gastroenterology. 1982;82:576–580. [PubMed] [Google Scholar]
- 155.Tabor E. Guillain-Barré syndrome and other neurologic syndromes in hepatitis A, B, and non-A, non-B. J Med Virol. 1987;21:207–216. doi: 10.1002/jmv.1890210303. [DOI] [PubMed] [Google Scholar]
- 156.Perseghin P, Balduini CL, Piccolo G, Bertolino G, Bellusci M, Scelsi R, et al. Guillain-Barré syndrome with autoimmune hemolytic anemia following acute viral hepatitis. Ital J Neurol Sci. 1985;6:447–450. doi: 10.1007/BF02331037. [DOI] [PubMed] [Google Scholar]
- 157.Goust JM, Chenais F, Carnes JE, Hames CG, Fudenberg HH, Hogan EL. Abnormal T cell subpopulations and circulating immune complexes in the Guillain-Barré syndrome and multiple sclerosis. Neurology. 1978;28:421–425. doi: 10.1212/wnl.28.5.421. [DOI] [PubMed] [Google Scholar]
- 158.Fujinami R, Oldstone M. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043–1045. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- 159.Zhou X, Huang F, Xu L, Lin Z, de Vrij FMS, Ayo-Martin AC, et al. Hepatitis E virus infects neurons and brains. J Infect Dis. 2017;215:1197–1206. doi: 10.1093/infdis/jix079. [DOI] [PubMed] [Google Scholar]
- 160.Shukla P, Nguyen HT, Faulk K, Mather K, Torian U, Engle RE, et al. Adaptation of a genotype 3 hepatitis E virus to efficient growth in cell culture depends on an inserted human gene segment acquired by recombination. J Virol. 2012;86:5697–5707. doi: 10.1128/JVI.00146-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dalton HR, Kamar N, van Eijk JJJ, Mclean BN, Cintas P, Bendall RP, et al. Hepatitis E virus and neurological injury. Nat Rev Neurol. 2015;12:77–85. doi: 10.1038/nrneurol.2015.234. [DOI] [PubMed] [Google Scholar]
- 162.Cronin S, McNicholas R, Kavanagh E, Reid V, O’Rourke K. Anti-glycolipid GM2-positive Guillain-Barre syndrome due to hepatitis E infection. Ir J Med Sci. 2011;180:255–257. doi: 10.1007/s11845-010-0635-7. [DOI] [PubMed] [Google Scholar]
- 163.Maurissen I, Jeurissen A, Strauven T, Sprengers D, De Schepper B. First case of anti-ganglioside GM1-positive Guillain–Barré syndrome due to hepatitis E virus infection. Infection. 2012;40:323–326. doi: 10.1007/s15010-011-0185-6. [DOI] [PubMed] [Google Scholar]
- 164.De Castro G, Bastos PG, Martinez R, Figueiredo JFDC. Episodes of Guillain-Barré syndrome associated with the acute phase of HIV-1 infection and with recurrence of viremia. Arq Neuropsiquiatr. 2006;64:606–608. doi: 10.1590/s0004-282x2006000400016. [DOI] [PubMed] [Google Scholar]
- 165.Keswani SC, Polley M, Pardo CA, Griffin JW, McArthur JC, Hoke A. Schwann cell chemokine receptors mediate HIV-1 gp120 toxicity to sensory neurons. Ann Neurol. 2003;54:287–296. doi: 10.1002/ana.10645. [DOI] [PubMed] [Google Scholar]
- 166.Nishijima T, Tsukada K, Takeuchi S, Chiba A, Honda M, Teruya K, et al. Antiretroviral therapy for treatment-naïve chronic HIV-1 infection with an axonal variant of Guillain-Barré syndrome positive for anti-ganglioside antibody: a case report. Intern Med. 2011;50:2427–2429. doi: 10.2169/internalmedicine.50.5883. [DOI] [PubMed] [Google Scholar]
- 167.Brannagan TH, Zhou Y. HIV-associated Guillain-Barré syndrome. J Neurol Sci. 2003;208:39–42. doi: 10.1016/s0022-510x(02)00418-5. [DOI] [PubMed] [Google Scholar]
- 168.Chen TY, Lee CT. Guillain-Barré syndrome following dengue fever. Ann Emerg Med. 2007;50:94–95. doi: 10.1016/j.annemergmed.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 169.Puccioni-Sohler M, Soares CN, Papaiz-Alvarenga R, Castro MJC, Faria LC, Peralta JM. Neurologic dengue manifestations associated with intrathecal specific immune response. Neurology. 2009;73:1413–1417. doi: 10.1212/WNL.0b013e3181bd8258. [DOI] [PubMed] [Google Scholar]
- 170.Lum LCS, Lam SK, Choy YS, George R, Harun F. Dengue encephalitis: A true entity? Am J Trop Med Hyg. 1996;54:256–259. doi: 10.4269/ajtmh.1996.54.256. [DOI] [PubMed] [Google Scholar]
- 171.Carod-Artal FJ, Wichmann O, Farrar J, Gascón J. Neurological complications of dengue virus infection. Lancet Neurol. 2013;12:906–919. doi: 10.1016/S1474-4422(13)70150-9. [DOI] [PubMed] [Google Scholar]
- 172.Oishi K, Saito M, Mapua CA, Natividad FF. Dengue illness: clinical features and pathogenesis. J Infect Chemother. 2007;13:125–133. doi: 10.1007/s10156-007-0516-9. [DOI] [PubMed] [Google Scholar]
- 173.Verma R, Sahu R, Holla V. Neurological manifestations of dengue infection: a review. J Neurol Sci. 2014;346:26–34. doi: 10.1016/j.jns.2014.08.044. [DOI] [PubMed] [Google Scholar]
- 174.Wielanek AC, De Monredon J, El Amrani M, Roger JC, Serveaux JP. Guillain-Barré syndrome complicating a Chikungunya virus infection. Neurology. 2007;69:2105–2107. doi: 10.1212/01.wnl.0000277267.07220.88. [DOI] [PubMed] [Google Scholar]
- 175.Tournebize P, Charlin C, Lagrange M. Neurological manifestations in Chikungunya: about 23 cases collected in Reunion Island. Rev Neurol (Paris) 2009;165:48–51. doi: 10.1016/j.neurol.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 176.Jaffar-Bandjee MC, Ramful D, Gauzere BA, Hoarau JJ, Krejbich-Trotot P, Robin S, et al. Emergence and clinical insights into the pathology of Chikungunya virus infection. Expert Rev Anti Infect Ther. 2010;8:987–996. doi: 10.1586/eri.10.92. [DOI] [PubMed] [Google Scholar]
- 177.Couderc T, Chrétien F, Schilte C, Disson O, Brigitte M, Guivel-Benhassine F, et al. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008;4:e29. doi: 10.1371/journal.ppat.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Metsky HC, Matranga CB, Wohl S, Schaffner SF, Freije CA, Winnicki SM, et al. Zika virus evolution and spread in the Americas. Nature. 2017;546:411–415. doi: 10.1038/nature22402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mlakar J, Korva M, Tul N, Popović M, Poljšak-Prijatelj M, Mraz J, et al. Zika virus associated with microcephaly. N Engl J Med. 2016;374:951–958. doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- 180.Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, et al. Zika Virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe. 2016;19:882–890. doi: 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Donald CL, Brennan B, Cumberworth SL, Rezelj VV, Clark JJ, Cordeiro MT, et al. Full genome sequence and sfRNA interferon antagonist activity of Zika virus from Recife, Brazil. PLoS Negl Trop Dis. 2016;10:e0005048. doi: 10.1371/journal.pntd.0005048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Meertens L, Labeau A, Dejarnac O, Cipriani S, Sinigaglia L, Bonnet-Madin L, et al. Axl mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep. 2017;18:324–333. doi: 10.1016/j.celrep.2016.12.045. [DOI] [PubMed] [Google Scholar]
- 183.Morris G, Barichello T, Stubbs B, Köhler CA, Carvalho AF, Maes M. Zika virus as an emerging neuropathogen: mechanisms of neurovirulence and neuro-immune interactions. Mol Neurobiol 2017;: 1–25. [DOI] [PubMed]
- 184.Bayless NL, Greenberg RS, Swigut T, Wysocka J, Blish CA. Zika virus infection induces cranial neural crest cells to produce cytokines at levels detrimental for neurogenesis. Cell Host Microbe. 2016;20:423–428. doi: 10.1016/j.chom.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cumberworth SL, Barrie JA, Cunningham ME, de Figueiredo DPG, Schultz V, Wilder-Smith AJ, et al. Zika virus tropism and interactions in myelinating neural cell cultures: CNS cells and myelin are preferentially affected. Acta Neuropathol Commun. 2017;5:50. doi: 10.1186/s40478-017-0450-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lucchese G, Kanduc D. Zika virus and autoimmunity: from microcephaly to Guillain-Barré syndrome, and beyond. Autoimmun. Rev. 2016;15:801–808. doi: 10.1016/j.autrev.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 187.Monel B, Compton AA, Bruel T, Amraoui S, Burlaud-Gaillard J, Roy N, et al. Zika virus induces massive cytoplasmic vacuolization and paraptosis-like death in infected cells. EMBO J. 2017;36:1653–1668. doi: 10.15252/embj.201695597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lessler J, Chaisson LH, Kucirka LM, Bi Q, Grantz K, Salje H, et al. Assessing the global threat from Zika virus. Science. 2016;353:aaf8160–aaf8160. doi: 10.1126/science.aaf8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gorman MJ, Poddar S, Farzan M, Diamond MS. The interferon-stimulated gene Ifitm3 restricts west nile virus infection and pathogenesis. J Virol. 2016;90:8212–8225. doi: 10.1128/JVI.00581-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Everitt AR, Clare S, Pertel T, John SP, Wash RS, Smith SE, et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature. 2012;484:519–523. doi: 10.1038/nature10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Li G, Poulsen M, Fenyvuesvolgyi C, Yashiroda Y, Yoshida M, Simard JM, et al. Characterization of cytopathic factors through genome-wide analysis of the Zika viral proteins in fission yeast. Proc Natl Acad Sci USA. 2017;114:E376–E385. doi: 10.1073/pnas.1619735114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Aid M, Abbink P, Larocca RA, Boyd M, Nityanandam R, Nanayakkara O, et al. Zika virus persistence in the central nervous system and lymph nodes of rhesus monkeys. Cell. 2017;169:610–620. doi: 10.1016/j.cell.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Flipse J, Wilschut J, Smit JM. Molecular mechanisms involved in antibody-dependent enhancement of dengue virus infection in humans. Traffic. 2013;14:25–35. doi: 10.1111/tra.12012. [DOI] [PubMed] [Google Scholar]
- 194.Paul LM, Carlin ER, Jenkins MM, Tan AL, Barcellona CM, Nicholson CO, et al. Dengue virus antibodies enhance Zika virus infection. Clin Transl Immunol. 2016;5:1–12. doi: 10.1038/cti.2016.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Freire CCM, Iamarino A, Neto DFL, Sall AA, Marinho P, Zanotto A. Spread of the pandemic Zika virus lineage is associated with NS1 codon usage adaptation in humans. bioRxiv. 2015;2015:1–8. [Google Scholar]
- 196.Sá MJ. Acute transverse myelitis: a practical reappraisal. Autoimmun Rev. 2009;9:128–131. doi: 10.1016/j.autrev.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 197.Kaplin AI, Krishnan C, Deshpande DM, Pardo CA, Kerr DA. Diagnosis and management of acute myelopathies. Neurologist. 2005;11:2–18. doi: 10.1097/01.nrl.0000149975.39201.0b. [DOI] [PubMed] [Google Scholar]
- 198.Komagamine T, Yuki N. Ganglioside mimicry as a cause of Guillain-Barré syndrome. CNS Neurol Disord Drug Targets. 2006;5:391–400. doi: 10.2174/187152706777950765. [DOI] [PubMed] [Google Scholar]
- 199.Baar I, Jacobs BC, Govers N, Jorens PG, Parizel PM, Cras P. Campylobacter jejuni-induced acute transverse myelitis. Spinal Cord. 2007;45:690–694. doi: 10.1038/sj.sc.3102012. [DOI] [PubMed] [Google Scholar]
- 200.Csábi G, Komáromy H, Hollódy K. Transverse myelitis as a rare, serious complication of Mycoplasma pneumoniae infection. Pediatr Neurol. 2009;41:312–313. doi: 10.1016/j.pediatrneurol.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 201.Renard JL, Guillamo JS, Ramirez JM, Taillia H, Felten D, Buisson Y. Acute transverse cervical myelitis following hepatitis B vaccination. Evolution of anti-HBs antibodies. Press Med. 1999;28:1290–1292. [PubMed] [Google Scholar]
- 202.Matsui M, Kakigi R, Watanabe S, Kuroda Y. Recurrent demyelinating transverse myelitis in a high titer HBs-antigen carrier. J Neurol Sci. 1996;139:235–237. [PubMed] [Google Scholar]
- 203.Zhang J, Vandevyver C, Stinissen P, Mertens N, van den Berg-Loonen E, Raus J. Activation and clonal expansion of human myelin basic protein-reactive T cells by bacterial superantigens. J Autoimmun. 1995;8:615–632. doi: 10.1016/0896-8411(95)90012-8. [DOI] [PubMed] [Google Scholar]
- 204.Hong SC, Waterbury G, Janeway CA. Different superantigens interact with distinct sites in the Vbeta domain of a single T cell receptor. J Exp Med. 1996;183:1437–1446. doi: 10.1084/jem.183.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Vanderlugt CL, Begolka WS, Neville KL, Katz-Levy Y, Howard LM, Eagar TN, et al. The functional significance of epitope spreading and its regulation by co-stimulatory molecules. Immunol Rev. 1998;164:63–72. doi: 10.1111/j.1600-065x.1998.tb01208.x. [DOI] [PubMed] [Google Scholar]
- 206.Reindl M, Linington C, Brehm U, Egg R, Dilitz E, Deisenhammer F, et al. Antibodies against the myelin oligodendrocyte glycoprotein and the myelin basic protein in multiple sclerosis and other neurological diseases: a comparative study. Brain. 1999;122((Pt 1)):2047–2056. doi: 10.1093/brain/122.11.2047. [DOI] [PubMed] [Google Scholar]
- 207.Mota MT, Estofolete CF, Zini N, Terzian ACB, Gongora DVN, Maia IL, et al. Transverse myelitis as an unusual complication of dengue fever. Am J Trop Med Hyg. 2017;96:380–381. doi: 10.4269/ajtmh.16-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Drulovic J, Dujmovic I, Stojsavlevic N, Tripkovic I, Apostolski S, Levic Z, et al. Transverse myelopathy in the antiphospholipid antibody syndrome: pinworm infestation as a trigger? J Neurol Neurosurg. Psychiatry. 2000;68:249. doi: 10.1136/jnnp.68.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.